

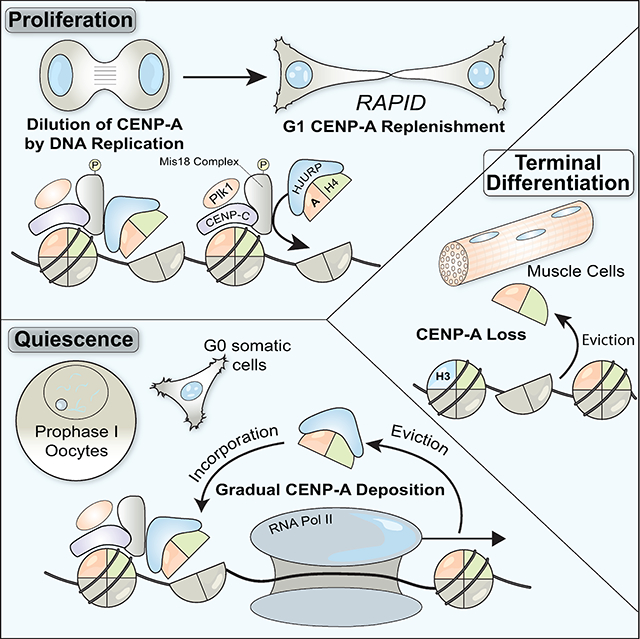
Summary
Centromeres provide a robust model for epigenetic inheritance as they are specified by sequence-independent mechanisms involving the histone H3-variant CENP-A. Prevailing models indicate that the high intrinsic stability of CENP-A nucleosomes maintains centromere identity indefinitely. Here, we demonstrate that CENP-A is not stable at centromeres, but is instead gradually and continuously incorporated in quiescent cells including G0-arrested tissue culture cells and prophase I-arrested oocytes. Quiescent CENP-A incorporation involves the canonical CENP-A deposition machinery, but displays distinct requirements from cell cycle-dependent deposition. We demonstrate that Plk1 is required specifically for G1 CENP-A deposition, whereas transcription promotes CENP-A incorporation in quiescent oocytes. Preventing CENP-A deposition during quiescence results in significantly reduced CENP-A levels and perturbs chromosome segregation following the resumption of cell division. In contrast to quiescent cells, terminally differentiated cells fail to maintain CENP-A levels. Our work reveals that quiescent cells actively maintain centromere identity providing an indicator of proliferative potential.
eTOC
Epigenetic marks must be retained during extended periods of quiescence to ensure the proper function of genomic loci. Although centromeric CENP-A nucleosomes were thought to be immobile, Swartz et al. identify gradual CENP-A deposition in quiescent cells and oocytes. CENP-A exchange is essential for faithful cell division following long arrests.
Graphical Abstract
Introduction
Heritable information is propagated by DNA sequence as well as via sequence-independent epigenetic marks that control the properties or activity of specific genomic loci. These marks include covalent changes to the DNA itself, such as methylation, post-translational modifications to histone proteins, and the incorporation of histone variants such as H2A.Z, macroH2A, and the histone H3 variant CENP-A. For an epigenetic mark to stably alter the behavior of a locus, it must be restricted to the correct location, propagated to new cells formed during cell division, and maintained under all circumstances where this information is needed to direct cellular behaviors. To understand the basis for epigenetic specification, it is critical to determine how these marks are stably maintained for extended periods. We sought to understand these requirements by focusing on the epigenetic specification that occurs at centromeres.
Inheritance of genetic information across cell divisions (to every cell in the body) and generations (from parent to progeny) requires the presence of the centromere at a single locus on each chromosome (McKinley and Cheeseman, 2016). Centromeres serve as the foundation for assembly of the macromolecular kinetochore structure that connects chromosomes to the spindle apparatus to direct their segregation (Cheeseman, 2014). Specific DNA sequences are neither necessary nor sufficient for centromere identity and function in most organisms, and instead centromeres are defined epigenetically by the presence of the histone H3-variant CENP-A. CENP-A is required for the localization of all known kinetochore components such that the loss of CENP-A results in a failure of kinetochore function, chromosome mis-segregation, and ultimately cell inviability or dysfunction. Thus, the central requirement for centromere identity and function is to specify, propagate, and maintain the presence of CENP-A nucleosomes at a single site on each chromosome.
Pre-existing CENP-A nucleosomes are required to direct new CENP-A deposition (McKinley and Cheeseman, 2016) and de novo centromere formation is exceptionally rare (Shang et al., 2013). As such, it is essential that this CENP-A mark be stably retained under all circumstances where subsequent division is required to ensure proper genome inheritance. Current models suggest that CENP-A is indefinitely stable at centromeres, with the replenishment of CENP-A nucleosomes following DNA replication restricted to the following G1 phase (Jansen et al., 2007). Prior work has proposed that pre-existing CENP-A nucleosomes are maintained through their immobility and stability conferred by its binding partners (Bodor et al., 2013; Cao et al., 2018; Guo et al., 2017; Jansen et al., 2007; McKinley and Cheeseman, 2016; Smoak et al., 2016). However, most work has focused on the mechanisms that propagate CENP-A in rapidly dividing mitotic cells. Much less is known about CENP-A maintenance at centromeres in the diverse metazoan cell types that exit the cell cycle for extended periods.
Given the critical role of quiescent cells in organismal development, homeostasis, and repair, it is critical to understand how centromere identity is maintained under diverse physiological situations, including during prolonged cell cycle arrest. Here, we explore CENP-A nucleosome dynamics in quiescent germline and somatic cells across animal species. In contrast to previous reports, we find that CENP-A levels are not indefinitely stable. Our work suggests that new CENP-A deposition requires that pre-existing nucleosomes at centromeres are destabilized in a Pol II-dependent manner and, in quiescent cells, CENP-A is gradually re-incorporated using its canonical deposition machinery to maintain steady state levels. We find that ongoing CENP-A deposition is essential to maintain centromere function and maintain proliferative capacity during quiescence. In contrast, terminally differentiated muscle cells fail to maintain CENP-A levels. This work defines a critical mechanism by which an essential epigenetic mark is maintained in quiescent cells to retain proliferative potential.
Results
CENP-A exchanges at centromeres in quiescent human cells
Prior work has analyzed CENP-A deposition in rapidly dividing cells, including human tissue culture cells. In contrast, little is known regarding CENP-A dynamics in non-dividing cells. Therefore, we first asked whether the levels of endogenous CENP-A remain constant in human hTERT RPE-1 (RPE-1) tissue culture cells induced to enter G0 by serum starvation and contact inhibition. Based on immunofluorescence against endogenous CENP-A, we found that CENP-A levels at centromeres remained constant across a 14-day time course (Fig. 1A,B). In contrast, the centromere protein CENP-T was strongly reduced at centromeres in quiescent cells (Fig. 1C,D; Dani Bodor and Lars Jansen, personal communication). This indicates that CENP-A is preferentially maintained in these arrested cells, consistent its essential role as an epigenetic mark.
Figure 1. CENP-A is gradually incorporated in quiescent human somatic cells.

(A) Immunofluorescence images showing endogenous CENP-A in quiescent RPE-1 cells at 0, 8, and 14 days after entering serum-starvation induced quiescence. (B) Fluorescence quantification reveals that CENP-A levels remain constant during quiescence. Points represent the sum of all centromeres from an individual cell relative to Day 0. Error bars represent the mean and standard deviation of 25 cells/time point. The differences between time points are not statistically different based on a Welch’s t test. (C) Immunofluorescence in RPE-1 cells for CENP-T and CENP-A in quiescence vs. proliferation. (D) Fluorescence quantification reveals a reduction of CENP-T at centromeres in quiescence, whereas CENP-A levels remain constant. Points represent the sum of all centromeres from an individual cell relative to Day 0. Error bars represent the mean and standard deviation of 31 cells (cycling) or 28 cells (quiescent). ****p<0.0001 by Mann Whitney test. (E) Halo-tag CENP-A incorporation assay in quiescent cells. Schematic indicates experimental conditions. Images - Top: Old HALO-CENP-A visualized with JF549. Middle: New CENP-A-Halo visualized with JF646. Bottom: Corresponding DNA staining. (F) Quantification of new CENP-A-Halo fluorescent intensity relative to pre-existing CENP-A (see Experimental Procedures). Points represent the sum of all centromeres of individual cells. Error bars represent the mean and standard deviation of at least 40 cells. Scale bars = 10 μm.
The persistence of CENP-A at centromeres in quiescent cells is consistent with either highly stable CENP-A nucleosomes, as suggested by prior work, or with an active maintenance program with balanced CENP-A loss and incorporation. To distinguish between these possibilities, we introduced a HaloTag heterozygously into the endogenous CENP-A locus in RPE-1 cells (see Star Methods, Fig. S1A). The HaloTag undergoes irreversible covalent conjugation to fluorescent small molecule derivatives, thereby enabling fluorescent pulse/chase-based assays to label and distinguish pre-existing versus newly deposited CENP-A (Fig. 1E; (Los et al., 2008)). We found that new CENP-A-Halo was gradually incorporated at centromeres in quiescent RPE-1 cells (Fig. 1E,F) despite the absence of division (Fig. S1B). This suggests that CENP-A is actively deposited in quiescent cells. However, it is conceivably possible that new fluorescent signal reflects incomplete stability of the HaloTag bond, enabling exchange of the old fluorophore for new fluorophore on the same CENP-A molecule. To test this, we reduced the expression of new CENP-A by treatment with siRNAs against CENP-A. CENP-A RNAi in quiescent cells substantially decreased new CENP-A-Halo incorporation, providing support that this assay monitors the deposition of newly synthesized protein (Fig. S2A,B). Although new CENP-A deposition is reduced, total centromeric CENP-A levels are only modestly decreased by CENP-A siRNA treatment (Fig. S2A,B). These results suggest that there is post-translational control to maintain a defined pool of soluble CENP-A nucleosomes. In this model, pre-existing CENP-A would be turned over when ongoing synthesis occurs, but stabilized when new CENP-A synthesis is blocked thereby maintaining constant levels. In support of this model, ectopic 3xGFP-CENP-A expression markedly reduces the levels of endogenous CENP-A (Fig. S2C). In contrast, depletion of the 3xGFP-CENP-A transgene partially reverses this down-regulation (Fig. S2C). Together, these results suggest that dynamic control mechanisms act to maintain steady state CENP-A levels in the cell and regulate its abundance at centromeres.
We next sought to evaluate the rate of CENP-A incorporation and loss in G0-arrested quiescent cells. By normalizing the level of new CENP-A relative to total CENP-A, we found that CENP-A deposition occurred at a rate of approximately 10% of total CENP-A per day, reaching an average of 68% of total CENP-A by day 7 (Fig. 1F). However, the extent of new incorporation varied substantially between cells (ranging from 25–100% of total CENP-A). Similar results were obtained when quiescent cells were grown continuously in the presence of STLC, an inhibitor of the mitotic kinesin Kif11, to prevent any rare mitotic divisions without affecting non-dividing cells (Fig. S1C). We also found that new histone H3.1A and H4 were gradually incorporated in quiescent RPE-1 cells throughout the chromosome based on HaloTag labeling (Fig. S1D, E; also see (Toyama et al., 2018)). To test whether a concomitant loss of old CENP-A occurs over time, we fluorescently labeled all CENP-A-HALO, and then allowed cells to divide to dilute out the soluble CENP-A pool prior to entering quiescence. After 11 days in quiescence, old CENP-A-HALO was reduced to ~30% of that on day 0 of quiescence (Fig. S1F), suggesting a loss of ~6% CENP-A per day. As with new CENP-A incorporation, there was a considerable variation for this loss ranging between 15 and 50% day 0 levels in individual cells. Given these results, and the finding that the average total level of CENP-A at centromeres remained unchanged during a 14-day quiescent arrest (Fig. 1A), we conclude that balanced loss and incorporation occurs at centromeres to maintain constant CENP-A levels in the absence of division.
CENP-A deposition in quiescent cells requires HJURP and the Mis18 complex
We next sought to define the requirements for CENP-A deposition and maintenance at centromeres in quiescent human cells. For these experiments, we used an inducible CRISPR/Cas9-based knockout system in RPE-1 cell lines (McKinley and Cheeseman, 2017; McKinley et al., 2015). Consistent with prior results, the inducible knockout of the CENP-A-specific chaperone HJURP or the Mis18 complex subunit Mis18β in cycling cells resulted in a dramatic reduction in centromeric CENP-A levels (Fig. S2D,E) and mitotic defects after 7 days of continuous proliferation. Furthermore, the HJURP inducible knockout resulted in a strong reduction of HJURP protein based on immunofluorescence (Fig. S2F). To test the role of HJURP and the Mis18 complex in CENP-A deposition during quiescence, we next induced the knockouts as cells entered quiescence (Fig. 2A). To ensure no further divisions occurred, the cells were cultured in the presence of STLC. Uninduced control cells retained CENP-A at centromeres throughout the time course (Fig. 2B–E). In contrast, in HJURP and Mis18β knockout cells, CENP-A centromere levels began declining at day 4 of their quiescent arrest, decreasing by an average of 35–45% at day 10 despite the absence of division (Fig. 2B–E). We note that this loss varies dramatically between cells (ranging from 0% to 100%), but that the average is smaller than that predicted by the ~65% increase in new CENP-A-HALO incorporation over a similar timescale (Fig. 1F). These differences may relate to the slow kinetics of knockdown under quiescent conditions, or to technical differences between these assays. As an orthologous metric for CENP-A loss after knockout, we performed Western blot for total CENP-A in quiescence following HJURP knockout induction. We found that total cellular CENP-A levels decreased slightly in control cells during a quiescence arrest, but were more substantially reduced in HJURP knockout cells dropping to ~26% of day 0 levels after 10 days in quiescence (Fig. 2F). We conclude that a substantial population of CENP-A nucleosomes are turned over at centromeres in non-dividing cells, in surprising contrast with previous models.
Figure 2. Gradual CENP-A incorporation in quiescent cells requires HJURP and the Mis18 complex.

(A) Schematic showing knockout strategy. Knockout of HJURP or Mis18β was induced in RPE-1 cells as they entered quiescence, which were then cultured in the presence of the KIF11-inhibitor STLC to ensure no further division. (B) Immunofluorescence for endogenous CENP-A showing a marked reduction in its centromere levels in HJURP knockout cells under quiescent conditions. Scale bar = 5 μm. (C) Fluorescence quantification of centromeric CENP-A in HJURP knockout cells relative to control levels. Each point represents the average of all centromeres within a cell. Error bars represent the mean and standard deviation (Day 0: control n=104, iKO n=81; Day 2: control n=83, iKO n=93; Day 4: control n=80, iKO n=86; Day 7: control n=81, iKO n=93; Day 10: control n=88, iKO n=87 cells). **** p < 0.0001 by two-tailed Mann-Whitney test. (D) Immunofluorescence for CENP-A in the Mis18β knockout, as in (B). (E) Quantification of CENP-A intensity for the Mis18β knockout, as for (C). Scale bar = 5 μm. (Day 0: control n=104, iKO n=95; Day 2: control n=98, iKO n=110; Day 4: control n=106, iKO n=113; Day 7: control n=62, iKO n=101; Day 10: control n=90, iKO n=109 cells). **** p < 0.0001 by two-tailed Mann-Whitney test. (F) Western blot for total CENP-A protein levels in quiescence in either control cells or with inducible HJURP knockout. CENP-A levels were quantified (Adobe Photoshop) and normalized to GAPDH for loading. Values represent the average of two independent experiments. (G) Immunofluorescence for microtubules and DNA showing anaphase chromosome segregation following release from quiescence by serum addition after either 2 days or 10 days in quiescence. The presence of lagging chromosomes and DNA bridges was scored as a rigorous measure of chromosome mis-segregation. ~100 anaphase cells were scored for each experimental replicate. Cycling cells, n = 5 replicates; Day 2: Control (Cas9), n = 3; HJURP iKO, n = 3; Mis18β iKO, n = 3; Day 10: Control (Cas9), n = 6; HJURP iKO, n = 3; Mis18β iKO, n = 4. Scale bar = 10 μm. * p = 0.0228, **** p < 0.0001 by Fisher’s exact test.
To assess the consequences of centromeric CENP-A reduction following knockout of deposition factors, we induced cell cycle reentry by serum addition and monitored chromosome segregation during anaphase in the first mitotic division following release, a rigorous measure of chromosome segregation fidelity. Prior work found that CENP-A levels at centromeres must be reduced substantially to observe a dramatic effect on chromosome segregation (Fachinetti et al., 2013). For cells released from quiescence after 2 days, HJURP and Mis18β knockout cells showed only modest chromosome mis-segregation. In contrast, when cells were released after 10 days in quiescence (when CENP-A levels are more substantially reduced; Fig. 2C,E), we found that HJURP, and to a lesser extent Mis18β inducible knockout cells displayed a significant increase in lagging chromosomes and chromosome bridges during anaphase (Fig. 2G). These chromosome segregation issues are unlikely to be related to DNA damage induced by the Cas9-based knockout strategy, as we did not detect substantial defects after 2 days in quiescence (Fig. 2G) and these cells did not display detectably increased gamma-H2AX (Fig. S2G). Thus, the presence of CENP-A nucleosomes at centromeres is maintained by an HJURP and Mis18 complex-dependent mechanism during quiescence in G0-arrested human cells, which is required for proper chromosome segregation following the return to growth.
CENP-A is gradually incorporated at centromeres in prophase I-arrested oocytes
The ongoing CENP-A incorporation that we observed in quiescent human tissue culture cells prompted us to test whether this behavior occurs in other cell types that undergo a prolonged cell cycle arrest. Oocytes remain arrested in prophase I of meiosis for extended periods until hormonal stimulus triggers their re-entry into meiosis. As fertilized oocytes divide to create an entire organism, it is critical to maintain centromere identity during this extended prophase I arrest to act as templates for all subsequent centromeres. To define the mechanisms that maintain CENP-A during quiescence, we analyzed oocytes from the starfish Patiria miniata, which provide a robust model for cell biological studies of meiosis and early development (Fig. 3A; (Borrego-Pinto et al., 2016a; Borrego-Pinto et al., 2016b). To assess centromere dynamics during the meiotic divisions, we identified starfish orthologues of CENP-A and the CENP-A-binding partner, CENP-N. When expressed as GFP fusions by mRNA injection into oocytes, both 3xGFP-CENP-A and 3xGFP-CENP-N localized specifically to centromeres in blastula-stage embryos in which cells undergo frequent divisions (Fig. S3A,B).
Figure 3. CENP-A is deposited in G1 following meiotic completion.

(A) Confocal z-projections of Patiria miniata oocytes in prophase I arrest (left) or meiosis II (right), with DlC images (top) and immunofluorescence for microtubules and DNA (bottom). Scale bar = 40 μm. (B) Schematic of meiotic progression and strategy to assess new centromere protein incorporation in starfish oocytes. (C) Localization dynamics for 3xGFP-CENP-N (middle row) and 3xGFP-CENP-A (bottom row). Counter staining for microtubules (scaled non-linearly) and DNA is provided on the top row, corresponding to CENP-N oocytes. CENP-A and CENP-N images were individually scaled linearly. Scale bar = 10 μm. (D) Quantification of relative CENP-N and CENP-A fluorescent intensity at centromeres throughout meiosis. Each time point represents the mean and standard deviation of at least 6 oocytes.
We next assessed the timing for the localization of newly synthesized centromere proteins in oocytes (Fig. 3B). Starfish oocytes remain arrested in prophase I of meiosis until hormonal stimulation. Upon hormonal stimulation, oocytes undergo two sequential rounds of chromosome segregation (meiosis I and II) without an intervening S-phase. Following mRNA injection of prophase I-arrested oocytes, newly expressed 3xGFP-CENP-N localized to centromeres within 24 hours following mRNA injection (Fig. 3C,D), indicative of rapid turnover. In contrast, new 3xGFP-CENP-A did not localize to centromeres in arrested oocytes (Fig. 3C,D), although soluble protein fluorescence was detected and Western blotting indicated the presence of 3xGFP-CENP-A (Fig. S3C). When meiosis was induced hormonally, we did not observe CENP-A incorporation until completion of meiosis and formation of the female pronucleus (Fig. 3C,D). This timing coincides with the first G1-like cell cycle state following the oocyte-to-embryo transition (Mori et al., 2006; Tachibana et al., 1997). Thus, rapid CENP-A deposition occurs following meiotic exit into G1 in starfish oocytes, similar to the G1 deposition observed in mitotically-dividing human tissue culture cells (Jansen et al., 2007).
As with mammalian oocytes that can remain arrested for decades, starfish oocytes are held in an extended prophase arrest for months until spawning. To assess CENP-A dynamics during the prophase I arrest, we developed a method to culture starfish oocytes in vitro using starfish coelomic fluid, which maintained their competency for meiosis and fertilization for several weeks. Although we did not detect new 3xGFP-CENP-A at centromeres at early time points, after 5 days we observed its incorporation at punctate foci (Fig. 4A,B; Fig. S4A). This newly deposited CENP-A was retained at centromeres after meiotic entry and co-localized with antibodies against the centromere protein CENP-C (Fig. 4A; Fig. S4A), indicating that it was incorporated specifically at centromeres. By quantifying 3xGFP-CENP-A levels incorporated during the arrest relative to the level of 3xGFP-CENP-A that is incorporated in the female egg G1 pronucleus, we found that 3xGFP-CENP-A intensity increased gradually at a rate of ~2% per day (Fig. 4B), consistent with a low level of ongoing deposition. New CENP-A incorporation occurred stochastically, with some centromeres in an individual oocyte incorporating >60% of the CENP-A levels deposited during the G1/pronucleus deposition event, but others incorporating relatively little (Fig. S4B). Thus, similar to the behavior of G0-arrested human cells, new CENP-A molecules are gradually incorporated in starfish oocytes during prophase I arrest.
Figure 4. CENP-A is gradually deposited in prophase arrested oocytes.

(A) Prophase-arrested chromatin (left) and meiosis I spindles (right) imaged 1 day or 10 days following expression of 3xGFP-CENP-A. 3xGFP-CENP-A images were scaled individually. (B) Quantification of 3xGFP fluorescence intensity incorporated in prophase relative to that deposited in G1 (see Experimental Procedures). Points show the mean of all centromeres from an individual oocyte. Error bars represent the mean and standard deviation of at least 16 oocytes. (C) Prophase incorporation of 3xGFP-CENP-A is reduced following Mis18BP1 knockdown or DN-Mis18 expression. To facilitate colabeling with CENP-C antibodies and eliminate background fluorescence from soluble 3xGFP-CENP-A in the bulk nucleoplasm, oocytes were induced to enter meiosis I to facilitate quantification. At this stage, centromeric CENP-A levels are unchanged relative to prophase I. (D) Quantification of 3xGFP-CENP-A fluorescence incorporation relative to control injected oocytes. Points represent the average of all centromeres from individual total oocytes. Error bars represent the mean and standard deviation of at least 29 oocytes. **** p < 0.0001 by Mann Whitney test. (E) Oocytes following 12 days culture while expressing DN-Mis18 or with control injection, stained for endogenous CENP-C, microtubules, and DNA. (F) Graph showing percent of oocytes with normal MI chromosome alignment at the indicated time points following a prophase I arrest. Points represent the mean and standard deviation for 3 trials for a total of at least 92 total oocytes per day. ****p < 0.0001 Fisher’s exact test. Scale bars = 5 μm. (G) Quantification of CENP-C immunofluorescence intensity at centromeres of aligned and misaligned chromosomes. Error bars represent the mean and standard deviation (Aligned, n = 267 centromeres; Misaligned, n = 54 centromeres; 10 total oocytes). ****p < 0.0001 unpaired 2-tailed t-test.
CENP-A deposition in prophase I arrested oocytes requires the Mis18 complex
We hypothesized that gradual CENP-A incorporation in starfish oocytes is required to maintain centromere identity, ensuring a precise first meiotic division after exiting prophase I arrest. To test this, we first sought to define the requirements for the deposition of new CENP-A nucleosomes in starfish. Depletion of the Mis18 complex subunit Mis18BP1, a canonical CENP-A deposition factor (McKinley and Cheeseman, 2016), by morpholino injection strongly reduced the G1 deposition of new CENP-A that normally occurs in the female egg pronucleus (Fig. S4C,D). Furthermore, expression of an N-terminally tagged mCherry-Mis18 (hereafter called DN-Mis18) acted in a dominant negative manner to block CENP-A deposition in the egg pronucleus (Fig. S4C,D), likely by preventing Mis18 holo-complex formation. Following fertilization, the resulting Mis18BP1 morphant or DN-Mis18-expressing embryos displayed severe chromosome mis-segregation (Fig. S4E), indicative of defective centromere and kinetochore function. These results highlight the important role for the Mis18 complex in maintaining centromere identity in rapidly dividing cells.
We next tested whether the Mis18 complex was required for the gradual CENP-A incorporation that occurs during an extended prophase I arrest. We first depleted arrested oocytes for Mis18BP1 by morpholino or expressed DN-Mis18, and then expressed 3xGFP-CENP-A (Fig. 4C). In contrast to controls, oocytes with Mis18BP1 knockdown or DN-Mis18 expression displayed substantially reduced CENP-A incorporation at centromeres during prophase I arrest (Fig 4C,D). These findings indicate that, in addition to the previously defined role for the Mis18 in cycling cells to promote rapid CENP-A replenishment during G1 (Jansen et al., 2007; McKinley and Cheeseman, 2016), the gradual CENP-A deposition that occurs during the prophase I arrest also requires the canonical CENP-A deposition machinery.
Prophase I CENP-A deposition is required for faithful meiotic chromosome segregation
The ongoing CENP-A deposition in prophase I-arrested oocytes suggests that active centromere maintenance is necessary to support the meiotic divisions. To test this, we inhibited CENP-A incorporation by expressing DN-Mis18 in prophase I-arrested oocytes. We then assessed meiotic chromosome segregation following hormonal stimulation at different time points. Meiosis proceeded normally at time points following up to 4 days of DN-Mis18 expression, indicating that centromeric CENP-A remained at sufficient levels over this arrest period (Fig. 4E,F). However, by 8 days following DN-Mis18 expression, we observed increased chromosome misalignment during meiosis I compared to controls (Fig. 4E,F). The prevalence of chromosome misalignment errors continued to increase with the length of the arrest period (Fig. 4E,F). Importantly, centromeres on misaligned chromosomes in DN-Mis18 embryos contained substantially reduced levels of the CENP-A binding protein CENP-C (Fig. 4E,G), suggesting a defect in centromere maintenance. Based on the ongoing CENP-A incorporation in prophase I-arrested oocytes (Fig. 4A,B) and the defective meiotic chromosome alignment that occurs when this deposition is prevented (Fig. 4E,F), we conclude that centromeric CENP-A is actively incorporated during the prophase I arrest in oocytes to maintain its full levels at centromeres. In contrast, a CENP-A translation-blocking morpholino failed to reduce endogenous CENP-A levels in prophase I-arrested oocytes after 10 days in culture, despite the fact that this morpholino potently reduced CENP-A levels after these cultured oocytes were then fertilized and allowed to develop for 18 hours (Fig. S4F). These results suggest that CENP-A protein is stable in oocytes (also see (Smoak et al., 2016), but that the centromere population is dynamically exchanged with a soluble pool to maintain its full levels at centromeres. Together, these findings demonstrate that CENP-A is exchanged in quiescent cells to maintain CENP-A levels and present a surprising contrast with previous models in which CENP-A is immobile at centromeres (Jansen et al., 2007).
Plk1 licensing distinguishes rapid G1 CENP-A deposition from gradual deposition during quiescence
The results described above suggest that there are two distinct, but related CENP-A deposition processes: 1) A rapid pulse of CENP-A deposition that occurs during G1 in cells undergoing mitotic divisions to replenish CENP-A levels following DNA replication, and 2) Gradual CENP-A deposition that occurs in non-dividing cells to maintain CENP-A levels. We next sought to define the basis for this substantial difference in behavior and the rate of CENP-A deposition by determining the requirements that distinguish G1 and G0 deposition. Our work demonstrates that the Mis18 complex is required for both modes of CENP-A deposition. However, our prior work additionally implicated the kinase Plk1 as a licensing factor for CENP-A deposition during G1 required to promote rapid CENP-A deposition (McKinley and Cheeseman, 2014). Surprisingly, in contrast to the clear requirement of Plk1 for the rapid pulse of CENP-A deposition that occurs following mitotic exit (Fig. 5A; (McKinley and Cheeseman, 2014), we found that treatment with the Plk1 inhibitor BI2536 did not prevent new CENP-A deposition in quiescent RPE-1 cells (Fig. 5B,C). Indeed, we were unable to detect Plk1 in G0-arrested cells (Fig. 5D; Fig. S5A,B), although this does not exclude the possibility of a small population of Plk1 in these cells. These findings indicate that gradual CENP-A deposition in quiescent cells requires the canonical CENP-A deposition machinery, but not licensing by Plk1, which may promote the more rapid CENP-A deposition observed in G1 cells.
Figure 5. Defining unique requirements for CENP-A deposition during G1 and quiescence.

(A) Plk1 inhibition blocks new CENP-A deposition during G1 (also see (McKinley and Cheeseman, 2014)). Top, schematic showing growth and Halo block conditions to test new CENP-A deposition in CENP-A-Halo Rpe1 cells. Bottom, immunofluorescence images showing new vs. old CENP-A staining either before division (18 hours following serum addition) or during G1 in either a DMSO control or in the presence of 100 nM of the Plk1 inhibitor BI2526. Scale bars = 10 μm. (B) Plk1 is not required for G1 CENP-A deposition in G0-arrested cells. Immunofluorescence images showing that treatment with 100 nM of the Plk1 inhibitor BI2536 does not prevent incorporation of new CENP-A-Halo in quiescent cells over a 7 day time course (as in Fig. 1E). Scale bar = 5 μm. (C) Quantification of new CENP-A-Halo fluorescent intensity relative to DMSO controls. Some data points from control cells are repeated from Fig. 1E,F. Each point represents the average of all centromeres in a cell. Error bars represent the mean and standard deviation for 62 cells/condition. (D) Plk1 is detectable at centromeres in G1, but not G0 quiescence by immunofluorescence. Scale bar = 10 μm.
Blocking transcription reduces new CENP-A deposition in arrested oocytes
The results described above suggest that CENP-A levels are maintained in quiescent cells through balanced loss and incorporation. As there do not appear to be gaps of naked DNA at centromeres (Dunleavy et al., 2011), new CENP-A deposition at centromeres would require the loss of a pre-existing nucleosome to create space for its incorporation. An important question is what forces evict CENP-A or histone H3-containing nucleosomes at centromeres, thereby facilitating new deposition. For canonical nucleosomes, prior work found that RNA polymerase transiently evicts nucleosomes during transcription (reviewed in (Venkatesh and Workman, 2015)). Centromeres are transcribed by RNA polymerase II in proliferating cells (Chan et al., 2012; Grenfell et al., 2016; Saffery et al., 2003), which could promote nucleosome displacement. Using a single molecule RNA-FISH-based approach to detect centromere transcripts, we found that centromere transcripts are also present in quiescent RPE-1 cells at similar levels as under proliferating conditions (Fig. 6A, B). In both cases, these transcripts were eliminated by treatment with the Cdk7 inhibitor THZ1 to block transcriptional elongation (Fig. 6A,B), suggesting that ongoing transcription is occurring at centromeres in quiescent cells.
Figure 6. CENP-A deposition in quiescence involves RNA transcription.

(A) Single molecule RNA fish showing the presence of centromeric alpha satellite-derived transcripts in RPE-1 cells. Scale bar = 5 μm. (B) Quantification of transcript numbers. Transcripts are approximately equal in cycling and G0 quiescent cells, but decrease when treated with the Cdk7 transcription inhibitor THZ1. Error bars represent the mean and standard deviation (cycling n = 267 nuclei; cycling + THZ1 n = 113; quiescent n = 269; quiescent + THZ1 n = 183). ****p<0.0001 by Mann Whitney test. (C) Immunofluorescence for RNA Pol II and detection of nascent RNA synthesis by EU incorporation reveals ongoing transcription in prophase I arrested starfish oocytes. Scale bar = 5 μm. (D) Pol II and nascent RNA (EU) are present in the vicinity of centromeres, visualized with 3xGFP-CENP-N in arrested oocytes. Pol II and EU images are scaled equivalently. (E) Treatment with the Pol II inhibitor Triptolide reduces Pol II localization and EU levels at centromeres. Individual points represent the average fluorescence at all centromeres. Error bars represent the mean and standard deviation (control n = 51 oocytes, triptolide n = 57 oocytes, measured for both Pol II and EU). ****p<0.0001 by Mann Whitney test. (F) Immunofluorescence showing new CENP-A incorporation in quiescent oocytes. Inhibition of Pol II with 10 μM of triptolide, or Cdk9 with 1 μM of LDC000067 reduces incorporation of new 3xGFP-CENP-A in prophase after 9 days in culture. Incorporation occurs in prophase I, but oocytes were stimulated to enter meiosis I for visualization to condense chromosomes for clarity. Scale bar = 5 μm. Metaphase 3xGFP-CENP-A images are scaled equivalently, and prophase images are linearly scaled individually (G) Quantification of new 3xGFP-CENP-A incorporation relative to DMSO control. Each point represents the mean of all centromeres from one oocyte. Error bars represent the mean and standard deviation (control, n = 58; triptolide, n = 47; Cdk9i, n = 51). ****p<0.0001 by Mann Whitney test. (H) Inhibition with 10 μM triptolide or 1 μM LDC000067 during prophase I arrest and in meiosis does not alter incorporation of new 3xGFP-CENP-A in G1. Scale bars = 5 μm. (I) Quantification of 3xGFP-CENP-A incorporation relative to DMSO control. Each point represents the mean of all centromeres from one oocyte. Error bars represent the mean and standard deviation (control n = 42, triptolide n = 20, Cdk9i n = 38). Significance determined by Mann Whitney test.
Therefore, we sought to test whether transcription is required for the ongoing deposition we observed in quiescent cells. Disrupting RNA Polymerase Il-dependent transcription in G0-arrested human tissue culture cells has severe consequences for cellular function, with cells losing viability after two days of treatment with triptolide or THZ1 (not shown; (Mak et al., 2009)). In contrast, fertilization-competent oocytes have substantial pre-existing mRNA pools such that they do not display a strict requirement for ongoing transcription (Bachvarova, 1985; Davidson, 1976). Indeed, we found that starfish oocytes could be cultured continuously in the presence of the RNA Pol II inhibitor Triptolide or the Cdk9 inhibitor LDC000067 without the loss of viability. In addition, inhibiting transcription did not prevent the translation of 3xGFP-CENP-A following mRNA injection based on cellular fluorescence, providing the ability to monitor new CENP-A deposition. Importantly, oocytes do display ongoing transcription as evidenced by RNA Polymerase II localization and the incorporation of EU into nascent transcripts (Fig. 6C), both of which are prevented by Triptolide treatment (Fig. 6D,E).
When transcription was inhibited in oocytes using Triptolide or LDC000067, we found that new CENP-A deposition (based on the incorporation of newly synthesized 3xGFP-CENP-A; see Fig. 4A) was reduced by an average of ~50% in prophase I-arrested oocytes over a 9-day time course (Fig 6F,G). Although inhibiting transcription disrupted new CENP-A incorporation, total centromere protein levels, as measured by staining for the CENP-A binding protein CENP-C, were not substantially altered (Fig. S6A). This suggests that transcription destabilizes pre-existing CENP-A, and that in the absence of transcription centromere proteins are able to avoid eviction. Similarly, we found that new histone H3.3 was gradually deposited on chromosomes in prophase I-arrested oocytes (Fig. S6B), and that that this incorporation was strongly reduced by Triptolide treatment (Fig. S6C,D). In contrast, when these oocytes were incubated in the presence transcription inhibitors for 9 days, but instead were allowed to exit meiosis and form the female egg pronucleus, they robustly deposited CENP-A in G1 to levels similar to controls (Fig 6H,I). Thus, transcription is required to allow new CENP-A deposition in prophase I-arrested oocytes, but is not required during the canonical G1 CENP-A deposition. In addition, these results indicate that transcription inhibitors did not grossly alter the presence or function of the deposition machinery based on the functional G1 deposition. Indeed, we found that the transcript levels of CENP-A, CENP-C, Mis18BP1, and Mis18 were unaffected by Triptolide treatment (Fig. S6E). Together, these data suggest that RNA Pol II transcription at the centromere provides at least part of the destabilizing force that evicts centromere-bound nucleosomes in quiescent cells, allowing their replacement by new CENP-A nucleosomes through a Plk1-independent mechanism.
Centromere identity is not maintained in terminally differentiated muscle cells
Our work demonstrates that two fundamentally different cell types that retain their proliferative potential - oocytes and serum-starved tissue culture cells- display a low rate of CENP-A deposition and exchange. To test whether ongoing CENP-A incorporation is a general feature of non-dividing cells, we next assessed CENP-A levels in terminally differentiated cells. We first tested the mouse C2C12 myoblast-like cell line as an in vitro model for terminal differentiation. C2C12 cells differentiate to form multi-nucleated myotubes upon serum starvation (Yaffe and Saxel, 1977). In contrast to cycling cells, the nuclei in the multi-nucleated myotubes were substantially depleted for both CENP-A and the CENP-A-binding protein CENP-C, with fewer centromere foci and less total fluorescence (Fig. 7A,B).
Figure 7. CENP-A levels decline in terminally differentiated muscle cells.

(A). Immunofluorescence images showing localization of CENP-A (left) and CENP-C (right) in mouse C2C12 cells that are either growing in culture (undifferentiated myoblasts) or were differentiated by serum starvation to form multi-nucleated muscle myotubes. Phalloidin staining is a marker for myotubes. Arrows indicate myotubule nuclei in the CENP-C experiments, whereas the other nuclei with CENP-C staining represents a quiescent, undifferentiated cell. Scale bar = 10 μm. (B) Quantification of CENP-A and CENP-C fluorescent intensity in either undifferentiated C2C12 cells or following myotube formation. (C) Immunofluorescence images of hepatocytes in post-natal day 0 or 8-week adult liver, showing CENP-A levels in vivo. Scale bar = 10 μm. Quantification of CENP-A fluorescence in hepatocytes normalized to ploidy as described in Experimental Procedures. Error bars represent the mean and standard deviation of N=40 post-natal hepatocytes, and N=41 adult hepatocytes. (D) Immunofluorescence images of cardiomyocytes in post-natal day 0 or 8-week adult heart, showing CENP-A levels in vivo. Scale bar = 10 μm. Quantification of CENP-A fluorescence in cardiomyocytes, where error bars represent the mean and standard deviation of N=40 post-natal cells, and N=33 adult cells. ****p<0.0001 by Mann Whitney test.
To determine whether a similar loss of CENP-A occurs in vivo, we conducted immunofluorescence on mouse tissue sections. The mammalian liver has remarkable regenerative capacity and quiescent hepatocytes retain the ability to proliferate into adulthood (Malato et al., 2011). We found that both neonatal and adult hepatocyte nuclei from unperturbed liver displayed equivalent and robust levels of CENP-A (Fig. 7C; Fig. S7). Cardiac muscle from neonatal mice, but not adult mice, is capable of self-repair (Porrello et al., 2011). Strikingly, we found that CENP-A is present at robust levels in neonatal cardiac muscle nuclei (postnatal day 0), but is substantially reduced in adult cardiac muscle nuclei with reduced CENP-A levels and fewer detectable centromere foci (Fig. 7D; Fig. S7). The presence of CENP-A in neonatal heart cells is consistent with their potential to undergo further cell division and suggests that they have not yet made the transition to irreversibly exit the cell cycle. We also found that adult skeletal muscle (gastrocnemius) had significantly reduced CENP-A levels relative to liver cells (Fig. S7). These results suggest that active centromere maintenance is not a default program of all cells, but might occur specifically in cells that need to divide again following quiescence.
Discussion
Here, we define a mechanism critical for ensuring the maintenance and inheritance of the epigenetically-defined centromere during extended periods of cellular quiescence. During development, subsets of cells within the body terminally differentiate and permanently exit the cell cycle, whereas slow-cycling or quiescent cells remain dormant, but poised to divide. Arrested cells require a mechanism to maintain the CENP-A epigenetic mark under all circumstances where future proliferation is required. Our results reveal that, in somatic human RPE-1 G0-arrested cells and prophase I-arrested oocytes, the centromere is actively maintained by a slow rate of ongoing CENP-A deposition. Prior studies in tissue culture cells have focused upon rapidly dividing cells in which gradual CENP-A incorporation was likely masked by the more rapid G1 deposition.
Our work raises an important distinction between the stability of CENP-A-H4 protein levels in the cell and its persistent localization at centromeres. Although CENP-A-H4 forms a highly stable protein complex whether at centromeres or in a soluble pool (this paper; (Smoak et al., 2016), centromere-bound CENP-A is subject to forces such as transcription that destabilize or evict these nucleosomes necessitating its replacement (Venkatesh and Workman, 2015). Displacement of CENP-A or centromere-localized canonical H3-containing nucleosomes would create gaps at centromeres that would then be filled through the action of the CENP-A deposition machinery. Based on the results described here, CENP-A nucleosomes must be continually incorporated at centromeres to maintain a constant net level with balanced loss and re-deposition.
The ability to maintain steady-state CENP-A levels through balanced loss and incorporation suggests the presence of a homeostatic control mechanism with important implications for epigenetic inheritance. In support of this model, we find that siRNA treatment to prevent new CENP-A synthesis only modestly reduced the pre-existing centromeric population (Figure S2A,B). In contrast, ectopic expression of CENP-A as a transgene leads to the downregulation of the endogenous copy (Fig. S2C). These results suggest that the pool of CENP-A is regulated post-translationally. This could be accomplished by a limiting amount of the CENP-A chaperone, HJURP, which would result in the instability of excess CENP-A molecules. The integration of balanced nucleosome loss and reincorporation, coupled to a regulated pool of soluble histones, provides a distinct paradigm for the long-term maintenance of this epigenetic mark from previous models of an immobile centromeric CENP-A.
Gradual CENP-A exchange provides a critical means to maintain centromere identity across diverse physiological states. Based on the requirement for an active program to maintain CENP-A at centromeres, we speculate that the presence and levels of CENP-A may provide a biomarker for the future proliferative potential of a cell, such as we observe for cardiac muscle and liver cells (Fig. 7). This gradual exchange is essential to support subsequent chromosome segregation following cell cycle reentry (Fig. 2F; Fig. 4E,F), whereas loss of centromeric CENP-A in terminally differentiated cell types could contribute to the block in their proliferation. Therefore, defects in CENP-A deposition in quiescent cells could result in subsequent chromosome segregation abnormalities contributing to tissue dysfunction, infertility/birth defects, or cancer.
Star Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Iain Cheeseman (icheese@wi.mit.edu). Cell lines, plasmids and antibodies are available upon request from the authors. Plasmids will additionally be deposited with Addgene.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Wild-caught Patiria miniata were provided by South Coast Bio Marine (http://scbiomarine.com/) and maintained in aquaria at 15°C. Oocytes and testes were extracted surgically by creating a small incision on the oral side of the animal and removing ovary pieces with forceps. Oocytes were then released by cutting the ovary fragments with scissors in filtered seawater, as described previously (Wessel et al., 2010).
C57BL/6J mice were purchased from the Jackson Laboratory. All mice were group-housed with a 12-hour light-dark cycle (light from 7 AM to 7 PM, dark from 7 PM to 7 AM) in a specific-pathogen-free animal facility with unlimited access to food and water. Liver, gastrocnemius, and heart were harvested from 8-week-old adult male mice or post-natal day 0. All animal procedures were approved by the Massachusetts Institute of Technology Committee on Animal Care.
Cell lines
C2C12 (female mouse derived) and hTERT RPE-1 (female human derived) cells were obtained from ATCC (CLR-1772 and CRL-4000, respectively). Inducible Cas9 RPE-1 cells (cTT33) were generated in (McKinley and Cheeseman, 2017).
METHOD DETAILS
Oocyte culture
For short-term culture (<18 hours) and rapid assessment of localization behavior (Fig. 3C), oocytes were kept in filtered seawater. For long term culture (up to 15 days; Fig. 4A,C), oocytes were kept in 6 or 12 well plates containing 0.2 μm filtered adult starfish coelomic fluid (CF) supplemented with antibiotics (a stock of 10 mg/ml trimethoprim, 50 mg/ml sulfamethoxazole prepared in DMSO diluted 1:1000 + 100 units/mL penicillin, 100 units/mL streptomycin final). CF was changed every 2–3 days. For transcription studies (Fig. 6), 10 μM triptolide (Pol II inhibitor, Sigma) or 1 μM LDC000067 (Cdk9 inhibitor, Selleck Chemicals) was added to CF and changed every 2 days. For analysis of nascent RNA, oocytes were cultured for 3 days in the presence of 1 mM EU (Click-iT RNA Imaging Kit, Invitrogen). For maturation, oocytes were transferred to filtered seawater and 1-methyladenine (Acros Organics) was added to a working concentration of 10 μM. For experiments in which embryos were analyzed, oocytes were fertilized at 60 minutes after 1-methyladenine addition. All oocyte and embryo culture was performed at 15°C.
Construct and antibody generation
Starfish homologs were identified using sequence analysis tools at echinobase.org and an ovary de novo transcriptome described in (Kudtarkar and Cameron, 2017; Reich et al., 2015). Full-length PmCENP-A and PmCENP-N were cloned as N-terminal 3xGFP fusions in PCS2+ (von Dassow et al., 2009). PmMis18 (DN-Mis18) was cloned as an N-terminal single mCherry fusion in PCS2+8 (Gokirmak et al., 2012). The first 68 amino acids of PmCENP-A, and the first 250 amino acids of PmCENP-C were expressed as a GST-fusions in BL21(DE3) E. coli. For mouse CENP-C, amino acids 88–232 were expressed as a GST fusion in pET3aTr-GST. Protein expression was induced with 0.1 mM IPTG overnight at 18° C. Protein was purified from 6 liters of bacterial culture with Glutathione-agarose (Sigma), eluted with reduced L-glutathione (Sigma), and purity was confirmed by SDS-PAGE analysis. Antibodies were then generated in rabbits (Covance). GST tags were removed using PreScission protease (GE Healthcare), and antigens were coupled to HiTrap NHS-activated columns according to the manufacturer’s instructions (GE Healthcare). Antibodies were then affinity purified using these columns and dialyzed into 50% glycerol-PBS.
Oocyte microinjection
For mRNA expression, plasmids were linearized with Notl and transcribed using mMessage mMachine SP6 followed by poly-A tailing (Ambion) and LiCl precipitation. Freshly extracted oocyte-follicle cell complexes were injected horizontally in Kiehart chambers with 2–4 picoliters of mRNA or morpholino solution in water. 3xGFP- CENPA mRNA was injected at 50–85 ng/μl, whereas 3xGFP-CENP-N and DN-Mis18 was injected at 500 ng/μl in water. Oocytes were then cultured as described in the previous section. Custom translation-blocking morpholino antisense oligos were synthesized to CENP-A and MIS18BP1 by Gene-Tools. The Gene-Tools standard control oligo targeting a human beta-globin intron mutation was used for all control injections. Morpholinos were injected at 500 μM in water.
Oocyte immunofluorescence and imaging
Oocytes were fixed for 3 hours to overnight in a buffer modified from (von Dassow et al., 2009) containing 2% paraformaldehyde, 0.1% Triton X-100, 100 mM Hepes, pH 7.0, 50 mM EGTA, 10 mM MgSO4, and 400 mM dextrose. Oocytes were blocked for 15 minutes in AbDil (3% BSA, 1 × TBS, 0.1% triton X-100, 0.1% Na Azide) and then stained with primary antibodies overnight at 4° C. Anti-CENP-C antibodies were used at 1 μg/ml. Pol II pSer5 antibody (ab5408, Abcam) was used at 1:1000. Microtubules were stained with 1:1000 DM1α (Sigma). GFP booster (Chromotek) was used at 1:500 to amplify the signal from GFP expression. DNA was visualized using Hoechst. Nascent RNA (EU) was detected as described by the manufacturer’s instructions (Click-iT RNA imaging kit, Invitrogen). Oocytes were imaged using a DeltaVision Core microscope (Applied Precision/GE Healthsciences) with a CoolSnap HQ2 CCD camera and a 100× 1.40 NA Olympus U- PlanApo objective. Confocal images (Fig. 3A) were collected on a Zeiss 710 confocal microscope.
Cell line generation
Inducible knockouts for Mis18β and HJURP in RPE-1 cells were created as described previously (McKinley and Cheeseman, 2017). Briefly, sgRNA sequences were cloned into pLenti (Wang et al., 2015), and used to generate lentiviruses for stable infection in RPE-1 cells harboring inducibly expressible Cas9 (cTT33). Cells were then selected with 10 μg/ml puromycin for 2 weeks. For endogenous tagging of CENP-A with the Halo tag (cKC288), RPE-1 cells were co-transfected with pX330 containing Cas9, an sgRNA targeting 3’ UTR of the genomic CENP-A locus (TGTCATCAGTGTTGCCATCG), and a repair CENP-A-Halo fusion-template pKC158, which harbors the HaloTag and a Neomycin resistance cassette derived from pL452 flanked by approximately 1 kb homology arms. The repair template is resistant to Cas9 by mutation of the PAM sequence TGG to TAA. Cells were selected with 1 mg/mL Geneticin (Gibco) before clonal cell line generation. The resulting cell line was heterozygously tagged as determined by Western blot for CENP-A (Fig. S1A) and DNA sequencing of the genomic locus. We note that we were unable to recover homozygously tagged cell lines, which could either reflect the poor efficiency of recombination in this cell line, or alternatively, incomplete functionality of the HaloTagged CENP-A allele. Endogenously tagged PLK1-NeonGreen cell line (cKC269) was generated as for the CENP-A-Halo cell line using a C-terminal targeting sgRNA expressed in pX330-BFP (McKinley and Cheeseman, 2014) and a TandemNeonGreen fusion-template with homology arms. To generate H3.1A-Halo (cLM26) and H4-Halo (cLM29) cell pools, RPE-1 cells were transfected with retrovirus carrying the transgenes (pLM32 and pLM33, respectively) and selected with 10 μg/ml Blasticidin (Life Technologies) (Cheeseman et al., 2004).
Cell culture
RPE-1 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) with 10% tetracycline-free fetal bovine serum (Gemini) (in the case of inducible knockout cell lines; standard FBS for all others), 100 units/mL penicillin, 100 units/mL streptomycin, and 2 mM L-glutamine (Complete Media) at 37°C with 5% CO2. For induction of quiescence, RPE-1-derived cell lines were allowed to reach confluence, and then shifted to Complete Media as above but with 0.1% fetal bovine serum. As shown in Fig. S1B, after cells entered confluence with this protocol, only a small fraction (2%) of cells undergo DNA replication over a 7 day time course as assessed continuous incubation with EdU. However, to ensure that no mitotic cells were present in the population, the media was supplemented with 10 μM STLC (inhibitor for the mitotic kinesin Kif11, Sigma) where indicated. This resulted in only 1% cells displaying EdU incorporation over a 7 day time course.
For knockout experiments under proliferating conditions (Fig. S2D), cells were seeded 1:10 onto uncoated glass coverslips, and doxycycline (DOX, Sigma) was added at 1 mg/L for 48 hours. The cells were then allowed to proliferate for 7 days before fixation. For knockout experiments in quiescent cells, 300,000 cells were seeded onto uncoated glass coverslips in 6-well plates. Knockout was induced 18 hours later by DOX addition (see scheme diagram in Fig. 2). Two days after plating, cells were shifted to 0.1% fetal bovine serum, with or without DOX, and 10 μM STLC. For all experiments in quiescence, the media containing 0.1% serum was changed daily. To induce DNA damage cells were incubated in 5 μM Etoposide (Calbiochem) for 24 h. To monitor DNA replication cells were incubated in 5 μM EdU (Invitrogen) for either 24 hours (proliferating cells) or 7 days (quiescent cells) with the media changed daily and fresh EdU added.
C2C12 myoblasts were cultured as previously described (Lawson and Purslow, 2000). Proliferating myoblasts were grown in in DMEM with 10% FBS and 1% penicillin/streptomycin. For immunofluorescence, myoblasts were seeded onto etched glass coverslips prepared as described by (Chaturvedi et al., 2015) and grown to confluence. To induce differentiation, the myoblasts were shifted to differentiation medium (DMEM with 10% horse serum or 5% FBS).
Halo tag pulse-chase detection
To assess CENP-A, H3.1A and H4 dynamics in quiescence, Histone-HaloTag cells were seeded onto uncoated coverslips and grown to confluence. The cells were then shifted to 0.1% serum-containing media and cultured for 5 days to ensure complete mitotic exit. The cells were then either left unblocked or blocked with 125 nM JF549-HaloTag ligand (gift from Luke Lavis; (Grimm et al., 2017; Grimm et al., 2015)). Coverslips were then taken at periodic intervals and fixed with 100% ice-cold methanol. New CENP-A was visualized by staining coverslips with 30 nM JF646-HaloTag ligand (gift from Luke Lavis; (Grimm et al., 2017)).
To detect the loss of old CENP-A-HaloTag at centromeres (e.g. Fig. S1E,F), CENP-A-HALO cells were blocked with 125 nM JF646-HaloTag ligand, then seeded onto glass bottom dishes such that they would be confluent after 3 days growth. These divisions post-block ensured that the soluble pool of CENP-A, which is also labeled by JF646-HaloTag ligand, is diluted through division and thus does not confound loss measurements. When confluent, cells were shifted to 0.1% serum-containing media supplemented with 10 μM STLC to arrest mitotic cells, after which the media was changed every day. Cells were imaged live at 37°C in CO2-independent media with 10% bovine serum and 100 ng/mL Hoechst at 0, 5, or 11 days after the beginning of serum starvation. Values were normalized to “old” CENP-A fluorescence at the beginning of quiescence, not when first incubated with the JF646-HaloTag ligand.
Knockdown of newly synthesized CENP-A by siRNA (Fig. S2A,B) was performed using a combination of two different siRNA sequences (Dharmacon) as described in (Black et al., 2007). RPE-1 cells with Halo-tagged CENP-A (cKC288) were grown to confluence and then shifted to 0.1% serum, after which media was changed every day. After 3 days in quiescence, the cells were blocked with 125 nM JF549, and transfected with 12nM each siRNA, or control using RNAiMAX (ThermoFisher). The cells were again treated with the same concentration of siRNA on day 4, and fixed at day 7 for analysis.
Immunofluorescence and live cell imaging of cultured cells
RPE-1 cells were fixed with 100% ice-cold methanol. Coverslips were washed with PBS containing 0.1% Triton X-100, then blocked with AbDil. Anti-CENP-A antibody (Abcam, ab13939) was used at 1:1000, anti-centromere antibodies (ACA) were used at 1:100 (Antibodies, Inc.), and anti-PLK1 antibodies (Santa Cruz) were used at 1:200. DNA was visualized with Hoechst (Sigma-Aldrich). Cells were imaged using a DeltaVision Core microscope (Applied Precision/GE Healthsciences) with a CoolSnap HQ2 CCD camera and a 60× 1.40 NA Olympus U- PlanApo objective. Anti-HJURP antibodies were generated in (Foltz et al., 2009). For immunofluorescence against HJURP, cells were pre-extracted with 0.1% Triton X-100 in PBS for 5 minutes, then fixed for 10 minutes with 4% formaldehyde in PBS. The cells were then quenched for 10 minutes in 100 mM Tris, pH 7.4. The cells were then blocked for 15 minutes in 2% BSA and 2% FBS in 0.1% Triton X-100. Anti-HJURP antibodies were diluted 1:500 in blocking solution, and incubated on the cells overnight at 4°C. The cells were then washed 3 times in PBS with 0.1% Triton X-100, and then stained with secondary antibodies for 1 hour.
For EdU detection, cells were stained following manufacturer instructions using the Click-iT™ EdU Alexa Fluor™ 488 Imaging Kit (Invitrogen). Images were acquired on a Nikon eclipse microscope equipped with a CCD camera (Clara, Andor) using a 20x Plan Fluor objective 0.1NA (Nikon) and appropriate fluorescence filters.
To visualize PLK1-NeonGreen in live cells, cells were imaged in CO2-independent media (Gibco) at 37°C with 10% bovine serum and 100 ng/mL Hoechst. Images were acquired as for EdU detection using a 40x Plan Fluor objective 1.3NA (Nikon).
C2C12 myoblasts and differentiated myotubes were fixed in methanol at −20° C for 20 minutes. Coverslips were washed three times in PBS, and then blocked and permeabilized in PBS containing 0.1% Triton X-100 and 2% BSA. Primary antibodies to mouse CENP-A (Cell Signaling Technology, C51A7 Rabbit mAb #2048; diluted 1:1000) or mouse CENP-C (Cheeseman lab) in block solution and incubated for 1 hr at room temperature. The coverslips were then washed three times in PBS and then incubated for 1 hr with anti-rabbit secondary antibodies. The cells were then counterstained with DAPI and phalloidin 688 (Thermo Fisher Scientific).
Plk1 inhibition studies
To test the requirement of Plk1 for CENP-A deposition, cells were synchronized by inducing quiescence as described above by 3 days culture in 0.1% FBS. Pre-existing CENP-A was blocked using 125 nM JF549-HaloTag ligand, then re-plated onto uncoated coverslips and returned to growth conditions in 10% FBS. After 24 hours (sufficient time to reenter the cell cycle), the cells were arrested in mitosis with 10 μM STLC overnight. The cells were released into complete media without STLC and, after 1.5 hours, were treated with either DMSO or 100 nM BI 2536 (VWR). The cells were then fixed in methanol and chased with 30 nM JF646-HaloTag ligand. In quiescence, 100 nM BI 2536 was added to 0.1% serum-containing media and changed daily. Coverslips were collected and analyzed as above.
Single molecule RNA fluorescence in situ hybridization (smFISH)
Custom Stellaris RNA FISH probes labeled with Quasar dyes (Quasar®570 or Quasar®670) were designed against consensus centromere alpha-satellite sequences (see Table S1 for oligo probe sequences; Karen Miga, personal communication) and purchased from Biosearch Technologies. RPE1 cells grown on uncoated coverslips in 6 well plates, washed with PBS, and fixed with 4% paraformaldehyde in 1X PBS containing Ribonucleoside Vanadyl Complex (Sigma Aldrich) for 10 minutes at room temperature (RT). After washing cells with 1X PBS, cells were permeabilized in 70% ethanol for at least 20 min at 4°C. Cells were pre-incubated with 2X SSC; 10% deionized formamide for 5 min, and incubated with hybridization mix (0.1 μM RNA FISH, 10% deionized formamide, in Hybridization Buffer (Biosearch Technologies)) overnight at 37°C in the dark. Finally, cells were washed twice with 10% deionized formamide in 2X SSC for 30 min at 37°C and once with Wash B (Bio search Technologies) for 5 min at RT. Coverslips were mounted on cells with Vectashield containing DAPI (VWR). Images were acquired within 48 hr of mounting. Specificity of the probes for RNA was confirmed using RNAse A treatment, which eliminated signal, and the lack of signal in mouse cells.
Mouse tissue immunofluorescence
Tissues were fixed in 4% paraformaldehyde in PBS at room temperature for 16–24 hours. Fixed tissues were washed with PBS, cryoprotected with 30% sucrose, and frozen in O. C. T. Compound (Tissue-Tek). Slides with 15 μm-thick sections were prepared using a cryostat. Slides were dried at room temperature for 4–24 hours. Slides were rehydrated in PBS for 5 minutes and incubated in sodium citrate buffer (10 mM trisodium citrate dihydrate, 0.05% Tween 20, pH 6.0) in a pressure cooker (Instant Pot) for 20 minutes at high pressure. Slides were washed with PBS, dried briefly, and sections outlined with a hydrophobic pen. Sections were incubated with extraction buffer (1% Triton X-100 in PBS) for 15 minutes followed by incubation in blocking solution (3% BSA, 0.3% Triton X-100 in PBS) for 1 hour at room temperature. Sections were incubated with primary antibodies (anti-mouse CENP-A; C51A7 from Cell Signaling) diluted in blocking solution at room temperature for 16–24 hours. Heart muscle was costained using directly conjugated antibodies to sarcomeric alpha-actinin (Abcam) to identify cardiomyocytes. Liver hepatocytes were co-stained for tubulin with directly conjugated FITC-DM1α (Sigma). Sections were washed three times with blocking solution for 10 minutes each and then incubated with secondary antibodies diluted in blocking solution with 5 μg/mL Hoechst 33342 (Thermo Fisher Scientific) at room temperature for 1–2 hours. Sections were washed with blocking solution for 5 minutes twice and once with PBS for 5 minutes. Sections were mounted with ProLong Gold Antifade Reagent (Life Technologies).
Western blots
For list of antibodies and the dilution used for Western blotting see Key Resource Table.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using Prism (GraphPad Software). Details of statistical tests and sample sizes are provided in the figure legends. For immunofluorescence images, DNA and microtubules were scaled individually and in some cases with gamma adjustment, for easier visualization. All images for centromere signals from human cells, mouse cells, and mouse tissues were scaled equivalently across samples for a condition. Unless otherwise specified, all centromere signals and histone incorporation in starfish oocyte images were scaled equivalently. In some cases (indicated in the figure legends), it was necessary to linearly scale centromere images individually for easier visualization, particularly for prophase arrested oocytes in which soluble GFP fluorescence in the germinal vesicle alters the background compared to oocytes that have entered meiosis. In all cases (including human cells, starfish oocytes, mouse cells, and mouse tissues, fluorescence intensity quantifications were performed using raw images with unaltered scaling).
Oocyte centromere measurements
Images were deconvolved and maximally projected. For image quantification, all images for comparison were acquired using the same microscope and acquisition settings. Centromere intensities for 3xGFP-CENP-A, 3xGFP-CENP-N, and CENP-C immunofluorescence were measured using Fiji (Schindelin et al., 2012). Individual centromeres were selected with 5 pixel diameter circles and the total integrated intensity was measured. Background correction was performed by selecting a nearby non-centromeric region of equal size for each centromere and subtracting its integrated intensity from that of the centromere region. The average of all centromeres was then determined per oocyte. For quantification of histone H3.3 incorporation, the Fiji selection brush tool was used to create a region of interest around the chromosomes in metaphase I from maximally-projected images. Signal intensity was then measured and divided by area. Background intensity was calculated by averaging 4 10×10 pixel regions near the selected chromatin divided by area, and subtracted from the chromatin ROI.
RPE1 and C2C12 myoblast centromere measurements
Centromere pixel intensities were quantified using Fiji (Schindelin et al., 2012). Z-stacks were maximally projected as single two-dimensional images and centromeres were identified using either the ACA or anti-CENP-A antibody signal. For the loss of old CENP-A-HALO, the CENP-A-HALO signal was used to identify centromeres. Individual centromeres within a nucleus were selected with 6 pixel diameter circles. At least 25 nuclei were analyzed for each time point and condition.
In the experiments measuring new CENP-A-HALO incorporation, background correction was performed by averaging 10 6 pixel diameter circles adjacent to centromeres, and subtracting that average from all centromere values. The background corrected centromere values were summed as one data point representing a single cell. To calculate the percent new CENP-A-HALO, the average JF646 fluorescence per nuclei in unblocked cells was used as total CENP-A. To account for natural differences in CENP-A levels between cells, the anti-CENP-A antibody fluorescence was measured for both blocked and unblocked cells at the same location as JF646 signal. The final equation used was as follows:
In the experiments measuring the loss of CENP-A following HJURP or Mis18β knockout, background correction was performed by selecting 6 pixel circles adjacent to each centromere and subtracting each background measurement from its related centromere measurement. The background corrected centromere values were averaged as one data point representing a single cell. To calculate the loss of CENP-A, the measured values at each timepoint were compared to the average value of the control cells collected and imaged at the same point.
In the experiments measuring the loss of blocked CENP-A-HALO and the loss of CENP-T over time, background correction was performed by selecting 6 pixel circles adjacent to each centromere and subtracting each background measurement from its related centromere measurement. The background corrected centromere values were summed as one data point representing a single cell. To calculate the loss over time, all measurements were compared to the average value of the relevant day 0 measurements.
RPE1 H3.1A/H4-Halo turnover measurements
Images were quantified using Fiji (Schindelin et al., 2012). 25 0.2pm Z-stacks were maximum signal projected to a single two-dimensional image. A 50 pixel diameter circle was placed manually within each nucleus to determine the average fluorescence. The background signal of each image was calculated by averaging the signal of 5 circles placed outside nuclei and subtracting that average from the average nuclear fluorescence. Over 100 nuclei were analyzed for each time point and condition. To calculate the percent new H3.1A/H4-HALO, the average JF646 fluorescence per nuclei in unblocked cells represented 100% signal.
Single molecule FISH quantification
ASAT RNA foci were counted per nucleus using CellProfiler (Carpenter et al., 2006).
Mouse tissue centromere measurements
For the analysis of CENP-A levels in tissue sections, the total level CENP-A in each nucleus was quantified in Fiji by drawing a circle around the entire nuclear volume, and then subtracting the background intensity as determined by the average of three smaller circles within the nucleus (not overlapping with centromeres). Liver cell nuclei can exist in either a diploid, tetraploid, or sometimes greater state. As nuclear diameter has been shown to correlate closely with ploidy (Knouse et al., 2014), CENP-A intensity was normalized to a diploid state based on the ploidy of a nucleus. In cases where the nuclear diameter was >10 μm, this was determined to be tetraploid, with CENP-A intensity divided by 2 for these cells. When the nuclear diameter was <9 μm, this was determined to be diploid and CENP-A levels are reported unchanged.
DATA AND CODE AVAILABILITY
Centromeric ASAT RNA foci measured by FISH were counted using a custom CellProfiler pipeline, available upon request from the Lead Contact, Iain Cheeseman (icheese@wi.mit.edu). No other datasets or code were generated or analyzed in this study.
Supplementary Material
Figure S1. Analysis of CENP-A and other histone incorporation. Related to Figure 1.
Figure S2. Characterization of Halo and inducible knockout cell lines. Related to Figure 2.
Figure S3. Localization of GFP fusions in oocytes and embryos. Related to Figure 3.
Figure S4. CENP-A protein is stable but gradually deposited in arrested oocytes. Related to Figure 4.
Figure S5. Plk1 levels are strongly reduced in quiescent human cells. Related to Figure 5.
Figure S6. Role of transcription in H3.3 incorporation and centromere identity. Related to Figure 6.
Figure S7. CENP-A levels declined in post-mitotic tissues. Related to Figure 7.
Table S1. Stellaris ASAT RNA FISH probe sequences. Related to Star Methods and Key Resources Table.
Highlights.
CENP-A nucleosomes are gradually incorporated in quiescent cells and oocytes.
CENP-A deposition during quiescence is required for future chromosome segregation.
RNA Polymerase transcription at centromeres promotes gradual CENP-A exchange.
Terminally differentiated muscle cells fail to retain CENP-A nucleosomes.
Acknowledgements
We thank the members of the Cheeseman laboratory for support, input, and critical reading of the manuscript, Terry Orr-Weaver, Peter Reddien, Priya Budde, and Kara McKinley for critical reading of the manuscript, and Luke D. Lavis for JF-Halo-reagents. We thank Peter Lenart for invaluable advice with starfish methodology, and Lars Jansen and Dani Bodor for sharing unpublished work. This work was supported by grants from The Harold G & Leila Y. Mathers Charitable Foundation, the NIH/National Institute of General Medical Sciences (GM088313, GM108718, and R35GM126930) to IMC, American Cancer Society post-doctoral fellowships to SZS and LB, an NIH Early Independence Award (DP5OD026369) to KAK, a National Science Foundation CAREER Award to P.S.M. (1652512), and support from the Scott Cook and Signe Ostby fund to KAK.
Footnotes
Declaration of Interests
The authors have no conflicts to declare.
Supplemental Information
The online supplemental information contains 7 figures and 1 table.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bachvarova R (1985). Gene expression during oogenesis and oocyte development in mammals. Dev Biol (N Y 1985) 1, 453–524. [DOI] [PubMed] [Google Scholar]
- Black BE, Brock MA, Bedard S, Woods VL Jr., and Cleveland DW (2007). An epigenetic mark generated by the incorporation of CENP-A into centromeric nucleosomes. Proc Natl Acad Sci U S A 104, 5008–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor DL, Valente LP, Mata JF, Black BE, and Jansen LE (2013). Assembly in G1 phase and long-term stability are unique intrinsic features of CENP-A nucleosomes. Mol Biol Cell 24, 923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrego-Pinto J, Somogyi K, Karreman MA, Konig J, Muller-Reichert T, Bettencourt-Dias M, Gonczy P, Schwab Y, and Lenart P (2016a). Distinct mechanisms eliminate mother and daughter centrioles in meiosis of starfish oocytes. J Cell Biol 212, 815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrego-Pinto J, Somogyi K, and Lenart P (2016b). Live Imaging of Centriole Dynamics by Fluorescently Tagged Proteins in Starfish Oocyte Meiosis. Methods Mol Biol 1457, 145–166. [DOI] [PubMed] [Google Scholar]
- Cao S, Zhou K, Zhang Z, Luger K, and Straight AF (2018). Constitutive centromere-associated network contacts confer differential stability on CENP-A nucleosomes in vitro and in the cell. Mol Biol Cell 29, 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FL, Marshall OJ, Saffery R, Kim BW, Earle E, Choo KH, and Wong LH (2012). Active transcription and essential role of RNA polymerase II at the centromere during mitosis. Proc Natl Acad Sci U S A 109, 1979–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi V, Dye DE, Kinnear BF, van Kuppevelt TH, Grounds MD, and Coombe DR (2015). Interactions between Skeletal Muscle Myoblasts and their Extracellular Matrix Revealed by a Serum Free Culture System. PLoS One 10, e0127675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM (2014). The kinetochore. Cold Spring Harb Perspect Biol 6, a015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Niessen S, Anderson S, Hyndman F, Yates JR III, Oegema K, and Desai A (2004). A conserved protein network controls assembly of the outer kinetochore and its ability to sustain tension. Genes Dev 18, 2255–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH (1976). Gene Activity in Early Development (Academic Press; ). [Google Scholar]
- Dunleavy EM, Almouzni G, and Karpen GH (2011). H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G(1) phase. Nucleus 2, 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Bailey AO, Yates JR 3rd, Bassett EA, Wood S, Black BE, and Cleveland DW (2009). Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell 137, 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokirmak T, Campanale JP, Shipp LE, Moy GW, Tao H, and Hamdoun A (2012). Localization and substrate selectivity of sea urchin multidrug (MDR) efflux transporters. J Biol Chem 287, 43876–43883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenfell AW, Heald R, and Strzelecka M (2016). Mitotic noncoding RNA processing promotes kinetochore and spindle assembly in Xenopus. J Cell Biol 214, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, Brown TA, English BP, Lionnet T, and Lavis LD (2017). Synthesis of Janelia Fluor HaloTag and SNAP-Tag Ligands and Their Use in Cellular Imaging Experiments. Methods Mol Biol 1663, 179–188. [DOI] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, et al. (2015). A general method to improve fluorophores for live-cell and single-molecule microscopy. Nature methods 12, 244–250, 243 p following 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo LY, Allu PK, Zandarashvili L, McKinley KL, Sekulic N, Dawicki-McKenna JM, Fachinetti D, Logsdon GA, Jamiolkowski RM, Cleveland DW, et al. (2017). Centromeres are maintained by fastening CENP-A to DNA and directing an arginine anchor-dependent nucleosome transition. Nat Commun 8, 15775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen LET, Black BE, Foltz DR, and Cleveland DW (2007). Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol 176, 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouse KA, Wu J, Whittaker CA, and Amon A (2014). Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci U S A 111, 13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudtarkar P, and Cameron RA (2017). Echinobase: an expanding resource for echinoderm genomic information. Database (Oxford) 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3, 373–382. [DOI] [PubMed] [Google Scholar]
- Mak DH, Schober WD, Chen W, Konopleva M, Cortes J, Kantarjian HM, Andreeff M, and Carter BZ (2009). Triptolide induces cell death independent of cellular responses to imatinib in blast crisis chronic myelogenous leukemia cells including quiescent CD34+ primitive progenitor cells. Molecular cancer therapeutics 8, 2509–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malato Y, Naqvi S, Schurmann N, Ng R, Wang B, Zape J, Kay MA, Grimm D, and Willenbring H (2011). Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J Clin Invest 121, 4850–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, and Cheeseman IM (2014). Polo-like Kinase 1 Licenses CENP-A Deposition at Centromeres. Cell 158, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, and Cheeseman IM (2016). The molecular basis for centromere identity and function. Nat Rev Mol Cell Biol 17, 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, and Cheeseman IM (2017). Large-Scale Analysis of CRISPR/Cas9 Cell-Cycle Knockouts Reveals the Diversity of p53-Dependent Responses to Cell-Cycle Defects. Dev Cell 40, 405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, Sekulic N, Guo LY, Tsinman T, Black BE, and Cheeseman IM (2015). The CENP-LN complex forms a critical node in an integrated meshwork of interactions at the centromere-kinetochore interface. Mol Cell 60, 886–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Hara M, Tachibana K, and Kishimoto T (2006). p90Rsk is required for G1 phase arrest in unfertilized starfish eggs. Development 133, 1823–1830. [DOI] [PubMed] [Google Scholar]
- Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, and Sadek HA (2011). Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich A, Dunn C, Akasaka K, and Wessel G (2015). Phylogenomic analyses of Echinodermata support the sister groups of Asterozoa and Echinozoa. PLoS One 10, e0119627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffery R, Sumer H, Hassan S, Wong LH, Craig JM, Todokoro K, Anderson M, Stafford A, and Choo KH (2003). Transcription within a functional human centromere. Mol Cell 12, 509–516. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang WH, Hori T, Martins NM, Toyoda A, Misu S, Monma N, Hiratani I, Maeshima K, Ikeo K, Fujiyama A, et al. (2013). Chromosome engineering allows the efficient isolation of vertebrate neocentromeres. Dev Cell 24, 635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoak EM, Stein P, Schultz RM, Lampson MA, and Black BE (2016). Long-Term Retention of CENP-A Nucleosomes in Mammalian Oocytes Underpins Transgenerational Inheritance of Centromere Identity. Curr Biol 26, 1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana K, Machida T, Nomura Y, and Kishimoto T (1997). MAP kinase links the fertilization signal transduction pathway to the G1/S-phase transition in starfish eggs. Embo J 16, 4333–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Arrojo EDR, Lev-Ram V, Ramachandra R, Deerinck TJ, Lechene C, Ellisman MH, and Hetzer MW (2018). Visualization of long-lived proteins reveals age mosaicism within nuclei of postmitotic cells. J Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, and Workman JL (2015). Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol 16, 178–189. [DOI] [PubMed] [Google Scholar]
- von Dassow G, Verbrugghe KJ, Miller AL, Sider JR, and Bement WM (2009). Action at a distance during cytokinesis. J Cell Biol 187, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel GM, Reich AM, and Klatsky PC (2010). Use of sea stars to study basic reproductive processes. Syst Biol Reprod Med 56, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D, and Saxel O (1977). Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270, 725–727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analysis of CENP-A and other histone incorporation. Related to Figure 1.
Figure S2. Characterization of Halo and inducible knockout cell lines. Related to Figure 2.
Figure S3. Localization of GFP fusions in oocytes and embryos. Related to Figure 3.
Figure S4. CENP-A protein is stable but gradually deposited in arrested oocytes. Related to Figure 4.
Figure S5. Plk1 levels are strongly reduced in quiescent human cells. Related to Figure 5.
Figure S6. Role of transcription in H3.3 incorporation and centromere identity. Related to Figure 6.
Figure S7. CENP-A levels declined in post-mitotic tissues. Related to Figure 7.
Table S1. Stellaris ASAT RNA FISH probe sequences. Related to Star Methods and Key Resources Table.
Data Availability Statement
Centromeric ASAT RNA foci measured by FISH were counted using a custom CellProfiler pipeline, available upon request from the Lead Contact, Iain Cheeseman (icheese@wi.mit.edu). No other datasets or code were generated or analyzed in this study.