

Abstract
There is growing evidence that noncanonical amino acids (ncAAs) can be utilized in the creation of biological therapeutics ranging from protein conjugates to cell-based therapies. However, when does genetically encoding ncAAs yield biologics with unique properties compared to other approaches? In this review, we attempt to answer this question in the broader context of therapeutic development, emphasizing advances within the past two years. In several areas, ncAAs add valuable routes to therapeutically relevant entities, but application-specific needs ultimately determine whether ncAA-mediated or alternative solutions are preferred. Looking forward, using ncAAs to perform “protein medicinal chemistry,” in which atomic-level changes to proteins dramatically enhance therapeutic properties, is a promising emerging area. Further upgrades to the performance of ncAA incorporation technologies will be essential to realizing the full potential of ncAAs in biological therapeutics.
Graphical abstract
Introduction
Genetically encoding noncanonical amino acids (ncAAs) enables powerful strategies for manipulating the properties of proteins, viruses, and cells. In addition to their rapidly expanding uses in basic research [1–3], ncAAs exhibit considerable potential in therapeutic applications (ncAAs, defined as amino acids beyond the 20 canonical amino acids (cAAs), are also referred to as unnatural amino acids (uAAs), nonstandard amino acids (nsAAs), and nonnatural amino acids (nAAs)). Two recent reviews summarized what therapeutic applications are feasible with ncAAs [4,5]. Here, we attempt to place these advances within a broader context: when does genetically encoding ncAAs yield biologics with unique properties compared to other approaches? And what opportunities lie ahead for utilizing ncAAs in therapeutic settings? Researchers are addressing these questions using several methods of genetically encoding ncAAs, including stop codon suppression in cells [2], residue-specific canonical amino acid replacement in cells [6], and drastic alterations to the genetic code in vitro [7].
The majority of this review focuses on three areas of application (Figure 1), drawing upon examples from the last two years whenever possible. In the areas of conjugates and limiting cell and viral replication, we highlight ways in which ncAA-mediated approaches provide valuable new routes for the creation of therapeutic candidates within these crowded spaces. Next, we show how the emerging concept of “protein medicinal chemistry” enables the use of atomic-level protein perturbations to drastically improve therapeutically relevant polypeptide properties; this area appears ripe for further exploitation. Finally, we emphasize the ongoing need to rigorously characterize and improve ncAA incorporation technologies, ranging from orthogonal translation systems (OTSs; Figure 1) to the translation apparatus and the genome itself [8–10]. Realizing nativelike translation efficiencies with alternative genetic codes will enable ncAAs to reach their full potential in therapeutic applications.
Figure 1.
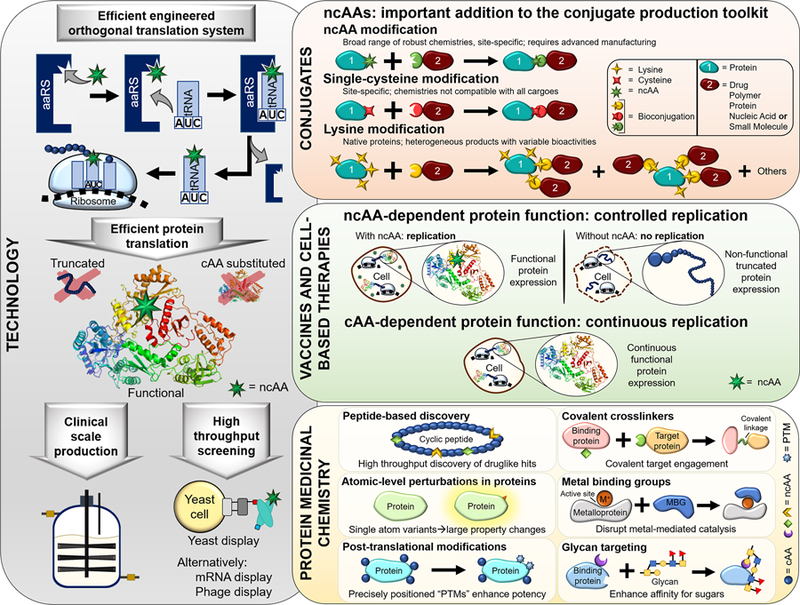
Overview of topics covered in this review. Technology: underlying successful applications of ncAAs in therapeutic settings are high efficiency, high fidelity platforms for genetically encoding ncAAs in proteins. The performance of orthogonal translation systems (OTSs), comprised of aminoacyl-tRNA synthetase (aaRS)/suppressor tRNA pairs, remains a limiting factor in many platforms. Conjugates: ncAA-mediated conjugations are important additions to the conjugate production toolkit. Vaccines and cell-based therapies: ncAA-dependent protein function allows for precise control over viral and cell replication. Protein medicinal chemistry: utilizing ncAAs to precisely alter protein structure and function provides many opportunities for discovering new classes of therapeutics. Protein structure taken from PDB ID 1DLO. Glycan structure taken from reference [61].
Conjugates
Selectively addressing individual chemical groups in proteins supports the generation of conjugates with therapeutic value. This includes utilizing ncAAs to link proteins to cytotoxic compounds, polymers, and additional classes of molecules via bioorthogonal functional groups including azides, alkynes, ketones, tetrazines, and cyclopropenes [11,12]. However, how do conjugates formed utilizing ncAA-mediated approaches compare to conjugates produced with alternative approaches? Comparisons between methods (Table 1) indicate that while the answer to this question is application-specific, careful exploitation of ncAAs significantly expands the range of therapeutically relevant conjugates.
Table 1.
Summary of advantages and limitations of selected conjugation strategies.
| Conjugation Strategy | Advantages | Limitations | ||
|---|---|---|---|---|
| Antibody Drug Conjugates (ADCs) | ncAA-mediated technologies | Strain-Promoted Cycloaddition: genetically encoded azide, exogenous strained alkyne | Reaction: fast kinetics; various payloads compatible | Manufacture: advanced translation engineering required (cell line/cell free system must support efficient protein translation with ncAAs) |
| Clinical: larger therapeutic window; improved stability | ||||
| Location: site-specific | ||||
| Copper-Catalyzed Cycloaddition: genetically encoded azide, exogenous terminal alkyne* | Reaction: see above | Reaction: complex kinetics | ||
| Clinical: see above | Manufacture: see above | |||
| Location: site-specific | ||||
| Oxime Ligation: genetically encoded ketone, exogenous hydroxylamine | Reaction: robust; compatible with large range of payloads | Reaction: slow reaction kinetics | ||
| Clinical: see above | Manufacture: see above | |||
| Location: site-specific | ||||
| Other competing technologies | Lysine Conjugation: surface-accessible lysine, exogenous amine-reactive compound | Manufacture: established production strategies | Clinical: lower bioactivity; low plasma stability | |
| Location: not site-specific | ||||
| Disulfide Conjugation: thiols from disulfide bond reduction, exogenous electrophile | Manufacture: established protein production strategies | Manufacture: carefully controlled reaction conditions | ||
| Location: not site-specific | ||||
| Single-Cysteine Conjugation: single engineered cysteine, exogenous thiol-reactive compound | Clinical: larger therapeutic window; improved stability | Reaction: careful control to avoid reduction of native disulfides; not compatible with some payloads | ||
| Manufacture: established protein production strategies | ||||
| Enzyme-Mediated Conjugation: encoded peptide substrate, exogenous compatible payload | Reaction: robust; compatible with large range of payloads | Reaction: conditions particular to enzyme and conjugation site | ||
| Location: site-specific | ||||
| Protein-Polymer Conjugates | ncAA | Copper-Catalyzed Cycloaddition | See above | See above |
| Oxime Ligation | See above | See above | ||
| Other | Lysine Conjugation | See above | See above | |
| N-terminal Amine Group Modification: free N-terminus, exogenous reactive compound | Location: site-specific | Reaction: tight control needed to avoid side products | ||
| PEG Mimetics: flexible, genetically encoded amino acid chains | Location: N- or C-terminus | Clinical: free terminus may be important for bioactivity | ||
| Bispecific Antibodies | ncAA | Strain-Promoted Cycloaddition | See above | See above |
| Other | Bispecific T-cell Engagers: genetically fused antibody fragments | Clinical: efficacy can be high at low concentrations | Clinical: some fusions have short half-lives | |
| Other Conjugates | ncAA | Antibody-Nucleic Acid: genetically encoded ketone, exogenous aminooxy-nucleic acid | See above | See above |
| Other | Lysine Conjugation | See above | See above |
The alkyne can also be genetically encoded and azide added exogenously.
Antibody-drug conjugates.
Utilizing antibodies to deliver cytotoxic compounds to specific cells is reaching maturity [11,13,14]. Four antibody-drug conjugates (ADCs) are clinically approved, and more than 175 clinical trials utilizing ADCs are ongoing [13]. At the present time, approximately 4% of clinical trials in this area involve ADCs produced using an ncAA-mediated conjugation strategy (NIH U.S. National Library of Science Clinical Trials Database; URL: https://www.clinicaltrials.gov). NcAAs appear to offer important, although nuanced, advantages for generating clinically relevant ADCs.
First-generation ADC chemistries targeting lysine residues or disulfide bonds in IgGs offer viable routes to ADCs (Table 1) [11,13]. Although these strategies have resulted in multiple clinically approved products, challenges with manufacturing, characterization, and product stability resulting from heterogeneous conjugation products have prompted intense research into site-specific conjugation approaches. ADCs with precisely positioned payloads exhibit comparable potency, improved pharmacokinetics, and an expanded therapeutic window relative to ADCs with heterogeneous payloads [15**-17**]. Table 1 depicts key technologies for producing homogeneous ADCs [18**]; each approach exhibits distinct advantages and limitations. For example, THIOMABs, which contain an unpaired cysteine residue for conjugation, lead to ADCs exhibiting excellent therapeutic properties [17**]. However, the site-dependent stability of thiol-reactive linkers in plasma requires careful conjugate engineering and selection of chemistry [11,19,20]. Enzyme-mediated installation of bioorthogonal groups at specific peptide sequences is another efficient route to site-specific ADCs, and resulting linkages do not typically exhibit site-dependent changes in stability [11]. A drawback of this approach is that scale-up of enzymatic transformations requires careful optimization [21,22]. These selected considerations highlight the tradeoffs inherent in choosing and ADC construction methodology.
NcAA-mediated approaches to ADC preparation (Table 1) [16] provide access to diverse, therapeutically relevant ADC structures, including some that are difficult to access with antibodies containing only cAAs [15]. Conjugation methods with ncAAs also avoid known challenges with other chemistries, such as additional reduction and oxidation steps to conjugate a payload at engineered cysteine residues [23], without sacrificing product yield [24]. Historically, challenges with large-scale production of proteins containing ncAAs has posed a significant barrier to clinical applications of ncAA-containing ADCs, but recent reports indicate that these challenges are being addressed [4,15**,24]. With clinical-scale production feasible, are there other potential benefits of utilizing ADCs formed with ncAAs, or are the benefits limited to distinct manufacturing routes? We are aware of only a single published study that directly addresses this question [15**]. Tian et al. used the same anti-Her2 antibody and payload to prepare ADCs via cysteine- or ncAA-mediated (ketone) chemistries at identical antibody locations and evaluate the resulting ADCs in preclinical mouse studies. Intriguingly, in both tumor control and pharmacokinetic studies, ADCs produced via the ncAA-mediated route exhibited improved potency and longer half-lives, respectively, in comparison to ADCs produced via cysteine modification. These tantalizing results indicate that simply changing chemistries to an ncAA-mediated approach led to improved ADC properties; such observations should prompt further study to evaluate the extent of their generality. Thus, even in the highly competitive area of ADCs, the distinct structures, manufacturing routes, and possibly enhanced efficacy, afforded by ncAA-mediated ADC production significantly expands the range of clinically relevant ADCs.
Protein-polymer conjugates.
Attaching polymers to proteins can enhance therapeutically relevant properties such as half-life and stability while reducing undesirable immunogenicity [25]. As a result, a number of protein-polymer conjugates are clinically approved [26]. Interestingly, lysine-mediated PEGylation strategies have led to several approved drugs, despite the range of bioactivities found in the heterogeneous conjugation products (Table 1) [25]. Site-specific conjugation strategies for producing protein-polymer conjugates extend beyond those used for ADCs to include N-terminal modifications and genetically encoded “PEG-like” stretches of cAAs (Table 1). Along with lysine- and cysteine-mediated approaches, these strategies have led to clinically approved products or candidates in ongoing clinical trials [27*-30]. All site-specific strategies, including ncAA-mediated approaches, are subtly distinct. N-terminal modifications and recombinant “polymers” enable modifications to native proteins, but are limited to positioning at protein termini; in some cases, biological activity can be abrogated upon modifying termini [30–32**]. In contrast, modifications utilizing cysteines or ncAAs enable access to a much larger range of sites [25]. Fibroblast growth factor 21 (FGF21) provides an interesting case study highlighting the advantages of this versatility and a basis for comparing cysteine- versus ncAA-mediated conjugation strategies [31,32**]. Both Xu et al. (cysteine) [31] and Mu et al. (ketone-containing ncAA) [32**] found that PEGylated FGF21 variants exhibited a range of activities that depended upon the location of the PEGylation site. While several substitution positions resulted in bioactive variants in each study, Xu et al. reported that some cysteine substitutions appeared to interfere with native disulfide bond formation, while the Mu et al. study did not report any such effects. These findings indicate, in this case, a distinct advantage for ncAA-containing FGF21 because ketones are not known to interfere with native disulfide bond formation. Clinical studies with PEGylated FGF21 emerging from Mu et al. (Pegbelfermin; BMS-986036) indicate favorable safety profiles and, in Phase II studies in the indication of type II diabetes, patients receiving the drug experienced improved metabolic parameters including reduced cholesterol and triglycerides [33]. These and other findings [34] validate the feasibility of large-scale manufacture and clinical administration of protein-polymer conjugates produced via an ncAA-mediated strategy (It is important to note that these findings do not rule out the development of an FGF21 conjugate produced via an alternative approach; in the PEGylated FGF21 space, development of BMS-986036 is simply the most advanced). The crowded nature of protein-polymer conjugate development indicates the need to carefully consider preferred technologies for lead candidate generation. As highlighted by FGF21 conjugates, ncAA-mediated approaches are particularly well-suited for preserving native disulfide bonding patterns and positioning of attachment sites away from protein termini.
Other types of conjugates.
Many additional well-defined conjugates are accessible with ncAA-mediated strategies that could be therapeutically relevant. For example, both bispecific antibodies [35] and antibody-nucleic acid conjugates can be produced utilizing ncAAs (Table 1) [36]. As discussed above, advantages of utilizing ncAAs to prepared these conjugates is likely to be nuanced. While numerous protein conjugates are formed by joining macromolecules or cytotoxic drugs to a protein, chemical introduction of small molecules into proteins with medicinal chemistry-like precision is an appealing but understudied area of opportunity. The Protein medicinal chemistry section below explores how ncAA-mediated approaches, which include conjugations, facilitate powerful, unique approaches to therapeutic discovery.
Vaccines and cell-based therapies
Researchers are exploring ways in which ncAA incorporation can augment the functions of vaccines and cell-based therapies. Both conjugation strategies and control over protein translation are possible with ncAAs in cells and viruses. To date, ncAA-mediated conjugation strategies in this area tend to focus on fundamental virology studies. Examples include utilizing fluorescent conjugates to investigate the roles of envelope glycoproteins in HIV-1 viral budding [37], and modifying adeno-associated viruses with different receptor targeting agents to infect distinct cell types [38,39]. Though intriguing, these approaches are not yet mature enough to have direct therapeutic applications. Thus, the remainder of this section focuses on using codon suppression with ncAAs to control virus or cell replication. Stringent regulation of protein synthesis is effective for controlling replication events in patients or the environment (biocontainment); ncAA-mediated methods offer unique advantages for doing so.
Limiting viral replication.
Although live or attenuated vaccines mimic pathogens and elicit robust immune responses, these vaccine formats do not completely eliminate the risk of viral replication. Conversely, synthetic vaccines based on conjugates, toxoids, subunits, or viral-like particles eliminate replication risks at the cost of native structure [40,41]. Genetic code expansion enables virus attenuation by making viral replication dependent on the incorporation of ncAAs in response to stop codons [38,42] or even four-base codons [43*]. For example, Chen et al. reported a genomically recoded HIV-1 that uses a quadruplet codon to encode Nε-(tert-butyloxy-carbonyl)-L-lysine (BocLys) in essential genes. The resulting engineered viruses require an OTS, a ncAA, and a successful frameshift to produce their full complement of proteins, providing extremely stringent regulation of translation. Control over protein synthesis via codon suppression can be defeated by aberrant misincorporation of cAAs [10,44**]. However, engineering higher fidelity OTSs or making protein function dependent on ncAA side chain structure can largely alleviate these concerns (see also below). Thus, the ability to produce native-like viral structures and carefully control replication with ncAAs appears to offer unique advantages in vaccine production over alternative methodologies.
Limiting cell replication.
Genetically modified microorganisms (GMMs) are an attractive platform for generating live bacterial vaccines and cell-based therapeutics [45]. Several complementary strategies for limiting the replication of GMMs now exist, including auxotrophy, gene circuits, unnatural nucleotides, and the use of ncAAs with OTSs. These biocontainment approaches all perform at similar levels, each reaching escape frequencies of 10−10 to 10−12 [45-47*]. However, with ncAAs an additional level of control is achievable: Mandell et al. redesigned enzymes in genomically recoded E. coli to require the incorporation of L-4,4′-biphenylalanine (bipA) to perform essential catalytic functions. Thus, not even cAA misincorporation in these proteins, which occurs with some OTSs, breaks containment [46*]. Finally, redundancy in biocontainment systems is important in ensuring that escape frequencies remain below detectable limits. Thus, ncAA-mediated biocontainment is a valuable addition to the biocontainment toolkit.
Protein medicinal chemistry
In principle, ncAA incorporation enables atom-by-atom control over protein function in ways that are not possible with cAAs. This precise control is routinely exploited in medicinal chemistry to identify small molecule drug leads [48]. There is growing evidence that “protein medicinal chemistry” supports the discovery of more druglike proteins and peptides (Figure 2) [4,7,49**]. Peptide-based discovery strategies (Figure 2a) already utilize this concept effectively [7], and basic studies with ncAAs in full-length proteins (Figure 2b) point the way to leveraging these concepts in larger structures [1,50]. Several additional areas (Figure 2c-f) appear ripe for further exploration of this concept. While examples below exploit functionalities found in genetically encoded ncAAs, conjugation strategies are also expected to support the practice of protein medicinal chemistry.
Figure 2.

Early examples of protein medicinal chemistry. (a) NcAA-containing macrocyclic peptide synthesis and screening. The portion of the genetic code encoding hydrophilic cAAs was reassigned to a set of hydrophobic ncAAs to make encoded peptides more druglike. A macrocyclic library generated with this altered genetic code was screened against the interleukin-6 receptor (IL6R) in mRNA display format. Identified hits exhibited hydrophobicities that would be extremely challenging to achieve utilizing only canonical amino acids [53**]. (b) A single-atom substitution at position 28 in the insulin B-chain significantly shortened the dissociation time of monomeric insulin (hexamer t1/2) while prolonging its shelf life (fibrillation lag time). Hzp: (4S)-hydroxyproline [49**]. (c) Precise control of posttranslational modifications using ncAA incorporation. Uniformly sulfated antibodies were produced by genetically encoding sulfotyrosine (sY) in response to a stop codon. By evaluating all possible sulfation patterns in an antibody region important for binding, combinations leading to the highest potencies were identified [58*]. (d) Two representative examples of ncAA-based, proximity-activated covalent crosslinkers that function in mammalian cell culture [68*,69*]. BetY: O-(2-bromoethyl)tyrosine; FSY: fluorosulfate-L-tyrosine. (e) The trimeric coiled-coil structure of the N-peptide of HIV surface protein gp41 (IZN17) can be stabilized by encoding a metal chelating ncAA within its structure. BpyAla: (2,2’-Bipyridin-5-yl)alanine [73*]. PDB: 2R3C. (f) The boronic acid group in 4-borono-L-phenylalanine (Bpa) is capable of forming a covalent bond with the diol structures found in many glycans.
Peptide-based discovery.
The unique properties of peptides enable interactions with traditionally “undruggable” features of therapeutic targets, but peptides containing only cAAs exhibit short circulating half-lives and proteolytic sensitivity. In vitro translation systems utilizing ncAAs support genetically encoded macrocyclic constraints, altered backbone structures, and altered side chain structures during discovery and optimization [51,52]. In particular, the use of mRNA display to screen libraries of up to 1013 ncAA-containing peptides has led to the identification of numerous leads that could not have been identified with other approaches. For example, Passioura et al. used a 23-amino acid genetic code containing a set of hydrophobic ncAA side chains to identify anti-interleukin-6 receptor macrocyclic peptide ligands (Figure 2a). The resulting hits exhibited more druglike hydrophobicities than hits from control cAA-containing libraries [53**]. This and other successes highlight the expanded range of properties accessible when ncAAs are incorporated into genetically encoded peptide discovery.
Atomic-level perturbations in proteins.
Historically, many studies employing ncAAs in full-length proteins have leveraged atomic-level changes to elucidate fundamental insights, especially in the area of membrane protein function [50]. For example, replacing aromatic cAAs with fluorinated aromatic analogs has validated numerous interactions between aromatic rings and positively charged chemical groups. These cation-pi interactions are strongly affected by the electron-withdrawing properties of even single-fluorine substitutions [54]. Subtle but powerful effects such as these lay the foundation for broader practices of protein medicinal chemistry.
Some work directly demonstrates how minute side chain alterations modulate properties of therapeutically relevant proteins. Using a model antibody fragment, Van Deventer et al. serendipitously discovered that replacing methionine residues with an azide-containing ncAA resulted in enhanced antigen binding [55]. This finding implies that ncAA side chains support altered modes of molecular recognition, which may be valuable in tailoring the interactions of proteins with therapeutic targets. In a groundbreaking study on insulin, Lieblich and Fang et al. discovered that a single proline-to-4S-hydroxyproline substitution facilitated more rapid monomer dissociation and slower fibrillation compared to the parent insulin molecule (Figure 2b); these enhancements are both highly clinically desirable [49**]. These studies clearly indicate the potential for ncAAs to support the discovery of more druglike proteins; the following subsections highlight additional areas ripe for exploitation.
Post-translational modifications.
Naturally occurring protein post-translational modifications (PTMs) can drastically alter protein properties. NcAA incorporation provides the means of producing proteins with precisely defined patterns of “post-translational” modifications [56]. Moreover, some PTMs modulate the functions of extracellular targets, raising the possibility of near-term therapeutic applications. For example, sulfation events at tyrosines play important roles in mediating extracellular protein-protein interactions [57,58*]. However, sulfation occurs when enzymes recognize amino acid motifs that may not be optimal for target recognition. Recent work by Li et al. used genetic code expansion in an anti-HIV antibody to quantify the potency of all combinations of tyrosine sulfations within an antibody region that mediates interactions with gp120 (Figure 2c) [58*]. This excellent work indicates clear opportunities for producing and screening proteins containing precisely defined sets of PTMs in search of proteins with more druglike properties. There are numerous additional ways in which naturally occurring PTMs could be harnessed in therapeutic applications. The reader is referred elsewhere for potential opportunities in areas including breaking immune tolerance [59], crosslinking (see also the following section) [60], glycosylation (requires chemical synthesis) [61,62], and intracellular PTMs (requires efficient intracellular protein delivery technologies) [63].
Covalent crosslinkers.
NcAAs also facilitate the presentation of crosslinkable groups within proteins [64,65]. Covalent therapeutic target engagement has long been a part of medicinal chemistry. Researchers have demonstrated several advantages of covalently bonding with active site residues. For example, covalent inhibitors support potent inactivation even during substrate buildup, which noncovalent inhibitors cannot [66,67]. However, covalent inhibitors can quickly become toxic if they engage off-targets. The exquisite specificities of antibodies and other binding proteins offer a potential solution to this challenge, provided that crosslinkable functionality can be presented within these scaffolds without the need for ultraviolet light or exogenous catalysts (Figure 2d) [68*,69*]. Several recent reports have demonstrated the use of ncAAs containing functional groups that undergo proximity-based crosslinking, but are not reactive enough to interfere with protein translation or other biochemical processes [68*,69*]. Expanding these approaches beyond model systems has strong therapeutic potential. In addition, proximity-based crosslinking enables formation of intramolecular crosslinks, leading to stabilized or constrained protein structures that may be more druglike [70,71].
Metal binding groups.
The introduction of metal-binding groups (MBGs) into proteins [72] is another enticing approach to enhancing therapeutic protein properties. This includes: 1) the creation of artificial metalloproteins with enhanced stability or nonnatural catalytic functions; and 2) the creation of metalloproteinase function-disrupting proteins by binding active site metal ions. In one strategy for enhancing stability, Luo et al. inserted an ncAA containing an MBG into the HIV gp41 trimeric coiled-coil (Figure 2e). Placement of the MBG at the N-terminus led to a highly stabilized trimer [73*], which could lead to viral inhibitors or vaccine components with better pharmacological properties.
Recent work in medicinal chemistry has led to the reemergence of MBGs as compelling elements of therapeutic molecules. In particular, fragment-based drug discovery efforts have revealed a diversity of MBGs that interfere with metalloprotein function [74,75*]. However, modulation of the specificities of these groups remains a significant challenge [76]. Introduction of MBGs within binding protein scaffolds could lead to inhibitors that eliminate the off-target effects of previous generations of metalloprotein inhibitors. Thus, judicious use of MBGs could lead to multiple new types of macromolecular therapeutics.
Glycan targeting.
Specific glycosylation patterns modulate important protein and cell regulation events [61,62]. Because these patterns are known to be disregulated in cancer and other diseases, glycan targeting is a tantalizing but largely unrealized therapeutic approach [61]. NcAAs offer potential ways to generate proteins that target specific glycan structures in ways that natural sugar-binding proteins cannot. Most notably, boronic acids, which covalently bind to diols in sugars, can be directly genetically encoded within proteins [77] (Figure 2f). Early work in this area includes the presentation of boronates within antibody variable regions [77] and other binding proteins [78*]. Realizing these approaches in therapeutic settings will require additional design and engineering to enhance the affinity and specificity of boron-containing proteins.
Underlying technologies for encoding ncAAs in proteins
The machinery required to genetically encode ncAAs must exhibit efficiencies and fidelities rivaling those of natural protein translation machineries in order to fully realize potential therapeutic applications. Some early successes include protein expression systems that support clinical-scale production of proteins containing a narrow range of ncAAs [4] and peptide-based discovery with mRNA display [7,51]. However, these examples are exceptional. In contrast, the integration of ncAAs with additional display technologies (yeast display [79], phage display [80], and bacterial display [55]) and selection-based approaches [71] remains in early stages.
We address here ongoing challenges in generating efficient genetic code expansion systems in cells. Examples of cell-free systems for efficient genetic code alteration and E. coli-based systems for residue-specific ncAA incorporation are described elsewhere [6,7]. The lack of efficient orthogonal translation systems (OTSs) is one of the largest barriers to establishing robust genetic code expansion. In addition, suppression events at noncognate codons tend to be inefficient compared to cognate codon readthrough. Characterizing the performance of engineered translation systems is crucial for understanding and improving all aspects of ncAA incorporation.
Quantifying stop codon readthrough efficiencies and fidelities with robust reporters is a critical but frequently overlooked aspect of evaluating genetic code manipulation systems. Reporters have the throughput needed to conduct the dozens of measurements of OTS performance needed to identify promising ncAA-containing protein expression conditions, and even the millions of measurements needed to screen for improved ncAA incorporation systems. These do not replace other important characterizations: soluble protein yields and mass spectrometry experiments on purified proteins are the ultimate proof of precisely defined ncAA substitution(s). Furthermore, enzymatic and structural characterizations of OTSs provide insights that may lead to OTSs with native-like properties. However, tuning the in vivo expression levels of aminoacyl-tRNA synthetases and tRNAs can result in efficient, high fidelity protein synthesis even when aaRS/tRNA properties are known to be poor [10,81]. Thus, reporters have an important role to play in evaluating ncAA incorporation events in cells.
Historically, single fluorescent protein reporters containing one or more stop codons have been detected in plate readers to characterize ncAA incorporation in cells [82]. However, these reporters ignore cell-to-cell variability and other potential changes in protein synthesis that accompany ncAA incorporation events. Advanced reporters that monitor both initiation of protein synthesis and codon readthrough events overcome limits of single-fluorescent protein reporters. Monk et al. utilized a dual fluorescent protein reporter in E. coli to measure ncAA incorporation efficiency and fidelity while controlling for cell-to-cell variability and cell viability [83**]. This work also established quantitative metrics of ncAA incorporation efficiency and fidelity that suit any dual reporter system. Beranek et al. implemented the use of a similar format in mammalian cells [84], although efficiency and fidelity metrics were not reported. Building off of Monk et al.’s approach, our lab recently described yeast-based measurements of ncAA incorporation in a dual reporter format [44**]. The yeast display-based reporter enabled flow cytometry-based evaluation of OTS performance on a single-cell basis while eliminating potential problems with fluorescent reporter folding kinetics. These types of reporters support evaluations of ncAA incorporation while controlling for stop codon position within a construct, nonsense-mediated decay (in eukaryotes), genome composition, and other factors.
Robust reporters can aid the discovery of improved ncAA incorporation platforms in engineering efforts ranging from OTS screening to genomic recoding. Many efforts to identify OTSs for new or improved ncAA incorporation are still conducted with selection systems where stringency is difficult to tune [10]. In contrast, screens utilizing fluorescence-activated cell sorting (FACS) provide quantitative cell-by-cell comparisons while readily supporting changes in sorting stringency. Even with single fluorescent protein reporters, FACS can lead to OTSs with excellent properties [85**,86]. Next-generation selection systems such as phage-assisted continuous evolution are also promising tools for evolving OTSs [87]. Finally, many aspects of the translation apparatus can be engineered to enhance ncAA incorporation efficiencies. The most sophisticated of these efforts have yielded E. coli in which the amber stop codon has been removed and recoded for a 21st amino acid [88] and E. coli with a third DNA base pair to provide additional codons [89]. Reporter systems are important for evaluating these impressive systems so that the process of ncAA incorporation rivals that of canonical amino acid incorporation.
Conclusions
There is no doubt that ncAAs will play important roles in future generations of therapeutically relevant proteins, viruses, and cells. NcAA-mediated approaches expand the routes available for discovery and development of entities including conjugates, vaccines, and cell-based therapies. Careful exploitation of ncAAs supports generation of a broader range of therapeutic leads, some which may even exhibit superior properties, compared to those produced with alternative methods. Integrating ncAAs into genetically encoded peptide discovery has yielded peptides with structures and functions that would be nearly impossible to duplicate with only canonical amino acids. Hints of the power of utilizing medicinal chemistry concepts in larger proteins, i.e. protein medicinal chemistry, are also coming into focus. However, the full potential of protein medicinal chemistry and other applications of ncAAs in biotherapeutics will be only realized with the availability of high-performing ncAA incorporation systems in bacterial and eukaryotic expression systems. Given the power of platforms for discovering biologics that utilize only canonical amino acids, providing these platforms with full access to the chemical cabinet is sure to lead to entirely new classes of therapeutics.
Highlights.
Use of noncanonical amino acid (ncAAs) in therapeutics requires careful evaluation.
NcAA-mediated strategies enable important routes to precisely defined conjugates.
Viral and cell replication can be controlled using ncAA-dependent protein function.
“Protein medicinal chemistry” with ncAAs can generate more druglike proteins.
Improved incorporation technologies will further advance therapeutic applications.
Acknowledgments
Research on noncanonical amino acids in the Van Deventer Lab is supported by the National Institutes of Health (R21CA214239), National Science Foundation (NSF1807415, NSF1815022), Army Research Office (W911NF-16–1-0175), Tufts University Faculty Research Fund, and Tufts University Startup Funds.
References
- 1.Chin JW: Expanding and reprogramming the genetic code. Nature 2017, 550:53–60. [DOI] [PubMed] [Google Scholar]
- 2.Wang L: Engineering the Genetic Code in Cells and Animals: Biological Considerations and Impacts. Acc Chem Res 2017, 50:2767–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ledbetter MP, Romesberg FE: Editorial overview: Expanding the genetic alphabet and code. Curr Opin Chem Biol 2018, 46:A1–A2. [DOI] [PubMed] [Google Scholar]
- 4.Kang M, Lu Y, Chen S, Tian F: Harnessing the power of an expanded genetic code toward next-generation biopharmaceuticals. Curr Opin Chem Biol 2018, 46:123–129. [DOI] [PubMed] [Google Scholar]
- 5.Huang Y, Liu T: Therapeutic applications of genetic code expansion. Synth Syst Biotechnol 2018, 3:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang KY, Lieblich SA, Tirrell DA: Incorporation of Non-Canonical Amino Acids into Proteins by Global Reassignment of Sense Codons. Methods Mol Biol 2018, 1798:173–186. [DOI] [PubMed] [Google Scholar]
- 7.Huang Y, Wiedmann MM, Suga H: RNA Display Methods for the Discovery of Bioactive Macrocycles. Chem Rev 2018. [DOI] [PubMed]
- 8.O’Donoghue P, Ling J, Wang YS, Soll D: Upgrading protein synthesis for synthetic biology. Nat Chem Biol 2013, 9:594–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arranz-Gibert P, Vanderschuren K, Isaacs FJ: Next-generation genetic code expansion. Curr Opin Chem Biol 2018, 46:203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vargas-Rodriguez O, Sevostyanova A, Soll D, Crnkovic A: Upgrading aminoacyl-tRNA synthetases for genetic code expansion. Curr Opin Chem Biol 2018, 46:115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agarwal P, Bertozzi CR: Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug Chem 2015, 26:176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devaraj NK: The Future of Bioorthogonal Chemistry. ACS Cent Sci 2018, 4:952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sau S, Alsaab HO, Kashaw SK, Tatiparti K, Iyer AK: Advances in antibody-drug conjugates: A new era of targeted cancer therapy. Drug Discov Today 2017, 22:1547–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schumacher D, Hackenberger CP, Leonhardt H, Helma J: Current Status: Site-Specific Antibody Drug Conjugates. J Clin Immunol 2016, 36 Suppl 1:100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian F, Lu Y, Manibusan A, Sellers A, Tran H, Sun Y, Phuong T, Barnett R, Hehli B, Song F, et al. : A general approach to site-specific antibody drug conjugates. Proc Natl Acad Sci U S A 2014, 111:1766–1771.** This work describes the ncAA-mediated preparation of antibody drug conjugates and directly compares the properties of the resulting ADCs with similar constructs prepared via a cysteine-mediated approach. Intriguingly, the ncAA-mediated route led to ADCs with improved pharmacokinetics and tumor control relative to the ADCs prepared via reactions with cysteine.
- 16.VanBrunt MP, Shanebeck K, Caldwell Z, Johnson J, Thompson P, Martin T, Dong H, Li G, Xu H, D’Hooge F, et al. : Genetically Encoded Azide Containing Amino Acid in Mammalian Cells Enables Site-Specific Antibody-Drug Conjugates Using Click Cycloaddition Chemistry. Bioconjug Chem 2015, 26:2249–2260. [DOI] [PubMed] [Google Scholar]
- 17.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al. : Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 2008, 26:925–932. * This paper is a powerful demonstration of the significant benefits of using site-specific conjugation techniques to construct antibody-drug conjugates. A single unpaired cysteine formed the basis for generating the well-defined ADC in this work.
- 18.Beck A, Goetsch L, Dumontet C, Corvaia N: Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov 2017, 16:315–337.** Thorough recent review of the preparation strategies and clinical applications of antibody-drug conjugates.
- 19.Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, et al. : Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol 2012, 30:184–189. [DOI] [PubMed] [Google Scholar]
- 20.Vollmar BS, Wei B, Ohri R, Zhou J, He J, Yu SF, Leipold D, Cosino E, Yee S, Fourie-O’Donohue A, et al. : Attachment Site Cysteine Thiol pKa Is a Key Driver for Site-Dependent Stability of THIOMAB Antibody-Drug Conjugates. Bioconjug Chem 2017, 28:2538–2548. [DOI] [PubMed] [Google Scholar]
- 21.Milczek EM: Commercial Applications for Enzyme-Mediated Protein Conjugation: New Developments in Enzymatic Processes to Deliver Functionalized Proteins on the Commercial Scale. Chem Rev 2018, 118:119–141. [DOI] [PubMed] [Google Scholar]
- 22.Rabuka D, Rush JS, deHart GW, Wu P, Bertozzi CR: Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat Protoc 2012, 7:1052–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhakta S, Raab H, Junutula JR: Engineering THIOMABs for site-specific conjugation of thiol-reactive linkers. Methods Mol Biol 2013, 1045:189–203. [DOI] [PubMed] [Google Scholar]
- 24.Thanos CD, McEvoy L, Yin G, Penta K, Baliga R, Bajad S, Pollitt S, Murray C, Steiner A, Gill A: Antibodies comprising site-specific non-natural amino acid residues, methods of their preparation and methods of their use US 9738724 B2; 2017.
- 25.Liu X, Sun J, Gao W: Site-selective protein modification with polymers for advanced biomedical applications. Biomaterials 2018, 178:413–434. [DOI] [PubMed] [Google Scholar]
- 26.Ekladious I, Colson YL, Grinstaff MW: Polymer-drug conjugate therapeutics: advances, insights and prospects. Nat Rev Drug Discov 2018. [DOI] [PubMed]
- 27.Schellenberger V, Wang CW, Geething NC, Spink BJ, Campbell A, To W, Scholle MD, Yin Y, Yao Y, Bogin O, et al. : A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat Biotechnol 2009, 27:1186–1190.* This is the first example of in vivo half-life extension of proteins using genetically encoded “PEG-like” sequences of canonical amino acids.
- 28.Podust VN, Balan S, Sim BC, Coyle MP, Ernst U, Peters RT, Schellenberger V: Extension of in vivo half-life of biologically active molecules by XTEN protein polymers. J Control Release 2016, 240:52–66. [DOI] [PubMed] [Google Scholar]
- 29.Pasut G, Veronese FM: State of the art in PEGylation: The great versatility achieved after forty years of research. Journal of Controlled Release 2012, 161:461–472. [DOI] [PubMed] [Google Scholar]
- 30.Dozier JK, Distefano MD: Site-Specific PEGylation of Therapeutic Proteins. Int J Mol Sci 2015, 16:25831–25864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J, Bussiere J, Yie J, Sickmier A, An P, Belouski E, Stanislaus S, Walker KW: Polyethylene glycol modified FGF21 engineered to maximize potency and minimize vacuole formation. Bioconjug Chem 2013, 24:915–925. [DOI] [PubMed] [Google Scholar]
- 32.Mu J, Pinkstaff J, Li Z, Skidmore L, Li N, Myler H, Dallas-Yang Q, Putnam AM, Yao J, Bussell S, et al. : FGF21 analogs of sustained action enabled by orthogonal biosynthesis demonstrate enhanced antidiabetic pharmacology in rodents. Diabetes 2012, 61:505–512.** This paper demonstrates attachment of PEG to several sites within fibroblast growth factor 21 via a genetically encoded, ketone-containing ncAA. Careful placement of the PEGylation site leads to conjugates that retain substantial bioactivity in preclinical evaluations.
- 33.Charles ED, Neuschwander-Tetri BA, Pablo Frias J, Kundu S, Luo Y, Tirucherai GS, Christian R: Pegbelfermin (BMS-986036), PEGylated FGF21, in Patients with Obesity and Type 2 Diabetes: Results from a Randomized Phase 2 Study. Obesity (Silver Spring) 2019, 27:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nairn NW, Bariola PA, Graddis TJ, VanBrunt MP, Wang A, Li G, Grabstein K: Cysteine as a Monothiol Reducing Agent to Prevent Copper-Mediated Oxidation of Interferon Beta During PEGylation by CuAAC. Bioconjug Chem 2015, 26:2070–2075. [DOI] [PubMed] [Google Scholar]
- 35.Cao Y, Axup JY, Ma JSY, Wang RSE, Choi S, Tardif V, Lim RKV, Pugh HM, Lawson BR, Welzel G, et al. : Multiformat T-Cell-Engaging Bispecific Antibodies Targeting Human Breast Cancers. Angewandte Chemie-International Edition 2015, 54:7022–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kazane SA, Axup JY, Kim CH, Ciobanu M, Wold ED, Barluenga S, Hutchins BA, Schultz PG, Winssinger N, Smider VV: Self-assembled antibody multimers through peptide nucleic acid conjugation. J Am Chem Soc 2013, 135:340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakin V, Hanne J, Dunder J, Anders-Osswein M, Laketa V, Nikic I, Krausslich HG, Lemke EA, Muller B: A Versatile Tool for Live-Cell Imaging and Super-Resolution Nanoscopy Studies of HIV-1 Env Distribution and Mobility. Cell Chem Biol 2017, 24:635–645 e635. [DOI] [PubMed] [Google Scholar]
- 38.Kelemen RE, Erickson SB, Chatterjee A: Synthesis at the interface of virology and genetic code expansion. Curr Opin Chem Biol 2018, 46:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelemen RE, Mukherjee R, Cao X, Erickson SB, Zheng Y, Chatterjee A: A Precise Chemical Strategy To Alter the Receptor Specificity of the Adeno-Associated Virus. Angew Chem Int Ed Engl 2016, 55:10645–10649. [DOI] [PubMed] [Google Scholar]
- 40.Jones LH: Recent advances in the molecular design of synthetic vaccines. Nat Chem 2015, 7:952–960. [DOI] [PubMed] [Google Scholar]
- 41.Symoniak MR, Farrokh P, Gandhi MA, Slish JC: Herpes zoster subunit vaccine for the prevention of herpes zoster. American Journal of Health-System Pharmacy 2018, 75:861–869. [DOI] [PubMed] [Google Scholar]
- 42.Si LL, Xu H, Zhou XY, Zhang ZW, Tian ZY, Wang Y, Wu YM, Zhang B, Niu ZL, Zhang CL, et al. : Generation of influenza A viruses as live but replication-incompetent virus vaccines. Science 2016, 354:1170–1173. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, Wan Y, Wang N, Yuan Z, Niu W, Li Q, Guo J: Controlling the Replication of a Genomically Recoded HIV-1 with a Functional Quadruplet Codon in Mammalian Cells. ACS Synth Biol 2018, 7:1612–1617.* This paper demonstrates the limitation of viral replication with the stringent requirements of an ncAA, orthogonal translation system, and successful frameshift event via four-base codon suppression.
- 44.Stieglitz JT, Kehoe HP, Lei M, Van Deventer JA: A Robust and Quantitative Reporter System To Evaluate Noncanonical Amino Acid Incorporation in Yeast. ACS Synth Biol 2018, 7:2256–2269.** This work utilizes an advanced reporter to quantitatively characterize stop codon-based ncAA incorporation in yeast. The yeast display-based system avoids potential issues with fluorescent reporter folding and supports flow cytometric analyses at both single-cell and population levels.
- 45.Lee JW, Chan CTY, Slomovic S, Collins JJ: Next-generation biocontainment systems for engineered organisms. Nat Chem Biol 2018, 14:530–537. [DOI] [PubMed] [Google Scholar]
- 46.Mandell DJ, Lajoie MJ, Mee MT, Takeuchi R, Kuznetsov G, Norville JE, Gregg CJ, Stoddard BL, Church GM: Biocontainment of genetically modified organisms by synthetic protein design. Nature 2015, 518:55–60.* This study reports the design of an essential protein in E. coli to function only when a specific ncAA is incorporated into its active site. This is a unique safety measure that renders the protein nonfunctional even when aberrant canonical amino acid incorporation results in a full-length protein.
- 47.Tack DS, Ellefson JW, Thyer R, Wang B, Gollihar J, Forster MT, Ellington AD: Addicting diverse bacteria to a noncanonical amino acid. Nat Chem Biol 2016, 12:138–140. [DOI] [PubMed] [Google Scholar]
- 48.Wermuth CG, Aldous D, Raboisson P, Rognan D (Ed): The Practice of Medicinal Chemistry Elsevier; 2015. [Google Scholar]
- 49.Lieblich SA, Fang KY, Cahn JKB, Rawson J, LeBon J, Ku HT, Tirrell DA: 4S-Hydroxylation of Insulin at ProB28 Accelerates Hexamer Dissociation and Delays Fibrillation. J Am Chem Soc 2017, 139:8384–8387.** In this study, precise addition of a single hydroxy group to insulin greatly improved the therapeutic properties of the resulting molecule. The replacement of proline by (4S)-hydroxyproline at position 28 of the B chain in this work is an exemplary demonstration of the power of “protein medicinal chemistry.”
- 50.Dougherty DA, Van Arnam EB: In vivo incorporation of non-canonical amino acids by using the chemical aminoacylation strategy: a broadly applicable mechanistic tool. Chembiochem 2014, 15:1710–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taylor RD, Rey-Carrizo M, Passioura T, Suga H: Identification of nonstandard macrocyclic peptide ligands through display screening. Drug Discov Today Technol 2017, 26:17–23. [DOI] [PubMed] [Google Scholar]
- 52.Richardson SL, Dods KK, Abrigo NA, Iqbal ES, Hartman MC: In vitro genetic code reprogramming and expansion to study protein function and discover macrocyclic peptide ligands. Curr Opin Chem Biol 2018, 46:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Passioura T, Liu W, Dunkelmann D, Higuchi T, Suga H: Display Selection of Exotic Macrocyclic Peptides Expressed under a Radically Reprogrammed 23 Amino Acid Genetic Code. J Am Chem Soc 2018, 140:11551–11555.** This recent extension of the in vitro Random nonstandard Peptides Integrated Discovery (RaPID) system demonstrated the reprogramming of the genetic code to a 23-amino acid set consisting of only hydrophobic residues. Screening with this altered code resulted in the discovery of cyclic peptides against interleukin-6 receptor with much higher hydrophobicities than typically accessed with peptides containing only canonical amino acids.
- 54.Davis MR, Dougherty DA: Cation-pi interactions: computational analyses of the aromatic box motif and the fluorination strategy for experimental evaluation. Phys Chem Chem Phys 2015, 17:29262–29270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Deventer JA, Yuet KP, Yoo TH, Tirrell DA: Cell surface display yields evolvable, clickable antibody fragments. Chembiochem 2014, 15:1777–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krall N, da Cruz FP, Boutureira O, Bernardes GJ: Site-selective protein-modification chemistry for basic biology and drug development. Nat Chem 2016, 8:103–113. [DOI] [PubMed] [Google Scholar]
- 57.Boyce M, Bertozzi CR: Bringing chemistry to life. Nat Methods 2011, 8:638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li X, Hitomi J, Liu CC: Characterization of a Sulfated Anti-HIV Antibody Using an Expanded Genetic Code. Biochemistry 2018, 57:2903–2907.* This work evaluates how sulfation patterns affect the properties of E51, an anti-HIV gp120 antibody. Genetically encoding sulfotyrosine and systematically evaluating sulfation patterns in a key region of the antibody revealed which sulfation events increase or decrease potency.
- 59.Gauba V, Grunewald J, Gorney V, Deaton LM, Kang M, Bursulaya B, Ou W, Lerner RA, Schmedt C, Geierstanger BH, et al. : Loss of CD4 T-cell-dependent tolerance to proteins with modified amino acids. Proc Natl Acad Sci U S A 2011, 108:12821–12826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hauf M, Richter F, Schneider T, Faidt T, Martins BM, Baumann T, Durkin P, Dobbek H, Jacobs K, Moglich A, et al. : Photoactivatable Mussel-Based Underwater Adhesive Proteins by an Expanded Genetic Code. Chembiochem 2017, 18:1819–1823. [DOI] [PubMed] [Google Scholar]
- 61.Pinho SS, Reis CA: Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 2015, 15:540–555. [DOI] [PubMed] [Google Scholar]
- 62.Poole J, Day CJ, von Itzstein M, Paton JC, Jennings MP: Glycointeractions in bacterial pathogenesis. Nat Rev Microbiol 2018, 16:440–452. [DOI] [PubMed] [Google Scholar]
- 63.Yamatsugu K, Kawashima SA, Kanai M: Leading approaches in synthetic epigenetics for novel therapeutic strategies. Curr Opin Chem Biol 2018, 46:10–17. [DOI] [PubMed] [Google Scholar]
- 64.Coin I: Application of non-canonical crosslinking amino acids to study protein-protein interactions in live cells. Curr Opin Chem Biol 2018, 46:156–163. [DOI] [PubMed] [Google Scholar]
- 65.Nguyen TA, Cigler M, Lang K: Expanding the Genetic Code to Study Protein-Protein Interactions. Angew Chem Int Ed Engl 2018, 57:14350–14361. [DOI] [PubMed] [Google Scholar]
- 66.Miller RM, Taunton J: Targeting protein kinases with selective and semipromiscuous covalent inhibitors. Methods Enzymol 2014, 548:93–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Cesco S, Kurian J, Dufresne C, Mittermaier AK, Moitessier N: Covalent inhibitors design and discovery. Eur J Med Chem 2017, 138:96–114. [DOI] [PubMed] [Google Scholar]
- 68.Wang N, Yang B, Fu C, Zhu H, Zheng F, Kobayashi T, Liu J, Li S, Ma C, Wang PG, et al. : Genetically Encoding Fluorosulfate-l-tyrosine To React with Lysine, Histidine, and Tyrosine via SuFEx in Proteins in Vivo. J Am Chem Soc 2018, 140:4995–4999.* This study demonstrated the genetic encoding of a ncAA capable of undergoing sulfur (VI) Fluoride Exchange (SuFEx), an emerging click chemistry reaction. Proof-of-concept experiments demonstrated proximity-based crosslinking with a range of nucleophilic canonical amino acids in cells.
- 69.Tang H, Dai Z, Qin X, Cai W, Hu L, Huang Y, Cao W, Yang F, Wang C, Liu T: Proteomic Identification of Protein Tyrosine Phosphatase and Substrate Interactions in Living Mammalian Cells by Genetic Encoding of Irreversible Enzyme Inhibitors. J Am Chem Soc 2018, 140:13253–13259.* This work identified a ncAA capable of inhibiting protein tyrosine phosphatase active sites via covalent interactions. The ncAA was positioned at a specific location of the HER2 receptor and used to identify members of the protein tyrosine phosphatase family that bind to this site.
- 70.Moore EJ, Zorine D, Hansen WA, Khare SD, Fasan R: Enzyme stabilization via computationally guided protein stapling. Proc Natl Acad Sci U S A 2017, 114:12472–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li JC, Liu T, Wang Y, Mehta AP, Schultz PG: Enhancing Protein Stability with Genetically Encoded Noncanonical Amino Acids. J Am Chem Soc 2018, 140:15997–16000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Almhjell PJ, Mills JH: Metal-chelating non-canonical amino acids in metalloprotein engineering and design. Curr Opin Struct Biol 2018, 51:170–176. [DOI] [PubMed] [Google Scholar]
- 73.Luo XZ, Wang TSA, Zhang Y, Wang F, Schultz PG: Stabilizing Protein Motifs with a Genetically Encoded Metal-Ion Chelator. Cell Chemical Biology 2016, 23:1098–1102.* This paper provides a good example of how increasing protein stability using metal binding groups can enhance the therapeutic properties of candidates for treating or preventing HIV infection.
- 74.Riccardi L, Genna V, De Vivo M: Metal-ligand interactions in drug design. Nature Reviews Chemistry 2018, 2:100–112. [Google Scholar]
- 75.Cohen SM: A Bioinorganic Approach to Fragment-Based Drug Discovery Targeting Metalloenzymes. Acc Chem Res 2017, 50:2007–2016.* This review describes recent advances in the discovery of metal binding groups that inhibit metalloproteinase activity. Such insights are important for leveraging metal binding groups in future protein medicinal chemistry efforts.
- 76.Murphy G: Riding the metalloproteinase roller coaster. Journal of Biological Chemistry 2017, 292:7708–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu CC, Mack AV, Brustad EM, Mills JH, Groff D, Smider VV, Schultz PG: Evolution of proteins with genetically encoded “chemical warheads”. J Am Chem Soc 2009, 131:9616–9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Edwardraja S, Eichinger A, Theobald I, Sommer CA, Reichert AJ, Skerra A: Rational Design of an Anticalin-Type Sugar-Binding Protein Using a Genetically Encoded Boronate Side Chain. ACS Synth Biol 2017, 6:2241–2247.* This study utilized genetically encoded boronate functionality within a binding protein scaffold to enable glycan targeting.
- 79.Van Deventer JA, Le DN, Zhao J, Kehoe HP, Kelly RL: A platform for constructing, evaluating, and screening bioconjugates on the yeast surface. Protein Eng Des Sel 2016, 29:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Feng T, Tsao ML, Schultz PG: A phage display system with unnatural amino acids. J Am Chem Soc 2004, 126:15962–15963. [DOI] [PubMed] [Google Scholar]
- 81.Guo LT, Wang YS, Nakamura A, Eiler D, Kavran JM, Wong M, Kiessling LL, Steitz TA, O’Donoghue P, Soll D: Polyspecific pyrrolysyl-tRNA synthetases from directed evolution. Proc Natl Acad Sci U S A 2014, 111:16724–16729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Des Soye BJ, Patel JR, Isaacs FJ, Jewett MC: Repurposing the translation apparatus for synthetic biology. Curr Opin Chem Biol 2015, 28:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Monk JW, Leonard SP, Brown CW, Hammerling MJ, Mortensen C, Gutierrez AE, Shin NY, Watkins E, Mishler DM, Barrick JE: Rapid and Inexpensive Evaluation of Nonstandard Amino Acid Incorporation in Escherichia coli. ACS Synth Biol 2017, 6:45–54.** This work in E. coli is the first report of a dual fluorescent protein reporter that enables measurement of ncAA incorporation efficiency and fidelity while taking into account cell-to-cell variability and alterations in cell viability.
- 84.Beranek V, Reinkemeier CD, Zhang MS, Liang AD, Kym G, Chin JW: Genetically Encoded Protein Phosphorylation in Mammalian Cells. Cell Chem Biol 2018, 25:1067–1074 e1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Amiram M, Haimovich AD, Fan C, Wang YS, Aerni HR, Ntai I, Moonan DW, Ma NJ, Rovner AJ, Hong SH, et al. : Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat Biotechnol 2015, 33:1272–1279.** This study utilized a fluorescent reporter containing multiple stop codons to discover orthogonal translation systems capable of supporting protein translation with up to 30 ncAAs in a single protein construct in genomically recoded E. coli. Also of note in this study is the use of chromosomally integrated OTSs during evolution and detailed characterizations of ncAA-containing proteins using top-down proteomic methodologies.
- 86.Kunjapur AM, Stork DA, Kuru E, Vargas-Rodriguez O, Landon M, Soll D, Church GM: Engineering posttranslational proofreading to discriminate nonstandard amino acids. Proc Natl Acad Sci U S A 2018, 115:619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bryson DI, Fan C, Guo LT, Miller C, Soll D, Liu DR: Continuous directed evolution of aminoacyl-tRNA synthetases. Nat Chem Biol 2017, 13:1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, et al. : Genomically recoded organisms expand biological functions. Science 2013, 342:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Ptacin JL, Fischer EC, Aerni HR, Caffaro CE, San Jose K, Feldman AW, Turner CR, Romesberg FE: A semi-synthetic organism that stores and retrieves increased genetic information. Nature 2017, 551:644–647. [DOI] [PMC free article] [PubMed] [Google Scholar]