

SUMMARY
Employing inducible genetically engineered and orthotopic mouse models, we demonstrate a key role for transcriptional regulator Yap in maintenance of Kras mutant pancreatic tumors. Integrated transcriptional and metabolomics analysis reveals that Yap transcribes Myc, and cooperates with Myc to maintain global transcription of metabolic genes. Yap loss triggers acute metabolic stress, which causes tumor regression while inducing epigenetic reprogramming and Sox2 upregulation in a subset of pancreatic neoplastic cells. Sox2 restores Myc expression and metabolic homeostasis in Yap-deficient neoplastic ductal cells, which gradually re-differentiate into acinar-like cells, partially restoring pancreatic parenchyma in vivo. Both the short-term and long-term effects of Yap loss in inducing cell death and re-differentiation, respectively, are blunted in advanced, poorly differentiated p53 mutant pancreatic tumors. Collectively, these findings reveal a highly dynamic and interdependent metabolic, transcriptional and epigenetic regulatory network governed by Yap, Myc, Sox2 and p53 that dictates pancreatic tumor metabolism, growth, survival, and differentiation.
Keywords: Yap, Kras, Myc, Sox2, p53, Tumor maintenance, Pancreatic Cancer
eTOC blurb
Murakami et al. demonstrate the stage-dependent requirement for Yap in PDAC maintenance. Yap knockout causes tumor regression and regeneration of pancreatic parenchyma in early stage PDAC, but only temporarily halts tumor growth in advanced p53 mutant PDAC tumors due to de-repression of Sox2, which substitutes Yap in maintaining metabolic homeostasis.
Graphical Abstract
INTRODUCTION
PDAC is the fourth leading cause of cancer-related deaths due to its aggressive nature and difficult to diagnose at the early stages (Ryan et al., 2014). Virtually all PDAC tumors carry activating KRAS mutations (Ryan et al., 2014), which are both necessary and sufficient for PDAC pathogenesis (Aguirre et al., 2003; Hingorani et al., 2003, 2005). Studies using sophisticated genetically engineered mouse models (GEMMs) have established that PDAC originates primarily from exocrine acinar cells (Friedlander et al., 2009; Guerra et al., 2007; Habbe et al., 2008), which upon acquiring Kras mutation undergo acinar to ductal metaplasia (ADM), followed by progression through low to high grades of pancreatic intraepithelial neoplasia (PanIN1–3) and eventually malignant PDAC.
Besides changes in cell differentiation, oncogenic Kras induces extensive metabolic reprogramming to meet the energetic and anabolic needs of sustained proliferation, to maintain redox balance, and to survive the environmental and nutrient stresses caused by the extensive fibrosis that is often associated with PDAC tumors (Halbrook and Lyssiotis, 2017). Oncogenic Kras increases glycolytic flux by upregulating Glut1 and other glycolytic enzymes to scavenge the limited extracellular glucose available in the pancreatic tumor microenvironment (Ying et al., 2012). Rather than feeding into the TCA cycle, Kras mutant PDAC cells divert glucose (Glc) carbon to the pentose phosphate pathway (PPP), hexosamine biosynthetic pathway and serine synthesis pathway (SSP) to support nucleotide biosynthesis and protein glycosylation (Maddocks et al., 2017; Ying et al., 2012). Furthermore, oncogenic Kras increases glutamine (Gln) metabolism to maintain redox homeostasis and nucleotide biosynthesis (Son et al., 2013). Kras-induced metabolic reprogramming in PDAC cells has been shown to be mediated in large part by Myc – a master transcription factor that coordinately regulates the expression of metabolic enzymes involved in glycolysis, glutaminolysis, nucleotide and lipid synthesis (Satoh et al., 2017; Stine et al., 2015; Ying et al., 2012). Myc blockade has been shown to cause complete tumor regression in Kras-driven lung cancer (Kortlever et al., 2017; Soucek et al., 2008, 2013). However, how oncogenic Kras modulates Myc expression and how Myc functionally interacts with other transcriptional regulators in maintaining PDAC tumors remain undefined.
We previously identified transcriptional regulator Yap as an essential driver of Kras-induced PDAC initiation in the presence or absence of mutant p53 (Zhang et al., 2014), and showed that increased YAP expression correlated with decreased survival in human PDAC (Murakami et al., 2017). These findings were subsequently corroborated by other groups (Gruber et al., 2016; Rozengurt et al., 2018; Zhao et al., 2017). Beyond its role in PDAC initiation, Yap amplification was identified as a bypass mechanism in Kras-ablated PDAC tumors (Kapoor et al., 2014). While these prior studies establish Yap as a critical mediator of PDAC initiation and resistance to Kras ablation, important questions remain regarding what role(s) Yap plays in the maintenance of PDAC and the functional connections between Yap and other transcription factors that have been implicated in PDAC survival such as Myc. Will Yap ablation merely slow growth or cause regression in established pancreatic tumors? Does Yap play a role in pancreatic tumor metabolism and if so, what is its relationship with Myc? Does Yap loss affect PDAC differentiation, and if so, how? Can PDAC cells adapt to Yap loss and if so, what are the underlying mechanisms? Finally, how do p53 status and/or disease stages influence the impacts of Yap ablation? This study seeks to address these important questions.
RESULTS
Ablation of Yap induces tumor regression and prolongs survival in mice bearing established Kras mutant pancreatic tumors
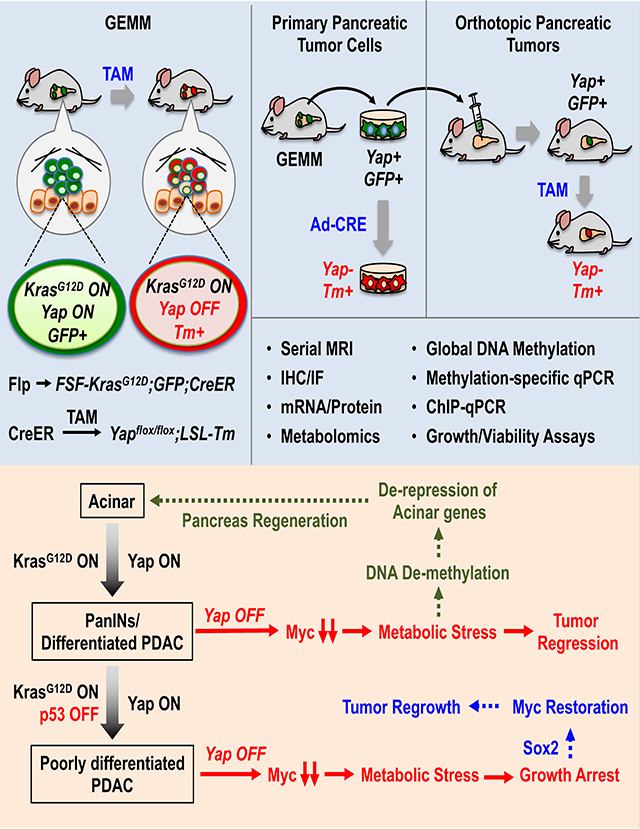
To determine the role of Yap in the maintenance of PDAC tumors, we developed a new inducible GEMM combining the Flp-FRT and Cre-loxP recombination systems, which allowed Yap to be switched off from spontaneously developed Kras mutant pancreatic tumors in immune competent mice (Figures 1A–1B). Also incorporated into the system is a dual-fluorescent reporter (R26dual), which marks the tumor cells according to their mutational statuses, enabling us to distinguish tumor cells from stromal cells and unrecombined normal tissues, and Yap competent from Yap deficient tumor cells (Figures 1A–1B). Two cohorts of mice, namely KF (FSF-KrasG12D/+;R26FSF-CreER/Dual;Yap+/+;Pdx1-Flp) and KYYF (FSF-KrasG12D/+;R26FSF-CreER/Dual;Yapflox/flox;Pdx1-Flp), are subjected to detailed analysis. In both cohorts, Flp-recombinase directed by the Pdx1 promoter (Pdx1-Flp) removes the FRT-flanked STOP cassettes from the FSF-KrasG12D, R26dual and R26FSF-CreER alleles in pancreatic progenitor cells, resulting in the expression of KrasG12D, EGFP and latent CreER throughout the pancreatic parenchyma (Figures 1A–1B) (Schönhuber et al., 2014). Without Tamoxifen (TAM) treatment, CreER remains inactive and cannot induce recombination in the Yapflox/flox alleles, and therefore Yap expression is maintained in both KF and KYYF mice (Figures 1A–1B).
Figure 1. Yap ablation induces tumor regression and prolongs survival in mice bearing Kras mutant pancreatic tumors.

(A) Genetic strategy to sequentially activate KrasG12D and delete Yap in the pancreas via the Flp-FRT and Tamoxifen (TAM)-induced Cre-loxP recombination systems. Note that FSF-KrasG12D and Yapflox/flox are under the separate controls of Flp and CreER, respectively. The R26dual reporter marks Flp-expressing KrasG12D cells with EGFP. Upon TAM-mediated CreER activation, the EGFP locus is removed, while tdTomato (Tm) is switched on.
(B) Illustration of the experimental design of animal studies. Mice were switched to TAM-containing diet only when the tumors become detectable via MRI.
KF: FSF-KrasG12D/+;R26FSF-CreER/Dual;Yap+/+;Pdx1-Flp.
KYYF: FSF-KrasG12D/+;R26FSF-CreER/Dual;Yapflox/flox;Pdx1-Flp.
(C) Representative images and tumor area quantification of sequential MRI of the pancreatic regions of KF and KYYF mice pre- or 3 months post- TAM treatment (top and middle panels) and representative photographs of pancreata resected from the same two mice after ~6 months of TAM treatment (bottom panel). Yellow dotted line marks the pancreas in each MRI image. Red arrows mark visible nodules on MRI images.
(D) Kaplan-Meier survival curve of KF (n = 10) and KYYF (n = 10) mice from the start of TAM treatment.
(E) Quantification of histopathological stages of KF and KYYF pancreata after being fed for indicated time periods with TAM-containing (+TAM) or regular diet (−TAM) starting from the time of detection of visible lesions via MRI. KF+TAM (1–3 months): n=12; KF+TAM (6–9 months): n=6; KYYF+TAM (1–3 months): n=12; KYYF+TAM (6–20 months): n=6; KF&KYYF-TAM (6–9 months): n= 5.
(F) Representative images and quantification of IF staining for Cleaved-Caspase 3 (Casp3), pH2AX or Ki67 (Green) in combination with tdTomato (red) and DAPI (blue) in KF and KYYF pancreata after ~1.5 month of TAM treatment. Scale bar =100 μm. n = 5.
*P < 0.05. **P < 0.005. ***P < 0.0005. Error bars indicate s.d.
We used periodic magnetic resonance imaging (MRI) to monitor disease progression over time, and confirmed through pilot studies that MRI-detectable lesions include both PanINs and PDAC with significant stromal buildup (Figure S1A). Only upon detection of multiple frank lesions via MRI, mice were switched to a TAM-containing diet to activate CreER, which induced LoxP-mediated recombination at the Yapflox/flox and R26dual alleles, resulting in simultaneous deletion of Yap and EGFP and activation of tdTomato (Tm) in the KrasG12D–expressing pancreatic neoplastic epithelial cells of KYYF mice (Figures 1A–1B and S1B). In contrast, Yap remained expressed in Tm+ tumor cells in KF mice (Figures 1B and S1B). Notably, as indicated by Tm and Yap staining, TAM-induced recombination is mosaic with the percentage of recombined (Tm+Yap−) cells gradually increases after extended treatment (Figure S1C–S1D). Despite the slow recombination rate, sequential MRI imaging indicated shrinkage of established pancreatic lesions (some to undetectable levels) in KYYF mice after three months of TAM treatment (Figure 1C). In contrast, existing lesions continued to grow while new nodules appearing in KF mice over the same time period under TAM treatment (Figure 1C). Consistent with findings from MRI, KYYF mice exhibited significantly prolonged survival compared to KF mice following TAM treatment (Figure 1D), which correlated with gradual decrease in advanced lesions (Figure 1E). These results demonstrate the requirement for Yap in maintenance of Kras mutant PDAC tumors.
Yap ablation triggers growth arrest and apoptosis in Kras mutant pancreatic tumor cells in vitro and in vivo
Corresponding to the shrinkage of frank lesions, the percentage of Cleaved-Caspase 3 (Casp3) positive apoptotic or pH2AX positive stressed cells increased dramatically within the Tm+Yap− population of KYYF pancreata but not in KF pancreata after 1.5 months of TAM treatment (Figures 1F and S1B). Conversely, the percentage of Ki67+ proliferating cells decreased significantly in KYYF relative to KF pancreata (Figures 1F and S1E). After extended TAM treatment, the vast majority of KYYF pancreata became completely quiescent except for a few remnant Tm+Yap+ ductal lesions that failed to undergo complete recombination (Figures S1E–S1F). Strikingly, the quiescent Tm+Yap− KYYF pancreatic cells remain positive for pERK and pS6 (Figure S1E), suggesting that Yap is not required for sustaining MAPK and mTOR signaling.
To elucidate the molecular functions of Yap in Kras mutant pancreatic tumor cells, we established primary culture from an invasive pancreatic tumor isolated from a KYYF mouse that did not undergo TAM treatment, and induced Yap knockout in these cells by infecting them with Ad-CRE or Ad-GFP as control (Figure 2A). While Ad-GFP treatment had no effect on Yap expression, cell proliferation or survival, Ad-CRE reduced cell proliferation associated with loss of Yap from 3 days post treatment, followed by a delayed buildup of cellular ROS and apoptotic makers (Figures 2B–2D). Similar to what was observed in vivo (Figure S1E), Yap loss in vitro also had little effect on pERK and pS6 levels in either low or high serum condition (Figures S1G–S1H), confirming that Yap controls the growth and survival of Kras mutant pancreatic tumor cells through mechanisms independent of the MAPK and mTOR pathways.
Figure 2. Yap functions as a master transcriptional regulator of multiple metabolic pathways that support nucleotide synthesis.

(A) Illustration of the experimental design of ex vivo studies. Primary pancreatic tumor cells were isolated from a tumor-bearing KYYF mouse that was not treated with TAM, and subsequently infected in vitro with Ad-CRE (CRE) to induce Yap deletion or Ad-GFP (GFP) as control.
(B-D) Relative cell growth rates (B), fold difference in median CellROX fluorescence (C), and percent of Annexin V positive cells (D) in Yap+ (GFP) and Yap+ (CRE) pancreatic tumor cells at 3 and 5 days post infection. n = 3.
(E) Log2 fold change (FC) in the levels of indicated metabolites as measured by LC-MS/MS in Yap− (CRE) relative to Yap+ (GFP) pancreatic tumor cells at 3 days post infection.
(F) P-value ranking of metabolic pathways significantly downregulated following Yap ablation based on targeted LC-MS/MS metabolomic analysis of Yap+ (GFP) and Yap− (CRE) pancreatic tumor cells at 3 days post infection. n = 4.
(G) Relative cell growth in KYYF cells treated with either GFP (−) or CRE (+) in the presence or absence of AICAR (12.5 μM) or nucleotide mix (125 μM). n = 3.
(H) Illustration of metabolic pathways associated with nucleotide synthesis. Metabolites are shown in black. Metabolic enzymes upregulated, downregulated or unchanged in response to Yap knockout are depicted in red, blue, and grey, respectively.
(I) Heatmap of relative mRNA levels of metabolic enzymes along indicated pathways in Yap+ (GFP) and Yap− (CRE) pancreatic tumor cells at 3 days post infection. n = 3.
*P < 0.05. **P < 0.005. ***P < 0.0005. ns: not significant. Error bars indicate s.d.
Yap maintains metabolic homeostasis in Kras mutant pancreatic tumor cells in vitro and in vivo
Oncogenic Kras-induced metabolic reprograming is essential for maintaining the growth and survival of pancreatic tumors (Son et al., 2013; Ying et al., 2012). Given that Yap has been recently implicated in direct regulation of enzymes involved in glucose and glutamine metabolism (Cox et al., 2016; Du et al., 2018; Edwards et al., 2017; Wang et al., 2015), we investigated the effects of Yap ablation on the metabolic state of Kras mutant PDAC cells by performing targeted liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of the aforedescribed Yap+ and Yap− primary pancreatic tumor cells at 3 days post GFP or CRE treatment. We selected this time point because Yap expression was sufficiently lost, yet oxidative stress and apoptosis had not initiated in CRE-treated Yap− cells (Figures 2B–D), thus precluding possible nonspecific effects of oxidative stress and cell death on cell metabolism at later times.
LC-MS/MS analysis revealed that Yap extinction caused significant downregulation of multiple metabolic pathways, the most significant of which were the pathways directly or indirectly associated with nucleotide biosynthesis, including virtually all the nucleobases, nucleosides and nucleotides as well as several key intermediate metabolites and amino acids that serve as the building blocks for pyrimidine and/or purine biosynthesis (Figures 2E–2F and S2A). Supplementing the cell culture medium with AICAR (an intermediate of purine synthesis) or nucleotide mixes partially rescued cell survival following Yap loss (Figure 2G), indicating that defects in nucleotide synthesis likely contributed to the growth arrest and cell death induced by Yap knockout.
To confirm that Yap ablation also blocks nucleotide synthesis in vivo, we injected the aforedescribed primary KYYF pancreatic tumor cells orthotopically into the pancreas of athymic mice. After allowing the tumors to develop without TAM treatment for approximately 3 weeks, they were either harvested or subjected to TAM treatment for additional 2 weeks (Figure S2B). As expected, TAM treatment induced mosaic Tm expression and Yap loss, similar to what we observed in the autochthonous model (Figures S1B, S1C and S2C). Despite the incomplete recombination and heavy stroma contamination, targeted LC/MS metabolomic analysis of TAM-treated and untreated pancreatic tumors showed significant downregulation of metabolites involved in purine and pyrimindine metabolism in TAM-treated tumors compared to untreated tumors (Figures S2C–S2E). These results demonstrate that Yap is also required for maintaining nucleotide synthesis in Kras mutant pancreatic tumors in vivo.
Yap functions as a master transcriptional regulator of multiple metabolic pathways that support nucleotide synthesis
Overexpression of a constitutive active yap mutant was recently reported to induce nucleotide synthesis in the zebrafish liver through transcriptional upregulation of glutamine synthetase (glul) (Cox et al., 2016). Contrary to this report, we observed slight upregulation in Glul mRNA levels following Yap ablation in Kras mutant pancreatic tumor cells (Figures 2H–2I). Re-examination of two published microarray datasets including one from our group (Shi et al., 2016; Yimlamai et al., 2014) showed that overexpression of Yap or knockout of Nf2 (a bona fide negative regulator of Yap) significantly downregulated Glul in the mouse liver (Figures S3A–S3B), again contradicting the above-mentioned zebrafish study.
Given that nucleotide biosynthesis depends on glycolysis, glutaminolysis, and serine/folate/glycine metabolism for key building blocks (Figure 2H), we next systematically examined how Yap loss may affect the expression of metabolic enzymes along these pathways. qRT-PCR analysis showed that Yap deletion caused rapid downregulation of numerous metabolic enzymes involved in glycolysis, serine/folate/glycine metabolism, and purine and pyrimidine synthesis (Figures 2H–2I), which was also confirmed by shRNA-mediated Yap knockdown in mouse and human PDAC cells (Figures S3C–S3E). On the other hand, Yap ablation had mixed effects on genes involved in glutamine metabolism, downregulating Gls and Gls2 while upregulating Glud1 and Glul without significantly affecting the expression of Slc1a5, Got1 or Got2 (Figures 2H–2I). These results reveal that rather than regulating a limited numbers of metabolic enzymes as previously reported (Cox et al., 2016; Du et al., 2018; Edwards et al., 2017; Wang et al., 2015), Yap systematically controls the transcriptional programs of multiple metabolic pathways to support nucleotide synthesis.
Yap partners with the TEAD family of transcription factors to directly transcribe the Myc gene to sustains nucleotide synthesis in vitro and in vivo
Myc is a bona fide master metabolic transcription factor and was previously implicated in metabolic reprogramming in Kras mutant PDAC (Stine et al., 2015; Ying et al., 2012). Yap was reported to promote Myc expression both at the transcriptional and posttranscriptional levels (Guo et al., 2017; Neto-Silva et al., 2010; Zanconato et al., 2015). A recent CHIP-seq study in the mouse liver showed that the Yap/Tead transcriptional complex cooperates in cis with Myc in promoting the transcription of genes important for growth and proliferation (Croci et al., 2017). We therefore investigated whether Yap functions through or in collaboration with Myc in maintaining the expression of the metabolic genes necessary for the survival of Kras mutant pancreatic tumor cells.
Without its own DNA binding domain (DBD), YAP partners with the TEAD family of transcription factors (TEAD1–4), which possess DBDs but no transaction domains, to regulate gene expression (Zanconato et al., 2016). Taking advantage of the publicly available ChIP-seq datasets from ENCODE (Encyclopedia of DNA Elements) and other sources (STAR Methods), we first examined the proximal MYC promoter for possible binding by TEAD. We found that TEAD1, TEAD3 and TEAD4 consistently bind across multiple cancer cell lines at three major sites (p1–3) along an approximately 4kb span from the transcription start site of the MYC gene, which overlap with the H3K27Ac active transcription marks (Figure 3A). We confirmed via ChIP that in primary murine pancreatic tumor cells, Tead3 binds to three conserved regions of the mouse Myc promoter corresponding to the TEAD-binding peaks in human cells, but not at the 3’ UTR (Figure 3B). We also performed Yap ChIP in Yap+ or Yap− pancreatic tumor cells, which showed specific enrichment of Yap antibody to the three TEAD-binding sites in Yap+ but not Yap− cells (Figure 3C). Finally, qRT-PCR, western blot and IF analyses showed that ablation of Yap or Teads from Kras mutant pancreatic tumor cells reduced both the mRNA and protein levels of Myc in vitro and in vivo (Figures 3D–3F and S3C-S3F), demonstrating that the Yap/Tead transcriptional complex directly promotes the expression of Myc.
Figure 3. The Yap/Tead complex directly transcribes Myc and cooperates with Myc in promoting the expression of metabolic enzymes that maintain growth and survival in Kras mutant pancreatic tumor cells.

(A) Graphic representation of ChIP-Seq data showing enrichment peaks of H3K27ac, TEAD1, TEAD3, and TEAD4 along the human MYC gene in HepG2, HCT-116, A549, MCF-7 and ECC-1 cells.
(B) ChIP and qRT-PCR analysis in pancreatic tumor cells with Tead3 antibody or IgG control using primers targeting regions on the mouse Myc promoter that correspond to the TEAD-binding peaks (p1–3) and a 3′UTR region as negative control (nc) as shown in A. n = 3.
(C) ChIP and qRT-PCR analysis in Yap null pancreatic tumor cells reconstituted with vector control (Yap−) or Flag-Yap (Yap+) using primers targeting regions on the mouse Myc promoter that correspond to the TEAD-binding peaks (p1–3) and a 3′UTR region as negative control (nc) as shown in A. n = 3.
(D) Relative endogenous Myc mRNA levels in KYYF cells at 3 and 5 days after GFP or CRE treatment. n = 3.
(E) Western blot analysis of indicated proteins in KYYF cells at different days post CRE treatment. Vinculin (Vinc) was used as the loading control. Shown is representative of at least three independent experiments.
(F) Representative images of IF staining for tdTomato (red), Myc (green), GFP (magenta) and DAPI (blue) in untreated (−TAM) or TAM-treated (+TAM) orthotopic pancreatic tumors. Scale bar represents 50 μm.
(G) Percent of proliferating cells as determined by EdU incorporation assay (left) or apoptotic cells as determined by TUNEL assay (right) in KYYF cells stably expressing vector control or human MYC at 5 days post infection with Ad-GFP (−) or CRE (+). n = 3.
(H) Heatmap of relative mRNA levels of indicated metabolic genes in KYYF cells stably expressing vector control or human MYC at 5 days post GFP or CRE treatment. n = 3.
(I) ChIP and qRT-PCR analysis in pancreatic tumor cells with rabbit IgG(r), rabbit MYC(r), Mouse IgG(m), or mouse Tead3(m) antibodies using primers targeting the promoters of indicated genes. n = 3.
*P < 0.05. **P < 0.005. ***P < 0.0005. ns: not significant. Error bars indicate s.d.
Yap and Myc cooperate at multiple levels to maintain the expression of metabolic genes required for pancreatic tumor cell proliferation and survival
To determine how downregulation of Myc contributes to the phenotypes induced by Yap loss, we generated KYYF lines stably expressing exogenous human MYC or vector control, and treated them with either Ad-GFP or Ad-CRE. Overexpression of either MYC prevented apoptosis and cell cycle arrest induced by Yap ablation (Figure 3G), confirming the inhibition of Myc as the major cause of cell death and growth arrest following Yap loss in Kras mutant pancreatic tumor cells.
Notably, even though MYC overexpression rescued the growth and survival of Yap-deleted PDAC, it did not fully over-write the inhibitory effects of Yap ablation on cell proliferation, as indicated by the significant reduced growth rates of CRE-treated relative to GFP-treated MYC overexpressing cells (Figure 3G). Correspondingly, while MYC overexpression rescued the expression of all the metabolic genes that were downregulated in control cells following Yap ablation, nearly half of those genes were expressed at significantly lower levels in CRE-treated versus GFP-treated MYC-overexpressing cells (Figure 3H), indicating that they are likely subjected to additional regulation by Yap independent of Myc.
To assess the functional hierarchy between the Yap/Tead transcriptional complex and Myc in regulating metabolic genes, we compared the occupancies of TEAD4 and MYC on the proximal promoters of all the Yap-regulated metabolic genes in matched TEAD4 and MYC ChIP-seq datasets from four different cancer cell lines (STAR Methods). Strikingly, in all the cell lines examined clear, TEAD4 was enriched at the active transcription sites (as indicated by H3K27Ac) of over half of these genes, all of which also exhibited robust MYC binding at overlapping or adjacent sites (Figure S3G). ChIP analysis confirmed that Myc and Tead also co-occupied the promoters of Ldha, Prps1, Tyms and Mthfd2 in primary murine pancreatic tumor cells (Figure 3I). In contrast, the Pgam1 promoter was bound by Myc but not by Tead3, whereas the canonical Tead target Cyr61 showed strong enrichment for Tead3 but not Myc (Figure 3I). Thus, the Yap/Tead transcription complex may function either in conjunction with or through Myc to regulate the transcription of metabolic genes (Figure S3H).
Upregulation of Sox2 compensates for Yap loss, restoring Myc expression, metabolic homeostasis and survival in a subset of Yap deficient pancreatic tumor cells
Despite the profound cell death and growth arrest induced by Yap ablation (Figures 2B–2D), a significant fraction of Kras mutant pancreatic tumor cells survived long term Yap loss, and over time regained ROS homeostasis (Figure S4A). This revival coincided with recovery in the expression of Myc and many of the metabolic enzymes downregulated following acute Yap loss (Figures 4A–4B), pointing to the existence of compensatory mechanism(s) that allow long-term (LT) surviving Yap− pancreatic tumor cells to restore Myc expression and Myc-controlled metabolic programs.
Figure 4. Upregulation of Sox2 compensates for Yap loss, restoring Myc expression, metabolic homeostasis and survival in a subset of Yap deficient pancreatic tumor cells.

(A) Heatmap of relative mRNA levels of indicated genes in Yap+ parental (P) KYYF cells or Ad-CRE-treated KYYF cells at day 3 (d3), day 5 (d5), and >2 weeks (long term, LT) post infection. n = 3.
(B) Western blot analysis of indicated proteins in Yap+ parental (P) and two long-term Yap-deleted KYYF lines (Yap− LT #1 and #2). Vinc was used as the loading control. Shown is representative of at least three independent experiments.
(C) Log2 FC in mRNA expression of indicated genes in two long-term Yap-deleted (Yap− LT #1 and #2) relative to Yap+ parental KYYF cells. n = 3.
(D) Representative IHC images of Sox2 in KF pancreata after ~1.5 months of TAM treatment and KYYF pancreata after ~1.5 or >6 months of TAM treatment. Scale bars represent 50 μm.
(E) Western blot analysis of Sox2 and Myc proteins in Yap− LT KYYF cells at 3 days post infection with lentivirus carrying vector control or two independent Sox2 shRNAs. Vinc was used as the loading control. Shown is representative of at least three independent experiments.
(F) Relative mRNA levels of indicated EMT genes as determined by qRT-PCR analysis in Yap− LT KYYF cells at 3 days post infection with lentivirus carrying vector control or two independent Sox2 shRNAs. n = 3.
(G-H) Percent of apoptotic cells as determined by TUNEL assay (G) or proliferating cells as determined by EdU assay (H) in Yap− LT KYYF cells at 5 days post infection with lentivirus carrying vector control or two independent Sox2 shRNAs. n = 3.
(I) Representative image (left) and quantification (right) of crystal violet staining of Yap− LT KYYF cells at 5 days post infection with lentivirus carrying vector control or two independent Sox2 shRNAs.
(J) Relative mRNA levels of indicated genes as determined by qRT-PCR analysis in Yap− LT KYYF cells at 3 days post infection with lentivirus carrying vector control or two independent Sox2 shRNAs. n = 3.
(K) ChIP and qRT-PCR analysis in Yap+ and Yap− murine pancreatic tumor cells with Sox2 antibody using primers targeting an enhancer (En), exon 1 (Ex1), exon 2 (Ex2) and 3-UTR (3utr) regions of the Myc gene, normalized to IgG control. n = 3.
*P < 0.05. **P < 0.005. ***P < 0.0005. ns: not significant. Error bars indicate s.d.
TAZ, a paralog of Yap, has been shown to be upregulated in response to Yap loss and compensate for its function (Moroishi et al., 2015). However, we did not observe any increase in Taz expression following Yap ablation in vitro or in vivo (Figures S4B-S4C). Knockdown of Taz also did not significantly impact the growth of pancreatic tumor cells in the presence or absence of Yap (Figure S4D), suggesting that Taz cannot functionally replace Yap in sustaining pancreatic tumor growth.
Overexpression of YAP has been previously shown to allow KRAS mutant colon cancer cell line HCT-116 to survive KRAS silencing by upregulating the epithelial-mesenchymal transition (EMT) program (Shao et al., 2014). Given the important roles for EMT-related genes in promoting stemness, heterogeneity and resistance in tumor cells and the numerous reports demonstrating YAP as a driver of EMT (Park et al., 2018; Zanconato et al., 2016; Zhang and Weinberg, 2018), we compared the expression of EMT-related genes in Yap+ and Yap− LT-surviving pancreatic tumor cells. Contrary to what we expected based on the existing literature, a number of canonical EMT genes including Sox2, Snai1, Zeb2 and Twist2 were significantly upregulated in Yap− cells compared to Yap+ cells, whereas Snai2 was significantly downregulated (Figures 4B–4C). The upregulation of Sox2 was also apparent in vivo in KYYF pancreata relative to KF pancreata after ~1.5 months of TAM treatment (Figure 4D). Despite the upregulation of several EMT genes, Yap− cells did not upregulate Vim or Zeb1 – two most widely accepted mesenchymal markers (Figure 4C). In agreement with these in vitro observations, Tm+Yap− Kras mutant neoplastic cells also maintained E-Cad expression and epithelial morphology in vivo (Figure S4E). These data suggest that Yap loss induces a partial but not overt EMT program in Kras mutant pancreatic tumor cells.
Sox2 has been previously shown to promote EMT and stemness in many types of tumor cells including human PDAC cells (Herreros-Villanueva et al., 2013; Singh et al., 2015; Wuebben and Rizzino, 2017). We therefore investigated whether the upregulation of Sox2 could be responsible for inducing the partial EMT program and allowing pancreatic tumor cells to survive Yap ablation. Knockdown of Sox2 with two independent shRNAs caused dose-dependent downregulation of Snai1, Twist2 and Zeb2 and induction of cell death and growth arrest in Yap null pancreatic tumor cells, which corresponded to significant reduction in the expression of Myc and Myc-regulated metabolic genes (Figures 4E–4J). Finally, we confirmed via ChIP-qPCR that Sox2 specifically binds to a previously reported enhancer region and exons 1 and 2 but not the 3’-UTR of the Myc gene in Yap null pancreatic tumor cells (Figure 4K) (Aksoy et al., 2013; cheloufi et al., 2015; Chronis et al., 2017; Deluz et al., 2016; King and Klose, 2017; Kwan et al., 2015). These results suggest that upregulation of Sox2 could compensate for Yap loss to rescue Myc expression and metabolic homeostasis, allowing pancreatic tumor cells to survive Yap ablation.
Yap loss induces gradual re-differentiation of Kras mutant neoplastic ductal cells into Amylase-secreting acinar-like cells and regeneration of pancreatic parenchyma
After establishing that a subset of Kras mutant pancreatic tumor cells are capable of surviving long term Yap loss in vitro and in vivo, we next examined the fates of surviving Yap-null pancreatic tumor cells in the autochthonous model (Figures 1A–1B). We found that following ~1.5 months of TAM treatment, KYYF pancreata exhibited a significant increase of hybrid neoplastic ductal structures containing a mixture of Amy+ (Amylase, acinar marker) and CK19+Sox9+ (ductal markers) cells (Figures 5A–5B and S4F–G), in addition to the afore-observed growth arrest and increased apoptosis (Figure 1F). Importantly, these Amy+ (including Amy+CK19+Sox9+) cells emerged within the KYYF neoplastic ductal structures were also Tm+, indicating that they derived from Yap-null pancreatic tumor cells (Figures 5A and S4G). In contrast, Amy+ cells were limited mostly to Tm−GFP− unrecombined normal tissues in TAM-treated KF pancreata (Figures 5A and S4G). Over time, KYYF pancreata underwent dramatic remodeling, characterized by a progressive increase in the numbers of Amy+Tm+Yap− acini-like structures dispersed through connective tissues, accompanied by gradual decreases in neoplastic ducts that were positive for Alcian blue, CK19 and Yap (Figures 5B, S1F and S5A). Finally, we confirmed that re-expression of Amy could also be detected in KYYF orthotopic tumors after TAM treatment (Figure S5B).
Figure 5. Yap loss induces gradual re-differentiation of Kras mutant neoplastic ductal cells into Amylase-secreting acinar-like cells and regeneration of pancreatic parenchyma.

(A) Representative images of IF staining for Amylase (Amy; red), CK19 (green), Tm (gray) and DAPI (blue) in KF and KYYF pancreata after ~1.5 months of TAM treatment. Yellow arrowheads indicate Amylase/CK19/Tm triple positive cells. Scale bars represent 500 μm (top) and 200 μm (bottom).
(B) Representative HE and IHC images of Amylase and Alcian Blue in KYYF pancreata before, after ~1.5 or >6 months of TAM treatment and KF pancreata after ~6 months of TAM. Yellow arrows mark the ductal/acinar transitional structures observed after short term TAM treatment. Scale bars represent 500 μm (top) and 25 μm (bottom).
Besides Amy, qRT-PCR and IHC analysis confirmed that extended TAM treatment induced the re-expression of acinar-specific transcription factors p48/Ptf1a and Mist1/Bhlha15 while downregulating ductal markers Sox9 and Krt19 in KYYF but not in KF pancreata (Figures S6A–S6C). Similar changes were also observed in long-term surviving Yap− cells compared to Yap+ pancreatic tumor cells in vitro (Figure S6D). Endocrine progenitor markers Onecut1, NeuroG3 and NeuroD1 were also upregulated in long-term surviving Yap− cells in vitro and in regenerated KYYF pancreata, although Hnf1a, a transcription factor necessary for maintaining the homeostasis of differentiated endocrine cells (Pontoglio et al., 1998), was largely unaffected (Figures S6B and S6D). Other differentiated endocrine markers such as Nkx6.1 and Pdx1 were undetectable in either Yap+ or Yap− cells. IF analysis with antibodies against Insulin (Ins, marker of β cells) and Glucagon (Gcg, marker of α cells) showed that there were no significant differences in the percentages of Ins+Tm+ and Gcg+Tm+ within the neoplastic ductal structures of KF and KYYF pancreata after TAM treatment (Figure S6E), suggesting that despite acquiring both acinar and endocrine progenitor makers, Yap− neoplastic ductal cells preferentially re-differentiate into acinar-like cells.
Metabolic-crisis-triggered epigenetic reprogramming drives Sox2 upregulation and lineage shift following Yap ablation in pancreatic tumor cells
Contrary to the rapid decrease in the expression of Myc and metabolic genes following Yap ablation (Figure 4A), the changes in the expression of lineage markers occurred slowly, and did not peak until more than one week after Yap was first deleted (Figure S6F). Even though the kinetics of the lineage shift closely followed that of Sox2 upregulation (Figure S6G), Sox2 knockdown had very little effects on the expression of these genes in Yap− cells in vitro (Figure S6H), and Sox2 expression eventually became barely detectable in regenerated acinar-like cells in KYYF pancreata after >6 months of TAM treatment (Figure 4D). These results suggest while Sox2 plays a critical role in rescuing Myc expression and cell survival upon Yap loss, it does not drive the re-expression of acinar lineage genes.
DNA-methylation-mediated gene silencing is critical for the establishment and maintenance of lineage commitment and cellular identity (Suelves et al., 2016). DNA methylation requires methyl groups to be donated through conversion of S-Adenosyl methionine (SAM) to S-Adenosyl homocysteine (SAH), both of which were significantly downregulated in response to Yap ablation in primary pancreatic tumor cells (Figure 2E). We therefore investigated whether DNA demethylation could be responsible for reactivating pancreatic lineage genes following Yap loss.
First, we confirmed via quantitative methylation-specific PCR (qMSP) that the promoters of acinar lineage genes Ptf1a and Bhlha15 became heavily methylated in KF pancreatic tumors in contrast to wild type (WT) pancreata, which was partially reversed in KYYF pancreata after TAM treatment (Figure S7A). To directly assess the effects of DNA de-methylation on the expression of pancreatic lineage genes, we treated Yap+ primary pancreatic tumor cells with vehicle control or DNA methylation inhibitor 5-azacytidine (5-Aza). Along with global DNA de-methylation (Figure S7B), 5-Aza treatment induced dose-dependent upregulation acinar and endocrine lineage genes as well as Sox2 (Figure 6A), demonstrating that DNA methylation is at least partially responsible for silencing these genes in Yap+ pancreatic tumor cells. Next, we measured the levels of global DNA methylation in primary KYYF pancreatic tumor cells following treatment with Ad-GFP or Ad-CRE, and confirmed that Yap ablation induced a rapid and significant decline in global DNA methylation, which partially recovered over time (Figure 6B). Given that the global DNA de-methylation coincided with the drop in SAM and SAH levels following Yap deletion (Figure 2E), we tested whether supplementing the growth medium with SAM/SAH after CRE treatment could prevent the reactivation of pancreatic lineage genes. Strikingly, addition of exogenous SAM/SAH, which did not prevent the proliferation decrease in CRE-treated KYYF cells (Figure S7C), caused complete or near complete silencing of pancreatic lineage genes including Ptf1a, Bhlha15, Amy2A, Onecut1, NeuroG3 and NeuroD1 and strongly suppressed Sox2 upregulation, but had no effect on the expression of Hnf1a (Figure 6C). Interestingly, SAM/SAH also caused downregulation in ductal markers Hes1, Sox9 and Krt19 to various degrees, suggesting that these genes may also subjected to some degree of regulation by methylation (Figure 6C).
Figure 6. Metabolic-stress-triggered epigenetic reprogramming drives Sox2 upregulation and lineage shift following Yap ablation in pancreatic tumor cells.

(A) Relative mRNA levels of indicated genes in KYYF cells treated with DMSO or 0.5, 2, 5 μM of 5-Azacytidine (5-Aza) for 3 days. n = 3.
(B) Percent of global DNA methylation in KYYF cells at 3 and 14 days post infection with Ad-GFP or CRE. n = 3.
(C) Relative mRNA levels of indicated genes in KYYF cells at 14 days post infection with Ad-GFP or Ad-CRE in the presence or absence of SAM/SAH supplement. nd: not detectable. n = 3.
(D) Experimental design of examining the effects of nutrient stress on KYYF cells.
(E) Percent of global DNA methylation in KYYF cells incubated for 2 days in normal or – Glc/Gln/Pyr medium, followed by recovery in normal growth medium for additional 8 days. n = 3.
(F) Relative mRNA levels of indicated genes in KYYF cells incubated for 2 days in normal or –Glc/Gln/Pyr medium, followed by recovery in normal growth medium for additional 12 days. n = 3.
(G) Relative mRNA levels of indicated genes in KYYF cells overexpressing MYC or vector control at 14 days post GFP or CRE treatment. n = 3.
(H) Schematic illustrating the proposed mechanisms of reactivation of Sox2 and acinar lineage genes following Yap ablation from pancreatic tumor cells based on results from this figure.
*P < 0.05. **P < 0.005. ***P < 0.0005. ns: not significant. Error bars indicate s.d.
To confirm that the metabolic stress was indeed the trigger of global DNA de-methylation and lineage shift in PDAC cells following Yap loss, we starved Yap+ pancreatic tumor cells for two days in growth media deprived of Glc, Gln and Pyr, followed by recover in normal growth media for addition 12 days (Figure 6D). As shown in Figures 6E–6F, temporary deprivation of carbon sources induced global demethylation accompanied by upregulation of Sox2 and acinar and endocrine lineage markers in Yap+ pancreatic tumor cells, recapitulating the effects of 5-Aza treatment (Figures 6A and S7B). Finally, we confirmed that overexpression of MYC, which prevented the short-term growth arrest and cell death induced by CRE treatment (Figure 3G), either completely or partially suppressed the acquisition of acinar and endocrine genes at 14 days after virus infection (Figure 6G).
Together, our data support a model in which Yap ablation from pancreatic tumor cells causes acute metabolic crisis, which triggers not only cell cycle arrest and apoptosis but also DNA de-methylation and epigenetic reprogramming, resulting in Sox2 upregulation that restores Myc expression and metabolic homeostasis, and de-repression of acinar lineage genes that gradually convert the surviving Yap-deficient neoplastic ductal cells into acinar-like cells (Figure 6H).
Yap ablation induces temporary growth inhibition but not apoptosis or acinar re-differentiation in advanced, poorly differentiated p53 mutant pancreatic tumors
Aside from KRAS, human pancreatic tumors frequently contain additional mutations in TP53, which can profoundly alter metabolic, differentiation, and metastatic states of PDAC cells (Bailey et al., 2016; Mueller et al., 2018; Schofield et al., 2018). P53 deficiency was recently reported to further activate Yap in Kras mutant PDAC through suppression of a negative regulator - Ptpn14 (Mello et al., 2017). We previously showed that deletion of Yap blocked PDAC initiation driven by either Kras and Kras:Trp53 mutations and knockdown or knockout of Yap reduced the proliferation of Kras:p53 mutant PDAC cells in vitro and in vivo (Zhang et al., 2014). qRT-PCR analysis confirmed that acute knockdown of YAP also reduced the expression of MYC and metabolic genes in primary and established p53 mutant human PDAC cells (Figures S7D–S7E). However, in contrast to p53 WT primary pancreatic tumor cells, p53 deficient primary pancreatic tumor cells did not exhibit significant increase in oxidative stress or apoptosis in response to Yap deletion, and quickly upregulated Sox2 and regained Myc expression and the ability to proliferate in vitro (Figures 7A–7D). Importantly, deletion of Sox2 restored Yap-deletion-induced apoptosis in p53 deficient pancreatic tumor cells (Figure 7E), demonstrating a critical role for Sox2 in maintaining pancreatic tumor cell survival upon Yap loss.
Figure 7. YAP loss does not induce apoptosis or acinar re-differentiation in poorly-differentiated, p53 deficient pancreatic tumor cells.

(A-B) Fold difference in median CellROX fluorescence (A) and percentage of apoptotic cells as determined by TUNEL assay (B) at 5 days post infection with Ad-GFP or Ad-CRE in KYY and KPYY pancreatic tumor cells. n = 3.
(C) Changes in the ratios of KPYY cells pretreated with GFP+ or Tm+ and co-cultured over indicated time and analyzed periodically by flow cytometry. n = 3.
(D) Western blot analysis of Sox2, Myc and Yap proteins in Yap− KPYY cells at indicated days post infection of Ad-GFP or Ad-CRE. Vinc was used as the loading control. Shown is representative of at least three independent experiments.
(E) Quantification and representative flow cytometry plots of Annexin-V/CYTOX-Blue staining of parental or crispr-Sox2-knockout (crSox2) KPYYF cells at 5 days post infection post infection with Ad-GFP or Ad-CRE. n = 3.
(F) Representative IHC images of Yap, Ki67, Casp3, Myc, Sox2, and Amy of orthotopic KPYY pancreatic tumors that were either untreated (−TAM) or treated with TAM for one week (ST) or > 2 months (LT). Scale bar = 100 μm.
**P < 0.005. ***P < 0.0005. ns: not significant. Error bars indicate s.d.
Consistent with these in vitro findings, we found that Yap ablation induced temporary Myc downregulation and growth inhibition but not apoptosis in orthotopic pancreatic tumors derived from p53 deficient primary pancreatic tumor cells; over time, Yap-deleted tumors regained Myc expression and resumed growth, which correlated with Sox2 upregulation (Figures 7C–7F). Interestingly, we found that Yap ablation failed to induce Amylase re-expression in these poorly differentiated, p53 deficient tumors even after >2 months of TAM treatment (Figure 7F). To test whether p53 deficiency or the advanced tumor stage prevents Yap null pancreatic tumor cells from re-differentiating into Amylase-expressing acinar like cells, we performed histopathological and IHC analysis of the pancreas from a 4-month-old KPYYF mouse that was treated for one month with TAM. As in the orthotopic model, we found that Yap ablation failed to reactivate Amylase in the poorly differentiated regions of the autochthonous tumors developed in the KPYYF mouse (Figure S7F). In contrast, in the well-differentiated tumor regions of the same pancreas, Yap loss readily induced Amylase re-expression along the neoplastic ducts (Figures S7F).
Together, these results suggest that while well-differentiated p53 deficient pancreatic tumors retain the ability to transdifferentiate into acinar-like cells upon Yap loss, inactivation of p53 promotes tumor de-differentiation and increases tumor plasticity, thereby blunting the impact of Yap loss on tumor growth, survival and differentiation.
DISCUSSION
Using the classic Cre-loxP–based KPC model, we were the first to report Yap as a key driver of PDAC initiation upon activation of oncogenic Kras (Zhang et al., 2014). Here by taking advantage of a next-generation inducible GEMM incorporating the Flp-FRT and Cre-loxP recombination platforms with a dual-fluorescence reporter (Schönhuber et al., 2014), we have successfully inactivated Yap in established pancreatic tumors and tracked the fates of Yap-deficient tumor cells over time. We show that Yap ablation, resembling previously reported Kras silencing, causes metabolic crisis and regression of PDAC tumors, which is followed by extensive tissue re-modeling cumulating in partial recovery of acinar parenchyma (Collins et al., 2012; Ying et al., 2012). Strikingly, tumor regression and tissue regeneration occurred despite the persistence of RAF/MEK/ERK and PI3K/AKT/mTOR signaling, thus establishing Yap as a central downstream effector of oncogenic Kras in pancreatic tumor maintenance.
Recent studies including our own have linked Yap to the transcription of metabolic enzymes Glut3, Slc1a5, Glul and Gls involved in glucose or glutamine metabolism (Cosset et al., 2017; Cox et al., 2016; Du et al., 2018; Edwards et al., 2017; Wang et al., 2015; White et al., 2019a). Here we show that in pancreatic tumor cells, Yap functions in tandem with Myc to regulate global transcription of metabolic enzymes along the glycolysis, glutaminolysis, serine/glycine/folate and nucleotide synthesis pathways. Knockout of Yap from Kras mutant pancreatic tumor cells induced profound Myc downregulation and metabolic collapse, resulting in oxidative stress, growth arrest, and apoptosis, which could be prevented by overexpression of Myc. Supplementing the cell culture medium with nucleotide mixtures also partially rescued the growth arrest caused by Yap loss, suggesting that a key role for Yap is to sustain the nucleotide pool necessary for the growth and survival of pancreatic tumors.
Yap has well-established functions in promoting EMT and stemness (Park et al., 2018; Zanconato et al., 2016). Unexpectedly, we found that a subset of Kras mutant pancreatic tumor cells were able to adapt to Yap loss by upregulating Sox2 and other known stem/EMT transcription factors. Intriguingly, we determined that following Yap ablation, DNA demethylation triggered by the acute metabolic stress was responsible for induction of Sox2, which in turn restores Myc expression and metabolic homeostasis in a subset of Yap deficient pancreatic tumor cells. These findings indicate that Kras mutant pancreatic tumor cells can activate EMT programs in the absence of Yap.
Beside upregulating Sox2 and other EMT/stemness genes, we find that metabolic-stress-induced DNA demethylation following Yap loss also triggers de-repression of acinar lineage genes. Through lineage tracing, we showed that the majority of the regenerated acinar-clusters in TAM-treated KYYF pancreata were derived from neoplastic tumor cells rather than due to incomplete recombination. Moreover, Yap loss also caused primary pancreatic tumor cells to reacquire the expression of acinar lineage genes in vitro and in vivo. Importantly, the re-activation of acinar markers in response to Yap loss could be blocked by overexpression of Myc that relieve metabolic crisis, or by addition of exogenous SAM/SAH to restore the methionine cycle. Conversely, temporary withdrawal of carbon sources or treatment with a DNA methylation inhibitor was sufficient to re-activate acinar lineage genes in Yap+ pancreatic tumor cells. Together, these results indicate that Yap suppresses the expression of acinar lineage genes by maintaining metabolic homeostasis and epigenetic balance in pancreatic tumor cells.
Yap and Taz, both highly expressed in ductal cells, were shown to function redundantly of each other in maintaining the expression of ductal genes (Gruber et al., 2016). Consistent with this study, we find that Yap deletion caused only slight downregulation of ductal genes in pancreatic tumor cells. In contrast, Yap loss profoundly inhibited the expression of Myc and other metabolic genes resulting in metabolic collapse, suggesting that PDAC cells rely primarily on Yap but not Taz for maintaining metabolic homeostasis. Indeed, while Yap ablaton dramatically inhibited cell proliferation and survival, silencing of Taz had minimal effects on the growth of PDAC cells in the presence or absence of Yap.
Over 70% of human PDACs contain TP53 mutations, which promote PDAC progression into a more aggressive and poorly differentiated basal-like/squamous subtype (Bailey et al., 2016; Hingorani et al., 2005; Vanderas and Papagiannoulis, 2002). We previously showed that Yap deletion completely blocks PDAC initiation driven by both Kras and p53 mutations (Zhang et al., 2014). Unexpectedly, we found that despite the downregulation of Myc and metabolic genes, Yap deficiency temporary slowed the growth but did not induce apoptosis in primary p53 deficient PDAC cells. A previous study showed that p53 suppresses EMT and de-differentiation of PDAC cells by transcribing miRNA-200c, which directly targets and inhibits Sox2 mRNA (Singh et al., 2015). Consistent with this report, we found that p53 deficiency accelerated the compensatory Sox2 upregulation upon Yap loss, and deletion of Sox2 restored apoptosis in response to Yap loss in p53 deficient PDAC cells. Besides promoting cell survival, our study suggests that p53 inactivation inhibits the re-differentiation of Yap deficient pancreatic tumor cells into acinar-like cells by promoting further de-differentiation.
In summary, our study establishes dual roles for Yap in pancreatic tumor maintenance: it sustains Myc expression and tumor metabolism, while preventing the re-differentiation of neoplastic ductal cells. Interestingly, we find that the second function of Yap in suppressing re-differentiation is dependent on its first function in maintaining metabolic homeostasis, which governs the epigenetic landscape of neoplastic ductal cells. Combined with our recent discovery of Yap as a critical inducer of tumor suppressive microenvironment in PDAC (Murakami et al., 2017; White et al., 2019b) and previous studies establishing Yap as a main resistance mechanism towards Kras inhibition (Kapoor et al., 2014; Shao et al., 2014), our study further ratifies Yap as a true achilles’ heel and compelling target for Kras mutant pancreatic tumors. Furthermore, our work illustrates the extraordinary capacity of PDAC cells to survive and adapt to cellular, environmental or nutrient stress especially in the presence of p53 mutations and highlights the profound and long-lasting influences of temporary metabolic changes on the epigenetic landscape and vice versa, and predicts that sustained tumor control in advance p53 mutant PDAC will likely require comprehensive targeting of a redundant transcriptional network including Yap, Myc, Sox2 and possibly other factors.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Chunling Yi (cy232@georgetown.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal studies
All animal studies were conducted in compliance with ethical regulations according to protocol #2016–1192 approved by the Institutional Animal Care and Use Committee at Georgetown University.
Genetically engineered mouse model of PDAC
Genetically engineered mouse strains Yapflox/flox, FSF-KrasG12D, R26CreER, R26Dual, and Pdx1-Flp were interbred to generate the experimental cohorts (Schönhuber et al., 2014; Zhang et al., 2010). TAM Diet (400 TC, 2016) purchased from Envigo RMS Inc, Indianapolis, IN was given in place of regular feed. To track pancreatic tumor growth, mice were subjected to longitudinal MRI every 3 months using a 7-Tesla horizontal Bruker spectrometer run by Paravision 5.1 as previously described (Sirajuddin et al., 2012).
Orthotopic mouse model of PDAC
5×104 primary KYYF and KPYYF PDAC cells infected with lentiviruses carrying a dual GFP-Luc reporter were injected into the pancreas of Athymic Nude mice (Charles River, Frederick MD) following surgical steps as previously described (Sirajuddin et al., 2012). The growth of orthotopic tumors was tracked by BLI using the IVIS Lumina Series III (PerkinElmer, Waltham, MA). For BLI, mice were administered D-luciferin (150 mg/kg; Thermo Fisher Scientific, Waltham, MA) via intraperitoneal injection immediately after anesthesia with inhalation of 2% isoflurane (Baxter Healthcare Corporation Deerfield, IL). TAM diet was initiated when the total flux from an orthotopic tumor reaches 1×108 p/s or above.
METHODS DETAILS
Generation of primary mouse PDAC cell culture
To establish primary PDAC cell culture, KYYF and KPYYF tumor-bearing mice were sacrificed and the pancreatic tumors were resected, minced, and digested with 6mg/ml Collagenase D (Sigma, St. Louis, MO), sorted by FACS for GFP fluorescence, plated on collagen-coated plates in DMEM medium (CORNING, Corning, NY) containing 10% fetal bovine serum. To induce Yap knockout, cells were incubated for 24 hrs in medium containing Ad-CMV-GFP or Ad-CMV-CRE (Iowa viral vector core, Iowa city, IA) before being switched back to normal growth medium with or without the following additives: 125 μM of nucleobase mixture including adenine (Sigma), cytosine (Thermo Fisher Scientific), guanine (Sigma), inosine (Thermo Fisher Scientific) and thymine (Alfa Aesar, Haverhill, MA); 12.5 mM of AICAR (Cayman Chemicals, Ann Arbor, MI). For assays using 96-well plates, 500 cells were plated into each of collagen-coated wells and allowed to attach overnight before viral infection. The total cell number in each well was estimated using CellTiter-Glo kit (Promega, Madison, WI) according to manufacturer’s instruction. For assays using 24-well plates, 30,000 cells were used.
Culture of primary and established human PDAC cells
Colo-357 cells and PANC-1 cells were obtained from ATCC (Manassas, VA) and cultured in DMEM medium containing 10% FBS. Human Primary PDAC cells were established from surgically resected primary pancreatic tumors. All patients were treated in the Department of Surgical Oncology at The Technical University of Munich, and gave informed consent prior to surgery and tumor collection. The tissue was cultured in RPMI-1640 with 5 mg/ml BSA (Sigma), 10 ng/ml EGF (Thermo Fisher Scientific), 2.5 mM L-glutamine (Thermo Fisher Scientific), Hydrocortison (Sigma), Insulin-Transferrin-NaSelenite Premix (Thermo Fisher Scientific), NaPyruvate (Thermo Fisher Scientific), and T3 (Sigma). All media were supplemented with 1% penicillin and streptomycin. The cells were maintained at 37°C in a humidified atmosphere of 5% CO2, and the medium was replaced every 3 days. Sanger sequencing was used to confirm the presence of KRAS and TP53 mutations.
Viral Production and infection
Lenti- or retro-viral plasmids pLKO.1 shSox2 HM a (#26353), pLKO.1 shSox2 3H b (#26352), pWZL-Blast-MYC (#10674), and pBABE-YAP (#15682) were purchased from Addgene, Cambridge, MA (Bass et al., 2009; Boehm et al., 2005; Overholtzer et al., 2006). Mouse Sox2 sgRNA sequences were inserted into LentiCrispr v2 (#52961, Addgene) (Ahnfelt-Ronne et al., 2012). Specific sgRNA sequences were listed in STAR Methods. Lentiviral and retroviral productions were performed as previously described (Zhang et al., 2014). To generate stable MYC or YAP overexpressing lines, primary PDAC cells were first selected with blasticidin or puromycin after infection with viruses carrying these genes.
Crystal Violet staining
Cells grown on collagen-coated 24-well plates were fixed in 4% paraformaldehyde, stained with crytal violet solution (0.1% crystal violet in 10% EtOH) (Alfa Aesar), washed with distilled water, and dried at room temperature. The absorbance at 595nm was measured using Synergy™ H4 Hybrid Multi-Mode Microplate Reader (BioTek, Winooski, VT).
EdU and TUNEL assays
Cells were plates on collagen-coated coverslips in 24 well plates, infected with viruses, and incubated for additional 5 days. To detect apoptotic cells, cells were fixed in 4% paraformaldehyde, permeabilized, and subjected to Click-iT® TUNEL Alexa Fluor 594 Imaging Assay according to the manufacturer’s instructions (Invitrogen). To detect proliferating cells, cells were treated with EdU for 30 min prior to fixing. EdU-positive cells were detected using the Click-iT EdU Alexa Fluor 594 Imaging Kit according to the manufacturer’s instructions (Invitrogen). Nuclei were counterstained with DAPI. Images were acquired in ix71 inverted epifluorescence microscope (Olympus, Tokyo, Japan) and, quantified using at least 5 randomly selected fields containing at least 100 cells using the Image J software.
Cell growth assay
Yap+ and Yap− PDAC cells were plated separately on 24 well at 1,000 cells per well and the cell numbers were counted every other day for 4–6 days using Beckman Coulter Z2 Particle Counter Particle Counter (Beckman Coulter, Brea, CA).
Alternatively, PDAC cells labeled with two different fluorescent reporters were mixed in approximately equal portions, and passaged together. The relative percentages of the two populations were tracked over time through sequential FACS.
5-Azacytidine treatment
PDAC cells were treated with 0.5, 2.5, and 5 μM of 5-Azacytidine (ApexBio, Houston, TX) for 3 days. Cells were harvested for RNA extraction. Total mRNA was purified with RNAeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Reverse transcription was performed with iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) and the cDNA products were amplified with iTaq Universal SYBR Green Supermix (Bio-Rad) in triplicates. Primers were listed in Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken polyclonal anti-GFP | Abcam | Cat# ab13970, RRID:AB_300798 |
| Goat polyclonal anti-tdTomato | LifeSpan | Cat# LS-C348313 |
| Mouse monoclonal Anti-Cadherin, E | BD Biosciences | Cat# 610182, RRID:AB_397581 |
| Mouse monoclonal anti-TEF-3 (Clone N-G2) | Santa Cruz Biotechnology | Cat# sc-101184, RRID:AB_2203086 |
| Mouse monoclonal anti-Vinculin (Clone 7F9) | Santa Cruz Biotechnology | Cat# sc-73614; RRID:AB_1131294 |
| Mouse monoclonal anti-β-Actin (Clone C4) | Santa Cruz Biotechnology | Cat# sc-47778; RRID:AB_2714189 |
| Mouse polyclonal anti-Mist1 | Santa Cruz Biotechnology | Cat# sc-80984, RRID:AB_2065216 |
| Rabbit anti-Cyclin D1/bcl-1 | Thermo Fisher Scientific | Cat# RB-9041-P1, RRID:AB_149865 |
| Rabbit monoclonal anti-alpha smooth muscle Actin | Abcam | Cat# ab124964, RRID:AB_11129103 |
| Rabbit monoclonal anti-c-Myc (D3N8F) | Cell Signaling Technology | Cat# 13987, RRID:AB_2631168 |
| Rabbit monoclonal anti-Histone H2A.X, phospho (Ser139) (Clone 20E3) | Cell Signaling Technology | Cat# 9718; RRID:AB_2118009 |
| Rabbit monoclonal anti-Ki67 (Clone SP6) | ThermoFisher Scientific | Cat# RM-9106; RRID:AB_2341197 |
| Rabbit monoclonal anti-Phospho-AKT (Ser473) (D9E) | Cell Signaling Technology | Cat# 4060; RRID:AB_2315049 |
| Rabbit monoclonal anti-Phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (Clone 20G11) | Cell Signaling Technology | Cat# 4376; RRID:AB_331772 |
| Rabbit monoclonal anti-Phospho-S6 Ribosomal Protein (Ser240/244) (Clone D68F8) | Cell Signaling Technology | Cat# 5364; RRID:AB_10694233 |
| Rabbit monoclonal anti-Phospho-Stat3 (Tyr705) (D3A7) | Cell Signaling Technology | Cat# 9145, RRID:AB_2491009 |
| Rabbit monoclonal anti-SOX2 (Clone D6D9) | Cell Signaling Technology | Cat# 5024, RRID:AB_1904142 |
| Rabbit monoclonal anti-Thymidylate Synthase (D5B3) | Cell Signaling Technology | Cat# 9045, RRID:AB_2797693 |
| Rabbit polyclonal anti-a-Amylase | Millipore Sigma | Cat# A8273, RRID: AB_258380 |
| Rabbit polyclonal anti-Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| Rabbit polyclonal anti-Glucagon | Cell Signaling Technology | Cat# 2760, RRID:AB_659831 |
| Rabbit polyclonal anti-Insulin | Cell Signaling Technology | Cat# 4590, RRID:AB_659820 |
| Rabbit polyclonal anti-p44/42 MAPK (ERK1/2) | Cell Signaling Technology | Cat# 9102; RRID:AB_330744 |
| Rabbit polyclonal anti-Sox9 | Millipore Sigma | Cat# AB5535, RRID:AB_2239761 |
| Rabbit polyclonal anti-Taz | Cell Signaling Technology | Cat# 4883; RRID:AB_1904158 |
| Rabbit polyclonal anti-Yap | Cell Signaling Technology | Cat# 4912; RRID:AB_2218911 |
| Rat monoclonal anti-Keratin, type I; cytokeratin 19 | DSHB | Cat# TROMA-III, RRID:AB_2133570 |
| Goat polyclonal anti-p48 | Dr. Wright Lab | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-Azacytidine | ApexBio | Cat#A1907; CAS: 320–67-2 |
| Adenine | Millipore Sigma | Cat# A2786;CAS: 73–24-5 |
| AICAR | ApexBio | Cat# A8184; CAS: 2627–69-2 |
| Cytosine | Thermo Fisher Scientific | Cat# A14731;CAS: 71–30-7 |
| Guanine | Millipore Sigma | Cat# 120250100;CAS: 73–40-5 |
| Inosine | Thermo Fisher Scientific | Cat# 122250100;CAS: 58–63-9 |
| S-(5’-Adenosyl)-L-methionine chloride (SAM) | Cayman Chemicals | Cat# 13956; CAS: 86867–01-8 |
| S-Adenosylhomocysteine (SAH) | Cayman Chemicals | Cat# 13603; CAS: 979–92-0 |
| Thymine | Alfa Aesar | Cat# T0895; CAS: 65–71-4 |
| Critical Commercial Assays | ||
| Annexin V | Biolegend | Cat# 640943 |
| CellROX™ Deep Red Reagent | Thermo Fisher Scientific | Cat# C10422 |
| CellTiter-Glo Luminescence Cell Viability assay | Promega | Cat# G7570 |
| Click-iT™ EdU Cell Proliferation Kit for Imaging | Thermo Fisher Scientific | Cat# C10339 |
| Click-iT™ TUNEL Alexa Fluor™ 594 Imaging Assay | Thermo Fisher Scientific | Cat# C10246 |
| Magne® Protein G Beads, 20% Slurry | Promega | Cat# G747A |
| EZ DNA methylation-Direct kit | ZYMO RESEARCH | Cat# D5020 |
| GoTaq® Green Master Mix | Promega | Cat# M7122 |
| ImmPACT DAB EqV HRP substrate | Vector Laboratories | Cat# SK-4103 |
| IMMPRESS HRP Polymer Detection Kit | Vector Laboratories | Cat# MP-7401, MP-7402, MP-7405 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708890 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat# 1725124 |
| MethylFlash Methylated DNA 5-mC Quantification Kit | EPIGENTEK | Cat# P-1034 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| PrepEase Genomic DNA isolation kit | Affymetrix | Cat# 78855 |
| Prepease® Genomic DNA Isolation Kit | Affymetrix | Cat# 78850 |
| RNAeasy Mini Kit | Qiagen | Cat# 74104 |
| Tyramide Signal Amplification (TSA) kit | Thermo Fisher Scientific | Cat# T20932, T20933, T20936 |
| Experimental Models: Cell Lines | ||
| Panc1 | ATCC | CRL-1469 |
| Primary hPDAC | This paper | N/A |
| Primary mPDAC (KYY) | This paper | N/A |
| Primary mPDAC (KPYY) | This paper | N/A |
| Experimental Models: Organizms/Strains | ||
| SCID beige (CB17.Cg-PrkdcscidLystbg-J/Crl) mice | Charles River Laboratories (Croy and Chapeau, 1990) | Strain code #250 |
| Genetically engineered mouse strains | (Schönhuber et al., 2014 (Zhang et al., 2010) | N/A |
| Deposited Data | ||
| HepG2_TEAD4_Chip-seq | Gertz et al., 2013 | GEO: GSM1010875 |
| K562_TEAD4_Chip-seq | Gertz et al., 2013 | GEO: GSM1010895 |
| HCT-116_TEAD4_Chip-seq | Gertz et al., 2013 | GEO: GSM1010772 |
| A549_TEAD4_Chip-seq | Gertz et al., 2013 | GEO: GSM1010868 |
| ECC-1_TEAD4_Chip-seq | Gertz et al., 2013 | GEO: GSM1010885 |
| MCF7_TEAD4_Chip-seq | Gertz et al., 2013 | GEO: GSM1010860 |
| HepG2_TEAD1_Chip-seq | The ENCODE Project Consortium, 2013 | GEO: GSE96195 |
| HepG2_TEAD3_Chip-seq | The ENCODE Project Consortium, 2013 | GEO: GSE96302 |
| A549_MYC_Chip-seq | Michael Snyder Lab | GEO: GSM1003607 |
| HCT-116_MYC_Chip-seq | Jian Yan Lab | GEO: GSM2065882 |
| HepG2_MYC_Chip-seq | Pope et al., 2014 | GEO: GSM822291 |
| K562_MYC_Chip-seq | Pope et al., 2014 | GEO: GSM822310 |
| NF2 knockout livers_Glul_Microarray | Yuhao et al., 2016 | GEO: GSE70742 |
| Yap overexpresing livers_Glul_Microarray | Yimlamai et al., 2014 | GEO: GSE55560 |
| Recombinant DNA | ||
| pHAGE-GFP-luciferase | Addgene | Cat# 46793 |
| pLKO.1 shSox2 HM a | Addgene | Cat# 26353 |
| pLKO.1 shSox2 3H b | Addgene | Cat# 26352 |
| pWZL-Blast-MYC | Addgene | Cat# 10674 |
| pBABE-YAP | Addgene | Cat# 15682 |
| LentiCrispr v2 | Addgene | Cat# 52961 |
| Ad-CMV-GFP | Iowa viral vector core | Cat# Iowa-1174 |
| Ad-CMV-CRE | Iowa viral vector core | Cat# Iowa-5 |
| Oligonucleotides | ||
| Mouse Yap_F qRT-PCR 5’ TACATAAACCATAAGAACAAGACCACA 3’ | This paper | N/A |
| Mouse Yap_R qRT-PCR 5’ GCTTCACTGGAGCACTCTGA 3’ | This paper | N/A |
| Mouse Cyr61_F qRT-PCR 5’ CTGCGCTAAACAACTCAACGA 3’ | This paper | N/A |
| Mouse Cyr61_R qRT-PCR 5’ GCAGATCCCTTTCAGAGCGG 3’ | This paper | N/A |
| Mouse Ctgf_F qRT-PCR 5’ AGCTGACCTGGAGGAAAACA 3’ | This paper | N/A |
| Mouse Ctgf_R qRT-PCR 5’ GACAGGCTTGGCGATTTTAG 3’ | This paper | N/A |
| Mouse Hk1_F qRT-PCR 5’ CGGAATGGGGAGCCTTTGG 3’ | This paper | N/A |
| Mouse Hk1_R qRT-PCR 5’ GCCTTCCTTATCCGTTTCAATGG 3’ | This paper | N/A |
| Mouse Hk2_F qRT-PCR 5’ TGATCGCCTGCTTATTCACGG 3’ | This paper | N/A |
| Mouse Hk2_R qRT-PCR 5’ AACCGCCTAGAAATCTCCAGA 3’ | This paper | N/A |
| Mouse Aldoa_F qRT-PCR 5’ CGTGTGAATCCCTGCATTGG 3’ | This paper | N/A |
| Mouse Aldoa_R qRT-PCR 5’ CAGCCCCTGGGTAGTTGTC 3’ | This paper | N/A |
| Mouse Gapdh_F qRT-PCR 5’ AGGTCGGTGTGAACGGATTTG 3’ | This paper | N/A |
| Mouse Gapdh_R qRT-PCR 5’ TGTAGACCATGTAGTTGAGGTCA 3’ | This paper | N/A |
| Mouse Pgk1_F qRT-PCR 5’ CTTGGACTGTGGTACTGAGAG 3’ | This paper | N/A |
| Mouse Pgk1_R qRT-PCR 5’ AGGCTTCCCATTCAAATACCC 3’ | This paper | N/A |
| Mouse Pgam1_F qRT-PCR 5’ TCTGTGCAGAAGAGAGCAATCC 3’ | This paper | N/A |
| Mouse Pgam1_R qRT-PCR 5’ CTGTCAGACCGCCATAGTGT 3’ | This paper | N/A |
| Mouse Ldha_F qRT-PCR 5’ TGTCTCCAGCAAAGACTACTGT 3’ | This paper | N/A |
| Mouse Ldha_R qRT-PCR 5’ GACTGTACTTGACAATGTTGGGA 3’ | This paper | N/A |
| Mouse Phgdh_F qRT-PCR 5’ ATGGCCTTCGCAAATCTGC 3’ | This paper | N/A |
| Mouse Phgdh_R qRT-PCR 5’ AGTTCAGCTATCAGCTCCTCC 3’ | This paper | N/A |
| Mouse Psat1_F qRT-PCR 5’ AAGCCACCAAGCAAGTGGTTA 3’ | This paper | N/A |
| Mouse Psat1_R qRT-PCR 5’ GATGCCGAGTCCTCTGTAGTC 3’ | This paper | N/A |
| Mouse Psph_F qRT-PCR 5’ AGGAAGCTCTTCTGTTCAGCG 3’ | This paper | N/A |
| Mouse Psph_R qRT-PCR 5’ GAGCCTCTGGACTTGATCCC 3’ | This paper | N/A |
| Mouse Shmt1_F qRT-PCR 5’ CAGGGCTCTGTCTGATGCAC 3’ | This paper | N/A |
| Mouse Shmt1_R qRT-PCR 5’ CGTAACGCGCTCTTGTCAC 3’ | This paper | N/A |
| Mouse Shmt2_F qRT-PCR 5’ TGGCAAGAGATACTACGGAGG 3’ | This paper | N/A |
| Mouse Shmt2_R qRT-PCR 5’ GCAGGTCCAACCCCATGAT 3’ | This paper | N/A |
| Mouse Mthfd1_F qRT-PCR 5’ GGGAATCCTGAACGGGAAACT 3’ | This paper | N/A |
| Mouse Mthfd1_R qRT-PCR 5’ TGAGTGGCTTTGATCCCAATC 3’ | This paper | N/A |
| Mouse Mthfd2_F qRT-PCR 5’ AGTGCGAAATGAAGCCGTTG 3’ | This paper | N/A |
| Mouse Mthfd2_R qRT-PCR 5’ GACTGGCGGGATTGTCACC 3’ | This paper | N/A |
| Mouse Prps1_F qRT-PCR 5’ ACTTATCCCAGAAAATCGCTGAC 3’ | This paper | N/A |
| Mouse Prps1_R qRT-PCR 5’ CCACACCCACTTTGAACAATGTA 3’ | This paper | N/A |
| Mouse Gart_F qRT-PCR 5’ TCCTCAGGTCAAGCAAGTGTT 3’ | This paper | N/A |
| Mouse Gart_R qRT-PCR 5’ TGGTCCGACAACTACGAGTTC 3’ | This paper | N/A |
| Mouse Pfas1_F qRT-PCR 5’ GACTCCAGCATCGACCAACAT 3’ | This paper | N/A |
| Mouse Pfas1_R qRT-PCR 5’ GAAAGTCGGTAACGTCGGGT 3’ | This paper | N/A |
| Mouse Ctps_F qRT-PCR 5’ GCAGTGTGGGCACAATACTTA 3’ | This paper | N/A |
| Mouse Ctps_R qRT-PCR 5’ GCGCTCCTTGTTAATGACGTA 3’ | This paper | N/A |
| Mouse Tyms_F qRT-PCR 5’ GGAAGGGTGTTTTGGAGGAGT 3’ | This paper | N/A |
| Mouse Tyms_R qRT-PCR 5’ GCTGTCCAGAAAATCTCGGGA 3’ | This paper | N/A |
| Mouse Myc_F qRT-PCR 5’ CAACGACAGCAGCTCGCCCA 3’ | This paper | N/A |
| Mouse Myc_R qRT-PCR 5’ AGCCCGACTCCGACCTCTTGG 3’ | This paper | N/A |
| Mouse Ccnd1_F qRT-PCR 5’ GTTCATTTCCAACCCACCCTC 3’ | This paper | N/A |
| Mouse Ccnd1_R qRT-PCR 5’ AGAAAGTGCGTTGTGCGGTAG 3’ | This paper | N/A |
| Mouse Cdkn1a_F qRT-PCR 5’ CCTGGTGATGTCCGACCTG 3’ | This paper | N/A |
| Mouse Cdkn1a_R qRT-PCR 5’ CCATGAGCGCATCGCAATC 3’ | This paper | N/A |
| Mouse Hes1_F qRT-PCR 5’ CCAGCCAGTGTCAACACGA 3’ | This paper | N/A |
| Mouse Hes1_R qRT-PCR 5’ AATGCCGGGAGCTATCTTTCT 3’ | This paper | N/A |
| Mouse Sox9_F qRT-PCR 5’ CAAGACTCTGGGCAAGCTCTG 3’ | This paper | N/A |
| Mouse Sox9R qRT-PCR 5’ TCCGCTTGTCCGTTCTTCAC 3’ | This paper | N/A |
| Mouse Ptfa1_F qRT-PCR 5’ TCCCATCCCCTTACTTTGATGA 3’ | This paper | N/A |
| Mouse Ptfa1_R qRT-PCR 5’ GTAGCAGTATTCGTGTAGCTGG 3’ | This paper | N/A |
| Mouse Bhlha15_F qRT-PCR 5’ GCTGACCGCCACCATACTTAC 3’ | This paper | N/A |
| Mouse Bhlha15_R qRT-PCR 5’ TGTGTAGAGTAGCGTTGCAGG 3’ | This paper | N/A |
| Mouse Amy2A_F qRT-PCR 5’ TTGCCAAGGAATGTGAGCGAT 3’ | This paper | N/A |
| Mouse Amy2A_R qRT-PCR 5’ CCAAGGTCTTGATGGGTTATGAA 3’ | This paper | N/A |
| Mouse Ngn3_F qRT-PCR 5’ CCAAGAGCGAGTTGGCACT 3’ | This paper | N/A |
| Mouse Ngn3_R qRT-PCR 5’ CGGGCCATAGAAGCTGTGG 3’ | This paper | N/A |
| Mouse NeuroD1_F qRT-PCR 5’ ATGACCAAATCATACAGCGAGAG 3’ | This paper | N/A |
| Mouse NeuroD1_R qRT-PCR 5’ TCTGCCTCGTGTTCCTCGT 3’ | This paper | N/A |
| Mouse Hnf1a_F qRT-PCR 5’ GTGGCGAAGATGGTCAAGTC 3’ | This paper | N/A |
| Mouse Hnf1a_R qRT-PCR 5’ GCGTGGGTGAATTGCTGAG 3’ | This paper | N/A |
| Mouse Cda_F qRT-PCR 5’ GATCTTCTCTGGGTGCAACATAG 3’ | This paper | N/A |
| Mouse Cda_R qRT-PCR 5’ CCTGAAATCCTTGTACCCTTCG 3’ | This paper | N/A |
| Mouse Me2_F qRT-PCR 5’ GGCTAAGAGCTGTTACCACTCC 3’ | This paper | N/A |
| Mouse Me2_R qRT-PCR 5’ CGTAAACGCCATTCCCTTGTT 3’ | This paper | N/A |
| Mouse Idh1_F qRT-PCR 5’ ATGCAAGGAGATGAAATGACACG 3’ | This paper | N/A |
| Mouse Idh1_R qRT-PCR 5’ GCATCACGATTCTCTATGCCTAA 3’ | This paper | N/A |
| Mouse Gls_F qRT-PCR 5’ TTCGCCCTCGGAGATCCTAC 3’ | This paper | N/A |
| Mouse Gls_R qRT-PCR 5’ CCAAGCTAGGTAACAGACCCT 3’ | This paper | N/A |
| Mouse Gls2_F qRT-PCR 5’ CGTCCGGTACTACCTCGGT 3’ | This paper | N/A |
| Mouse Gls2_R qRT-PCR 5’ TGTCCCTCTGCAATAGTGTAGAA 3’ | This paper | N/A |
| Mouse Sox2_F qRT-PCR 5’ CGGCACAGATGCAACCGAT 3’ | This paper | N/A |
| Mouse Sox2_R qRT-PCR 5’ CCGTTCATGTAGGTCTGCG 3’ | This paper | N/A |
| Mouse Zeb1_F qRT-PCR 5’ GCTGGCAAGACAACGTGAAAG 3’ | This paper | N/A |
| Mouse Zeb1_R qRT-PCR 5’ GCCTCAGGATAAATGACGGC 3’ | This paper | N/A |
| Mouse Zeb2_F qRT-PCR 5’ ATTGCACATCAGACTTTGAGGAA 3’ | This paper | N/A |
| Mouse Zeb2_R qRT-PCR 5’ ATAATGGCCGTGTCGCTTCG 3’ | This paper | N/A |
| Mouse Sani1_F qRT-PCR 5’ CACACGCTGCCTTGTGTCT 3’ | This paper | N/A |
| Mouse Snai1_R qRT-PCR 5’ GGTCAGCAAAAGCACGGTT 3’ | This paper | N/A |
| Mouse Sani2_F qRT-PCR 5’ TGGTCAAGAAACATTTCAACGCC 3’ | This paper | N/A |
| Mouse Snai2_R qRT-PCR 5’ GGTGAGGATCTCTGGTTTTGGTA 3’ | This paper | N/A |
| Mouse Twist1_F qRT-PCR 5’ GGACAAGCTGAGCAAGATTCA 3’ | This paper | N/A |
| Mouse Twist1_R qRT-PCR 5’ CGGAGAAGGCGTAGCTGAG 3’ | This paper | N/A |
| Mouse Twist2_F qRT-PCR 5’ CGCTACAGCAAGAAATCGAGC 3’ | This paper | N/A |
| Mouse Twist2_R qRT-PCR 5’ GCTGAGCTTGTCAGAGGGG 3’ | This paper | N/A |
| Mouse Ezh2_F qRT-PCR 5’ AGTGACTTGGATTTTCCAGCAC 3’ | This paper | N/A |
| Mouse Ezh2_R qRT-PCR 5’ AATTCTGTTGTAAGGGCGACC 3’ | This paper | N/A |
| Mouse Smarcb1_F qRT-PCR 5’ TCCGAGGTGGGAAACTACCTG 3’ | This paper | N/A |
| Mouse Smarcb1_R qRT-PCR 5’ CAGAGTGAGGGGTATCTCTTGT 3’ | This paper | N/A |
| Mouse Tead1-F qRT-PCR 5’ AAGCTGAAGGTAACAAGCATGG 3’ | This paper | N/A |
| Mouse Tead1_R qRT-PCR 5’ GCTGACGTAGGCTCAAACCC 3’ | This paper | N/A |
| Mouse Tead3_F qRT-PCR 5’ CAACCAGCACAATAGCGTCCA 3’ | This paper | N/A |
| Mouse Tead3_R qRT-PCR 5’ CTGAAAGCTCTGCTCGATGTC 3’ | This paper | N/A |
| Mouse Tead4_F qRT-PCR 5’ CAACCTGGAACATCCCACGAT 3’ | This paper | N/A |
| Mouse Tead4_R qRT-PCR 5’ GAAAGCCGAGAACTCCAACAT 3’ | This paper | N/A |
| Mouse Myc_s1_F ChIP 5’ GCACTACAAACCCTGAAGCA 3’ | This paper | N/A |
| Mouse Myc_s1_R ChIP 5’ AAAGGACAGGAAAGCCACAA 3’ | This paper | N/A |
| Mouse Myc_s2_F ChIP 5’ TTGGGATTCAAGGCATTCAT 3’ | This paper | N/A |
| Mouse Myc_s2_R ChIP 5’ CTGCTTACGAATTGGGTGGT 3’ | This paper | N/A |
| Mouse Myc_s3_F ChIP 5’ GGGGTCGTTCTGGAAAGAAT 3’ | This paper | N/A |
| Mouse Myc_s3_R ChIP 5’ CGCTCAGTGTGTGGAGTGAT 3’ | This paper | N/A |
| Mouse nc (3utr)_F ChIP 5’ AAGGGTTCTTGCTGGGTTTT 3’ | This paper | N/A |
| Mouse nc (3utr)_R ChIP 5’ AGACCACGAAAACGGTCTTG 3’ | This paper | N/A |
| Mouse Sox2_s1_F ChIP 5’ CGTGGGAGGGAGTTTGTG 3’ | This paper | N/A |
| Mouse Sox2_s1_R ChIP 5’ GCATCAACGGAAAGAAGCTC 3’ | This paper | N/A |
| Mouse Sox2_s2_F ChIP 5’ GGAAAGGAGCTGTCGTCTTG 3’ | This paper | N/A |
| Mouse Sox2_s2_R ChIP 5’ GCTGGGGAACCTTTGTATCC 3’ | This paper | N/A |
| Mouse Ldha_F ChIP 5’ AGTTTTCCGGTGAAGGAGGC 3’ | This paper | N/A |
| Mouse Ldha_R ChIP 5’ ACACCCCAAAGAAAGGCCAA 3’ | This paper | N/A |
| Mouse Tyms_F ChIP 5’ CCATTAGCACCCCCATCCTC 3’ | This paper | N/A |
| Mouse Tyms_R ChIP 5’ CAAGAGGCTGTTGAGGAGCA 3’ | This paper | N/A |
| Mouse Mthfd2_F ChIP 5’ TTCAGGGGTTCTCAGCACAC 3’ | This paper | N/A |
| Mouse Mthfd2_R ChIP 5’ CTGAAGGAGTCAAGCCCCTG 3’ | This paper | N/A |
| Mouse Prps1_F ChIP 5’ GCGTCGACTGGTGTACATCA 3’ | This paper | N/A |
| Mouse Prps1_R ChIP 5’ CCAAGACCAAACCAGAACGC 3’ | This paper | N/A |
| Mouse Pgam1_F ChIP 5’ TACCGCTACGTACAACGCAT 3’ | This paper | N/A |
| Mouse Pgam1_R ChIP 5’ TTTCCACTCGCTCGACAGAC 3’ | This paper | N/A |
| Mouse Cyr61_F ChIP 5’ ATTGTGGACACACAGGCA 3’ | This paper | N/A |
| Mouse Cyr61_R ChIP 5’ CTCCTCCCTCCCTCCTTT 3’ | This paper | N/A |
| Mouse Myc_enhancer_F ChIP 5’ AGACAGATGCCGCCCAAAAG 3’ | This paper | N/A |
| Mouse Myc_enhancer_R ChIP 5’ CAGTGCTTCAAAGGCTTCGTT 3’ | This paper | N/A |
| Mouse Myc_exon1_F ChIP 5’ GTACCTCGTCCGATTCCACG 3’ | This paper | N/A |
| Mouse Myc_exon1_R ChIP 5’ GGGTAGCTTACCAGAGTCGC 3’ | This paper | N/A |
| Mouse Myc_exon2_F ChIP 5’ GGAAGGACTATCCAGCTGCC 3’ | This paper | N/A |
| Mouse Myc_exon2_R ChIP 5’ TGTCCGCCTCTTGTCGTTTT 3’ | This paper | N/A |
| Human YAP_F qRT-PCR 5’ CAGGTTGGGAGATGGCAAAG 3’ | This paper | N/A |
| Human YAP_R qRT-PCR 5’ TGTTGTCTGATCGATGTGATTTAAGA 3’ | This paper | N/A |
| Human MYC_F qRT-PCR 5’ AATGAAAAGGCCCCCAAGGTAGTTATCC 3’ | This paper | N/A |
| Human MYC_R qRT-PCR 5’ GTCGTTTCCGCAACAAGTCCTCTTC 3’ | This paper | N/A |
| Human PGK1_F qRT-PCR 5’ CTTTCATGTGGAGGAAGAAG 3’ | This paper | N/A |
| Human PGK1_R qRT-PCR 5’ TAGCTTGGAAAGTGAAGCTC 3’ | This paper | N/A |
| Human LDHA_F qRT-PCR 5’ TATGGAGTGGAATGAATGTTGC 3’ | This paper | N/A |
| Human LDHA_R qRT-PCR 5’ CCCTTAATCATGGTGGAAACT 3’ | This paper | N/A |
| Human PHGDH_F qRT-PCR 5’ CTGCGGAAAGTGCTCATCAGT 3’ | This paper | N/A |
| Human PHGDH_R qRT-PCR 5’ TGGCAGAGCGAACAATAAGGC 3’ | This paper | N/A |
| Human PSAT1_F qRT-PCR 5’ TGCCGCACTCAGTGTTGTTAG 3’ | This paper | N/A |
| Human PSAT1_R qRT-PCR 5’ GCAATTCCCGCACAAGATTCT 3’ | This paper | N/A |
| Human MTHFD2_F qRT-PCR 5’ CTGCGACTTCTCTAATGTCTGC 3’ | This paper | N/A |
| Human MTHFD2_R qRT-PCR 5’ CTCGCCAACCAGGATCACA 3’ | This paper | N/A |
| Human TYMS_F qRT-PCR 5’ CTGCTGACAACCAAACGTGTG 3’ | This paper | N/A |
| Human TYMS_R qRT-PCR 5’ GCATCCCAGATTTTCACTCCCTT 3’ | This paper | N/A |
| Human PRPS1_F qRT-PCR 5’ ATCTTCTCCGGTCCTGCTATT 3’ | This paper | N/A |
| Human PRPS1_R qRT-PCR 5’ TGGTGACTACTACTGCCTCAAA 3’ | This paper | N/A |
| Human GART_F qRT-PCR 5’ GGAATCCCAACCGCACAATG 3’ | This paper | N/A |
| Human GART_R qRT-PCR 5’ AGCAGGGAAGTCTGCACTCA 3’ | This paper | N/A |
| Human CTPS_F qRT-PCR 5’ CAGTGTGGGCACAATACTCAA 3’ | This paper | N/A |
| Human CTPS_R qRT-PCR 5’ CGCTCATAGTTACCCAGGTCA 3’ | This paper | N/A |
| Mouse mBhlha15_M_ F qMSP 5’ TTTTATTGTTTTATTTTGTTTTCGT 3’ | This paper | N/A |
| Mouse mBhlha15_M_ R qMSP 5’ ACCATAAACCCCTTCTAATTACGTT 3’ | This paper | N/A |
| Mouse mBhlha15_UM_ F qMSP 5’ TTTTATTGTTTTATTTTGTTTTTGT 3’ | This paper | N/A |
| Mouse mBhlha15_UM_ R qMSP 5’ ACCATAAACCCCTTCTAATTACATT 3’ | This paper | N/A |
| Mouse mPtfa1_M_F qMSP 5’ GAGAGGTAGATTAATTTTTGGTGAC 3’ | This paper | N/A |
| Mouse mPtfa1_M_R qMSP 5’ TCAAAAATTCTTACAAATTTACGAA 3’ | This paper | N/A |
| Mouse mPtfa1_UM_F qMSP 5’ GAGGTAGATTAATTTTTGGTGATGT 3’ | This paper | N/A |
| Mouse mPtfa1_UM_R qMSP 5’ TCAAAAATTCTTACAAATTTACAAA 3’ | This paper | N/A |
| Mouse mox2_gRNA_#1-f Crispr 5’ GACCGGGTGGGCGAGCCGTTCATGT 3’ | This paper | N/A |
| Mouse Sox2_gRNA_#1-r Crispr 5’ AAACACATGAACGGCTCGCCCACCC 3’ | This paper | N/A |
| Mouse Sox2_gRNA_#2-f Crispr 5’ GACCGGCTACGCGCACATGAACGGC 3’ | This paper | N/A |
| Mouse Sox2_gRNA_#2-r Crispr 5’ AAACGCCGTTCATGTGCGCGTAGCC 3’ | This paper | N/A |
| Software and Algorithms | ||
| Graphpad Prism 6.0 | Graphpad | N/A |
| Limma | (Ritchie et al., 2015) | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Gene Set Enrichment Analysis | (Subramaniana et al., 2005) (Mootha et al., 2003) | http://software.broadinstitute.org/gsea/index.jsp |
| NDP.view2 Viewing software | Hamamatsu Photonics | http://www.hamamatsu.com/all/en/U12388–01.html |
| Image J software version 2.0.0-rc-49/1.51e | NIH | http://wsr.imagej.net/distros/ |
SAM/SAH treatment
PDAC cells were treated with 200 μM of S-Adenosylmethionine (SAM; Cayman) and 0.4 μM of S-Adenosylhomocysteine (SAH; Cayman) for 12 days. Growth medium was changed every 2 days.
Nutrient Starvation
Cells were grown in DMEM without FBS, glucose, glutamine and pyruvate for 2 days and then changed to normal medium to recover from nutrient stress for 12 days.
LC-MS/MS
3 days post infection with Ad-CRE or Ad-GFP, PDAC cells were washed twice with ice-cold PBS, drained, snapped-frozen in liquid nitrogen and stored at −80°C until extraction using 50% methanol, 30% ACN, and 20% water (Mackay et al., 2015). After addition of extraction solution (1 mL/1×106 cells), samples were vortexed for 5 min at 4°C, and then centrifuged at 16,000 g for 15 minutes at 4°C. The supernatants were collected and separately by liquid chromatography–mass spectrometry using SeQuant ZIC-pHilic column (Millipore). The aqueous mobile-phase solvent was 20 mM ammonium carbonate plus 0.1% ammonium hydroxide solution and the organic mobile phase was acetonitrile. The metabolites were separated over a linear gradient from 80% organic to 80% aqueous for 15 min. The column temperature was 48 °C and the flow rate was 200 μl/min. The metabolites were detected across a mass range of 75–1,000 m/z using the Q-Exactive mass spectrometer at a resolution of 35,000 (at 200 m/z) with electrospray ionization and polarity switching mode (Mackay et al., 2015). Lock masses were used to insure mass accuracy below 5 ppm. The peak areas of different metabolites were determined using Thermo TraceFinder software using the exact mass of the singly charged ion and known retention time on the HPLC column. Data analysis was performed in the Ingenuity Pathway Analysis software package.
IHC/IF
Mouse pancreas was fixed in 10% buffered formalin and paraffin-embedded sections were used for all IHC and IF. For both IHC and IF, unstained slides were deparaffinized and heated in antigen retrieval buffer as indicated (IHC-Tek™ Epitope Retrieval Solution, IHC World LLC, Woodstock, MD; or 10mM Tris Base, 1mM EDTA Solution, 0.05% Tween 20, pH 9.0) for 30 min at 95°C. After the slides cool down to RT, they were washed 2 times with PBST (PBS + 0.1% Tween-20), blocked with 3% H2O2 for 15 min, then with 5% normal horse serum for 30 min, followed by incubation overnight with primary antibodies at 4°C. Next day, slides were washed for 6 times with PBST. For IHC, slides were incubated with corresponding IMMPRESS HRP POLYMER REAGENTS (Vector Laboratories, Burlingame, CA) for 1 hour at RT. After washing with PBST, staining was visualized using the ImmPACT DAB EqV HRP substrate (Vector Laboratories) according to the manufacturer’s instructions. The IHC slides were scanned using the Hamamatsu NanoZoomer slide scanning system (the Alafi Neuroimaging Laboratory, St. Louis, MO) and images were analyzed by NDP.view2 Viewing software (Hamamatsu Photonics, Shizuoka, Japan). IF was performed as previously described using fluorescein-conjugated secondary antibodies or Tyramide Signal Amplification (TSA) kit (Thermo Fisher Scientific) (Murakami et al., 2017). Confocal fluorescence images were obtained with the Leica SP8 confocal microscope, and analyzed using Image J software version 2.0.0-rc-49/1.51e. All the quantification from IHC and IF was performed using ImageJ and data from at least 5 slides/fields were averaged. All antibodies were listed in Key Resources Table.
Flow cytometry
For measurement of cellular ROS, cells were incubated CellRox Deep Red (Thermo Fisher Scientific) at a final concentration of 5 μM for 30 min before harvest. Cells were trypsinized, resuspended in DMEM, filtered through a 70 μm strainer and analyzed for fluorescence at 660 nm by flow cytometry. For measurement of apoptosis, cells were trypsinized, resuspended in 100 μL annexin-binding buffer containing 5 μL of Annexin V-647, incubated for 15 min in the dark, filtered through a 70 μm strainer and analyzed immediately by flow cytometry (Biolegend, San Diego, CA).
qRT-PCR
Total mRNA was purified from PDAC cells and pancreatic tissues from WT, KF, and KYYF mice with RNAeasy Mini kit (Qiagen) according to the manufacturer’s instructions. Reverse transcription was performed with iScript cDNA Synthesis Kit (Bio-Rad) and the cDNA products were amplified with iTaq Universal SYBR Green Supermix (Bio-Rad) in triplicates. Fold change of gene expression was calculated as a unit value of 2−ΔCt=2−[Ct(HPRT)-Ct(Gene of Interest)]]. Data is represented by mean of all replicates. Primer sequences of the genes analyzed are listed in Key Resources Table.
Chromatin immunoprecipitation (ChIP)
ChIP analysis was performed as previously described (Murakami et al., 2017). PDAC cells were plated and fixed with 1% (v/v) formaldehyde and stopped crosslink with 2M Glycine. Cells were washed with ice-cold PBS, lysed with ice-cold nuclear isolation buffer (50 mM Tris-HCl, 60 mM KCl, 0.5% NP40, 10mM DTT) and resuspended in ice-cold nuclear lysis buffer (50 mM Tris-HCl, 1% SDS, 10 mM EDTA). After sonication for 25 min, protein-DNA complexes were diluted in ChIP dilution buffer (16.7 mM Tris-HCl, 0.01% SDS, 1.2 mM EDTA, 1.1% Triton X-100, 167 mM NaCl), and incubated overnight with primary antibodies at 4°C. Next day, the complexes were incubated with Magne protein G beads (Promega) for 1 hour, and washed with low salt buffer (20 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 2 mM EDTA, 1% Triton X-100), high salt buffer (20 mM Tris-HCl, 500 mM NaCl, 0.1% SDS, 2 mM EDTA, 1% Triton X-100), LiCl buffer 10 mM Tris-HCl, 250 mM LiCl, 1% sodium deoxycholate, 1% NP-40, 1 mM EDTA), and TE buffer. Immunoprecipitates were eluted in 1% SDS/100 mM NaHCO3 and reversed cross-linked overnight at 65°C. Immunoprecipitated DNA was eluted and analyzed by qPCR with primer pairs flanking different enhancer, promoter and gene body regions of the indicated mouse genes that contain putative Tead, Myc and Sox2 binding sites. The antibodies and primer sequences used in the study are listed in Key Resources Table.
Western Blotting
Whole cell extracts were prepared with urea buffer (9.5 M urea, 2% CHAPS) and subjected to SDS-PAGE and western blot as previously described (Murakami et al., 2017). Briefly, after wash with cold PBS, cells were lysed in urea buffer (9.5 M urea, 2% CHAPS), adjusted to similar concentrations, mixed with 6x SDS loading dye, heated at 95°C for 10 minutes, and subjected to SDS-polyacrylamide gel electrophoresis and semi-dry transferring to PVDF membranes. All primary antibodies are listed in Key Resources Table.
Global DNA methylation assay
Genomic DNA was extracted from freshly cultured cells using PrepEase Genomic DNA isolation kit (usb, cleavland, OH) according to the manufacture’s instruction. 200 ng of genomic DNA were used to analyze global DNA methylation analysis with MethylFlash Methylated DNA 5-mC Quantification Kit (EPIGENTEK, Farmingdale, NY) according to the manufacture’s instruction.
Bisulfate Conversion and quantitative Methylation-Specific PCR (qMSP)
Genomic DNA was purified from the pancreatic tissues of WT, KF, and KYYF using PrepEase Genomic DNA isolation kit (Affymetrix, Cleveland, Ohio) according to the manufacture’s instruction. 400 ng of genomic DNA were converted with sodium bisulfate using EZ DNA methylation-Direct kit (ZYMO RESEARCH, Irvine, CA) according to the manufacture’s instruction. Bisulfate converted DNA were used for qMSP. PCR primers were designed for specific for methylated gDNA (M), or unmethylated gDNA (UM). The percentage of methylation was calculated as [100/1+2^-(CTum-CTm)]. Data is represented by mean of all replicates. Primer sequences of the genes analyzed are listed in Key Resources Table.
Re-analysis of published microarray and ENCODE data
Raw intensity data from previously published microarray studies listed in STAR Methods were background corrected and normalized in R using the oligo (for Affymetrix data) or limma (for Illumina data) package. The enrichment peaks of H3K27Ac, TEAD1, TEAD3, TEAD4 and MYC along the human MYC and other metabolic genes were visualized using the WashU genome browser from publically available ChIP-seq datasets listed in Key Resources Table.
Quantification and Statistical Analysis
The GraphPad Prism software was used for statistical analysis. Minimum of three mice or independent samples were used for all the experiments unless otherwise indicated. Kaplan-Meier survival curve was used to estimate the lifespan of the mice. Student’s t-test was used to determine significance unless otherwise indicated. Significance is defined as a p value of 0.05 or less. Error bars on all graphs are standard error of the mean or standard deviation as indicated.
Data Availability
The authors declare that all the data supporting the findings of this study are available within the paper [and its supplementary information files] or all the source data are available from the corresponding author upon reasonable request.
Supplementary Material
Highlights.
Yap maintains the global metabolic transcriptional program in conjunction with Myc
Yap ablation triggers metabolic crisis and regresses early stage pancreatic tumors
Yap ablation de-represses Sox2 and acinar lineage genes via DNA demethylation
Sox2 restores Myc and metabolic homeostasis in Yap null pancreatic tumor cells
ACKNOWLEDGEMENT
We thank Drs. T Jacks (MIT) and DJ Pan (UT Southwestern) for sharing some of the mouse strains used in this study, and Dr. C Wright (Vanderblit) for providing the p48/Ptf1a antibody. We are grateful for the helpful comments of our manuscript from Dr. Anton Wellstein (Georgetown). We appreciate the technical assistance from Max Kushner, the Flow Cytometry and Cell Sorting Shared Resource (FCSR), Microscopy & Imaging Shared Resource (MISR), Histopathology & Tissue Shared Resource (HTSR), and Genomics & Epigenomics Shared Resource (GESR) at Georgetown Lombardi Cancer Center. We thank the staff of the Division of Comparative Medicine (DCM) at Georgetown for support in our animal studies. This project is supported by NIH (R01CA187090), Sher Grant and Cancer Center Support Grant (CA051008).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, and DePinho RA (2003). Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 17, 3112–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnfelt-Ronne J, Jorgensen MC, Klinck R, Jensen JN, Fuchtbauer E-M, Deering T, MacDonald RJ, Wright CVE, Madsen OD, and Serup P (2012). Ptf1a-mediated control of Dll1 reveals an alternative to the lateral inhibition mechanism. Development 139, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy I, Jauch R, Chen J, Dyla M, Divakar U, Bogu GK, Teo R, Leng Ng CK, Herath W, Lili S, et al. (2013). Oct4 switches partnering from Sox2 to Sox17 to reinterpret the enhancer code and specify endoderm. EMBO J. 32, 938–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, Miller DK, Christ AN, Bruxner TJC, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. [DOI] [PubMed] [Google Scholar]
- Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, et al. (2009). SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 41, 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm JS, Hession MT, Bulmer SE, and Hahn WC (2005). Transformation of Human and Murine Fibroblasts without Viral Oncoproteins. Mol. Cell. Biol. 25, 6464–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- cheloufi S, Elling U, Hopfgartner barbara, Jung YL, murn J, Ninova maria, Hubmann maria, badeaux A, Euong Ang, cheen, Tenen danielle, et al. (2015). The histone chaperone CAF-1 safeguards somatic cell identity. Nature 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, Ernst J, and Plath K (2017). Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Bednar F, Zhang Y, Brisset J-C, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, and Pasca di Magliano M (2012). Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Invest. 122, 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosset É, Ilmjärv S, Dutoit V, Elliott K, von Schalscha T, Camargo MF, Reiss A, Moroishi T, Seguin L, Gomez G, et al. (2017). Glut3 Addiction Is a Druggable Vulnerability for a Molecularly Defined Subpopulation of Glioblastoma. Cancer Cell 32, 856–868.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AG, Hwang KL, Brown KK, Evason KJ, Beltz S, Tsomides A, O’Connor K, Galli GG, Yimlamai D, Chhangawala S, et al. (2016). Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat. Cell Biol. 18, 886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croci O, De Fazio S, Biagioni F, Donato E, Caganova M, Curti L, Doni M, Sberna S, Aldeghi D, Biancotto C, et al. (2017). Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genes Dev. 31, 2017–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deluz C, Friman ET, Strebinger D, Benke A, Raccaud M, Callegari A, Leleu M, Manley S, and Suter DM (2016). A role for mitotic bookmarking of SOX2 in pluripotency and differentiation. Genes Dev. 30, 2538–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, Dalton GD, Thelen E, Rizi BS, Jung Y, et al. (2018). Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 154, 1465–1479.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DN, Ngwa VM, Wang S, Shiuan E, Brantley-Sieders DM, Kim LC, Reynolds AB, and Chen J (2017). The receptor tyrosine kinase EphA2 promotes glutamine metabolism in tumors by activating the transcriptional coactivators YAP and TAZ. Sci. Signal 10, eaan4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander SYG, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, Vasile E, DePinho RA, and Jacks T (2009). Context-Dependent Transformation of Adult Pancreatic Cells by Oncogenic K-Ras. Cancer Cell 16, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber R, Panayiotou R, Nye E, Spencer-Dene B, Stamp G, and Behrens A (2016). YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-regulation of JAK–STAT3 Signaling. Gastroenterology 151, 526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, and Barbacid M (2007). Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 11, 291–302. [DOI] [PubMed] [Google Scholar]
- Guo S, Contratto M, Miller G, Leichman L, and Wu J (2017). Immunotherapy in pancreatic cancer: Unleash its potential through novel combinations. World J. Clin. Oncol 8, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD, et al. (2008). Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc. Natl. Acad. Sci 105, 18913–18918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbrook CJ, and Lyssiotis CA (2017). Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 31, 5–19. [DOI] [PubMed] [Google Scholar]
- Herreros-Villanueva M, Zhang J-S, Koenig A, Abel EV, Smyrk TC, Bamlet WR, de Narvajas AA-M, Gomez TS, Simeone DM, Bujanda L, et al. (2013). SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis 2, e61–e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. (2003). Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu C-J, Sadanandam A, Hu B, et al. (2014). Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 158, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King HW, and Klose RJ (2017). The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, Littlewood TD, and Evan GI (2017). Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 171, 1301–1315.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Shen J, and Corey DP (2015). C-MYC transcriptionally amplifies SOX2 target genes to regulate self-renewal in multipotent otic progenitor cells. Stem Cell Reports 4, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay GM, Zheng L, van den Broek NJF, and Gottlieb E (2015). Analysis of Cell Metabolism Using LC-MS and Isotope Tracers. In Methods in Enzymology, pp. 171–196. [DOI] [PubMed] [Google Scholar]
- Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, Van Den Broek NJF, Mackay GM, Labuschagne CF, Gay D, Kruiswijk F, et al. (2017). Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nat. Publ. Gr 544. [DOI] [PubMed] [Google Scholar]
- Mello SS, Valente LJ, Raj N, Seoane JA, Flowers BM, McClendon J, Bieging-Rolett KT, Lee J, Ivanochko D, Kozak MM, et al. (2017). A p53 Super-tumor Suppressor Reveals a Tumor Suppressive p53-Ptpn14-Yap Axis in Pancreatic Cancer. Cancer Cell 32, 460–473.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, Taniguchi K, Yu FX, Karin M, Pan D, et al. (2015). A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 29, 1271–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S, Engleitner T, Maresch R, Zukowska M, Lange S, Kaltenbacher T, Konukiewitz B, Öllinger R, Zwiebel M, Strong A, et al. (2018). Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 554, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Shahbazian D, Surana R, Zhang W, Chen H, Graham GT, White SM, Weiner LM, and Yi C (2017). Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 36, 1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neto-Silva RM, de Beco S, and Johnston LA (2010). Evidence for a Growth-Stabilizing Regulatory Feedback Mechanism between Myc and Yorkie, the Drosophila Homolog of Yap. Dev. Cell 19, 507–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng C-X, Brugge JS, and Haber DA (2006). Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci 103, 12405–12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Shin JE, and Park HW (2018). The Role of Hippo Pathway in Cancer Stem Cell Biology. Mol. Cells [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontoglio M, Sreenan S, Roe M, Pugh W, Ostrega D, Doyen A, Pick AJ, Baldwin A, Velho G, Froguel P, et al. (1998). Defective insulin secretion in hepatocyte nuclear factor 1alpha-deficient mice. J. Clin. Invest. 101, 2215–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozengurt E, Sinnett-Smith J, and Eibl G (2018). Yes-associated protein (YAP) in pancreatic cancer: at the epicenter of a targetable signaling network associated with patient survival. Signal Transduct. Target. Ther. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DP, Hong TS, and Bardeesy N (2014). Pancreatic adenocarcinoma. N. Engl. J. Med 371, 1039–1049. [DOI] [PubMed] [Google Scholar]
- Satoh K, Yachida S, Sugimoto M, Oshima M, Nakagawa T, Akamoto S, Tabata S, Saitoh K, Kato K, Sato S, et al. (2017). Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl. Acad. Sci. U. S. A. 114, E7697–E7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield HK, Zeller J, Espinoza C, Halbrook CJ, Del Vecchio A, Magnuson B, Fabo T, Daylan AEC, Kovalenko I, Lee H-J, et al. (2018). Mutant p53R270H drives altered metabolism and increased invasion in pancreatic ductal adenocarcinoma. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönhuber N, Seidler B, Schuck K, Veltkamp C, Schachtler C, Zukowska M, Eser S, Feyerabend TB, Paul MC, Eser P, et al. (2014). A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat. Med. 20, 1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, et al. (2014). KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Bollam SR, White SM, Laughlin SZ, Graham GT, Wadhwa M, Chen H, Nguyen C, Vitte J, Giovannini M, et al. (2016). Rac1-Mediated DNA Damage and Inflammation Promote Nf2 Tumorigenesis but Also Limit Cell-Cycle Progression. Dev. Cell 39, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Chen N-M, Hessmann E, Siveke J, Lahmann M, Singh G, Voelker N, Vogt S, Esposito I, Schmidt A, et al. (2015). Antithetical NFATc1–Sox2 and p53–miR200 signaling networks govern pancreatic cancer cell plasticity. EMBO J. 34, 517–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirajuddin P, Das S, Ringer L, Rodriguez OC, Sivakumar A, Lee Y-C, Üren A, Fricke ST, Rood B, Ozcan A, et al. (2012). Quantifying the CDK inhibitor VMY-1–103’s activity and tissue levels in an in vivo tumor model by LC-MS/MS and by MRI. Cell Cycle 11, 3801–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, and Evan GI (2008). Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, Swigart LB, and Evan GI (2013). Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev 27, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, and Dang CV (2015). MYC, Metabolism, and Cancer. Cancer Discov. 5, 1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suelves M, Carrió E, Núñez-Álvarez Y, and Peinado MA (2016). DNA methylation dynamics in cellular commitment and differentiation. Brief. Funct. Genomics [DOI] [PubMed] [Google Scholar]
- Vanderas AP, and Papagiannoulis L (2002). Multifactorial analysis of the aetiology of craniomandibular dysfunction in children. Int. J. Paediatr. Dent 12, 336–346. [DOI] [PubMed] [Google Scholar]
- Wang W, Xiao Z-D, Li X, Aziz KE, Gan B, Johnson RL, and Chen J (2015). AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol 17, 490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SM, Avantaggiati ML, Nemazanyy I, Di Poto C, Yang Y, Pende M, Gibney GT, Ressom HW, Field J, Atkins MB, et al. (2019a). YAP/TAZ Inhibition Induces Metabolic and Signaling Rewiring Resulting in Targetable Vulnerabilities in NF2-Deficient Tumor Cells. Dev. Cell 49, 425–443.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SM, Murakami S, and Yi C (2019b). The complex entanglement of Hippo-Yap/Taz signaling in tumor immunity. Oncogene 38, 2899–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuebben EL, and Rizzino A (2017). The dark side of SOX2: cancer - a comprehensive overview. Oncotarget 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, and Camargo FD (2014). Hippo Pathway Activity Influences Liver Cell Fate. Cell 157, 1324–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, and Piccolo S (2015). Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol 17, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, and Piccolo S (2016). YAP/TAZ at the Roots of Cancer. Cancer Cell 29, 783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Weinberg RA (2018). Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front. Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, and Pan D (2010). The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev. Cell 19, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G, Gupta S, Vietsch EE, Laughlin SZ, Wadhwa M, et al. (2014). Downstream of Mutant KRAS, the Transcription Regulator YAP Is Essential for Neoplastic Progression to Pancreatic Ductal Adenocarcinoma. Sci. Signal 7, ra42–ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Wang X, Fang L, Lan C, Zheng X, Wang Y, Zhang Y, Han X, Liu S, Cheng K, et al. (2017). A combinatorial strategy using YAP and pan-RAF inhibitors for treating KRAS-mutant pancreatic cancer. Cancer Lett. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all the data supporting the findings of this study are available within the paper [and its supplementary information files] or all the source data are available from the corresponding author upon reasonable request.