

Abstract
Nuclear translocation of mothers against decapentaplegic homolog 2/3 (SMAD2/3), core transcription factors of transforming growth factor β (TGF‐β) signaling, is critical for hepatic stellate cell (HSC) differentiation into metastasis‐promoting myofibroblasts. SMAD2/3 have multiple coactivators, including WW domain‐containing transcription regulator protein 1 (WWTR1 or TAZ) and p300 acetyltransferase. In the nucleus, TAZ binds to SMAD2/3 to prevent SMAD2/3 nuclear export. However, how TAZ and SMAD2/3 enter the nucleus remains poorly understood because neither contains a nuclear localization signal (NLS), an amino acid sequence tagging proteins for nuclear transport. p300 is an NLS‐containing large scaffold protein, so we hypothesized that SMAD2/3 and TAZ may undergo nuclear import through complexing with p300. Coimmunoprecipitation, immunofluorescence, and nuclear fractionation assays revealed that TGF‐β1 promoted binding of SMAD2/3 and TAZ to p300 and that p300 inactivation disrupted TGF‐β1‐mediated SMAD2/3 and TAZ nuclear accumulation. Deleting the p300 NLS blocked TGF‐β1‐induced SMAD2/3 and TAZ nuclear transport. Consistently, p300 inactivation suppressed TGF‐β1‐mediated HSC activation and transcription of genes encoding tumor‐promoting factors, such as connective tissue growth factor, Tenascin C, Periostin, platelet‐derived growth factor C, and fibroblast growth factor 2, as revealed by microarray analysis. Chromatin immunoprecipitation‐real‐time quantitative PCR showed that canonical p300‐mediated acetylation of histones also facilitated transcription in response to TGF‐β1 stimulation. Interestingly, although both TGF‐β1‐mediated and stiffness‐mediated HSC activation require p300, comparison of gene expression data sets revealed that transcriptional targets of TGF‐β1 were distinct from those of stiffness‐p300 mechanosignaling. Lastly, in tumor/HSC coinjection and intrasplenic tumor injection models, targeting p300 of activated‐HSC/myofibroblasts by C646, short hairpin RNA, or cre‐mediated gene disruption reduced tumor and liver metastatic growth in mice. Conclusion: p300 facilitates TGF‐β1‐stimulated HSC activation by both noncanonical (cytoplasm‐to‐nucleus shuttle for SMAD2/3 and TAZ) and canonical (histone acetylation) mechanisms. p300 is an attractive target for inhibiting HSC activation and the prometastatic liver microenvironment.
Abbreviations
- α‐SMA
α‐smooth muscle actin
- ChIP‐qPCR
chromatin immunoprecipitation‐real‐time quantitative PCR
- coIP
coimmunoprecipitation
- CTGF
connective tissue growth factor
- DMSO
dimethyl sulfoxide
- FGF2
fibroblast growth factor 2
- HSC
hepatic stellate cell
- IF
immunofluorescence
- LLCs
Lewis lung carcinoma cells
- NLS
nuclear localization signal
- PDGFR‐α
platelet‐derived growth factor receptor α
- RNA‐seq
RNA sequencing
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- SMAD2/3
mothers against decapentaplegic homolog 2/3
- TGF‐β
transforming growth factor β
- WB
western blot analysis
- wt
wild‐type
- YAP1
Yes‐associated protein 1
Transforming growth factor β (TGF‐β) within the hepatic tumor microenvironment induces hepatic stellate cell (HSC) differentiation into myofibroblasts. Myofibroblasts, in return, promote liver metastasis through paracrine mechanisms, such as release of growth factors, cytokines, extracellular matrix proteins, and matrix metalloproteinases.1 TGF‐β activates HSCs by inducing intracellular signaling events, including ligation of TGF‐β receptor I and TGF‐β receptor II on the plasma membrane, phosphorylation and nuclear translocation of mothers against decapentaplegic homolog 2/3 (SMAD2/3), and gene transcription in the nucleus.2 Better understanding of how these signaling events are regulated may lead to novel targets to inhibit HSC activation and the prometastatic liver microenvironment.
TGF‐β‐mediated gene transcription is critically dependent on activation of SMAD2/3; however, how SMAD2/3 enter the nucleus is incompletely understood. Canonically, nuclear transport of proteins larger than 45 kDa, including SMAD2/3, requires a nuclear localization signal (NLS), a short amino acid sequence that tags a cargo protein for nuclear import.3 The NLS motif is usually recognized by a complex containing importin α and β, which facilitates nuclear transport. Importin α and β function as a heterodimer, with importin α containing an NLS‐binding site and importin β mediating the docking of the importin‐cargo complex to the cytoplasmic side of the nuclear pore for subsequent transport through the pore.3 Because neither SMAD2 nor 3 contains a classical NLS motif,4, 5 three models have been proposed for their nuclear transport: (a) an NLS‐like basic motif 40‐KKLKK‐44 may function as an alternative NLS to mediate SMAD3/importinβ binding,4, 5 (b) SMAD2/3 are transported into the nucleus by binding to CAN/Nup214 nucleoporin,6 and (c) SMAD2/3 are transported into the nucleus by importin 7 and 8.7 Given the critical role of SMAD2/3 for TGF‐β signaling, identifying novel mechanisms governing SMAD2/3 nuclear transport would provide unique mechanisms for targeting HSC activation.
Yes‐associated protein 1 (YAP1) and WW domain‐containing transcription regulator protein 1 (WWTR1 or TAZ) are transcriptional coactivators that promote transcriptional enhancer factor TEF (TEAD/TEF) dependent gene transcription.8, 9, 10 YAP1 activity contributes to not only the initiation, progression, and metastasis of cancer but also HSC activation and liver fibrosis.11, 12, 13 However, luminescence‐based mammalian interaction mapping identified that TAZ, but not YAP1, bound to SMAD2/3,14 suggesting that TAZ may participate in TGF‐β1/SMAD‐mediated HSC activation as a coactivator of SMAD2/3. Indeed, in human embryonic stem cells, TAZ, retained in the nucleus by ARC105, provided a nuclear anchor for SMAD2/3 to prevent their nuclear export so as to promote TGF‐β1 signaling.14 As both TAZ and YAP1 lack an NLS motif,9 understanding how TAZ, YAP1, and SMAD2/3 enter the nucleus would provide novel mechanistic insight into HSC activation.
p300 acetyltransferase is a large scaffold protein that shuttles between the cytoplasm and nucleus of the cell for its N‐terminal NLS (11‐PSAKRPK‐17). p300 promotes DNA binding and the transcriptional activity of SMAD2/3 by epigenetic mechanisms, such as acetylation of SMAD2/3 and histones.15, 16, 17 p300 also critically mediates collagen synthesis in fibroblasts18 and myofibroblastic activation of HSCs induced by stiffness.19 Because p300 interacts with SMAD2/3,16 we hypothesized that TGF‐β‐mediated SMAD2/3 and TAZ nuclear transport may be facilitated by p300. Using coimmunoprecipitation (coIP), we found that TGF‐β1 induced a p300/SMAD2/3/TAZ protein complex. Immunofluorescence (IF) and nuclear fractionation revealed that p300 knockdown or inhibition of p300 acetyltransferase by C646 reduced nuclear accumulation of SMAD2/3 and TAZ and myofibroblastic activation of HSCs induced by TGF‐β1. Additionally, overexpression of a p300 NLS‐deletion mutant led to retention of SMAD2/3 and TAZ in the cytoplasm and blocked their nuclear transport induced by TGF‐β1. Functionally, inactivation of p300 abrogated tumor‐promoting effects of HSCs in vitro and in tumor implantation mouse models. Furthermore, chromatin immunoprecipitation‐real‐time quantitative PCR (ChIP‐qPCR) demonstrated that p300 also facilitated gene transcription by a canonical epigenetic mechanism, such as acetylating histones. Thus, p300 plays both noncanonical and canonical roles for HSC activation by functioning as a cytoplasm‐to‐nucleus shuttle for SMAD2/3 and TAZ and by epigenetically promoting gene transcription through histone acetylation and chromatin remodeling.
Materials and Methods
Cell Culture
Murine HSCs were isolated from p300F/F mice as described.19 Primary human HSCs were purchased from ScienCell Research Laboratories (#5300; Carlsbad, CA). HT29 human colorectal cancer cells and Lewis lung carcinoma cells (LLCs) were purchased from American Type Culture Collection (Manassas, VA). L3.6 human pancreatic cancer cells were provided by Dr. Raul Urrutia (Medical College of Wisconsin, Wauwatosa, WI),20 and LX2 human hepatic stellate cells were from Dr. Scott Friedman (Icahn School of Medicine at Mount Sinai, New York, NY). Cells were authenticated by Genetica. Cells were regularly monitored for mycoplasma contamination by a MycoAlert detection kit (Lonza, Basel, Switzerland).
Transfection of Plasmid or siRNA and VIral Transduction of Cells
FLAG‐tagged p300 complementary DNA (cDNA) was from Dr. Makiko Fujii.21 p300 NLS‐deletion mutant with 11‐PSAKRPK‐17 removed was generated using a Q5 Site‐Directed Mutagenesis kit (New England Biolabs, Ipswich, MA). Details regarding control, TAZ and YAP1 small interfering RNA (siRNA), p300 short hairpin RNA (shRNA) constructs, protocols of transfection, retro‐ or lentiviral packaging, and viral transduction22, 23, 24 may be found in the Supporting Materials and Methods.
Nuclear Fractionation, coIP, and Western Blot Analysis (WB)
Cell nuclei were isolated using CelLytic NuCLEAR Extraction Kit (NXTRACT; MilliporeSigma). Details about nuclear fractionation and use of isolated nuclei for coimmunoprecipitation (coIP) and WB may be found in the Supporting Materials and Methods.
Microarray and RNA Sequencing (RNA‐seq)
RNeasy Plus Universal Mini Kit was used to isolate RNA. gDNA eliminator was added to remove contaminating DNA. Microarray analysis, RNA‐seq, and bioinfomatics were done by the University of Minnesota Genomic Center. In brief, after samples were quantitated by nanodrop and RNA integrity was assessed by capillary electrophoresis, quality RNA samples were hybridized with human HT‐12 v4 Expression BeadChip (Illumina, San Diego, CA) or converted to Illumina sequencing libraries for sequencing using HiSeq2500.19 Data are in Gene Expression Omnibus (GSE116509 and GSE127964).
HSC/Tumor Coculture, HSC/Tumor Coinjection, and Liver Metastasis Mouse Models
Details about HSC/tumor coculture may be found in the Supporting Materials and Methods. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Mayo Clinic or the University of Minnesota. Subcutaneous HSC/tumor injection into nude mice and in vivo Xenogen imaging were performed as described.23, 25 LLCs were implanted into p300F/Fcre mice by intrasplenic injection,20 and L3.6 cells were injected into severe combined immunodeficient (SCID) mice by portal vein injection.23, 24 Details about control and p300F/Fcre mice as well as tumor injection protocols may be found in the Supporting Materials and Methods.
Statistics
Data are expressed as mean ± SEM and subjected to statistical analysis by Student t test or analysis of variance followed by a posthoc test using the GraphPad Prism 5 software. P < 0.05 was considered statistically different. Details regarding IF, WB, reagents and antibodies, ChIP‐qPCR, and other in vitro and animal studies are located in the Supporting Materials and Methods.
Results
TGF‐β1 Induces Binding of p300 to SMAD2/3 and TAZ in HSCs
SMAD2/3 and TAZ interact in the nucleus and are involved in TGF‐β‐mediated signaling14; however, it is unclear how SMAD2/3 and TAZ enter the nucleus because neither contains an NLS. Because p300 binds to SMAD2/3 and contains an NLS,16 we hypothesized that p300 may act as a cytoplasm‐to‐nucleus shuttle for SMAD2/3 and TAZ. To test this, we performed coIP to study if TGF‐β1 induced p300 binding to SMAD2/3, TAZ, or the TAZ‐related protein YAP1. Human primary HSCs stimulated with TGF‐β1 (5 ng/mL) were collected for nuclear fractionation and coIP, which revealed that TGF‐β1 indeed promoted binding of p300 to SMAD2/3 and TAZ in HSCs (Fig. 1A). In contrast, p300 did not bind to YAP1 (Fig. 1A), which led us to focus on SMAD2/3 and TAZ in this study, rather than YAP1. IF confirmed that TGF‐β1 induced coaccumulation of SMAD2, TAZ, and p300 proteins in the nucleus (Fig. 1B). Thus, TGF‐β1 induces a p300/SMAD2/3/TAZ protein complex that may mediate SMAD2/3 and TAZ nuclear transport.
Figure 1.
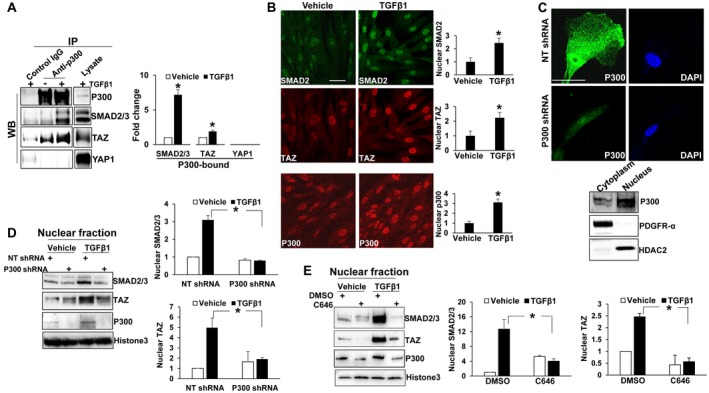
TGF‐β1 induces p300 binding to SMAD2/3 and TAZ, and p300 inactivation suppresses SMAD2/3 and TAZ nuclear accumulation induced by TGF‐β1. (A) Human primary HSCs stimulated with TGF‐β1 (5 ng/mL) for 15 minutes were harvested for nuclear fractionation and coIP. *, P < 0.05 by t test, n = 3. (B) IF revealed that TGF‐β1 increased nuclear p300, SMAD2, and TAZ. *, P < 0.05 by t test, n > 14 cells per group. (C) Upper: HSCs transduced with NT shRNA (control) or p300 shRNA lentiviruses were harvested for IF. Nuclei were counterstained by DAPI. p300 was in both cytoplasm and nucleus of HSCs. Lower: Subcellular fractionation confirmed cytoplasmic and nuclear p300 of control HSCs. PDGFRα and HDAC2 were used as the marker of cytoplasmic and nuclear fraction, respectively. (D,E) Control, p300 knockdown, or C646‐preincubated HSCs were stimulated with TGF‐β1 and collected for nuclear fractionation. Densitometry data are shown on the right. p300 shRNA or C646 reduced SMAD2/3 and TAZ nuclear accumulation induced by TGF‐β1. *, P < 0.05 by analysis of variance, n = 3. Bars in B and C, 50 µm. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; HDAC2, histone deacetylase 2; NT, nontargeting.
p300 Inactivation Suppresses SMAD2/3 and TAZ Nuclear Accumulation Induced by TGF‐β1
Because p300 is the only NLS‐containing protein in this complex, we tested if SMAD2/3 and TAZ nuclear transport by TGF‐β1 was indeed mediated by p300. IF and subcellular fractionation showed that p300 protein was distributed to both the nucleus and cytoplasm of human primary HSCs (Fig. 1C). To assess whether p300 was critical for SMAD2/3 and TAZ nuclear import, we disrupted p300 using two mechanisms: shRNA‐mediated knockdown of p300 and inhibition of p300 acetyltransferase activity using the compound C646. As revealed by nuclear fractionation assay, TGF‐β1 increased nuclear SMAD2/3 and TAZ protein levels in control cells, and this effect of TGF‐β1 was reduced in p300 knockdown or C646‐treated HSCs (Fig. 1D,E; P < 0.05). IF confirmed that p300 knockdown or C646 inhibited SMAD2/3 nuclear accumulation induced by TGF‐β1 (Fig. 2A,B; P < 0.05). p300 knockdown appeared to specifically affect SMAD2/3 nuclear transport, as TGF‐β1 induction of SMAD2 phoshorylation and total protein levels of TGF‐β receptor I, TGF‐β receptor II, and YAP1/TAZ were unaffected (Supporting Fig. S1A,B). Thus, nuclear transport of SMAD2/3 and TAZ induced by TGF‐β1 in HSCs requires p300.
Figure 2.
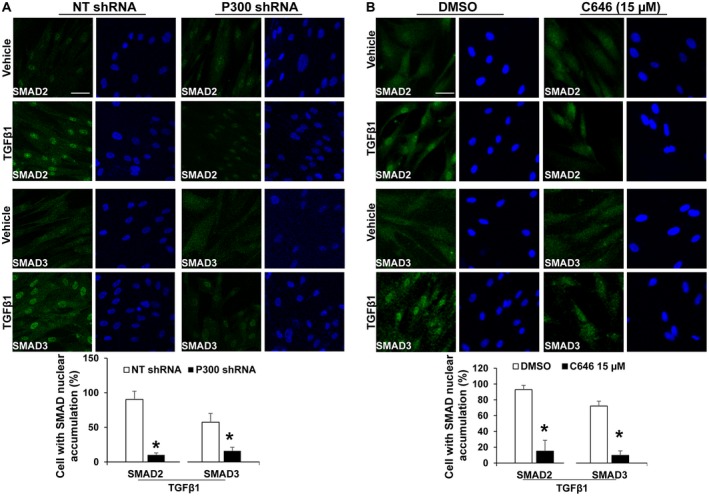
p300 inactivation inhibits SMAD2/3 nuclear accumulation induced by TGF‐β1. IF revealed that TGF‐β1‐induced SMAD2/3 nuclear accumulation was inhibited by p300 knockdown (A) or C646 (B). *, P < 0.05 by t test, n > 100 cells per group. Bar, 50 µm. Abbreviation: NT, nontargeting.
Deleting NLS of p300 Abolishes SMAD2/3 and TAZ Nuclear Transport Induced by TGF‐β1
To understand the mechanisms of SMAD2/3 and TAZ nuclear import by p300, we generated an NLS‐deletion mutant (FLAG‐p300NLS‐Del). A construct encoding wild‐type (wt) p300 (FLAG‐p300wt) was used as a control. Because p300 cDNA was too large to be packaged into a retrovirus or lentivirus, we performed transient plasmid transfection of LX2 cells and identified transfected cells by IF. As revealed by FLAG IF (green, Fig. 3A), p300wt was expressed strongly in the nucleus of LX2 cells and weakly in the cytoplasm. In contrast, p300NLS‐Del mutant was predominantly in the cytoplasm. WB confirmed similar protein levels of full‐length p300wt and p300NLS‐Del proteins generated by plasmid transfection (Fig. 3A). Double IF revealed that in nontransfected cells (asterisks, Fig. 3B), SMAD2/3 were transported into the nucleus by TGF‐β1. Overexpression of FLAG‐p300NLS‐Del mutant blocked TGF‐β1‐induced SMAD2/3 nuclear transport (arrowheads, Fig. 3B; P < 0.05). Interestingly, FLAG‐p300wt led to SMAD2/3 nuclear targeting even under vehicle condition (arrows, Fig. 3B) and promoted SMAD2/3 nuclear accumulation after TGF‐β1 stimulation (P < 0.05). Similarly, FLAG‐p300NLS‐Del mutant colocalized with and retained TAZ in the cytoplasm under both vehicle and TGF‐β1‐stimulated conditions (arrowheads, Fig. 4A), whereas FLAG‐p300wt increased nuclear TAZ in both vehicle and TGF‐β1‐stimulated conditions compared with nontransfected cells (arrows vs. asterisks, Fig. 4A; P < 0.05). Thus, the NLS of p300 is indeed required for SMAD2/3 and TAZ nuclear import.
Figure 3.
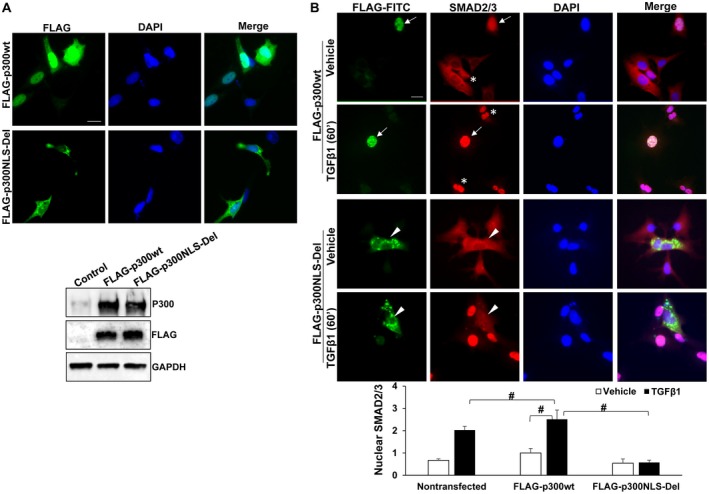
p300NLS‐Del mutant blocks SMAD2/3 nuclear transport induced by TGF‐β1. (A) Upper: LX2 cells transfected with FLAG‐p300wt or FLAG‐p300NLS‐Del were collected for IF for FLAG tag. Lower: Full‐length wild‐type p300 and p300NLS‐Del proteins were detected by WB. (B) LX2 transfected with FLAG‐p300wt or FLAG‐p300NLS‐Del were stimulated with TGF‐β1 and collected for IF. Nuclear SMAD2/3 IF was quantitated by the Image J software. FLAG‐p300wt promoted whereas FLAG‐p300NLS‐Del blocked SMAD2/3 nuclear transport by TGF‐β1. #, P < 0.05 by analysis of variance, n = 5‐8 cells per group. Bars in A and B, 20 µm. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; FITC, Fluorescein isothiocyanate.
Figure 4.
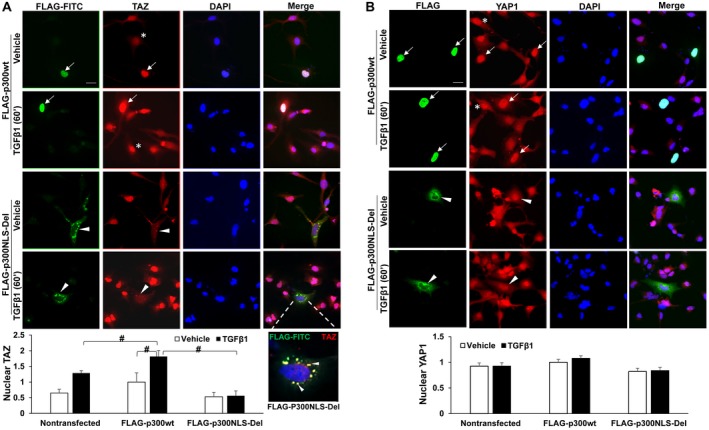
p300NLS‐Del mutant blocks TGF‐β1‐mediated TAZ nuclear transport. (A) LX2 cells treated as described in Fig. 3B were collected for TAZ IF. FLAG‐p300wt promoted whereas FLAG‐p300NLS‐Del mutant inhibited TAZ nuclear transport by TGF‐β1. An enlarged view showing that TAZ and FLAG‐p300NLS‐Del colocalized at cytoplasmic puncta (yellow). #, P < 0.05 by analysis of variance, n = 6‐8 cells per group. (B) LX2 treated as described in Fig. 3B were collected for YAP1 IF. YAP1 nuclear localization was not influenced by TGF‐β1 and slightly affected by p300NLS‐Del mutant. n = 8‐14 cells per group. Bars, 20 µm. Abbreviation: DAPI, 4′,6‐diamidino‐2‐phenylindole; FITC, Fluorescein isothiocyanate.
To confirm that this mechanism is specific for SMAD2/3 and TAZ, we investigated the role of the FLAG‐p300NLS‐Del mutant on YAP1 because YAP1 did not bind to p300 (Fig. 1A). In contrast to SMAD2/3 and TAZ, YAP1 nuclear localization was unaffected by TGF‐β1 or FLAG‐p300wt (Fig. 4B, rows 1 and 2; P > 0.05) and only slightly reduced by the p300NLS‐Del mutant (Fig. 4B, arrowheads). We also obtained NIH3T3 fibroblasts expressing either a FLAG‐TAZ‐4SA or FLAG‐YAP1‐5SA mutant under the control of doxycycline‐inducible Tet‐On system.26 Because of S‐A mutations, these mutants, once imported into the nucleus, could not be phosphorylated and exported out of the nucleus.26 IF and nuclear fractionation (Supporting Figs. S2 and S3) revealed that whereas FLAG‐TAZ‐4SA nuclear localization was significantly inhibited by C646 or p300 knockdown (P < 0.05), FLAG‐YAP1‐5SA nuclear localization was not (P > 0.05). Thus, TAZ and YAP1 enter the nucleus through distinct mechanisms.
Perturbation of p300/SMAD2/3/TAZ Complex Suppresses TGF‐β‐Induced Activation of HSCs into Tumor‐Promoting Myofibroblasts
p300 and TAZ are coactivators of SMAD2/3, so we assessed whether inactivation of p300 or TAZ could disrupt HSC activation by TGF‐β1. Primary murine HSCs, isolated from mice harboring conditional p300 knockout alleles (p300F/F),19 were transduced with adenoviruses encoding lacZ (control) or cre. WB revealed that TGF‐β1‐induced up‐regulation of α‐smooth muscle actin (α‐SMA) and fibronectin, markers of HSC activation, was suppressed by cre‐mediated p300 gene disruption (Fig. 5A; P < 0.05). Next, human primary HSCs expressing p300 shRNA or incubated with C646 were collected for WB and IF. WB showed that TGF‐β1‐induced up‐regulation of α‐SMA and fibronectin was consistently inhibited by three different p300 shRNAs. α‐SMA IF revealed that 48% of control HSCs were differentiated into myofibroblasts by TGF‐β1 whereas only 3% of p300 knockdown HSCs were differentiated (Fig. 5B; P < 0.05). Consistently, TGF‐β1‐stimulated HSC activation was inhibited by C646 (Fig. 5C,D; P < 0.05). Thus, perturbation of p300 in murine and human HSCs inhibited TGF‐β1‐stimulated HSC activation in vitro.
Figure 5.
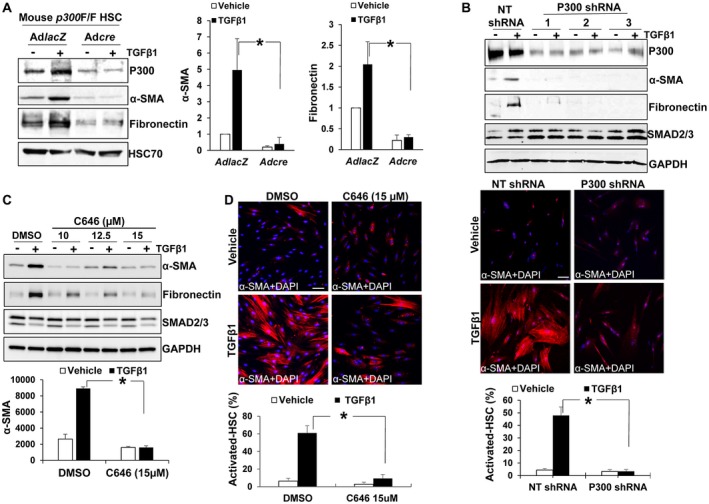
p300 inactivation suppresses TGF‐β1‐mediated myofibroblastic activation of HSCs. (A) HSCs isolated from p300F/F mice were transduced with lacZ or cre adenoviruses. Cells were stimulated with TGF‐β1 for 24 hours and collected for WB. Adcre‐mediated p300 disruption inhibited α‐SMA and fibronectin up‐regulation induced by TGF‐β1. *, P < 0.05 by analysis of variance (ANOVA), n = 3. (B) Upper: HSCs transduced with NT shRNA or p300 shRNA lentiviruses were stimulated with TGF‐β1 and collected for WB. TGF‐β1 up‐regulation of α‐SMA and fibronectin was consistently inhibited by three different p300 shRNAs. Lower: α‐SMA IF (red) showed that p300 knockdown suppressed TGF‐β1‐mediated myofibroblastic activation of HSCs. *, P < 0.05 by ANOVA, n = 5 randomly picked microscopic fields, each containing 50‐150 cells. (C,D) WB and IF showed that C646 suppressed TGF‐β1 activation of HSCs. *, P < 0.05 by ANOVA, n = 3 for WB and n = 5 randomly picked microscopic fields, each containing 50‐150 cells, for IF. Bars in B and D, 50 µm. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; NT, nontargeting.
We next transfected control or TAZ siRNA into HSCs and assessed myofibroblastic activation of HSCs. Suprisingly, TAZ knockdown alone could not influence α‐SMA and fibronectin expression in TGF‐β1‐stimulated HSCs (Supporting Fig. S4A). This led us to speculate that YAP1 may crosstalk with the TGF‐β signaling to compensate for TAZ. Thus, we cotransfected TAZ siRNA and YAP1 siRNA into human primary HSCs, followed by TGF‐β1 stimulation. WB revealed that TAZ/YAP1 knockdown significantly reduced up‐regulation of α‐SMA and fibronectin by TGF‐β1. IF confirmed that more than 50% of control HSCs were differentiated into myofibroblasts by TGF‐β1 whereas only 3% of TAZ/YAP1 knockdown HSCs were differentiated (Fig. 6A; P < 0.05). Thus, disrupting the coactivators of SMAD2/3 inhibits TGF‐β1‐mediated activation of HSCs into myofibroblasts.
Figure 6.
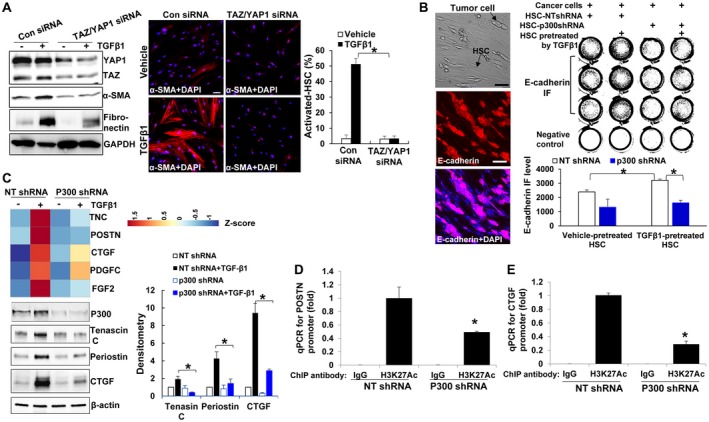
p300 epigenetically facilitates HSC to express tumor‐promoting factors. (A) WB and IF revealed that knockdown of TAZ and YAP1 by siRNA suppressed TGF‐β1‐stimulated HSC activation. *, P < 0.05 by analysis of variance (ANOVA), n = 5 randomly picked microscopic fields, each containing 50‐150 cells. Bar, 50 µm. (B) L3.6 and HSCs were cocultured in serum‐free medium for 3 days and collected for E‐cadherin IF (red). Tumor‐promoting effects of HSCs were abolished by p300 knockdown. *, P < 0.05 by ANOVA, n = 3. Bar, 100 µm. (C) Microarray analysis and WB demonstrated that TGF‐β1 increased transcription of tumor‐promoting factors in control but not in p300 knockdown HSCs. *, P < 0.05 by ANOVA, n = 3. (D,E) ChIP was performed using anti‐ histone H3‐acetyl K27 (H3K27Ac) antibody, and qPCR was done using primers specific for POSTN or CTGF promoter. p300 knockdown reduced H3K27Ac on the promoter of POSTN and CTGF in TGF‐β1‐stimulated HSCs. *, P < 0.05 by ANOVA, n = 3. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; H3K27Ac, acetylation at the lysine residue 27 (K27) of histone 3 protein; IgG, immunoglobulin G; NT, nontargeting.
Myofibroblasts are well documented to promote tumorigenesis. So we assessed the function of the TGF‐β‐p300/SMAD2/3/TAZ signaling of HSCs for tumors using a tumor/HSC coculture model. HSCs seeded onto a cell culture plate were pretreated with or without TGF‐β1 and cells were grown to confluence. L3.6 human pancreatic cancer cells, in a single‐cell suspension, were seeded on the top of HSCs so that L3.6/HSCs were cocultured in serum‐free medium. IF for E‐cadherin, an epithelial cell marker, was performed to identify and quantitate cancer cell proliferation 3 days later (Fig. 6B). As shown in Fig. 6B, TGF‐β1‐pretreated control HSCs promoted L3.6 proliferation compared with vehicle‐pretreated HSCs (P < 0.05), and this tumor‐promoting effect of HSCs was blocked by p300 knockdown (P < 0.05). Thus, TGF‐β1 potentiation of the tumor‐promoting effect of HSCs can be inhibited by knockdown of p300.
p300 Acetylates Histones to Facilitate HSC to Express Tumor‐Promoting Factors
To better understand how p300/SMAD2/3/TAZ influence the tumor‐promoting effect of HSCs, we collected control and p300 knockdown HSCs, with or without TGF‐β1 stimulation, for microarray analysis, and data were deposited to Gene Expression Omnibus (GSE116509). Using 1.5‐fold increase as a cutoff, more than 700 genes were identified as TGF‐β inducible genes (GSE116509, 22 representative genes are listed in Supporting Fig. S5A). Importantly, TGF‐β1 promoted transcripts of genes encoding tumor‐promoting factors, (Genes: TNC, POSTN, CTGF, PDGFC, and FGF2),27, 28, 29, 30, 31 and this effect of TGF‐β1 was abolished by p300 knockdown (Fig. 6C). WB confirmed that TGF‐β1 increased protein levels of tenascin C, periostin, and CTGF through p300 (Fig. 6C). Consistently, up‐regulation of these tumor‐promoting factors by TGF‐β1 was abrogated by silencing both TAZ and YAP1 by siRNA (Supporting Fig. S4B). To assess whether these proteins are responsible for the tumor‐promoting effects of HSCs, HSC conditioned media were collected and used to stimulate HT29 human colorectal cancer cell proliferation in vitro.32 As revealed by MTS proliferation assay, the tumor‐promoting effect of HSCs was reduced by p300 knockdown and the impaired tumor‐promoting effect of p300 knockdown HSCs was partially rescued by recombinant CTGF (Supporting Fig. S5B). Thus, p300 potentiates tumor‐promoting effects of HSCs by regulating expression of paracrine factors, including CTGF.
Canonically, p300 modulates gene transcription by an acetylation‐dependent mechanism. We next used ChIP‐qPCR19 to test if p300 facilitated transcription of POSTN or CTGF by acetylating histone 3 at lysine 27 associated with their gene promoters. As revealed by ChIP‐qPCR, acetylation levels of H3K27 on the promoter of POSTN or CTGF after TGF‐β1 stimulation were indeed lower in p300 knockdown HSCs than in control cells (Fig. 6D,E; P < 0.05). Consistent data were obtained with HSCs treated with C646 (Supporting Fig. S5C). Thus, in addition to noncanonical transport of SMAD2/3 and TAZ into the nucleus, p300 also acetylates histones and remodels chromatins to facilitate TGF‐β1‐mediated gene transcription.
Targeting p300 in HSCs Suppresses Tumor Growth in Mice
We next tested if inhibiting p300 in HSCs could inhibit tumor growth in mice. To this end, we mixed HT29 with HSCs and coinjected them into nude mice subcutaneously.23, 25 Seven days later, 28 tumor nodules resulting from HT29/HSC coinjection were divided symmetrically into two groups; one group received intratumoral injection of C646 at 20 µM and another received dimethyl sulfoxide (DMSO). Twenty micromole of C646 was used because C646 at this dose did not influence HT29 proliferation (Supporting Fig. S6A). Tumor size measurement revealed that C646 significantly reduced tumor growth in mice compared with DMSO (Fig. 7A; P < 0.05). IF and WB showed that compared with DMSO, C646 reduced myofibroblast densities and protein levels of α‐SMA, CTGF, tenascin C, and periostin of the tumors, which are TGF‐β/SMAD transcriptional targets (Fig. 7B,C; Supporting Fig. S7A; P < 0.05). Consistent with our in vitro model, targeting p300 by C646 reduced nuclear SMAD2/3 and TAZ of activated‐HSC/myofibroblasts of the tumors (Supporting Fig. S6B‐D; P < 0.05). Thus, C646 inhibits tumor growth in a preclinical mouse model.
Figure 7.
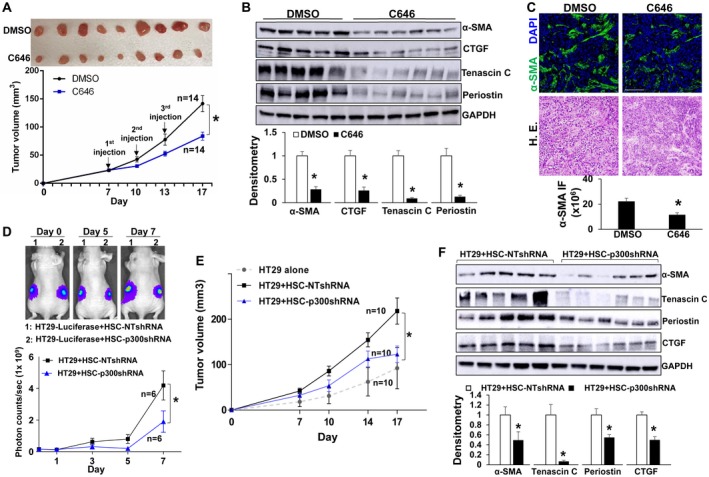
p300 inactivation in activated‐HSC/myofibroblasts suppresses tumor growth in mice. (A) HT29 cells were mixed with HSCs and coinjected into nude mice subcutaneously. Seven days later, 28 size‐comparable tumor nodules were divided into two groups for control and C646 treatment. Tumor nodules at the endpoint are shown. C646 reduced HT29 tumor growth in mice. *, P < 0.05 by analysis of variance (ANOVA), n = 14 per group. (B) C646 reduced protein levels of α‐SMA, CTGF, tenascin C, and periostin of HT29 tumors. *, P < 0.05 by t test, n = 5, 6. (C) C646 reduced myofibroblast densities of tumors. *, P < 0.05 by t test, n = 4 per group. Bar, 100 µm. (D) HT29 tagged by firefly luciferase were mixed with HSCs and coinjected into nude mice subcutaneously. HT29 bioluminescence was measured by Xenogen live imaging. p300 knockdown HSCs had a reduced effect on HT29 implantation compared with control HSCs. *, P < 0.05 by ANOVA, n = 6. (E) Tumor nodules were measured by a caliper. p300 knockdown HSCs were less effective than control HSCs at promoting HT29 growth in mice. *, P < 0.05 by ANOVA, n = 10. (F) Tumors arising from HT29/HSC‐p300shRNA coinjections contained reduced α‐SMA, tenascin C, periostin, and CTGF compared with tumors arising from control coinjections. *, P < 0.05 by t test, n = 5, 6. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HE, hematoxylin and eosin; NT, nontargeting.
To confirm that C646 inhibited tumor growth through the effect on HSCs, we next performed coinjection of HT29 cells with either control or p300 knockdown HSCs. HT29 cells were tagged by firefly luciferase before injection, and Xenogen live imaging of HT29 bioluminescence demonstrated that p300 knockdown HSCs were less effective than control HSCs at promoting HT29 implantation in mice at days 5 and 7 after coinjection (Fig. 7D; P < 0.05). Tumor size measurement showed that p300 knockdown HSCs were also less effective than control HSCs at promoting HT29 growth in mice (Fig. 7E; P < 0.05). In addition, protein levels of α‐SMA, CTGF, tenascin C, periostin, and myofibroblast densities in tumors arising from HT29/HSC‐p300shRNA coinjections were significantly lower compared with tumors arising from control coinjections (Fig. 7F; Supporting Fig. S7B; P < 0.05). Thus, targeting HSC p300 selectively inhibits HSC activation and tumor growth in mice.
Nuclear SMAD2/3 and TAZ in Activated‐HSC/Myofibroblasts of Murine Liver Metastases are Down‐Regulated by cre‐Mediated p300 Gene Disruption
To translate our findings to liver metastases, we used a murine model of liver metastasis where L3.6 pancreatic cancer cells were injected into the portal vein of SCID mice (Fig. 8A). Liver metastases were harvested and assessed for SMAD2/3 and TAZ localization. Nuclear SMAD2/3 and TAZ were indeed more increased in activated‐HSC/myofibroblasts of liver metastases (arrows) than in quiescent HSCs (arrowheads; Fig. 8B,C; Supporting Fig. S8A; P < 0.05), similar to p300, which we showed previously.19 To assess whether p300 mediated SMAD2/3 and TAZ nuclear translocation in HSCs in this model, we used HSC/myofibroblast‐specific p300 knockout mice (p300F/Fcre) developed by crossing p300F/F mice with collagen1A1‐cre transgenic mice. We showed that MC38 colorectal cancer cells formed fewer liver metastases in p300F/Fcre mice compared with control mice after portal vein injection.19 In this study, we injected LLCs into the spleen of mice (Fig. 8D) because this intrasplenic tumor injection model mirrored many clinical conditions of patients with metastatic liver disease. Similar to MC38 cells, LLCs formed fewer liver metastases in p300F/Fcre mice than in p300+/+cre mice, and p300F/Fcre liver metastases contained reduced protein levels of α‐SMA, periostin, and CTGF (Fig. 8D,E; P < 0.05). Additionally, IF revealed that nuclear SMAD2/3 and TAZ were significantly lower in p300F/Fcre myofibroblasts (arrows) than in p300+/+cre myofibrolasts (arrowheads; Fig. 8F; Supporting Fig. S8B; P < 0.05). Thus, p300 is indeed responsible for nuclear accumulation of SMAD2/3 and TAZ in the activated‐HSC/myofibroblasts of liver metastases.
Figure 8.
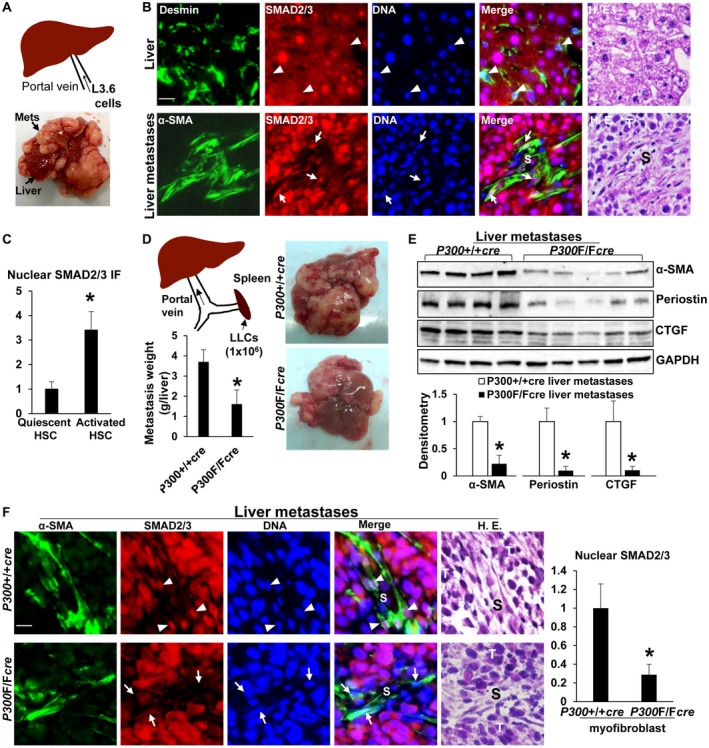
Nuclear SMAD2/3 in activated‐HSC/myofibroblasts of liver metastases are down‐regulated by cre‐mediated p300 gene disruption. (A) Illustration of portal vein injection of L3.6 cells. (B,C) SMAD2/3 were assessed in liver and isolated liver metastases using IF simultaneously with antibodies against Desmin (marker for quiescent HSCs) or α‐SMA (marker for activated HSCs). Nuclear SMAD2/3 were more increased in activated‐HSC/myofibroblasts (arrows) than in HSCs (arrowheads). *, P < 0.05 by t test, n > 26 cells per group. (D) Illustration of intrasplenic injection of LLCs. LLCs formed fewer liver metastases in p300F/Fcre mice than in p300+/+cre mice. *, P < 0.05 by t test, n = 5, 5. (E) Isolated liver metastases were lysed for WB. p300F/Fcre liver metastases contained reduced α‐SMA, periostin, and CTGF protein levels. *, P < 0.05 by t test, n = 4, 5. (F) Nuclear SMAD2/3 were reduced in p300F/Fcre myofibroblasts than in p300+/+cre myofibroblasts. *, P < 0.05 by t test, n > 40 cells per group. Bars in B and F, 20 µm. Abbreviations: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HE, hematoxylin and eosin; S, stroma; T, tumor.
Tumor‐Promoting Factors Induced by TGF‐β1‐p300/SMAD2/3/TAZ Signaling are Distinct From those Induced by Stiffness‐p300 Mechanosignaling
We have reported that a stiff substrate targeted p300 to the nucleus by activating a RhoA‐Akt mechanosignaling and led to epigenetic modifications that promoted HSC activation.19 RNA‐seq (GSE101343) revealed that the stiffness‐p300 signaling increased transcription of a panel of tumor‐promoting factors, including chemokine (C‐X‐C motif) ligand 12 (CXCL12). Because p300 is a common mediator for both TGF‐β1‐mediated and stiffness‐mediated HSC activation, we performed RNA‐seq for TGF‐β1‐stimulated HSCs and compared two gene expression data sets (GSE101343 and GSE127964). Surprisingly, among 23 tumor‐promoting targets of stiffness identified by RNA‐seq,19 only interleukin (IL) 11, SERPINE1, THBS1, FGF2, and CTGF were enhanced by TGF‐β1 (Supporting Fig. S9A). Conversely, among TNC, POSTN, CTGF, FGF2, and PDGFC, five TGF‐β1 targets identified by this study, only CTGF and FGF2 were turned on by stiffness‐p300 mechanosignaling (Supporting Fig. S9B). Thus, the distinct gene targets induced by TGF‐β1 or stiffness‐mediated HSC activation suggest that stiffness may induce yet unidentified transcription factors, in addition to SMAD2/3, to promote HSC activation.
Discussion
This study revealed that p300 plays a dual role in HSC differentiation into myofibroblasts in response to TGF‐β1. First, p300 acts as a cytoplasm‐to‐nucleus shuttle to transport SMAD2/3 and TAZ into the nucleus of HSCs. This reveals a function for p300 in mediating nuclear import of proteins that lack an NLS. Second, p300 acetylates histones and remodels chromatins to facilitate expression of TGF‐β1 targets. Moreover, inactivation of p300 suppresses myofibroblastic activation of HSCs and tumor‐promoting effects of HSCs in vitro and in a tumor implantation mouse model. Thus, p300 plays both noncanonical (cytoplasm‐to‐nucleus shuttle) and canonical (histone acetylation) roles during TGF‐β1‐mediated activation of HSCs into tumor‐promoting myofibroblasts.
Tumors are stiffer than normal tissues because of excessive extracellular matrix deposition by myofibroblasts. Our prior study demonstrated that tumor stromal stiffness targeted p300 to the nucleus of HSCs where p300 epigenetically turned on transcription of a panel of tumor‐promoting factors, including CXCL12, IL11, IL6, PDGFA and B, and vascular endothelial growth factor A, and activated HSCs into tumor‐promoting myofibroblasts.19 Thus, p300 represents a common mediator for both TGF‐β‐mediated and stiffness‐mediated HSC activation. Because of this, we compared two RNA‐seq data sets (GSE127964 and GSE101343) and interestingly found that the tumor‐promoting factors induced by TGF‐β1 and stiffness were relatively distinct but contained a very small overlap that included CTGF, a known transcriptional target of TAZ. This finding suggests that in the setting of stiffness‐induced HSC activation, unidentified transcription factors, in addition to SMAD2/3, are activated by p300‐mediated acetylation or p300‐driven nuclear transport. Together, these data highlight p300 as an attractive target for suppressing HSC activation, liver fibrosis, and the prometastatic liver microenvironment.
The notable finding of this study was the identification of p300 as a protein scaffold and cytoplasm‐to‐nucleus shuttle for SMAD2/3 and TAZ. Interestingly, C646, an inhibitor targeting p300 acetyltransferase activity, also inhibited TGF‐β1‐mediated nuclear transport of SMAD2/3 and TAZ (Figs. 1E, 2B). These data suggest that both the protein scaffolding function and acetyltransferase activity of p300 are required for nuclear transport of SMAD2/3 and TAZ. Indeed, both SMAD2/3 and importin α have been reported as targets of acetylation.15, 33 Thus, p300‐mediated acetylation is not unique to proteins involved in gene transcription, such as transcription factors, coactivators, and histones. In addition to gene transcription, p300‐mediated acetylation also participates in numerous biological processes, including protein nuclear import. Identification of additional acetylation targets of p300 may reveal further roles for p300 in regulating protein function as well as gene transcription.
YAP1/TAZ transcriptional coactivators play important roles in organ fibrogenesis, including lung and liver fibrosis.11, 12, 13, 26 How YAP1 and TAZ enter the nucleus is intriguing because neither contains an NLS motif.9 Here we demonstrated that in HSCs and NIH3T3 mouse embryonic fibroblasts, nuclear transport of TAZ was dependent on p300 whereas nuclear transport of YAP1 was not, supporting distinct mechanisms by which TAZ and YAP1 entered the nucleus of fibroblasts. Stiffness targets both p300 and TAZ to the nucleus, so TAZ nuclear transport induced by stiffness may be also facilitated by p300. Although the current study focuses on TAZ rather than YAP1, the role of YAP1 in HSC biology should not be overlooked. YAP1 is in fact a critical proliferative factor for HSCs in a murine liver injury model,34 and it modulates the myocontractility of activated‐HSC/myofibroblasts at the downstream of β1‐integrin signaling.12 Hedgehog‐YAP1 signaling promotes glutaminolysis to metabolically reprogram activated‐HSC/myofibroblasts.13 Additionally, targeting YAP1 led to suppression of HSC activation and liver fibrosis in mice.11, 12 These data support that YAP1 participates in profibrotic signaling pathways, other than TGF‐β/SMAD signaling, to modulate HSC activation and liver fibrosis, and further studies are needed to understand the mechanisms of YAP1 nuclear import.
In conclusion, p300 promotes TGF‐β‐stimulated HSC activation by both noncanonical and canonical mechanisms; it acts as a cytoplasm‐to‐nucleus shuttle for SMAD2/3 and TAZ, and it also epigenetically modifies histones to support transcription of TGF‐β targets. These data highlight p300 as a target for suppressing HSC activation and the prometastatic liver microenvironment. Because a desmoplastic tumor stroma contributes to not only metastatic growth but also impaired drug delivery and resistance to chemotherapy and radiation therapy, targeting HSC p300 in combination with conventional cancer therapies may improve the clinical outcome of patients with metastatic liver disease.
Supporting information
Acknowledgment
We thank Elyse Froehling, Aaron Becker, and Juan Abrahante, Ph.D., at the University of Minnesota Genomic Center for performing microarray, RNA‐seq, and data analysis; Dr. Makiko Fujii at Aichi Cancer Center Research Institute, Japan, for providing p300 plasmid; and Dr. Tatiana Kisseleva at UCSD for collagen1A1‐cre transgenic mouse line.
Supported by NIH grants R01 CA160069 to N.K., R01 AA021171 to V.H.S., and K01 DK112915 to J.L.M. C.D., Q.L., and Z.L. are funded by China Scholarship Council. D.L. and Y.C. are funded by their hospitals in P. R. China.
Microarray data: Gene Expression Omnibus GSE116509. RNA sequencing data: Gene Expression Omnibus GSE127964.
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Kang N, Gores GJ, Shah VH. Hepatic stellate cells: partners in crime for liver metastases? Hepatology 2011;54:707‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shi Y, Massague J. Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell 2003;113:685‐700. [DOI] [PubMed] [Google Scholar]
- 3. Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH. Classical nuclear localization signals: definition, function, and interaction with importin alpha. J Biol Chem 2007;282:5101‐5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xiao Z, Liu X, Lodish HF. Importin beta mediates nuclear translocation of Smad 3. J Biol Chem 2000;275:23425‐23428. [DOI] [PubMed] [Google Scholar]
- 5. Xiao Z, Liu X, Henis YI, Lodish HF. A distinct nuclear localization signal in the N terminus of Smad 3 determines its ligand‐induced nuclear translocation. Proc Natl Acad Sci U S A 2000;97:7853‐7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xu L, Alarcon C, Col S, Massague J. Distinct domain utilization by Smad3 and Smad4 for nucleoporin interaction and nuclear import. J Biol Chem 2003;278:42569‐42577. [DOI] [PubMed] [Google Scholar]
- 7. Yao X, Chen X, Cottonham C, Xu L. Preferential utilization of Imp7/8 in nuclear import of Smads. J Biol Chem 2008;283:22867‐22874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev 2008;22:1962‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 2014;94:1287‐1312. [DOI] [PubMed] [Google Scholar]
- 10. Zhao B, Li L, Lei Q, Guan KL. The Hippo‐YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 2010;24:862‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF, et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol 2015;63:679‐688. [DOI] [PubMed] [Google Scholar]
- 12. Martin K, Pritchett J, Llewellyn J, Mullan AF, Athwal VS, Dobie R, et al. PAK proteins and YAP‐1 signalling downstream of integrin beta‐1 in myofibroblasts promote liver fibrosis. Nat Commun 2016;7:12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska‐Syn M, et al. Hedgehog‐YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology 2018;154:1465‐1479.e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Varelas X, Sakuma R, Samavarchi‐Tehrani P, Peerani R, Rao BM, Dembowy J, et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem‐cell self‐renewal. Nat Cell Biol 2008;10:837‐848. [DOI] [PubMed] [Google Scholar]
- 15. Inoue Y, Itoh Y, Abe K, Okamoto T, Daitoku H, Fukamizu A, et al. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene 2007;26:500‐508. [DOI] [PubMed] [Google Scholar]
- 16. Janknecht R, Wells NJ, Hunter T. TGF‐beta‐stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes Dev 1998;12:2114‐2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen X, Hu PP, Liberati NT, Datto MB, Frederick JP, Wang XF. TGF‐beta‐induced phosphorylation of Smad3 regulates its interaction with coactivator p300/CREB‐binding protein. Mol Biol Cell 1998;9:3309‐3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhattacharyya S, Ghosh AK, Pannu J, Mori Y, Takagawa S, Chen G, et al. Fibroblast expression of the coactivator p300 governs the intensity of profibrotic response to transforming growth factor beta. Arthritis Rheum 2005;52:1248‐1258. [DOI] [PubMed] [Google Scholar]
- 19. Dou C, Liu Z, Tu K, Zhang H, Chen C, Yaqoob U, et al. p300 acetyltransferase mediates stiffness‐induced activation of hepatic stellate cells into tumor‐promoting myofibroblasts. Gastroenterology 2018;154:2209‐2221.e2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiang X, Wang Y, Zhang H, Piao J, Muthusamy S, Wang L, et al. Vasodilator‐stimulated phosphoprotein promotes liver metastasis of gastrointestinal cancer by activating a β1‐integrin‐FAK‐YAP1/TAZ signaling pathway. NPJ Precis Oncol 2018;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fujii M, Toyoda T, Nakanishi H, Yatabe Y, Sato A, Matsudaira Y, et al. TGF‐beta synergizes with defects in the Hippo pathway to stimulate human malignant mesothelioma growth. J Exp Med 2012;209:479‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kang N, Yaqoob U, Geng Z, Bloch K, Liu C, Gomez T, et al. Focal adhesion assembly in myofibroblasts fosters a microenvironment that promotes tumor growth. Am J Pathol 2010;177:1888‐1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu C, Billadeau DD, Abdelhakim H, Leof E, Kaibuchi K, Bernabeu C, et al. IQGAP1 suppresses TbetaRII‐mediated myofibroblastic activation and metastatic growth in liver. J Clin Invest 2013;123:1138‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Decker NK, Abdelmoneim SS, Yaqoob U, Hendrickson H, Hormes J, Bentley M, et al. Nitric oxide regulates tumor cell cross‐talk with stromal cells in the tumor microenvironment of the liver. Am J Pathol 2008;173:1002‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tu K, Li J, Verma VK, Liu C, Billadeau DD, Lamprecht G, et al. VASP promotes TGF‐β activation of hepatic stellate cells by regulating Rab11 dependent plasma membrane targeting of TGF‐β receptors. Hepatology 2015;61:361‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu F, Lagares D, Choi KM, Stopfer L, Marinkovic A, Vrbanac V, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol 2015;308:L344‐L357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoshida T, Akatsuka T, Imanaka‐Yoshida K. Tenascin‐C and integrins in cancer. Cell Adh Migr 2015;9:96‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gonzalez‐Gonzalez L, Alonso J. Periostin: a matricellular protein with multiple functions in cancer development and progression. Front Oncol 2018;8:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim H, Son S, Shin I. Role of the CCN protein family in cancer. BMB Rep 2018;51:486‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anderberg C, Li H, Fredriksson L, Andrae J, Betsholtz C, Li X, et al. Paracrine signaling by platelet‐derived growth factor‐CC promotes tumor growth by recruitment of cancer‐associated fibroblasts. Cancer Res 2009;69:369‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maehara O, Suda G, Natsuizaka M, Ohnishi S, Komatsu Y, Sato F, et al. Fibroblast growth factor‐2‐mediated FGFR/Erk signaling supports maintenance of cancer stem‐like cells in esophageal squamous cell carcinoma. Carcinogenesis 2017;38:1073‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu C, Li J, Xiang X, Guo L, Tu K, Liu Q, et al. PDGF receptor alpha promotes TGF‐beta signaling in hepatic stellate cells via transcriptional and post transcriptional regulation of TGF‐beta receptors. Am J Physiol Gastrointest Liver Physiol 2014;307:G749‐G759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bannister AJ, Miska EA, Gorlich D, Kouzarides T. Acetylation of importin‐alpha nuclear import factors by CBP/p300. Curr Biol 2000;10:467‐470. [DOI] [PubMed] [Google Scholar]
- 34. Konishi T, Schuster RM, Lentsch AB. Proliferation of hepatic stellate cells, mediated by YAP and TAZ, contributes to liver repair and regeneration after liver ischemia‐reperfusion injury. Am J Physiol Gastrointest Liver Physiol 2018;314:G471‐G482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials