

Summary
A hallmark of neurodegenerative disease is focal onset of pathological protein aggregation, followed by progressive spread of pathology to connected brain regions. In amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), pathology is often associated with aggregation of TAR DNA Binding protein 43 (TDP-43). Although aggregated TDP-43 protein moves between cells, it is not clear whether and how this movement propagates the degeneration. Here we have established a Drosophila model of human TDP-43 in which we initiated toxic expression of human TDP-43 focally within small groups of glial cells. We found that this focal onset kills adjacent neurons. Surprisingly, we show that this spreading death is caused by an endogenous retrovirus within the glia, which leads to DNA damage and death in adjacent neurons. These findings suggest a possible mechanism by which human retroviruses such as HERV-K might contribute to TDP-43 mediated propagation of neurodegeneration.
In Brief
Chang and Dubnau show that initiation of toxic levels of TDP-43 within small groups of glial cells causes death of adjacent neurons. This propagating neuronal death is triggered by RTE/ERV activated within the glia. Silencing of the ERV in glia is sufficient to rescue nearby neurons, suggesting that the ERV contributes to propagation of toxicity.
Graphical Abstract
Introduction
One feature that is shared across a diversity of neurodegenerative diseases (NDs) is that onset typically involves localized accumulation of pathological protein aggregation followed by progressive spread, of both the protein aggregation pathology and the cellular degenerative effects [1–4]. For example, cytoplasmic aggregation of TDP-43, a normally nuclear RNA and DNA binding protein, is a central pathological hallmark in a suite of disorders, including ALS and FTD [5–7]. TDP-43, and many other aggregation prone RNA binding proteins, contain low complexity domains (LCDs) that undergo phase transitions to form liquid droplets under conditions of cellular stress. Over time, residence in this state is thought to drive fibrilization [7]. For many LCD proteins, including TDP-43, the tendency to undergo such phase separation can be increased by disease causing amino acid substitutions, but also can occur spontaneously in wild type protein when the concentration is increased [8–16]. Importantly, such conditions may occur in diseased tissue because nuclear TDP-43 protein normally exerts a feedback inhibition that keeps TDP-43 levels in check. Thus, the initiation of cytoplasmic inclusions causes clearance from the nuclear compartment, thereby disrupting feedback inhibition of TDP-43 expression [17].
Most animal models of NDs rely on transgenic expression to mimic this effect [18]. Although over-expression based approaches do not fully recapitulate the conditions seen in sporadic ALS (sALS), such expression can drive aggregation of TDP-43 protein in cytoplasmic inclusions, leading also to loss of nuclear localization. TDP-43 pathology in animal models is now understood to cause global alterations in mRNA stability and splicing, biogenesis of some microRNAs (miRNAs), de-repression of retrotransposons (RTEs) and endogenous retroviruses (ERVs), defects in nucleo-cytoplasmic transport, and activation of DNA damage response pathways [5, 7, 14, 15, 19–22]. Most of these phenomena are observed not only in animal models, but also in post mortem tissues from human subjects [21, 23–27].
The cellular toxicity that results from TDP-43 aggregation pathology is thought to initiate a cascade of downstream toxic effects that not only occur within the initiating cell, but also can be spread between vulnerable cell types and to other regions of the nervous system [1–4, 28, 29]. This transmission through the tissue may involve prion-like properties of TDP-43, in which pathologically misfolded protein templates the propagation of aggregation pathology [1, 2, 30, 31]. A series of studies also support the idea that TDP-43 proteinopathy causes glial cells to become toxic to neurons. This is seen in a rat transgenic model in which TDP-43 expression is restricted to astrocytes [28] and also is seen in primary astrocyte-motor neuron co-culture experiments from sALS [29]. But there are as yet no in vivo models to investigate the mechanisms mediating cell-to-cell spread of toxicity, or even to establish whether focal onset of pathological TDP-43 can cause propagation of toxic effects to adjacent cells leading to their demise. We previously demonstrated that in Drosophila, either pan-glial or pan-neuronal human TDP-43 (hTDP-43) over expression disrupts endogenous genome surveillance systems that normally stifle expression of RTEs and ERVs [32]. The resulting expression of the gypsy ERV contributes causally to the toxicity of glial expression of hTDP-43 because blocking gypsy expression was sufficient to extend lifespan of the animals expressing transgenic hTDP-43 in glia [32]. These data are parsimonious with the observation that HERV-K in particular [24, 33] and RTEs/ERVs in general are expressed in post mortem brain tissues from ALS patients [23, 25, 26]. HERV-K also is demonstrated to be neurotoxic in culture and leads to MND like phenotypes in a rodent model [24]. More generally, a growing body of evidence has implicated RTEs/ERVs as potentially important actors in neurons and glia [19, 34, 35].
Here, we used a Drosophila model to initiate toxic expression of hTDP-43 either within all glia or focally within small groups of different glial subtypes in the adult brain. Such focal expression in several glial sub-types, including astrocyte-like glia, is sufficient to cause both cell autonomous toxicity leading to glial cell death, and non-autonomous toxicity that kills adjacent neurons. We further demonstrate that both autonomous and non-autonomous effects involve a cascade of events that include reactivation of gypsy RTE/ERV, DNA damage and cell death. Expression of gypsy RTE/ERV and DNA damage signaling within glia are required to produce the secreted toxic factor(s) that ultimately causes DNA damage and death in adjacent neurons. Our data therefore provides a mechanism linking the TDP-43 proteinopathy to propagation of NDs.
Results
Glial Expression of hTDP-43 Leads to Gypsy ERV Replication, DNA Damage and Apoptosis
We have previously demonstrated elevated levels of a panel of RTEs/ERVs, including gypsy, in response to TDP-43 proteinopathy [32]. To investigate the mechanisms by which gypsy ERV contributes to TDP-43 toxicity in glia, we first monitored the replication of a transgenic copy of this ERV in response to pan-glial expression in the Drosophila model. To accomplish this, we developed Cellular Labeling of Endogenous Retrovirus Replication of gypsy (gypsy-CLEVR in Figure 1A; see also [36]), a novel approach to mark cells in which the gypsy ERV has replicated through an RNA intermediate to cDNA and been integrated back into host’s genome. The gypsy-CLEVR construct commandeers the gypsy ERV’s own replication machinery to fuse a Gal4 responsive UAS enhancer element to a dual fluorescent reporter, Watermelon (WM, a membrane myr-GFP and nuclear H2B-mCherry fusion gene with P2A self-cleaving peptide). While the replication of gypsy-CLEVR is independent of Gal4, the expression of the reporter only occurs in the presence of the Gal4 protein.
Figure 1. Gypsy ERV Replication and DNA Damage Are Seen in Glia after hTDP-43 Induction.

(A) Schematic of the gypsy-CLEVR replication-based reporter. Gypsy-CLEVR carries a Gal4-responsive UAS regulatory element with no reporter in the U5 domain of the 5’-LTR and the dual-color WM reporter without a promoter in the U3 domain of 3’-LTR. Retrovirus replication creates hybrid LTRs after re-integration. This process places the WM dual color reporter under the UAS element control within each of the two LTRs. After reintegrating into the host genome, the rearranged WM reporter and UAS element within gypsy-CLEVR will response to Gal4 activity.
(B) Temporal control of hTDP-43 induction in specific cell types via temperature-shift to disrupt the Gal80ts repressor function. A temperature shift after development releases Gal4 to induce hTDP-43 under the UAS control.
(C) Post induction of glial hTDP-43 using the repo-Gal4 yields toxicity that shortens lifespan. Survival curves are shown for control animals with nls-LacZ and the CPV reporter (repots>nls-LacZ+CPV) or hTDP-43 with the CPV reporter (repots>hTDP-43+CPV). All animals were shifted to 29°C immediately after eclosion. The median survival post induction was 23 days in repots>nls-LacZ+CPV (n=78) and 7 days in repots>hTDP-43+CPV (n=81). ****p<0.0001 (Log-rank test) and ####p<0.0001 (Gehan-Breslow-Wilcoxon test).
(D) Gypsy-CLEVR replication over a time-course (Day 2 - Day 5) after induction of nls-LacZ control (repots>nls-LacZ+gypsy-CLEVR) or hTDP-43 (repots>hTDP-43+gypsy-CLEVR). Glial nuclei were labeled with the Repo antibody (White) and gypsy-CLEVR positive nuclei are revealed with the H2B-mCherry (Red) from the WM reporter in the gypsy-CLEVR construct. Scale bar=20 μm. See also Figure S1 and S2A.
(E) Total gypsy-CLEVR positive nuclei (mean ± SEM) from (D). The mean numbers of CLEVR-labeled nuclei in repots>nls-LacZ+gypsy-CLEVR were 20.4±4.9 (Day 2, n=8), 58.2±7.7 (Day 3, n=9), 63.0±8.9 (Day 4, n=10) and 102.2±11.1 (Day 5, n=9). Total numbers in repots>hTDP-43+gypsy-CLEVR were 170.9±16.9 (Day 2, n=10), 324.8±22.9 (Day 3, n=10), 634.3±38.8 (Day 4, n=8) and 801.8±35.2 (Day 5, n=12). *p<0.05, **p<0.01, ****p<0.0001, n.s., no significant difference (Unpaired t-test). ###p<0.001, ####p<0.0001 (Two-way ANOVA).
(F) Total glial number from (D) shown for control and hTDP-43 groups (mean ± SEM). The mean numbers in repots>nls-LacZ+gypsy-CLEVR were 1154±28.0 (Day 2, n=8), 1198±36.9 (Day 3, n=9), 1124±28.0 (Day 4, n=10) and 1181±31.3 (Day 5, n=9). Total numbers in repots>hTDP-43+gypsy-CLEVR were 1165±30.6 (Day 2, n=10), 1137±29.4 (Day 3, n=10), 463.5±66.7 (Day 4, n=8) and 25.4±6.5 (Day 5, n=12). ****p<0.0001, n.s., no significant difference (Unpaired t-test). ####p<0.0001, no significant difference (Two-way ANOVA).
(G) Increased numbers of DNA damage foci (detected with γH2AV antibody) were seen over a time-course (Day 3 - Day 5) after induction of glial hTDP-43 (repots>hTDP-43+gypsy-CLEVR), which co-localized with gypsy-CLEVR positive glial nuclei. Such DNA damage induction was not observed within control (repots>nls-LacZ+gypsy-CLEVR) brains. Glial cell nuclei were independently labeled with Repo antibody (Green), gypsy-CLEVR positive nuclei are revealed by H2B-mCherry (Red) and DNA damage was marked with γH2AV antibody (White). Scale bar=10 μm. See also Figure S2B.
(H) Total numbers of γH2AV positive nuclei from (G) comparing control (nls-LacZ) and hTDP-43 groups are shown (mean ± SEM). The average numbers in repots>nls-LacZ+gypsy-CLEVR were 0.3±0.7 (Day 3, n=9), 0.4±0.8 (Day 4, n=8) and 0.8±1.0 (Day 5, n=10). Total numbers in repots>hTDP-43+gypsy-CLEVR were 8.7±5.5 (Day 3, n=7), 70.0±10.9 (Day 4, n=10) and 170.4±28.5 (Day 5, n=12). *p<0.05, ***p<0.001, ****p<0.0001, n.s., no significant difference (Unpaired t-test). #p<0.05, ####p<0.0001, no significant difference (Two-way ANOVA).
We compared the number of glial cells expressing the of gypsy-CLEVR replication reporter expression in glial cells in the presence of either hTDP-43 [32, 37] or a nls-LacZ control transgene. Glia in nls-LacZ control (repo>nls-LacZ+gypsy-CLEVR) animals exhibited a modest age-dependent increase in the numbers of glia showing gypsy ERV replication, with a relatively small fraction of glia in the central brain labeled with the H2B-mCherry nuclear reporter at 2 or 5–7 days post eclosion (Figure S1, see also [36]). In the presence of pan-glial hTDP43 expression (repo>hTDP-43+gypsy-CLEVR), by contrast, we observed a striking increase in the number of H2B-mCherry labeled glia either at 2 or 5–7 days post eclosion (Figure S1). This gypsy-CLEVR reporter expression was reduced in glia when we used an RNAi transgene (gypsy-IR) targeting gypsy transcripts to disrupt gypsy ERV expression [32, 36, 38], consistent with the conclusion that the appearance of the H2B-mCherry label was dependent upon prior gypsy expression (Figure S1, see also [36]). A small decrease in total number of glia was detected in these panglial hTDP-43 expressing animals (repo>hTDP-43) at day 2, consistent with the possibility that constitutive expression of hTDP-43 throughout development might impact glial differentiation or survival (Figure S4B). To prevent glial loss during development from impacting results, we repeated the experiments using a temporal and regional gene expressing targeting (TARGET) inducible system that relies on a temperature sensitive Gal80 repressor (tub-Gal80ts) [39].
When incubated at the permissive temperature (21°C) during development, the Gal80ts transgene blocks the Gal4-mediated expression of hTDP-43 within glial cells by silencing repo-Gal4 activity (here after repo-Gal4 combined with tub-Gal80ts is abbreviated as repots in the text and figures). By shifting the animals to the non-permissive temperature after development is complete (Figure 1B, see also [39]), the hTDP-43 transgene becomes expressed in target cells within adult animals. We used this approach to keep the nls-LacZ control or hTDP-43 transgenes off during development, and then switched the transgenes on after eclosion (Figure 1B). When the hTDP-43 transgene (repots>hTDP-43+CPV) was induced post development in glial cells using repots, it resulted in robust toxicity associated with dramatically shortened lifespan compared to the nls-LacZ control (repots>nls-LacZ+CPV) group (Figure 1C). Importantly, this TARGET inducible system provides the potential to examine the kinetics of downstream molecular effects and cellular events in a population of glial cells that are at the same ‘stage’ in their response to pathological accumulation of hTDP-43.
To explore the mechanisms of hTDP-43 toxicity in glia, we examined the molecular events occurring at defined time windows after hTDP-43 induction and before the animals began to die. We first used this strategy to examine the kinetics of gypsy ERV replication using the gypsy-CLEVR reporter in response to glial induction of either hTDP-43 or nls-LacZ as a control. In parallel, the glial nuclear marker Repo was used to monitor changes in glial cell number. Consistent with the observations from constitutive nls-LacZ expression described above (Figure S1), there is a gradual age-dependent increase (from Day 2–5 post induction) in numbers of gypsy-CLEVR labeled glia with post-development induction of nls-LacZ (repots>nls-LacZ+gypsy-CLEVR) (Figure 1D, 1E and S2A). The age dependent increase of gypsy ERV expression and replication is also consistent with our prior reports for neural tissue [32, 36, 40] and similar studies of other tissues [41–44]. In contrast with this gradual and modest increase in number of gypsy-CLEVR labeled glia with induced expression of nls-LacZ, we detected a dramatic increase in numbers of glia labeled with this same reporter in response to induction of hTDP-43 (Figure 1D, 1E and S2A). By 2–3 days after induction of hTDP43 in glia, the majority of Repo+ glial cell nuclei also were labeled by the H2B-mCherry signal from the gypsy-CLEVR reporter. We noted, however, that this increase in number of H2B-mCherry labeled glia over time after induction of hTDP-43 was correlated with a loss in the total number of glial nuclei labeled with the nuclear marker, Repo (Day 4–5) (Figure 1D, 1F and S2A). This dramatic reduction in the number of glial cells was not observed in control flies that expressed nls-LacZ (Figure 1D, 1F and S2A). Together, the above findings demonstrate that the toxicity of hTDP-43 expression in glia unleashes replication of the gypsy ERV and leads to loss of glial cells.
Expression of RTEs and ERVs have myriad negative effects on cell homeostasis, including from toxicity of the RTE/ERV encoded proteins, RNAs, and extrachromosomal cDNAs, from activation of inflammatory responses, and from the impact of DNA damage caused by attempts at re-insertion of replicated copies [20, 34, 45–47]. Loss of nuclear TDP-43 protein and accumulation of pathological inclusions in cytoplasm also has been associated with DNA damage signaling [21, 32, 48]. We investigated whether hTDP-43 toxicity in fly glia caused DNA damage, and whether this correlated with gypsy ERV replication on a cell-by-cell level. To visualize DNA double strand break (DSB), we used the γH2AV (mammalian yH2AX equivalent) antibody, which detects a histone variant that is recruited to sites of DNA damage [49]. In order to determine if any DNA damage foci were present within glia after hTDP-43 induction, we made use of repots TARGET inducible system to inducibly drive hTDP-43. We also monitored gypsy replication via the nuclear H2B-mCherry readout of the gypsy-CLEVR reporter. The glial nuclear protein, Repo and γH2AV DNA damage markers also were independently labeled in order to detect glial cells with DNA damage. In brains of control (repots>nls-LacZ+gypsy-CLEVR) animals, we did not observe any γH2AV labeling during a time-course of 5 days after nls-LacZ induction, and only a small number of H2B-mCherry labeled (gypsy-CLEVR) glia were observed (Figure 1G–H and S2B). In contrast, we observed a progressive increase in γH2AV signal between Day 3 and 5 after induction of the hTDP-43 transgene (repots>hTDP-43+gypsy-CLEVR) (Figure 1G–H and S2B). Remarkably, these γH2AV positive glia were almost always labeled with nuclear H2B-mCherry from the gypsy-CLEVR reporter (yellow arrow in Figure 1G). We also noted that between 4 and 5 days post induction of hTDP-43, some of gypsy-CLEVR positive glial nuclei exhibited morphological changes that were reminiscent of late stages of apoptotic cell death (Day4 and Day5 in Figure S2C). Activation of Caspase-3, a key executive Caspase in the apoptotic program, was also detected within these gypsy-CLEVR positive apoptotic cells using an antibody against the cleaved (activated) form of Caspase-3 (Figure S2C). Taken together, the above findings demonstrate that expression of hTDP-43 within glia leads to expression and replication of the gypsy ERV (Figure 1, S1 and S2, see also [32]), which is correlated at a cell-by-cell level with accumulation of DNA damage, activation of Caspase-3, and loss of glial marker detection.
The accumulation of cleaved Caspase-3 is consistent with an active apoptotic signaling cascade. To confirm this finding and to test whether Caspase-3 is functionally active, we next used a Gal4-driven genetic reporter, UAS-CD8::PARP::Venus (UAS-CPV), of Caspase-3 functional activity [50]. This Caspase-3 reporter makes use of CD8 to tether a Venus fluorescent protein to the membrane. A Caspase-3 cleavage target from PARP is positioned between the CD8 membrane tether and the Venus reporter. This CPV reporter reveals functional Caspase-3 activity in two ways (Figure S3A). First, Caspase-3-mediated cleavage uncovers an epitope in the PARP motif that can be detected with a cleaved PARP (cPARP) antibody. Second, the Caspase-3 mediated cleavage of the CPV reporter releases the C-terminal Venus fluorescent protein from the membrane into the cytosol (Figure S3A). We used the repots TARGET system to express the CPV transgene so that we could monitor Caspase-3 activity, along with expression of either hTDP-43 or nls-LacZ as a control. To independently mark nuclei from glia and neurons, we co-labeled with antibodies against the Repo and Elav nuclear proteins that mark glia and neurons respectively. In order to facilitate quantification, we analyzed signals in a defined region of the dorsal protocerebrum across all brains (yellow rectangle in Figure 2A, see also STAR METHODS). In the control (repots>nls-LacZ+CPV) animals, we observed very limited signal from either of the two outputs of the CPV reporter over a time-course between Days 2–5 after induction of nls-LacZ (Figure 2A–D). We also quantified the total number of glia and neurons, by counting the numbers of Repo and Elav labeled nuclei, and found no significant change in total glial or neuronal numbers over this time period in brains from the control animals (Figure 2A–B and 2E–F). We next examined the effects on the CPV reporter of induction of hTDP-43 (repots>hTDP-43+CPV) and found a sharp increase in Caspase-3 functional activation between days 2 and 3 after induction (Figure 2A–D). Notably, there was no detectable signal for either of the CPV reporter outputs (PARP antibody and cytosolic Venus) at 2 days post induction of hTDP-43 (Figure 2A–D), and no change in total glial and neuronal cell counts were detected at this time point (Figure 2A–B and 2E–F). This result highlights the effectiveness of the repots TARGET approach to efficiently prohibit glial loss during development. In contrast, by 3 days after hTDP-43 induction, both the cPARP and Venus reporters of Caspase-3 activity dramatically increased (Figure 2A–D). This increase in the Caspase-3 functional reporter in glia was followed by a marked loss of glial number at day 4 to day 5 (Figure 2A–B and 2E). It is worth noting that although the hTDP-43 transgene was exclusively expressed in glia, there was a significant reduction in total neuron number beginning at Day 4, which is after the wave of Caspase-3 activity observed in glia at Day 3 (Figure 2A–B and 2F). Such neuronal loss was typically seen within large vacuole-like areas with no labeled nuclei, but with faint residual Venus signal (Figure 2B). Importantly, this non-cell autonomous neuronal loss was seen throughout the adult brain (Figure S3B) and was not restricted to the specific region where cell number was quantified (Figure 2). Given the widespread glial death, the observed neuronal loss could derive either from deficit in homeostatic roles of the glia and/or from release of toxic factors from the glia (see below). Taken together, the above findings demonstrate that pan glial expression of hTDP-43 leads to replication of the gypsy ERV within a subset of glial cells. Accumulation of DNA damage is also is seen within many glia that express hTDP-43. And finally, such glial hTDP-43 toxicity results in widespread apoptotic signaling in glia, and a concomitant loss of neighboring neurons. Importantly, these molecular and cellular events initiate prior to decline in the animals survival curve (Figure 1C) and prior to decrement in locomotor activity [32].
Figure 2. Induction of hTDP-43 in Glia Activates Caspase-3 and Causes Glial and Neuronal Cell Loss.

(A) CPV reporter was used to trace Caspase-3 activity after induction of nls-LacZ (repots>nls-LacZ+CPV) vs hTDP-43 (repots>hTDP-43+CPV) in glia over a time-course post induction (Day 2 – Day 5). The glia and neurons were independently labeled with cell type specific markers, Repo (Red) and Elav (Blue), and the Venus and cPARP reporters from CPV are shown with Green and Cyan. Cells located within the region highlighted with a dashed yellow rectangle were quantified. The total numbers of each marker are shown in (C-F). Scale bar=20 μm. See also Figure S3.
(B) Higher magnification from (A). Scale bar=10 μm.
(C) Total numbers of cPARP positive cells from repots>nls-LacZ+CPV and repots>hTDP-43+CPV were quantified. cPARP positive numbers from each time-point are shown (mean ± SEM). Average cPARP numbers in repots>nls-LacZ+CPV were 0.1±0.1 (Day 2, n=10), 0.9±0.4 (Day 3, n=12), 1.3±0.3 (Day 4, n=13) and 1.3±0.4 (Day 5, n=7). cPARP numbers in repots>hTDP-43+CPV were 3.8±0.7 (Day 2, n=12), 24.6±1.3 (Day 3, n=11), 7.6±1.1 (Day 4, n=12) and 5.4±0.9 (Day 5, n=7). ****p<0.0001, n.s., no significant difference (One-way ANOVA). #p<0.05, ####p<0.0001, n.s., no significant difference (Two-way ANOVA).
(D) Total numbers of Venus positive cells from repots>nls-LacZ+CPV and repots>hTDP-43+CPV. Venus were quantified (mean ± SEM) for each time-point. Average numbers of Venus positive cells in repots>nls-LacZ+CPV were 0.1±0.1 (Day 2, n=10), 0.9±0.4 (Day 3, n=12), 1.3±0.3 (Day 4, n=13) and 1.3±0.4 (Day 5, n=7). Venus numbers in repots>hTDP-43+CPV were 3.8±0.7 (Day 2, n=12), 24.6±1.3 (Day 3, n=11), 7.6±1.1 (Day 4, n=12) and 5.4±0.9 (Day 5, n=7). ****p<0.0001, n.s., no significant difference (One-way ANOVA). #p<0.05, ####p<0.0001, n.s., no significant difference (Two-way ANOVA).
(E) Total numbers of glia labeled with the pan-glial marker, Repo are shown from repots>nls-LacZ+CPV and repots>hTDP-43+CPV at different time points. Average numbers of Repo-labeled glia numbers are shown (mean ± SEM). Glial numbers in repots>nls-LacZ+CPV were 37.0±1.2 (Day 2, n=10), 39.5±1.3 (Day 3, n=12), 43.3±2.1 (Day 4, n=13) and 37.9±1.8 (Day 5, n=7). Glial numbers in repots>hTDP-43+CPV were 40.0±1.5 (Day 2, n=12), 38.8±2.7 (Day 3, n=11), 9.4±2.8 (Day 4, n=12) and 2.9±1.3 (Day 5, n=7). ****p<0.0001, n.s., no significant difference (One-way ANOVA). ####p<0.0001, n.s., no significant difference (Two-way ANOVA).
(F) Total numbers of neurons (mean ± SEM) labeled with the pan-neuronal marker, Elav, are shown from repots>nls-LacZ+CPV and repots>hTDP-43+CPV during over a time-course. Total neuronal numbers in repots>nls-LacZ+CPV were 384.8±8.1 (Day 2, n=10), 413.9±16.5 (Day 3, n=12), 396.1±21.4 (Day 4, n=13) and 359.9±16.6 (Day 5, n=7). Neuronal numbers in repots>hTDP-43+CPV were 379.4±8.0 (Day 2, n=12), 389.4±5.7 (Day 3, n=11), 274.0±12.9 (Day 4, n=12) and 241.6±22.2 (Day 5, n=7). ****p<0.0001, n.s., no significant difference (One-way ANOVA). ###p<0.001, ####p<0.0001, n.s., no significant difference (Two-way ANOVA).
Glial hTDP-43 Pathology Triggers Gypsy-ERV Replication and Leads to DNA Damage and Apoptotic Cell Death in Nearby Neurons
In the above experiments, the CPV reporter of Caspase-3 activity only provided the means to assign Caspase-3 signaling to the glial cells in which the reporter and hTDP-43 were expressed. In order to assay the Caspase-3 signaling with cell-by-cell resolution in both neurons and glia, we used a well-established active Caspase-3 antibody, which probes activation of endogenous Caspase-3. This nuclear accumulation of cleaved Caspase-3 also allowed for meaningful colabeling with the nuclear H2B-mCherry protein in the gypsy-CLEVR replication reporter and with glial (Repo) and neuronal (Elav) markers.
We first used the activated Caspase-3 antibody with the gypsy-CLEVR reporter to label brains of nls-LacZ control (repots>nls-LacZ+gypsy-CLEVR) flies versus those expressing hTDP-43 (repots>hTDP-43+gypsy-CLEVR). In addition to the H2B-mCherry reporter of gypsy replication and the activated Caspase-3 antibody, we also used antisera against the Repo and Elav proteins that marked glial and neuronal nuclei. Consistent with findings reported above (Figure 1, S1 and S2) there were very few H2B-mCherry labeled glial nuclei in control brains (Figure 3). Labeling with the activated Caspase-3 antibody was similarly extremely sparse in adult brains from these control animals (Figure 3A–C and S3C–D). In contrast, activated Caspase-3 was detected within many nuclei in brains expressing hTDP-43 in glia (repots>hTDP-43+gypsy-CLVER) starting at day 3 (red arrow in Figure 3D). Strikingly, the active Caspase-3 was almost always detected within the same subset of glial nuclei that exhibited positive signal for the gypsy-CLEVR replication reporter (Figure 3D). Thus, as with DNA damage, the Caspase-3 label co-occurred in the same subset of glia in which our ERV reporter cassette was replicating. We also noted that some neurons also exhibited activated Caspase-3 label, and these labeled neurons were almost always adjacent to Caspase-3 marked glia that also expressed the reporter of gypsy-CLEVR (blue arrow in Figure 3D). By days 4–5 post induction of hTDP-43, there were a greater number of neurons labeled with active Caspase-3 antibody (blue arrow in Figure 3E–F and S3C). The above findings indicate that after induction of hTDP-43 in glia, progressively larger numbers of neurons appear to have activated apoptotic signaling, and such neurons are closely associated with glial cells which themselves exhibit evidence of gypsy ERV replication. And when the highly stable Venus reporter from CPV was used in glia, the areas with large numbers of lost neurons typically were nearby to residual Venus from apoptotic glia (red star in Figure S3D). These remaining neurons within the Venus vacuoles were almost invariably labeled with active Caspase-3 (Figure S3D). Here too, this phenomenon was not observed in (repots>nls-LacZ+CPV) control brains, although rare sporadic Caspase-3 labeled cells were seen (Figure S3D). A similar phenomenon in which pan-glial hTDP-43 caused apoptotic signaling in neighboring neurons was also observed when hTDP-43 was continuingly expressed in glia throughout developmental rather than induced post development (Figure S4A–D).
Figure 3. Glial Induction of hTDP-43 Causes DNA Damage and Activated Caspase-3 within Neurons.

(A-C) Gypsy-CLEVR replication and activated Caspase-3 are shown in repots>nls-LacZ+gypsy-CLEVR during a time course after nls-LacZ induction: (Day 3(A), Day 4 (B) and Day 5 (C)). The glia and neurons were independently labeled with their nuclear markers, Repo (Green) and Elav (Blue). The nuclear H2B-mCherry (Red) and membrane myr-GFP (Green) from gypsy-CLEVR and activated Caspase-3 (White) are also shown. Scale bar=5μm.
(D-F) Gypsy-CLEVR replication and activated Caspase-3 are shown in repots>hTDP-43+gypsy-CLEVR during a time-course after hTDP-43 induction: Day 3(D), Day 4 (E) and Day 5 (F). The glia and neurons were independently labeled with their nuclear markers, Repo (Green) and Elav (Blue). The nuclear H2B-mCherry (Red) and membrane myr-GFP (Green) from gypsy-CLEVR and activated Caspase-3 (White) are also shown. Scale bar=5μm.
(G-I) Gypsy-CLEVR replication and DNA damage marker, γH2AV, are shown in repots>nls-LacZ+gypsy-CLEVR during a time course after induction of nls-LacZ: (Day 3(G), Day 4 (H) and Day 5 (I)). The glia and neurons were independently labeled with their nuclear markers, Repo (Green) and Elav (Blue). The nuclear H2B-mCherry (Red) and membrane myr-GFP (Green) from gypsy-CLEVR and γH2AV (White) are also shown. Scale bar=5μm.
(J-L) Gypsy-CLEVR replication and DNA damage marker, γH2AV, are shown in repots>hTDP-43+gypsy-CLEVR during a time-course after induction of hTDP-43: Day 3(J), Day 4 (K) and Day 5 (L). The glia and neurons were independently labeled with their nuclear markers, Repo (Green) and Elav (Blue). The nuclear H2B-mCherry (Red) and membrane myr-GFP (Green) from gypsy-CLEVR and γH2AV (White) are also shown. Scale bar=5μm.
See also Figures S3 and S4.
Thus, glial hTDP-43 toxicity results in gypsy ERV replication in a subset of glia (Figure 1, 3 and S1–3), and these gypsy ERV active glia are associated with markers of DNA damage (Figure 1G and S2B) and of apoptotic signaling (Figure 2, 3D–F and S2C). Because Caspase-3 signaling also is initiated within neurons nearby to gypsy ERV active glia, we wondered whether these nearby neurons also would exhibit DNA damage. To address this, γH2AV antibody was used to detect DSB while simultaneously labeling with the nuclear H2B-mCherry reporter from the gypsy-CLEVR in order to reveal which glial cells exhibited gypsy ERV replication. In brains from control (repots>nls-LacZ+gypsy-CLEVR) animals, no γH2AV positive signal was detected in either glia or neurons during the time course of observation (Day 3–5) (Figure 3G–I). In contrast, in brains where hTDP-43 was induced in glia (repots>hTDP-43+gypsy-CLEVR), we observed progressive increases in γH2AV signal over a several day period (days 3–5) after induction (Figure 3J–L). When DNA damage foci were detected in glial nuclei, they were almost always co-localized with the H2B-mCherry marker of gypsy-CLEVR reporter (red arrow in Figure 3J). Glia in which gypsy ERV replication was undetected almost never exhibited evidence of DNA damage (green arrow in Figure 3J). Remarkably, we also detected DNA damage foci, labeled with γH2AV (blue arrow in Figure 3K–L), in some neurons that were adjacent to glial cells in which the gypsy ERV had replicated (red arrow in Figure 3K–L). Taken together, these findings demonstrate that glial hTDP-43 induces DNA damage to the subset of glia in which the gypsy ERV is induced to replicate, but also causes DNA damage to neurons that are nearby to the subset of glia in which gypsy ERV replicates.
Glial hTDP-43 Toxicity to Neurons Is Mediated by the Gypsy ERV and by DNA Damage Signaling
We have previously shown that expression of the gypsy ERV contributes causally to the toxicity of hTDP-43 at the organismal level [32]. Given the potential of viral elements to cause DNA damage and to spread from cell-to-cell, we wondered whether the gypsy ERV activation in glia might contribute causally to the observed apoptotic cell death both within glial cells and their neighboring neurons. To address this question, we used a dsRNA transgene (UAS-gypsy-IR), which is sufficient to knock down gypsy expression by approximately 50% [32, 38]. As a control, we used a dsRNA transgene targeting GFP (UAS-GFP-IR). When hTDP-43 was co-expressed in glia along with the control GFP-IR (repots>hTDP-43+GFP-IR), active Caspase-3 was detected in glia and neighboring neurons at Day 3 (Figure 4A and 4I–J; see also Figure 3D–F and S3D). And by Day 4, many glia and neurons were lost (Figure 4D and 4G–H). When the gypsy-IR was used to knock down gypsy levels within hTDP-43 expressing glia (repots>hTDP-43+gypsy-IR), we saw significantly fewer Caspase-3 labeled glia and also fewer Caspase-3 labeled neurons at Day 3 after hTDP-43 induction (Figure 4B and 4I–J). Notably, the loss of neurons and glial cells by Day 4 also was reduced in the presence of the gypsy-IR transgene (Figure 4E and 4G–H). This supports the conclusion that the expression of the gypsy-ERV in glia contributes to both the cell autonomous toxicity of hTDP-43 in glia and to the non-cell autonomous effects of glial hTDP-43 proteinopathy on neuronal survival.
Figure 4. Knock-Down of Gypsy-ERV or DNA Damage Signaling in Glia that Express Toxic hTDP-43 Rescues both Autonomous Glial and Non-autonomous Neuronal Cell Death.

(A-F) Activated Caspase-3 (White) is detected inside glial nuclei (Red) and neuronal nuclei (Blue) when toxic levels of hTDP-43 are induced in glia along with a control RNAi, GFP-IR (repots>hTDP-43+GFP-IR) (A,D). In contrast, knock down of gypsy-ERV (repots>hTDP-43+gypsy-IR) (B,E) or chk2/loki (repots>hTDP-43+loki-IR) (C,F) are sufficient to greatly reduce the numbers of glia and neurons exhibiting activated caspase-3 at Day 3 (A-C) and Day 4 (D-E) post induction. Scale bar=10μm.
(G) Quantification of glial numbers from (A-F), mean ± SEM. Average numbers of Repo-labeled glial cells were 32.1±1.0 (Day 3, n=8) and 8.0±1.1 (Day 4, n=11) in repots>hTDP-43+GFP-IR, 37.1±1.0 (Day 3, n=7) and 36.9±1.6 (Day 4, n=9) in repots>hTDP-43+gypsy-IR, and 38.4±0.9 (Day 3, n=14) and 37.4±1.1 (Day 4, n=10) in repots>hTDP-43+loki-IR. ****p<0.0001, n.s., no significant difference (Unpaired t-test). ##p<0.01, ####p<0.0001, n.s., no significant difference (Two-way ANOVA).
(H) Total neuronal numbers were from (A-F) (mean ± SEM). Average neuronal numbers were 388.5±8.8 (Day 3, n=8) and 292.8±10.0 (Day 4, n=11) in repots>hTDP-43+GFP-IR, 401.7±8.0 (Day 3, n=7) and 381.3±11.4 (Day 4, n=9) in repots>hTDP-43+gypsy-IR, and 389.1±6.9 (Day 3, n=14) and 372.3±9.4 (Day 4, n=10) in repots>hTDP-43+loki-IR. ****p<0.0001, n.s., no significant difference (Unpaired t-test). ####p<0.0001, n.s., no significant difference (Two-way ANOVA).
(I) Total number of Repo-labeled glia that also were Caspase-3 positive from (A-F) (mean ± SEM). Average numbers were 17.8±1.1 (Day 3, n=8) and 7.1±0.8 (Day 4, n=11) in repots>hTDP-43+GFP-IR, 7.9±0.7 (Day 3, n=7) and 7.8±0.9 (Day 4, n=9) in repots>hTDP-43+gypsy-IR, and 9.4±1.0 (Day 3, n=14) and 8.4±0.7 (Day 4, n=10) in repots>hTDP-43+loki-IR. ****p<0.0001, n.s., no significant difference (Unpaired t-test). ####p<0.0001, n.s., no significant difference (Two-way ANOVA).
(J) Total number of Elav-labeled neurons that also were Caspase-3 positive from were (A-F) and (mean ± SEM). Average numbers were 14.3±1.0 (Day 3, n=8) and 26.9±1.7 (Day 4, n=11) in repots>hTDP-43+GFP-IR, 5.9±1.3 (Day 3, n=7) and 7.3±1.5 (Day 4, n=9) in repots>hTDP-43+gypsy-IR, and 7.5±1.2 (Day 3, n=14) and 10.1±0.9 (Day 4, n=10) in repots>hTDP-43+loki-IR. ****p<0.0001, n.s., no significant difference (Unpaired t-test). ##p<0.01, ####p<0.0001 (Two-way ANOVA).
See also Figure S4.
As shown above, we observe physical co-localization of gypsy ERV replication and DNA damage within the same glial nuclei (Figure 1 and 3). DNA damage signaling and RTE/ERV replication in fact share an intimate relationship, with the potential for DNA damage to induce RTE/ERV replication and with RTEs/ERV replication having the potential to generate DNA breaks to complete their integration [34, 45, 47, 51]. One of the most striking observations is that knock down of loki, the Drosophila ortholog of checkpoint kinase 2 (chk2), is sufficient to greatly reduce the toxicity caused by RTEs in germline tissue [52]. In the absence of Chk2/loki, DSBs repair is left intact, but signaling to the machinery for DNA damage mediated apoptosis is disrupted.
We recently demonstrated that loki/chk2 knock down in fly brain is also sufficient to dramatically reduce the impact on lifespan of the animals when hTDP-43 is expressed in either neurons or glial cells [32]. We used the same approach to knock down loki/chk2 in glial cells in which hTDP-43 also was expressed, and assayed the impact on Caspase-3 activation in neurons and glia. We found that knocking down loki/chk2 within glial cells was sufficient to reduce the number of glial cells labeled with Caspase-3 at both 3 and 4 days post induction of hTDP-43 (repots>hTDP-43+loki-IR), and also reduced the number of neighboring neurons that were labeled with Caspase-3 (Figure 4C, 4F and 4I–J). Notably, loki/chk2 knock down also prevented the loss of the glial cell marker, Repo, consistent with the conclusion that apoptotic cell death was blocked (Figure 4C and 4F–G). Thus when hTDP-43 is expressed in glia, DNA damage is detected within a subset of glial cells in which the gypsy-ERV replicates, and in nearby neurons. And blocking either the expression of the gypsy ERV or of DNA damage mediated signaling to apoptosis are each sufficient to reduce the number of Caspase-3 labeled glial cells and the number of Caspase-3 labeled neurons nearby. Similar rescue effects were also observed when silencing gypsy ERV expression or DNA damage signaling in glia expressing hTDP-43 through development (Figure S4E–H).
Focal Expression of hTDP-43 in Small Subsets of Glia Kills nearby Neurons via the Gypsy-ERV
The non-cell autonomous effects on neurons from pan-glial induction of TDP-43 protein could result either from release of a toxic factor or from loss of important homeostatic functions that glia normally provide for neuronal survival [53, 54]. To distinguish between these possibilities, we used two different strategies to create conditions in which hTDP-43 pathology was initiated focally in just a small group of glial cells. We then assayed the impacts on activation of Caspase-3 in those glial cells and in adjacent neurons.
The first strategy, mosaic clone analysis [55], was accomplished using FLP-specific recombinase to delete a Gal80 cassette flanked by FRT recombination sites, resulting in activation of Gal4-mediated expression of hTDP-43 in those glial cells along with a dual fluorescent reporter (UAS-WM, see also [36]) to label the nuclei (H2B-mCherry) and the membranes (myr-GFP) of the individual glia in which recombination took place (Figure 5A, see STAR METHODS). By inducing the recombinase briefly and late in development, we were able to generate only a few small mosaic “clones” per brain. We were able to detect single clones with focal hTDP-43 expression that belonged to each of the known major glial subtypes in adult Drosophila central brain [53, 56], which we identified based on morphology and location (Figure 5B–D). Perineurial glia (PNG) and subperineurial glia (SPG) form a contiguous structure that together wraps the surface of the adult central brain. These glial cells are thought to play a role similar to the mammalian blood-brain barrier (BBB) [53, 56]. Cortex glia (CG) have their cell bodies interspersed with those of most of the neurons within the superficial layer under the BBB. The CG have honey-comb like membranous structures that contact with most of the neuronal cell bodies. Astrocyte-like glia (ALG) exhibit a morphology and marker gene expression reminiscent of that of mammalian astrocytes, and they are located in neuropil regions nearby to synaptic connections. And finally, ensheathing glia (EG) form a barrier between neuropil regions [53, 56].
Figure 5. Focal Induction of Toxic Levels of hTDP-43 in Several Glial Sub-types Causes Apoptotic Cell Death in Adjacent Neurons.

(A) Schematic the dual label WM reporter to simultaneously mark glial nuclei (with H2B-mCherry) and glial membranes (with myr-GFP). This allowed identification of glial subtypes by morphology and location. Blue indicated neurons and grey represented unmarked glial cells.
(B) Co-induction of hTDP-43 and the dual WM reporter was stochastically initiated in glial cells via a transient induction of the hs-FLP recombinase and a FRT recombination sites flanked “FLP-Off-Gal80” cassette. The FLP-Off Gal80 was used in combination with the glial expressing repo-Gal4, along with UAS-driven hTDP-43 and WM. The hTDP-43 expressing clones were visualized with the nuclear H2B-mCherry (Red) and membrane myr-GFP (Green) from the WM reporter. The dual WM reporter allowed identification of the glial subtype for each clone. Apoptotic cell death signaling was monitored via an activated Caspase-3 antibody (White) and each neuronal nucleus was labeled with antibodies against the Elav marker (Blue). Perineurial glia (PNG), Subperineurial glia (SPG), Astrocyte-like glia (ALG), Cortex glia (CG) and Ensheathing glia (EG) were separately detected by this method. Scale bar=10μm. (See also STAR METHODS for detailed genotypes). We detect Caspase-3 labeled neurons adjacent to the majority of PNG, SPG and ALG clones expressing hTDP-43. This is not seen with clonal induction of pro-apoptotic genes (See Figure S5A).
(C) Gypsy-ERV expression levels were knocked down within hTDP-43 expressing glial clones via co-expression of the gypsy-IR. The WM dual reporter was used as in (A), to mark hTDP-43 expressing glia. All neuronal nuclei were labeled with an antibody against the Elav marker (Blue) and cell death signaling is revealed with an antibody against activated Caspase-3 (White). Knocking down gypsy-ERV expression significantly reduced the numbers of Caspase-3 labeled neurons (E). Perineurial glia (PNG), Subperineurial glia (SPG) and Astrocyte-like glia (ALG) were shown. Scale bar=10μm. (See also STAR METHODS for detail genotype).
(D) Chk2/Loki expression levels were knocked down within hTDP-43 expressing glial clones via co-expression of the Loki-IR. The WM dual reporter was used as in (A), to mark hTDP-43 expressing glia. All neuronal nuclei were labeled with an antibody against the Elav marker (Blue) and cell death signaling is revealed with an antibody against activated Caspase-3 (White). Knocking down Chk2/Loki expression significantly reduced the numbers of Caspase-3 labeled neurons (E). Perineurial glia (PNG), Subperineurial glia (SPG) and Astrocyte-like glia (ALG) were shown. Scale bar=10μm. (See also STAR METHODS for detail genotype).
(E) Quantification of the fraction of glial clones of each glial cell type for which nearby neurons are labeled with activated Caspase-3 from (B-D). Cases where we could not unambiguously distinguish PNG from SPG were grouped.
(F) Effects of post development induction of hTDP-43 in SPGs was examined on Caspase-3 signaling in nearby neurons. The hTDP-43 or nls-LacZ (control) were co-induced with WM in SPG glia using the moodyts TARGET method. Activated cell death signaling was visualized with an antibody against Caspase-3 (White). SPG nuclei were labeled with H2B-mCherry (Red) and membranes with myr-GFP (Green) from the WM reporter. All neuronal nuclei were labeled with an antibody against the Elav marker (Blue). In addition to Caspase-3 activation in nearby neurons, we also observed Caspase-3 label in PNG nuclei (White arrowhead), which lie just superficial to the SPG. Caspase-3 label was not observed in neurons with expression of the nls-LacZ control (moodyts>nls-LacZ+WM). In contrast, expression of hTDP-43 (moodyts>hTDP-43+WM) in SPG was associated with induction of activated Caspase-3 signaling in adjacent neurons. Direct activation cell death via induction of the pro-apoptotic gene rpr (moodyts>rpr+WM) within SPG did not cause cell death signaling within adjacent neurons. Scale bar=20μm. See also Figure S5.
All together, we analyzed Caspase-3 signal surrounding 95 clones of glia expressing hTDP-43 (Figure 5B and 5E). In these experiments, all neuronal nuclei were also labeled with Elav marker. In the majority of cases where hTDP-43 was expressed in small clusters of PNG, SPG or ALG, there were surrounding neurons displaying activated Caspase-3. Thus TDP-43 pathology in these three glial cell types appeared capable of inducing apoptotic signaling in adjacent neurons (Figure 5B and 5E). This effect was seen for 11/11 PNG clones, for 15/28 SPG clones, for 24/24 cases where SPG and PNG could not be unambiguously distinguished, and for 9/18 ALG clones. In contrast, we did not detect Caspase-3 label in neighboring neurons for the cases where hTDP-43 was expressed in CG or EG (Figure 5B and 5E), although the number of CG clones that we were able to generate was quite small (N=3). These results strongly support the conclusion that the induction of focal hTDP-43 toxicity in PNG, SPG or ALG can trigger nonautonomous effects on nearby neurons, leading to activation of apoptotic signaling. Because the induction of hTDP-43 was in most cases restricted to just a single or a few glial cells, this finding also strongly argues against the possibility that the effects on neurons are an indirect result of loss of homeostatic support functions of glia. To further rule this possibility out, we also generated clones of glia in which the pro-apoptotic head involution defective (hid) cDNA was expressed instead of hTDP-43 via the same mosaic clonal analysis approach. Expression of hid is sufficient to cause apoptotic cell death. Although we were not able to generate surviving hid expressing ALG clones, we were able to analyze 10 cases where hid expression had been induced in PNG or SPG glia (Figure S5A). In these cases, we were able to detect Caspase-3 within the (H2B-mCherry labeled) nuclei of the glial clones, and membrane architectures of such glia were often disrupted indicating that the hid expression was sufficient to induce apoptosis as expected. But we saw no cases where Caspase-3 activation could be detected in surrounding neurons (Figure S5A). Thus, the effects on surrounding neurons observed with focal induction of hTDP-43 are not seen when glial apoptotic death is induced by another means.
In the context of pan-glial hTDP-43 induction, effects on surrounding neurons could be ameliorated by knock down of gypsy ERV expression levels within the glia, or by knockdown of the loki/chk2 DNA damage signaling molecule. To test whether this also was the case in the context of focal onset of hTDP-43 toxicity, we generated glial clones by mosaic analysis in which focal expression of hTDP-43 was co-induced with the gypsy-IR or the chk2/loki-IR transgenes. Here too, we found that the impact of glial hTDP-43 on apoptotic signaling in nearby neurons was dependent on expression within the glial cells of gypsy ERV and on DNA damage response signaling to the apoptotic machinery. With gypsy-IR and with chk2/loki-IR, the percentage of PNG, SPG and ALG clones with detectable Caspase-3 activation in adjacent neurons was significantly reduced or eliminated (Figure 5C–E). These findings highlight the role of the gypsy ERV and of DNA damage signaling within glia in transmitting a toxic effect from hTDP-43 expressing glia to surrounding neurons.
As a second strategy to test the idea that gypsy-ERV expression and DNA damage in glia contribute to spreading death, we used the SPG specific moody-Gal4 driver [57] in combination with tub-Gal8ts (here after moody-Gal4+tub-Gal80ts is abbreviated as moodyts in text and figures) to induce hTDP-43 expression only in this glial subtype, after development was complete. The SPG glia form a surface structure that surrounds the cortical layer in which most of the neuronal cell bodies reside, and therefor provide an easily visualized spatial relationship to neuronal cell bodies that lie just under the surface or to ones that lie deeper and therefore farther from the SPG glia. Such post-development induction of hTDP-43 by the TARGET method in just the SPG glial subtype had a significant impact on the lifespan of the animals (Figure S5B), which is consistent with the observed impact on cell death signaling seen in the clonal analysis above. Perhaps not surprisingly, the effect on lifespan with just SPG expression of hTDP-43 is not as dramatic as that observed with pan-glia induction (Figure S5B).
To test the effects of focal expression of hTDP-43 in SPG, we co-expressed the dual UAS-WM reporter in order to reveal the location of SPG nuclei and the morphology of their membranes. Co-expression of nls-LacZ was used as a control for effects of hTDP-43, and active Caspase-3 antibody was used as a readout of apoptotic signaling. In brains from control animals that express nls-LacZ in SPG (moodyts>nls-LacZ+WM), we did not observe activated Caspase-3 in the glia or in neurons (Figure 5F). In contrast, when we induced focal hTDP-43 within SPG (moodyts>hTDP-43+WM), we detected activated Caspase-3 in many neuronal nuclei (Figure 5F), including neurons that were a few cell diameters from the labeled glia. We also observed activated Caspase-3 in some of the overlying PNG glia close to the hTDP-43 expressing SPGs (white arrow in Figure 5F), indicating that glial-to-glial spread may also be at play. The WM dual reporter permitted us to label both the nuclei and membranes of the hTDP-43 expressing SPG, which revealed an apparently normal morphology that was indistinguishable from SPG expressing nls-LacZ as a control (Figure 5F and S5C). Thus as with the clonal analysis described above, the focal induction of hTDP-43 pathology in this subset of glia is sufficient to drive apoptotic signaling within the glial cells, but also in nearby neurons and in other glia. Here too, we tested whether direct induction of apoptosis in these glia without hTDP-43 expression could cause the same effects, and it could not (Figure 5F). We used the same moodyts TARGET approach to induce expression of the pro-apoptotic gene, reaper (rpr), in SPG. As with induction of hid in the context of clonal analysis above, induction of rpr in SPG (moodyts>rpr+WM) did not yield detectable active Caspase-3 within neighboring neurons (Figure 5F). These convergent findings, from induction of hTDP-43 pathology in all glia, in just the SPG subset, or with focal onset of hTDP-43 pathology within recombinant clones, all support the hypothesis that glial cells become actively toxic to neighboring neurons. This glial toxicity involves activation of the loki/chk2 DNA damage sensing pathway in glia, and is mediated at least in part by expression and/or replication of the gypsy-ERV.
With Pathological Expression of hTDP-43, Glia Become Actively Toxic to nearby Neurons in vivo
Above, we demonstrated that in PNG, SPG and ALG, hTDP-43 protein pathology results in three observed cellular phenomena: expression and replication of the gypsy ERV, accumulation of DNA damage and activation of Caspase-3 signaling. In addition, toxic over-expression of hTDP-43 in glia initiates production of non-cell autonomous factor(s) that impact neurons. This non-cell autonomous effect requires DNA damage signaling and gypsy expression within the glia. To test whether the apoptotic signaling pathway itself might also contribute to this toxic spread, we used expression of the anti-apoptotic proteins, p35 and DIAP-1, which directly bind to and inhibit Caspase-3 [58]. Two main possibilities occurred to us, and could be distinguished by this series of experiments. First, if the glial apoptotic process itself produced some toxic factor(s) that acted on nearby neurons, we would expect inhibitors of apoptosis in glia to rescue the glial loss and provide a concomitant rescue of neuronal death. We viewed this possibility as unlikely given that we do not observe non-cell autonomous neuronal death when we induce apoptosis directly, without hTDP-43 expression, either by rpr or hid expression in small groups of glia. The alternative possibility, however, is that an impact of hTDP-43 pathology in glia is to cause production of a toxic factor from glia independent of glial apoptosis. In this case the glial death might actually be neuroprotective by removing a source of toxic factor(s). This would predict that blocking apoptotic activity in TDP-43 expressing glia would exacerbate the fallout in neurons from glial hTDP-43 pathology.
We first tested the effects of inhibiting Caspase-3 on glial hTDP-43 overexpression by examining the organismal effects on lifespan. We used the repots TARGET approach to induce hTDP-43 (or nls-LacZ as a control) in glia after development, and tested the impact of co-expressing either p35 or DIAP-1 to block Caspase-3 activity. In the context of control flies that express nls-LacZ (repots>2xnls-LacZ), inhibition of Caspase-3 by co-expression of p35 (repots>nls-LacZ+p35) or DIAP-1 (repots>nls-LacZ+DIAP-1) prolonged the lifespan of these otherwise wild-type flies (Figure S6A). This in itself may be interesting in that it suggests that glial cell death may contribute to the normal constraints on lifespan. More striking, however, was the finding that inhibiting apoptotic cell death signaling (repots>hTDP-43+p35 and repots>hTDP-43+DIAP-1) actually exacerbated the effects of glial hTDP-43 (repots>hTDP-43+nls-LacZ) on lifespan (Figure S6A). Under conditions in which the repots temperature switch is fully “induced” (30°C), the relatively high levels of hTDP-43 are toxic enough that the impact of expressing Caspase-3 inhibitors is significant, but subtle due to a floor effect on lifespan. However, by using intermediate the temperatures, we were able to titrate the levels of hTDP-43 (and p35 or DIAP-1) expression. With lower levels of hTDP-43, the survival curves are shifted to the right, allowing a larger window to observe impacts from p35 and DIAP-1 in either direction. Under these conditions, we were able to reveal a more dramatic impact (30°C vs 29°C vs 27°C in Figure S6A). In each of these conditions, blocking apoptosis by expression of either of two inhibitors was sufficient to significantly exacerbate the toxic effects of hTDP-43 on lifespan. We next examined the impact on cell death in glia and adjacent neurons using immunofluorescence to detect activated Caspase-3.
In brains from animals that co-express hTDP-43 in glia along with nls-LacZ as a control (repots>hTDP-43+nls-LacZ), most of the glia were still detectable with the glial Repo marker at Day 3 post induction, but many of them were Caspase-3 positive (Figure 6A and 6I). At the same time, neighboring neurons were labeled with active Caspase-3 (Figure 6A and 6J). By Day 4 post induction, the total number of glia had declined (Figure 6D and 6G, see also Figure 1–2), and the remaining glia were Caspase-3 positive (Figure 6D and 6I). Importantly, the number of Caspase-3 labeled neurons increased between Day 3 and Day 4 (Figure 6A, 6D and 6J), even while the total number of neurons had decreased (Figure 6H). When we over-expressed either the p35 (repots>hTDP-43+p35) or DIAP-1 (repots>hTDP-43+DIAP-1) inhibitors of Caspase-3 function, it was sufficient to protect the glia from dying. Glial numbers in these animals were stable between Day 3 and 4 (Figure 6G), even while Caspase-3 was still detectable as expected (Figure 6B–C, 6E–F and 6I). Thus inhibiting Caspase-3 function could rescue the glia. On the other hand, the numbers of nearby neurons that were labeled with Caspase-3 actually was higher with either p35 or DIAP-1 co-expression (Figure 6A–F and 6J), and the total numbers of surviving neurons also was actually lower at Day 4 (Figure 6H). Thus, co-expression in glia of inhibitors of Caspase-3 was sufficient to protect those glia from hTDP-43, but the effects on nearby neurons was more severe.
Figure 6. Blocking Cell Death in hTDP-43 Expressing Glia Exacerbates Non-Autonomous Neuronal Toxicity.

(A-F) Distribution of activated Caspase-3 (White) was monitored after pan-glial but post development induction of hTDP-43 at Day 3 (A-C) and Day 4 (D-E) post induction. All glial and neuronal nuclei were co-labeled with antibodies against the Repo (Red) and Elav (Blue) markers. Effects of hTDP-43 on Caspase-3 were monitored when it was co-induced with either nls-LacZ (A, D) control (repots>hTDP-43+nls-LacZ), vs p35 (repots>hTDP-43+p35) or DIAP-1 (repots>hTDP-43+DIAP-1), inhibitors of Caspase-3 function (B,E and C,F, respectively). Scale bar=10μm.
(G) Quantification of total glial numbers from a defined brain region (mean ± SEM). Total glial numbers were 30.5±2.3 (Day 3, n=8) and 8.8±0.9 (Day 4, n=13) in repots>hTDP-43+nls-LacZ, 36.3±1.4 (Day 3, n=16) and 33.5±1.5 (Day 4, n=8) in repots>hTDP-43+p35, and 32.8±1.3 (Day 3, n=15) and 25.1±1.8 (Day 4, n=12) in repots>hTDP-43+DIAP-1. **p<0.01, ****p<0.0001, n.s., no significant difference (Unpaired t-test). ####p<0.0001, n.s., no significant difference (Two-way ANOVA).
(H) Quantification of total neuronal numbers from a defined brain region (mean ± SEM). Total neuronal numbers were 374.4±8.9 (Day 3, n=8) and 311.0±7.2 (Day 4, n=13) in repots>hTDP-43+nls-LacZ, 361.5±9.4 (Day 3, n=16) and 209.3±17.4 (Day 4, n=8) in repots>hTDP-43+p35, and 379.6±6.3 (Day 3, n=15) and 191.6±11.6 (Day 4, n=12) in repots>hTDP-43+DIAP-1. ****p<0.0001 (Unpaired t-test). ####p<0.0001, n.s., no significant difference (Two-way ANOVA).
(I) Quantification of total numbers of Caspase-3 positive glia from a defined brain region (mean ± SEM). Total Caspase-3 positive glial numbers were 17.3±1.0 (Day 3, n=8) and 4.9±0.7 (Day 4, n=13) in repots>hTDP-43+nls-LacZ, 16.3±1.2 (Day 3, n=16) and 22.0±2.1 (Day 4, n=8) in repots>hTDP-43+p35, and 15.9±1.2 (Day 3, n=15) and 11.6±1.1 (Day 4, n=12) in repots>hTDP-43+DIAP-1. *p<0.05, **p<0.01, ****p<0.0001 (Unpaired t-test). ##p<0.01, ####p<0.0001, n.s., no significant difference (Two-way ANOVA).
(J) Quantification of total numbers of Caspase-3 positive neuron (mean ± SEM). Total Caspase-3 positive neuronal numbers were 9.5±1.5 (Day 3, n=8) and 26.5±1.9 (Day 4, n=13) in repots>hTDP-43+nls-LacZ, 18.5±1.3 (Day 3, n=16) and 43.3±3.2 (Day 4, n=8) in repots>hTDP-43+p35, and 19.4±1.6 (Day 3, n=15) and 45.5±2.8 (Day 4, n=12) in repots>hTDP-43+DIAP-1. ****p<0.0001 (Unpaired t-test). #p<0.05, ####p<0.0001 (Two-way ANOVA).
Given the protective effect from knocking down chk2, it is tempting to propose a model in which hTDP-43 proteinopathy leads to RTE/ERV expression, that in turn generates DNA damage, activation of chk2, leading to apoptosis. But this idea is at odds with the finding that blocking Caspase-3 function in glia exacerbates the spreading toxicity, i.e. the opposite to what we observe with knockdown of chk2. An examination of the impacts on gypsy-ERV expression levels adds some clarity. We used a monoclonal antibody against the Env glycoprotein encoded by the gypsy [32, 40, 59] to quantify the Env immunofluorescence within brain region (Figure S6C, see STAR METHODS). As reported previously [32], Env expression is increased in response to glial expression of hTDP-43 (repots>hTDP-43+nls-LacZ) (Figure S6B–C). When we inhibited Caspase-3 function, either by p35 (repots>hTDP-43+p35) or DIAP-1 (repots>hTDP-43+DIAP-1) co-expression, Env levels were significantly increased (Figure S6B–C), consistent with the fact that glia are prevented from undergoing apoptosis and therefor survive longer in the tissue, thereby producing more gypsy-ERV encoded protein. By contrast, when we knock down loki/chk2 (repots>hTDP-43+loki-IR), thereby reducing DNA-damage mediated signaling to apoptotic machinery, the levels of expression is lower. This is seen both via RT-qPCR mediated detection at the RNA level for gypsy ORF2 and ORF3 (Figure S7A), and at the protein level in situ using a monoclonal antibody against the gypsy ORF3 encoded protein, Env (Figure S7B–C). These findings are consistent with a model in which DNA damage is caused by TDP-43 protein mislocalization [21, 48] and acts as an inducer of gypsy-ERV expression via the loki/chk2 pathway (Figure 7).
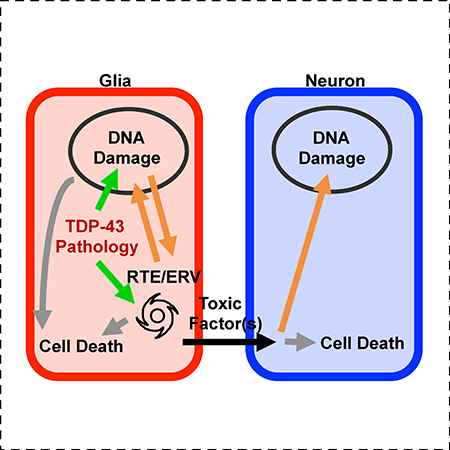
Figure 7. TDE Feedback Amplification Loop for Non-Cell Autonomous Propagation from Glia to Neurons.

TDP-43 proteinopathy (T) and loss of nuclear TDP-43 within glia leads activation of RTEs/ERVs (E), likely through two mechanisms. First, via activation of DNA damage signaling (D) and second via disruption to small RNA mediated silencing. RTEs/ERVs may also feed-back to amplify DNA damage signal. DNA damage signaling and the expression and/or replication of ERVs is toxic within glia, but also produces a non-cell autonomous signal that causes a recurrent activation of DNA damage signaling and cell death signaling within nearby neurons. RTEs/ERVs may contribute to toxicity by initiating inflammatory responses that may influence nearby neurons. It also is possible that ERVs contribute to non-cell autonomous toxicity via viral spread within the tissue. The relationship with transfer of TDP-43 seeding activity is not clear.
Discussion
Focal onset followed by propagation through neural tissue is a fundamental feature of most NDs. This likely involves both neuron-to-neuron and glial-to-neuron effects [1, 2, 5]. But little is known about cellular mechanisms by which local onset of ND pathology in one neuron or glial cell causes the demise of neurons in distal but connected regions of tissue. A key insight into non-cell autonomous spread of ND stems from the observation that many of the implicated proteins exhibit prion like properties misfolding properties and the ability to spread intracellularly [30, 31, 60–64]. Although the transfer of misfolded seeds of TDP-43 between cells has not been shown to underlie propagation of cellular toxicity itself, the finding that TDP-43 seeding activity can move between cells is compelling.
We established an in vivo system in Drosophila to initiate focal onset of hTDP-43 overexpression in glial cells and used this system to investigate mechanisms of non-cell autonomous spread of toxic cellular outcomes into surrounding wild-type neurons (Figure 7). In our in vivo system also observed cell-lethal propagation from TDP-43 over-expressing glia to wild type neurons, and we determined that this effect is mediated by DNA damage signaling and by expression of gypsy, a Drosophila ERV, within the glial cells.
Glial TDP-43 Pathology Induces DNA Damage and Apoptosis in Glia and in Adjacent Neurons
Although research related to TDP-43 function has focused on the role of its RNA binding properties, TDP-43 also has DNA binding properties [24, 65] and is normally recruited to DNA DSB as a component of the DNA repair machinery in motor neurons [21]. Loss of nuclear TDP-43, which is seen both in patient tissue and with over-expression, has been shown to induce DSB accumulation [21, 48]. We have shown previously that neurodegenerative impact of hTDP-43 transgenic expression in Drosophila glia and neurons each involves DNA damage signaling through chk2/loki [32]. We demonstrate here that DNA damage plays a key role in the propagation of TDP-43 mediated cell death from glia to neurons.
We found that glial hTDP-43 toxicity results in accumulation of DNA damage foci both in the glial cells and in adjacent neurons. And blocking chk2/loki signaling within the glia is sufficient to prevent DNA damage in neurons and to rescue the nearby neurons from cell death. This suggests for the first time that DNA damage signaling is a component of the non-cell autonomous propagation in vivo. Our observations with the gypsy-ERV suggest the idea that one means by which DNA damage in glia impacts propagation to neurons is via induced expression and replication of ERVs.
TDP-43 Pathology Induces RTEs/ERVs
A growing body of literature supports the conclusion that TDP-43 proteinopathy and/or loss of nuclear function interferes with the normal systems that silence RTEs and ERVs, leading to their expression [23, 24, 26, 27, 32, 33, 66]. Indeed, expression of the HERV-K Env gene is toxic to neurons in culture, and causes motor neuron degeneration in transgenic mice, suggestive of a causal impact [24]. We have previously demonstrated in a Drosophila transgenic model, that the effect of hTDP-43 pathology on mobile elements and includes not only ERVs, but also a panel of LINE-like elements and non-autonomous RTEs as well [32]. A similarly broad suite of RTE/ERV expression may be at play in C9orf72 ALS [25], which also involves TDP-43 proteinopathy. More recently, we confirmed that broad RTE/ERV activation is consistently observed in motor cortex from approximately 20% of samples from among a large cohort of 77 ALS subjects [26]. The subjects with high levels of RTE/ERV levels also exhibited the greatest alterations in expression of confirmed TDP-43 target genes, and the highest levels of phospho-TDP-43 in paraffin sections. It also is worth note that RTE/ERV induction has been reported in Drosophila tau ND models as well as in Alzheimer’s disease patient tissue [67, 68].
RTE/ERV expression is canonically seen as highly detrimental. Expression of these selfish genetic elements can cause insertional mutations, activation of inflammatory responses to both the RNAs and cDNAs that they produce, and genomic stress from DNA damage [19, 20, 45, 46]. In this study, we used our novel CLEVR reporter [36] to provide a cell-by-cell readout of gypsy-ERV replication in response to induced pan-glial expression of hTDP-43. Remarkably, we see that the ERV replication co-localizes cell-by-cell with the appearance of DNA damage foci and activation of death Caspase signaling. In addition, neurons that are adjacent to these ERV active glia also exhibit DNA damage and activation of the Caspase-3 cell death marker. Importantly, we observe this spreading death phenomenon both in the context of pan-glial hTDP-43 expression and with focal onset of hTDP-43 in small clusters of several types of glia, including astrocyte like glia. In the case of pan-glial hTDP-43 expression, we see stochastic activation of gypsy-ERV replication, and a tight correlation of this with DNA damage and death signaling within those glia and in nearby neurons. And knocking down either the gypsy-ERV or chk2/loki signaling within the glia blocks the non-cell autonomous effects.
Several findings support the hypothesis that DNA damage lies upstream of ERV activation. First, UV and X-irradiation are established drivers of RTE/ERV activation in mammalian and Drosophila cells [69–71]. Second, TDP-43 nuclear loss has been shown to induce DNA damage, and TDP-43 is normally recruited to sites of DNA damage [21, 48]. Finally, we find that knocking down chk2/loki signaling significantly reduces the expression of the gypsy-ERV, indicating that DNA damage signaling activates or sustains gypsy-ERV levels. Because RTEs/ERVs also create DNA damage as part of their replication cycle, this suggests the possibility of a positive feedback loop in which loss of TDP-43 nuclear function induces DNA damage and activates RTEs/ERVs, and RTEs/ERVs in turn generate more DNA damage.
TDP-43 – DNA damage – ERV -- Feedback Model of Non-Cell Autonomous Spread
We have discovered a cellular mechanism by which focal onset of TDP-43 pathology can impact surrounding cells via a feedback amplification involving DNA damage and ERV expression and replication (Figure 7). TDP-43 protein pathology likely activates RTEs and ERVs in two ways. First, loss of nuclear localization that accompanies the formation of cytoplasmic inclusions causes DNA damage [21, 32, 48], leading to activation of chk2/loki signaling. Chk2/loki activation in turn drives ERV expression (Figure S7). Second, TDP-43 dysfunction also disrupts small RNA silencing systems that normally help to stifle RTE/ERV expression [32, 66]. Together, these forces act to induce a lethal wave of ERV activation. RTE/ERV replication, or even abortive attempts at replication, may generate additional chromosomal DNA damage, acting as a feed back amplification loop. Such accumulating DNA damage within the glia would almost certainly drive cell death via activation of death Caspases. But this feedback cycle between TDP-43 toxicity, DNA damage, and ERV activation also contributes to the destruction of nearby neurons via release of a toxic factor or factors.
Although we cannot rule out the possibility that loss of glial support functions contribute to neuronal death, a convergence of three lines of evidence argue that an actively toxic effect of glia also is at play. First, induction of TDP-43 toxicity even in a few glial cells is associated with spreading death to nearby neurons (Figure 5). Second, direct activation of apoptotic signaling in equivalent sized clones of glia does not cause death of surrounding neurons (Figure 5F and S5A). And third, blocking cell death via expression of inhibitors of apoptosis can rescue death of the hTDP-43 expressing glial cells, but actually increases their toxicity to nearby neurons (Figure 6). Our findings support the conclusion that DNA-damage signaling and gypsy-ERV expression in glia mediate the spreading toxicity of those glia to nearby neurons. It is not clear whether chk2 and ERV expression contribute to TDP-43 protein movement and/or produce an additional toxic factor. But because the levels of gypsy-ERV are dependent on chk2/loki signaling, it seems likely that the effects of chk2 on spreading death are mediated by the gypsy-ERV. Although we do not know how chk2 signaling impacts ERV expression, there is some reason to suspect that this could be mediated by effects of p53, a target of chk2 [72, 73].
There are several ways in which RTE/ERV expression may contribute to non-cell autonomous toxicity. First, these genotoxic factors can cause apoptotic cell death, which in some contexts is known to produce a spreading death phenomenon known as apoptosis-induced-apoptosis [74]. But we do not observe spreading death when we directly induce canonical apoptosis in clusters of glia. A second potential non-cell autonomous impact of RTEs/ERVs is via activation of inflammatory signaling. An intriguing possibility is that RTE/ERV activation drives the senescence associated secretory pathway (SASP) [75], leading to release of inflammatory cytokines. Finally, we cannot rule out the possibility that the spread of toxicity between cells may involve the movement of viral-like particles that can be produced by ERVs such as gypsy and HERV-K [76–79]. Given the literature demonstrating that a similar phenomenology of RTE/ERV activation is at play in affected brain tissue from ALS subjects [23–27, 33], it will be important to investigate whether HERV-K or other human RTEs/ERVs may also contribute to the propagation of TDP-43 pathology during disease progression.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Josh Dubnau (ioshua.dubnau@stonybrook.edu). All plasmids and Drosophila strains will be made readily available upon request to the lead contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fly Stocks and Culturing Conditions
All Drosophila transgenic lines used in this study are listed in Key Sources Table. To prevent influence of genetic background, all transgenic flies used in this study were backcrossed to our laboratory wild-type strain, Canton-S derivative w1118 (isoCJ1), for at least five generations. Male flies were chosen as the experimental subjects throughout the study, other than the assay of single glial subtype effect (Figures 5A–D and S5A) where only female could fully satisfy the specific genetic combination required in these experiments. In these cases, female flies were kept in the same culture condition as males.
In general, flies for TARGET temperature-shift experiments (repo-Gal4 or moody-Gal4 combined with tub-Gal80ts) were raised in a 21°C incubator from embryonic stage until eclosion. TARGET flies were immediately shifted to desired permissive temperature (27, 29 or 30°C) incubators after eclosion for induction of transgenes in specific target cells. These TARGET flies then were either dissected at different time points for each experimental assay or incubated in the same temperature for lifespan analysis. For constitutive expression of genes in glial cells, all flies were kept in a 25°C incubator throughout their lifespans. For single glial subtype Flip-clonal analysis, the desired flies were maintained and raised in a 25°C incubator until late pupal sta ges. In order to obtain as fewer glial clones within each brain, flies were given a short 30 second heat shock in a 37°C water bath and then returned to the 25° C incubator. After eclosion the next day, flies with desired genetic combination were collected and maintained in a new vial in a 25°C incubator until the time point for analysis. The genotypes for Flip-mediated clonal analysis were hsp70-FLP/αTub84B>FRT-Gal80>FRT; UAS-WM/+; repo-Gal4/UAS-hTDP-43 (Figure 5B), hsp70-FLP/αTub84B>FRT-Gal80>FRT; UAS-WM/UAS-gypsy-IR; repo-Gal4/UAS-hTDP-43 (Figure 5C), hsp70-FLP/αTub84B>FRT-Gal80>FRT; UAS-WM/UAS-loki-IR; repo-Gal4/UAS-hTDP-43 (Figure 5D), and hsp70-FLP/αTub84B>FRT-Gal80>FRT; UAS-WM/UAS-hid; repo-Gal4/+ (Figure S5A).
METHODS DETAILS
Immunofluorescent Staining of Fly Brains
Adult brains were dissected at specific time points and immunofluorescent staining was performed as previously described [32, 36] with minor modifications to optimize the signal-to-noise for a given antibody. In brief, adult fly brains were dissected in ice-cold phosphate-buffered-saline (PBS) and then transferred into 4% paraformaldehyde (Electron Microscopy Sciences) PBS solution (1XPBS with 4% paraformaldehyde and 0.2% Triton-X-100 (SIGMA-ALDRICH)) and incubated for 30 minutes with vacuum 2X to remove air from trachea. After fixation, dissected brains were washed three times 10 minutes with 1XPBST wash solution (1XPBS with 1% Triton-X-100). Brain sample were then incubated in blocking solution (1XPBST with 10% normal horse serum) overnight at 4°C on a nutator. After blocking, dissected brains were transferred into primary antibodies with the following dilutions: Repo (1:10, Developmental Studies Hybridoma Bank 8D12), Elav (1:10, Developmental Studies Hybridoma Bank 7E8A10 or 9F8A9), cleaved Caspase-3 (1:100, Cell Signaling Technology 9661), cleaved PARP1 (1:100, abcam ab2317), γH2AV (1:150, Rockland Immunochemicals 600–401-914), Env (1:100) in 1XPBST washing solution with 10% normal horse serum. Primary antibody incubation was overnight at 4°C. Brains were then washed four times 15 minutes each with 1XPBST washing solution. Fly brains were incubated within secondary antibody solution (1XPBST washing solution with 10% normal horse serum) at 4°C overnight. Brains were washed 4X 15 minutes with 1XPBST washing solution and then mounted in FocusClear (CelExplorer). In the experiments with gypsy-CLEVR, modifications were made in order to optimize signal from the WM reporter. In these cases, fixed brains were blocked, washed and incubated with primary and secondary antibodies in 1XPBST-20 washing solution (1XPBS with 0.1% of Tween-20 (BIO-RAD)) with same 10% normal horse serum. The dilution of each antibody were the same as above. Brain samples were imaged using a Zeiss LSM 800 with Airyscan mode and acquired images were processed by Zeiss Zen software package.
Quantification of Molecular Markers in Brain Images
To compare the change of different molecular markers, a defined 7688pm2 (124pm x 62pm) area (yellow dash rectangle in Figure 2A) of each brain was chosen to calculate the total number of cells with specific markers detected. To standardize brain region between brain samples, the rectangle was placed at the connection point between central brain and optic lobe at the same section number along the imaging axis.
Survival Curve Assay
Male flies were collected immediately after eclosion for survival analysis at desired temperatures as in previous studies [32, 40]. 10 flies of a given genotype were housed within one vial and more than 70 flies in total of each genotype within the same experimental group used. Flies in each vial were flipped into fresh vials with fresh fly food every other day, and flies that escaped during the time window were removed from the final survival curve analysis. The Log-Rank (Mantel-Cox) test and the Gehan-Breslow-Wilcoxon test were used to compare the survival curves.
Single Glial Subtype Analysis by FLP-Mediated Clonal Analysis
Fly central brains with desired genotypes were subjected to immunolabeling as above. After images were acquired, brains with too many glial clones were excluded from analysis. In order to obtain the pure effect from one glial subtype at a time, only those clones with pure single glial subtypes within the same imaging regions were selected to further analyze.
Quantification of gypsy ORF3 (Env) Intensity
The Env intensity (shown in Figure S6B–C and S7B–C) from each pixel2 of central brains were transformed and plotted based on the Env level gained from that particular area by MetaMorph as in a previous report [80]. To compare the relative of Env intensities from different groups, the average Env intensity from each central brain (yellow dashed circle in Figure S6C) was extracted by MetaMorph as the previous report [80]. Ten random 20×20 pixel2 areas (red solid square in Figure S6C) without positive Env signaling were selected within each brain and averaged to yield background intensity. The final average Env intensity of each brain was obtained by subtracting background intensity (value from red solid square) from the average value (yellow dashed circle) gained from central brain. Three representative brains from each groups were selected and averaged to compare the relative Env intensity change between groups (Figure S6C and S7C).
RNA preparation and RT-qPCR for gypsy Expression
Total RNA was isolated from 50–70 fly heads (per biological replicate) as previously described [32, 40]. Fly head total RNAs were extracted by using Trizol reagent (Invitrogen), and the total RNA extractions were treated with RQ1 DNase (Promega) before performing reverse transcription. 0.5μg of total RNA from each sample was used to synthesize cDNA with SuperScript IV VILO Master Mix (Invitrogen). qPCR was then performed using TaqMan Fast Advanced Master Mix (Applied Biosystems) on an Applied Biosystems StepOnePlus Real Time PCR System. All TaqMan assays used FAM Reporters and MGB Quencher with assay IDs as following, gypsy ORF2 (AI5106V, probe: 5’-AAGCATTTGTGTTTGATTTC-3’), gypsy ORF3 (AID1UHW, probe: 5’-CTCTAGGATAGGCAATTAA-3’) and Act5C (Dm02361909_s1) [32]. To obtain the relative fold changes of gypsy ORF2 and gypsy ORF3 (Figure S7A), values from each groups were further normalized to the levels in repots>hTDP-43+GFP-IR at Day 3 in order to obtain the relative levels.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
All of statistical analyses were performed with GraphPad Prism V6.01. Details of the tests used and results (shown as mean ± SEM) were outlined within each figure legend. Two-tailed Unpaired t-test, One-Way ANOVA, Two-way ANOVA, Log-Rank (Mantel-Cox) test and Gehan-Breslow-Wilcoxon test were used to compare the differences between groups depending on the experiment.
DATA AND CODE AVAILABILITY
This study did not generate/analyze datasets/code.
Supplementary Material
Highlights.
Glial Expression of hTDP-43 Causes Gypsy-ERV Replication, DNA Damage and Apoptosis
Glial hTDP-43 Pathology Triggers DNA Damage and Apoptosis in Nearby Neurons
Glial hTDP-43 Toxicity to Neurons Is Mediated by Gypsy-ERV and DNA Damage Signaling
With Pathological hTDP-43, Glia Become Actively Toxic to nearby Neurons in vivo
Acknowledgments
We thank Eli Arama (Weizmann Institute of Science, Israel) for the UAS-CPV transgenic flies and Bing Zhang (Baylor College of Medicine, USA) for the aTub84B>FRT-Gal80>FRT flies. We thank Jennifer Beshel (Loyola University Chicago) for the UAS-hid transgenic flies. We thank Anthony Zador and Roger Sher for helpful comments and discussions. This work was supported by grants to J.D. from NINDS (R01NS091748), the NIA (RF1AG057338), and the Ride For Life.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jucker M, and Walker LC (2018). Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21, 1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brettschneider J, Del Tredici K, Lee VM, and Trojanowski JQ (2015). Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ravits J, Laurie P, Fan Y, and Moore DH (2007). Implications of ALS focality: rostral-caudal distribution of lower motor neuron loss postmortem. Neurology 68, 1576–1582. [DOI] [PubMed] [Google Scholar]
- 4.Ravits J, Paul P, and Jorg C (2007). Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 68, 1571–1575. [DOI] [PubMed] [Google Scholar]
- 5.Ling SC, Polymenidou M, and Cleveland DW (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanden Broeck L, Callaerts P, and Dermaut B (2014). TDP-43-mediated neurodegeneration: towards a loss-of-function hypothesis? Trends Mol Med 20, 66–71. [DOI] [PubMed] [Google Scholar]
- 7.Taylor JP, Brown RH Jr., and Cleveland DW (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conicella AE, Zerze GH, Mittal J, and Fawzi NL (2016). ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 24, 1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, et al. (2017). TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 95, 808–816 e809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt HB, and Rohatgi R (2016). In Vivo Formation of Vacuolated Multi-phase Compartments Lacking Membranes. Cell Rep 16, 1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, and Taylor JP (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiang S, Kato M, Wu LC, Lin Y, Ding M, Zhang Y, Yu Y, and McKnight SL (2015). The LC Domain of hnRNPA2 Adopts Similar Conformations in Hydrogel Polymers, Liquid-like Droplets, and Nuclei. Cell 163, 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH, et al. (2015). ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 88, 678–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fratta P, Sivakumar P, Humphrey J, Lo K, Ricketts T, Oliveira H, Brito-Armas JM, Kalmar B, Ule A, Yu Y, et al. (2018). Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White MA, Kim E, Duffy A, Adalbert R, Phillips BU, Peters OM, Stephenson J, Yang S, Massenzio F, Lin Z, et al. (2018). TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat Neurosci 21, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang JC, and Morton DB (2017). Drosophila lines with mutant and wild type human TDP-43 replacing the endogenous gene reveals phosphorylation and ubiquitination in mutant lines in the absence of viability or lifespan defects. PLoS One 12, e0180828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koyama A, Sugai A, Kato T, Ishihara T, Shiga A, Toyoshima Y, Koyama M, Konno T, Hirokawa S, Yokoseki A, et al. (2016). Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res 44, 5820–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dawson TM, Golde TE, and Lagier-Tourenne C (2018). Animal models of neurodegenerative diseases. Nat Neurosci 21, 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubnau J (2018). The Retrotransposon storm and the dangers of a Collyer’s genome. Curr Opin Genet Dev 49, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kury P, Nath A, Creange A, Dolei A, Marche P, Gold J, Giovannoni G, Hartung HP, and Perron H (2018). Human Endogenous Retroviruses in Neurological Diseases. Trends Mol Med 24, 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitra J, Guerrero EN, Hegde PM, Liachko NF, Wang H, Vasquez V, Gao J, Pandey A, Taylor JP, Kraemer BC, et al. (2019). Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HJ, and Taylor JP (2017). Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron 96, 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Jin Y, Prazak L, Hammell M, and Dubnau J (2012). Transposable elements in TDP-43-mediated neurodegenerative disorders. PLoS One 7, e44099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W, Lee MH, Henderson L, Tyagi R, Bachani M, Steiner J, Campanac E, Hoffman DA, von Geldern G, Johnson K, et al. (2015). Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med 7, 307ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prudencio M, Gonzales PK, Cook CN, Gendron TF, Daughrity LM, Song Y, Ebbert MTW, van Blitterswijk M, Zhang YJ, Jansen-West K, et al. (2017). Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum Mol Genet 26, 3421–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tam OH, Rozhkov NV, Shaw R, Kim D, Hubbard I, Fennessey S, Propp N, Fagegaltier D, Ostrow LW, Phatnani H, et al. (2019). Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. bioRxiv, 574509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu EY, Russ J, Cali CP, Phan JM, Amlie-Wolf A, and Lee EB (2019). Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep 27, 1409–1421 e1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tong J, Huang C, Bi F, Wu Q, Huang B, Liu X, Li F, Zhou H, and Xia XG (2013). Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J 32, 1917–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, et al. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29, 824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, Yoshida M, Murayama S, Mann DM, Akiyama H, et al. (2013). Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 4, 124–134. [DOI] [PubMed] [Google Scholar]
- 31.Porta S, Xu Y, Restrepo CR, Kwong LK, Zhang B, Brown HJ, Lee EB, Trojanowski JQ, and Lee VM (2018). Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat Commun 9, 4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krug L, Chatterjee N, Borges-Monroy R, Hearn S, Liao WW, Morrill K, Prazak L, Rozhkov N, Theodorou D, Hammell M, et al. (2017). Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet 13, e1006635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Douville R, Liu J, Rothstein J, and Nath A (2011). Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol 69, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bodea GO, McKelvey EGZ, and Faulkner GJ (2018). Retrotransposon-induced mosaicism in the neural genome. Open Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singer T, McConnell MJ, Marchetto MC, Coufal NG, and Gage FH (2010). LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci 33, 345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang YH, Keegan RM, Prazak L, and Dubnau J (2019). Cellular labeling of endogenous retrovirus replication (CLEVR) reveals de novo insertions of the gypsy retrotransposable element in cell culture and in both neurons and glial cells of aging fruit flies. PLoS Biol 17, e3000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miguel L, Frebourg T, Campion D, and Lecourtois M (2011). Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in Drosophila models of TDP-43 proteinopathies. Neurobiol Dis 41, 398–406. [DOI] [PubMed] [Google Scholar]
- 38.Tan H, Qurashi A, Poidevin M, Nelson DL, Li H, and Jin P (2012). Retrotransposon activation contributes to fragile X premutation rCGG-mediated neurodegeneration. Hum Mol Genet 21, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGuire SE, Le PT, Osborn AJ, Matsumoto K, and Davis RL (2003). Spatiotemporal rescue of memory dysfunction in Drosophila. Science 302, 1765–1768. [DOI] [PubMed] [Google Scholar]
- 40.Li W, Prazak L, Chatterjee N, Gruninger S, Krug L, Theodorou D, and Dubnau J (2013). Activation of transposable elements during aging and neuronal decline in Drosophila. Nat Neurosci 16, 529–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones BC, Wood JG, Chang C, Tam AD, Franklin MJ, Siegel ER, and Helfand SL (2016). A somatic piRNA pathway in the Drosophila fat body ensures metabolic homeostasis and normal lifespan. Nat Commun 7, 13856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wood JG, Jones BC, Jiang N, Chang C, Hosier S, Wickremesinghe P, Garcia M, Hartnett DA, Burhenn L, Neretti N, et al. (2016). Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc Natl Acad Sci U S A 113, 11277–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sousa-Victor P, Ayyaz A, Hayashi R, Qi Y, Madden DT, Lunyak VV, and Jasper H (2017). Piwi Is Required to Limit Exhaustion of Aging Somatic Stem Cells. Cell Rep 20, 2527–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savva YA, Jepson JE, Chang YJ, Whitaker R, Jones BC, St Laurent G, Tackett MR, Kapranov P, Jiang N, Du G, et al. (2013). RNA editing regulates transposon-mediated heterochromatic gene silencing. Nat Commun 4, 2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hancks DC, and Kazazian HH Jr. (2016). Roles for retrotransposon insertions in human disease. Mob DNA 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crow YJ, and Manel N (2015). Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15, 429–440. [DOI] [PubMed] [Google Scholar]
- 47.Kazazian HH Jr., and Moran JV (2017). Mobile DNA in Health and Disease. N Engl J Med 377, 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hill SJ, Mordes DA, Cameron LA, Neuberg DS, Landini S, Eggan K, and Livingston DM (2016). Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc Natl Acad Sci U S A 113, E7701–E7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jang JK, Sherizen DE, Bhagat R, Manheim EA, and McKim KS (2003). Relationship of DNA double-strand breaks to synapsis in Drosophila. J Cell Sci 116, 3069–3077. [DOI] [PubMed] [Google Scholar]
- 50.Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, and Truman JW (2006). Local caspase activity directs engulfment of dendrites during pruning. Nat Neurosci 9, 1234–1236. [DOI] [PubMed] [Google Scholar]
- 51.Yu Y, Pham N, Xia B, Papusha A, Wang G, Yan Z, Peng G, Chen K, and Ira G (2018). Dna2 nuclease deficiency results in large and complex DNA insertions at chromosomal breaks. Nature 564, 287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y, Pane A, and Schupbach T (2007). Cutoff and aubergine mutations result in retrotransposon upregulation and checkpoint activation in Drosophila. Curr Biol 17, 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Freeman MR (2015). Drosophila Central Nervous System Glia. Cold Spring Harb Perspect Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ben Haim L, and Rowitch DH (2017). Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci 18, 31–41. [DOI] [PubMed] [Google Scholar]
- 55.Gordon MD, and Scott K (2009). Motor control in a Drosophila taste circuit. Neuron 61, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kremer MC, Jung C, Batelli S, Rubin GM, and Gaul U (2017). The glia of the adult Drosophila nervous system. Glia 65, 606–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwabe T, Bainton RJ, Fetter RD, Heberlein U, and Gaul U (2005). GPCR signaling is required for blood-brain barrier formation in drosophila. Cell 123, 133–144. [DOI] [PubMed] [Google Scholar]
- 58.Meier P, Silke J, Leevers SJ, and Evan GI (2000). The Drosophila caspase DRONC is regulated by DIAP1. EMBO J 19, 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song SU, Kurkulos M, Boeke JD, and Corces VG (1997). Infection of the germ line by retroviral particles produced in the follicle cells: a possible mechanism for the mobilization of the gypsy retroelement of Drosophila. Development 124, 2789–2798. [DOI] [PubMed] [Google Scholar]
- 60.Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM, Li D, Thal DR, Walther P, Ludolph AC, et al. (2015). TDP-43 is intercellularly transmitted across axon terminals. J Cell Biol 211, 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shimonaka S, Nonaka T, Suzuki G, Hisanaga S, and Hasegawa M (2016). Templated Aggregation of TAR DNA-binding Protein of 43 kDa (TDP-43) by Seeding with TDP-43 Peptide Fibrils. J Biol Chem 291, 8896–8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, and Duff K (2012). Trans-synaptic spread of tau pathology in vivo. PLoS One 7, e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RA, et al. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19, 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smethurst P, Newcombe J, Troakes C, Simone R, Chen YR, Patani R, and Sidle K (2016). In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol Dis 96, 236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, and Gaynor RB (1995). Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69, 3584–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saldi TK, Ash PE, Wilson G, Gonzales P, Garrido-Lecca A, Roberts CM, Dostal V, Gendron TF, Stein LD, Blumenthal T, et al. (2014). TDP-1, the Caenorhabditis elegans ortholog of TDP-43, limits the accumulation of double-stranded RNA. EMBO J 33, 2947–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guo C, Jeong HH, Hsieh YC, Klein HU, Bennett DA, De Jager PL, Liu Z, and Shulman JM (2018). Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep 23, 2874–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun W, Samimi H, Gamez M, Zare H, and Frost B (2018). Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat Neurosci 21, 1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stein B, Kramer M, Rahmsdorf HJ, Ponta H, and Herrlich P (1989). UV-induced transcription from the human immunodeficiency virus type 1 (HIV-1) long terminal repeat and UV-induced secretion of an extracellular factor that induces HIV-1 transcription in nonirradiated cells. J Virol 63, 4540–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stein B, Rahmsdorf HJ, Steffen A, Litfin M, and Herrlich P (1989). UV-induced DNA damage is an intermediate step in UV-induced expression of human immunodeficiency virus type 1, collagenase, c-fos, and metallothionein. Mol Cell Biol 9, 5169–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Faure E, Best-Belpomme M, and Champion S (1996). X-irradiation activates the Drosophila 1731 retrotransposon LTR and stimulates secretion of an extracellular factor that induces the 1731-LTR transcription in nonirradiated cells. J Biochem 120, 313–319. [DOI] [PubMed] [Google Scholar]
- 72.Wylie A, Jones AE, D’Brot A, Lu WJ, Kurtz P, Moran JV, Rakheja D, Chen KS, Hammer RE, Comerford SA, et al. (2016). p53 genes function to restrain mobile elements. Genes Dev 30, 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pisanic TR 2nd, Asaka S, Lin SF, Yen TT, Sun H, Bahadirli-Talbott A, Wang TH, Burns KH, Wang TL, and Shih IM (2019). Long Interspersed Nuclear Element 1 Retrotransposons Become Deregulated during the Development of Ovarian Cancer Precursor Lesions. Am J Pathol 189, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perez-Garijo A, Fuchs Y, and Steller H (2013). Apoptotic cells can induce non-autonomous apoptosis through the TNF pathway. Elife 2, e01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, et al. (2019). L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim A, Terzian C, Santamaria P, Pelisson A, Purd’homme N, and Bucheton A (1994). Retroviruses in invertebrates: the gypsy retrotransposon is apparently an infectious retrovirus of Drosophila melanogaster. Proc Natl Acad Sci U S A 91, 1285–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Song SU, Gerasimova T, Kurkulos M, Boeke JD, and Corces VG (1994). An env-like protein encoded by a Drosophila retroelement: evidence that gypsy is an infectious retrovirus. Genes Dev 8, 2046–2057. [DOI] [PubMed] [Google Scholar]
- 78.Dewannieux M, Harper F, Richaud A, Letzelter C, Ribet D, Pierron G, and Heidmann T (2006). Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res 16, 1548–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee YN, and Bieniasz PD (2007). Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog 3, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chang YH, and Sun YH (2014). Carrier of Wingless (Cow), a secreted heparan sulfate proteoglycan, promotes extracellular transport of Wingless. PLoS One 9, e111573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.