

Abstract
Aims
Drug disposition in children may vary from adults due to age‐related variation in drug metabolism. Microdose studies present an innovation to study pharmacokinetics (PK) in paediatrics; however, they should be used only when the PK is dose linear. We aimed to assess dose linearity of a [14C]midazolam microdose, by comparing the PK of an intravenous (IV) microtracer (a microdose given simultaneously with a therapeutic midazolam dose), with the PK of a single isolated microdose.
Methods
Preterm to 2‐year‐old infants admitted to the intensive care unit received [14C]midazolam IV as a microtracer or microdose, followed by dense blood sampling up to 36 hours. Plasma concentrations of [14C]midazolam and [14C]1‐hydroxy‐midazolam were determined by accelerator mass spectrometry. Noncompartmental PK analysis was performed and a population PK model was developed.
Results
Of 15 infants (median gestational age 39.4 [range 23.9–41.4] weeks, postnatal age 11.4 [0.6–49.1] weeks), 6 received a microtracer and 9 a microdose of [14C]midazolam (111 Bq kg−1; 37.6 ng kg−1). In a 2‐compartment PK model, bodyweight was the most significant covariate for volume of distribution. There was no statistically significant difference in any PK parameter between the microdose and microtracer, nor in the area under curve ratio [14C]1‐OH‐midazolam/[14C]midazolam, showing the PK of midazolam to be linear within the range of the therapeutic and microdoses.
Conclusion
Our data support the dose linearity of the PK of an IV [14C]midazolam microdose in children. Hence, a [14C]midazolam microdosing approach may be used as an alternative to a therapeutic dose of midazolam to study developmental changes in hepatic CYP3A activity in young children.
Keywords: cytochrome P450, drug metabolism, paediatrics
What is already known about this subject
Microdose studies with radioactive labelled ([14C]) substrates present an innovation to study pharmacokinetics of drugs in paediatrics
Dose linearity between the pharmacokinetics of a microdose and therapeutic dose is an important prerequisite
Dose linearity of the pharmacokinetics of a microdose of [14C]midazolam has not been established in children
What this study adds
The pharmacokinetics of a microdose of [14C]midazolam are dose linear to therapeutic doses in children
As midazolam is a well‐established marker for the developmentally regulated CYP3A enzyme, a [14C]midazolam microdosing approach is a minimal risk strategy to study hepatic CYP3A activity variability in children
1. INTRODUCTION
Drug disposition in children may vary from adults due to age‐related variation in the processes governing absorption, distribution, metabolism and excretion.1, 2 This variation is largest in the first years of life and is not directly proportionate to size.3, 4 However, in daily clinical practice, drug dosing in paediatrics is often based on bodyweight based corrections, which, because of variation arising from development, can result in subtherapeutic or toxic drug exposure in certain subgroups.2 Hence, doses used for children cannot simply be extrapolated from adults using a simple bodyweight‐based correction.
Phenotyping studies, in which model drugs representative for a certain pathway are studied across the paediatric age range, can be used to elucidate the age‐related variation in drug disposition pathways in vivo.5 However, these studies are faced with ethical, practical and scientific challenges. Children are vulnerable, and so exposing them to (almost) therapeutic doses of drugs for a nontherapeutic reason, as in a phenotyping study, may not be ethically acceptable. Moreover, blood sampling for pharmacokinetic (PK) analyses in children is challenging because of the burden for the individual child, the smaller blood volume that can be taken, as well as the technical difficulties associated with sampling.
Microdosing studies present an attractive alternative to overcome the ethical and analytical challenges of phenotyping studies.6 A microdose is a very small, subtherapeutic dose of a drug (<1/100th of the therapeutic dose or <100 μg) that is unlikely to result in pharmacological effects or adverse events.7, 8 A radioactive label [14C] allows ultrasensitive quantification of extremely low plasma concentrations by accelerator mass spectrometry (AMS) for which only 10–15 μL plasma is required.9, 10 The radiation dose associated with a [14C]microdose is safe as it is below 1 μSievert. This is much lower than yearly background exposure (2.5 mSv year−1 in The Netherlands), a computed tomography scan of the head (1200 μSievert), or chest X‐ray (12 μSievert).6
Microdosing studies can provide unique information on the PK of drugs in children, and with that valuable information on developmental changes in drug metabolism pathways, as shown successfully before.6, 11, 12, 13 Importantly, a prerequisite is that the PK of a microdose is linear to the PK of a therapeutic dose.14, 15 Lack of linearity may occur for example, when a therapeutic dose saturates drug metabolism pathways, plasma protein binding and/or active transporters, which may result in altered PK when studying a microdose.15 A very elegant approach to study dose linearity is by comparing the PK parameters of an isolated [14C]microdose with the PK parameters of a [14C]microtracer, where the labelled microdose is administered concurrently or even mixed with a therapeutic drug dose.12
Cytochrome P450 (CYP) 3A is a developmentally regulated drug metabolizing enzyme that is abundant in the liver and accounts for nearly 46% of the oxidative metabolism of clinically relevant drugs.1, 2, 16, 17, 18, 19, 20, 21 As midazolam is a well‐established model substrate for CYP3A activity, this drug may be used for phenotyping studies using a microdosing approach to elucidate developmental changes in CYP3A.5, 22, 23, 24, 25 To the best of our knowledge, dose linearity of the PK of a microdose to those of a therapeutic dose of midazolam has been established in adults,14, 26, 27 but not in children. However, the results in adults cannot simply be extrapolated to children due to the development of drug metabolism, hepatic blood flow, protein binding and drug transport.
We therefore aimed to study the dose linearity of the PK of a [14C]midazolam microdose in children, by studying the PK parameters of midazolam when given as an intravenous (IV) [14C]microdose, and as a [14C]microtracer given simultaneously with a therapeutic midazolam dose.
2. METHODS
2.1. Study design
This study was part of the ERA‐NET PRIOMEDCHILD project Paediatric Accelerator Mass Spectrometry Evaluation Research Study (PAMPER). The 2 units participating in this study were the Alder Hey Children's NHS Foundation Trust, Liverpool, UK and the Liverpool Women's NHS Foundation Trust, Liverpool, UK. Children were recruited on the paediatric intensive care wards of these units. Ethical approval was obtained from the Research Ethics Committees for the hospitals where patients were enrolled. All parents or an adult who carried parental responsibility provided written informed consent for their child to be included prior to any study‐specific procedures. No radioactive substance administration approval was required as the administered radioactive dose was below 1 μSievert, the UK Administration of Radioactive Substances Advisory Committee (ARSAC) exemption level.
2.2. Subjects
Children were eligible to be included in this study from birth up to age 2 years, when they had intravenous lines in place for intravenous administration, and had suitable vascular access for blood sampling. Exclusion criteria were serious hepatic impairment (defined by aspartate aminotransferase [ASAT] and alanine aminotransferase [ALAT] > 200 U L−1) or renal impairment (defined by plasma creatinine >150 μmol), haemofiltration, peritoneal/haemodialysis or extracorporeal membrane oxygenation.
2.3. Study procedures
A single [14C]midazolam (111 Bq kg−1; 37.6 ng kg−1) dose was administered IV either as a microtracer during therapeutic midazolam infusion or as an isolated microdose (Figure 1). The microtracer was mixed with the first therapeutic loading dose of midazolam given by the treating physician for sedation, and was administered over 30 min. The microdose was administered with a similar infusion rate to ensure similar exposure to [14C]levels. The IV therapeutic midazolam dose was prescribed by the treating physician for clinical purposes according to British National Formulary for Children dosing guidelines. Blood samples were taken before and up to 36 hours after administration of the [14C]midazolam microtracer or microdose. The time points for blood sampling were based on the PK of midazolam in paediatric ICU patients where a median half‐life of 5.5 hours was found.28 To ensure complete sampling of a single dose, at least 5 times the half‐life was taken. Moreover, to capture the distribution, metabolism and elimination phases, the sampling times were set at predose, and 0.17, 0.5, 1, 2, 4, 6, 10, 24 and 36 hours post‐IV dose. The maximum number of study specific blood samples was limited to 6 per subject. The specific time points for each patient were decided based on discussion between the research team, clinical team and parents to ensure cares were coordinated at this time and with minimal disruption to the patients' routine. The maximum amount of blood could not exceed the guidelines by European Medicines Agency (up to 1% of calculated circulating blood volume).29 The blood samples were centrifuged and plasma was stored at −80°C until analysed.
Figure 1.
Explanation of the terms intravenous microdose and microtracer midazolam
2.4. Radiopharmaceutical preparation
[14C]midazolam was synthesized by Selcia Ltd. (Ongar, Essex, UK) at a specific activity of 1072 MBq mmol−1 (equal to 2.95 MBq mg−1). The chemical name is 8‐chloro‐6‐(2‐fluorophenyl)‐1‐methyl‐4H‐[1‐14C] imidazo[l,5‐a][l,4]benzodiazepine hydrochloride. In the Radiopharmacy Department, Addenbrookes Hospital, Cambridge, UK under aseptic conditions, [14C]midazolam was brought in ethanol 96% solution, the activity was measured and the solution was further diluted 10 000 fold in 5% w/v dextrose solution to the required concentration. The final solution was filter sterilized (pore size 0.2 μm) and batched for intravenous injection. The final [14C]midazolam concentration was 500 Bq mL−1.
2.5. [14C]midazolam and [14C]1‐hydroxy‐midazolam plasma concentration analysis
2.5.1. Plasma sample extraction and ultraperformance liquid chromatography separation
Methanol (10 μL) was added to plasma samples in order precipitate proteins and to extract the test substance using protein precipitation plates. Each run consisted of samples measured once and eight calibrator levels in duplicate plus 3 different Quality control (QC) levels in duplicate. The extract was evaporated to dryness, re‐dissolved and analysed using ultraperformance liquid chromatography. The fraction where midazolam and 1‐hydroxy‐midazolam eluted from the column was collected for each sample, transferred to a tin foil cup, evaporated to dryness and subsequently analysed using Combustion‐CO2‐AMS.
2.5.2. AMS analysis
[14C] levels were quantified as described before.13, 30 The ultraperformance liquid chromatography and AMS qualification were performed in accordance with the recommendation of the European Bioanalytical Forum.31 The tin foil cups (see 2.5.1) were combusted on an elemental analyser (Vario Micro; Elementar, Langenselbold, Germany). Generated CO2 was transferred to a home‐built gas interface, composed of a zeolite trap and syringe.30 CO2 was adsorbed to the trap on the interface; and after heating of the trap, the CO2 was transferred to a vacuum syringe using helium. A final CO2/helium mixture of 6% was directed to the AMS ion source, at a pressure of 1 bar and a flow of 60 μL min−1. A 1‐MV Tandetron AMS (High Voltage Engineering Europe B.V., Amersfoort, The Netherlands)32 was used. The lower limit of quantification was 0.31 mBq mL−1.
2.6. Patient characteristics
Patient characteristics (age, weight) and patient laboratory values (creatinine, total bilirubin, ASAT, ALAT) were described using standard statistics, and data was presented as median (range). Microtracer and microdosing groups were compared using Mann–Whitney test, as data were not distributed normally.
2.7. PK analysis
2.7.1. Exploration of the data
The data were first explored by visualization of time–concentration profiles of [14C]midazolam and [14C]1‐hydroxy‐midazolam (GraphPad Prism 5). Next, their area under the curve (AUC) and the ratio AUC [14C]1‐hydroxy‐midazolam/[14C]midazolam were estimated using a log‐linear noncompartmental model (Excel PKSolver add‐in software33) and compared between microdose and microtracer administration using Mann–Whitney U test.
2.7.2. Nonlinear mixed effects modelling
[14C]midazolam concentration–time data were analysed using the nonlinear mixed effects modeling software NONMEM version 7.4 (ICON; Globomax LLC, Ellicott, MD). Model development was in 4 steps: (i) selection of a structural model; (ii) selection of an error model; (iii) covariate analysis, and (iv) internal validation of the model.
For model selection, we used the objective function value (OFV) and standard goodness of fit plots. For the OFV, a drop of more than 3.84 points between nested models was considered statistically significant, which corresponds to P < .05 assuming a χ2 distribution.34, 35 For the structural and error models, a decrease in OFV of 3.84 points was considered statistically significant (P < .05). For the structural model, 1‐, 2‐ and 3‐compartment models were tested. Inclusion of log‐normally distributed interindividual variability (IIV) was tested on all model parameters. For the residual unexplained variability additive, proportional and a combination of additive and proportional error model were tested. The continuous covariates evaluated were postnatal age, postmenstrual age, bodyweight, creatinine, ALAT, ASAT and total bilirubin. Categorical covariates included treatment arm (i.e. microdosing or microtracer administration) only. All covariates were tested on all model parameters. Potential covariates were evaluated using forward inclusion and backward elimination with a level of significance of <.005 (ΔOFV < −7.9 points) and <.001 (ΔOFV > 10.8 points), respectively. In addition, inclusion of a covariate in the model had to result in a decline in unexplained IIV and/or improved goodness of fit plots before it was included in the final model.36 Next, the model was internally validated using bootstrap analysis in Perl‐speaks‐NONMEM.
2.8. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,37 and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18.38
3. RESULTS
3.1. Subjects and data
Fifteen infants (gestational age 39.4 [23.9–41.4] weeks, postnatal age 11.4 [0.6–49.1 weeks]) were included in the study, of whom 9 received a microdose and 6 a microtracer of [14C]midazolam. See Table 1 for the patient characteristics. There were no significant differences found between characteristics of the microdose and microtracer group.
Table 1.
Characteristics of patients that participated in the study and received a microdose or microtracer [14C]midazolam. Data are presented as median (range)
| Total | Microdose | Microtracer | Mann–Whitney U microdose vs microtracer group (P‐value) | |
|---|---|---|---|---|
| Number of patients | 15 | 9 | 6 | ‐ |
| Number of samples | 67 | 37 | 30 | ‐ |
| Samples per patient (n) | 5 (2–5) | 5 (2–5) | 5 (5–5) | ‐ |
| Gestational age (wk) | 39.4 (23.9–41.4) | 39.4 (23.9–41.4) | 38.4 (26.7–41.0) | .15 |
| Postnatal age (weeks) | 11.4 (0.6–49.1) | 11.4 (0.6–49.1) | 13.4 (2.6–42.3) | .39 |
| Weight (kg) | 3.6 (2.6–8.9) | 3.5 (2.7–8.9) | 3.8 (2.6–6.0) | 1.00 |
| Plasma creatinine (μmol L−1) | 35 (20–51) | 41 (29–51) | 33 (20–36) | .07 |
| Total bilirubin (μmol L−1) | 9 (2–274) | 9 (5–274) | 9 (2–146) | .46 |
| ASAT (U L−1) | 42 (12–93) | 41 (12–93) | 57 (25–85) | .39 |
| ALAT (U L−1) | 17 (7–68) | 15 (7–43) | 23 (16–68) | .09 |
The complete dataset included data on 67 blood samples. Eight measurements had [14C]midazolam concentrations under the AMS detection limit and were not included in the analysis.39
3.2. Exploration of the data
The time–concentration profiles of [14C]midazolam and [14C]1‐hydroxy‐midazolam of the individual subjects are depicted in Figure 2 and 3. In Table 2 the individual AUCs and ratio AUC0–t [14C]1‐hydroxy‐midazolam/[14C]midazolam of the microdose and microtracer are presented. There were no significant differences found between the 2 groups.
Figure 2.
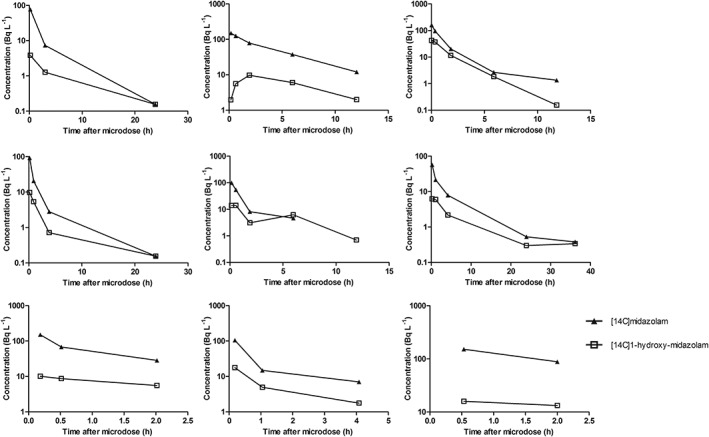
Individual (n = 9) semilog plasma concentration–time profiles of [14C]midazolam and [14C]1‐hydroxy‐midazolam after administration of a [14C]midazolam microdose
Figure 3.
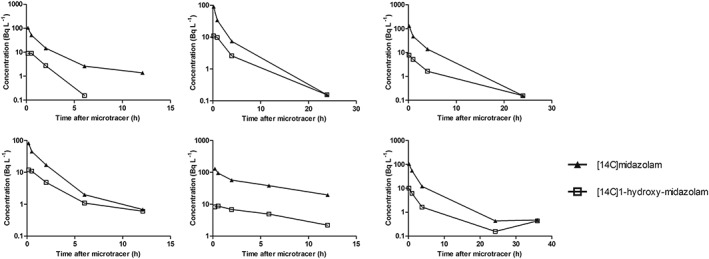
Individual (n = 6) semilog plasma concentration–time profiles of [14C]midazolam and [14C]1‐hydroxy‐midazolam after administration of a [14C]midazolam microtracer
Table 2.
Area under the curve (AUC) of [14C]midazolam and [14C]1‐hydroxy‐midazolam after administration of a microdose or microtracer [14C]midazolam, presented as median (range)
| Total (n = 15) | Microdose (n = 9) | Microtracer (n = 6) | Mann–Whitney U microdose vs microtracer group (P‐value) | |
|---|---|---|---|---|
| [ 14 C]midazolam | ||||
| AUC0–t (ng L−1 *h) | 46.77 (32.42–196.77) | 46.77 (32.42–196.77) | 48.28 (39.17–81.40) | .86 |
| AUC0–∞ (ng L−1 *h) | 48.90 (34.15–218.80) (n = 14a) | 48.90 (34.15–218.80) (n = 8a) | 49.11 (39.75–82.45) | .66 |
| [ 14 C]1‐hydroxy‐midazolam | ||||
| AUC0–t (ng L−1 *h) | 10.89 (5.28–24.21) | 10.19 (5.28–24.21) | 11.20 (5.84–19.93) | .86 |
| AUC0–∞ (ng L−1 *h) | 12.39 (5.99–26.41) (n = 14a) | 13.14 (7.40–26.41) (n = 8a) | 12.39 (5.99–26.27) | .95 |
| [ 14 C]1‐hydroxy‐midazolam/ [ 14 C]midazolam | ||||
| AUC0–t ratiob | 0.23 (0.11–0.51) | 0.23 (0.11–0.49) | 0.21 (0.13–0.51) | .69 |
for 1 subject this parameter could not be established as there were only 2 plasma samples available.
AUC0–t ratio = [14C]1‐hydroxy‐midazolam AUC0–t/[14C]midazolam AUC0–t
3.3. Nonlinear mixed effects modelling
A 2‐compartment model described the PK of [14C]midazolam best. Inclusion of IIV for clearance improved the model statistically significantly. A combined error model was superior over a proportional error model or an additive error model.
Bodyweight was a significant predictor for the central volume of distribution and was therefore included in the model. After inclusion of bodyweight, age and other tested covariates were not found to be statistically significant. There was a trend for a relation between bodyweight and clearance, but this did not reach statistical significance (∆OFV ‐4.38).
Inclusion of the covariate treatment (e.g. microtracer or microdose) upon inclusion on any of the PK parameters was found to not statistically significantly influence the model fit (∆OFV >0.01).
The PK parameter estimates of the final model and the bootstrap results are presented in Table 3. Most RSE values of the parameter estimates are well below 50%, suggesting that the estimates are precise. Mean bootstrap values are close to model estimates and 0 is not in the 95% bootstrap interval, meaning the model is robust. Figure 4 shows the diagnostic plots for the final model and illustrates the predictive value of the model for both the microtracer and microdose group. The figure shows no bias, suggesting that concentrations for both the microdose and the microtracer are accurately predicted by this model, supporting dose linearity of the microdose.
Table 3.
Parameter estimates of the pharmacokinetic model for IV [14C]midazolam
| Parameter | Estimate (RSE%) | Bootstrap median (2.5th to 97.5th bootstrap percentile) |
|---|---|---|
| Clearance | ||
| CL (L h−1) | 2.06 (24) | 2.23 (1.57–3.23) |
| Intercompartmental clearance | ||
| Q (L h−1) | 0.79 (44) | 0.90 (0.60–2.45) |
| Volume of distribution | ||
| V1i = V14kg * (WT/4)k1 | ||
| - V14kg (L) | 3.81 (8) | 3.75 (3.07–4.66) |
| - k1 | 1.36 (10) | 1.34 (0.68–1.68) |
| V2 (L) | 3.19 (18) | 3.30 (2.64–6.41) |
| Interindividual variability | ||
| ω2 CL | 0.73 (42) | 0.62 (0.13–1.41) |
| Residual error | ||
| Proportional error | 0.09 (24) | 0.08 (0.05–0.14) |
| Additional error | 0.08 (50) | 0.07 (0.01–0.20) |
CL = population predicted clearance; Q = intercompartmental clearance; V1i = individual predicted volume of distribution in the central compartment for individual i; V14kg = population value for volume of distribution in the central compartment at 4 kg; WT = body weight; k1 = exponent to relate body weight to volume of distribution; V2 = volume of distribution in the peripheral compartment; ω2 = variance for the interindividual variability of the parameter mentioned. The bootstrap was based on 50 resampled datasets.
Figure 4.
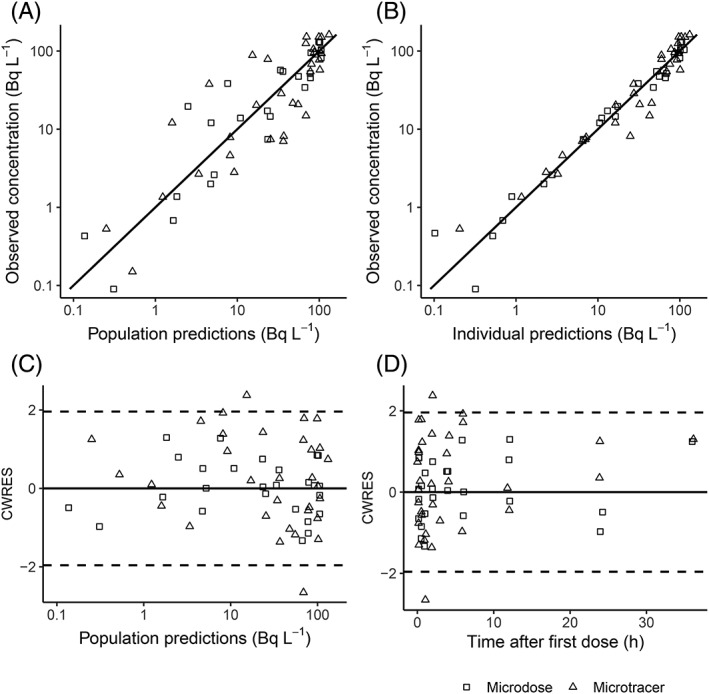
Diagnostic plots for [14C]midazolam pharmacokinetic model, using different symbols for the different treatments. (A) Observed vs population predicted [14C]midazolam concentrations. (B) Observed vs individually predicted [14C]midazolam concentrations. (C) Weighted residuals vs population predicted [14C]midazolam concentration. (D) Weighted residuals vs time. Solid lines represent the line of unity in A and B, and a value of 0 in C and D. dotted line represent ±1.96 standard deviation, representing the interval in which 95% of the conditional weighted residual (CWRES) values are expected
4. DISCUSSION
Our study shows dose linearity of the PK of a [14C]midazolam microdose to the therapeutic dose in children, by the finding that none of the PK parameters of midazolam were influenced by the treatment group, i.e. microdose or microtracer [14C]midazolam. A lack of difference in AUC values for [14C]midazolam and [14C]1‐hydroxy‐midazolam further supports that there is no difference between the PK of a microtracer and microdose.
These results are in line with the findings in adults (n = 6), where dose linearity of a 100 μg [14C]midazolam microdose was assessed in a cross‐over design with 3 treatment regimens.14 The subjects were administered (i) an oral microdose, (ii) an IV microdose or (iii) a simultaneous dose of an IV microtracer with a therapeutic nonradiolabelled oral dose. Like our results, no difference in IV disposition of midazolam was found when given as a microdose alone or in presence of a therapeutic dose.
Previously, studies have reported the midazolam PK in paediatrics after a single IV administration.40, 41, 42 Clearance in our study was found to be 2.06 L h−1 for an infant of 4 kg (equal to 8.6 mL kg−1 min−1). In preterm infants, the clearance was reported to be lower (median 1.8 [range 0.7–6.7] mL kg−1 min−1)40 reflecting that CYP3A activity is less mature in preterm infants than in an infant of 4 kg. A study with critically ill children reported a clearance of 1.11 L h−1 for an infant of 5 kg (equal to 3.7 mL kg−1 min−1),43 which is lower than in our population. This paper concludes that inflammation (reflected by high C‐reactive protein concentrations) and/or number of failing organs influenced midazolam clearance, possibly as a result of reduced CYP3A activity.43 The lower clearance can probably be explained by the fact that this study included patients with a higher inflammation‐state and/or more failing organs, as subjects in the current study were only eligible when renal‐ or hepatic failure was absent. This is further evidenced by 2 studies investigating a 0.15 mg kg−1 dose in healthy children, where clearance was found to be similar (3–10 year old, clearance mean ± SD 9.11 ± 1.21 mL kg−1 min−1)42 and slightly higher (0.5–2 year, clearance 11.3 ± 6.3 mL kg−1 min−1)41 than in our population.
Regulatory authorities have indicated that microdose studies with radioactive labelled compounds are an acceptable component of drug development.7, 44 However, to the best of our knowledge, this approach has not been used during paediatric drug development, despite this study and previous other studies illustrating feasibility and ethical acceptance in that population.11, 12, 13 For paracetamol, the dose linearity of an oral and IV microdose was successfully assessed in paediatrics.12 A slightly different approach was taken to study developmental changes in oral disposition of paracetamol and metabolites when an oral microtracer of [14C]paracetamol was administered together with a therapeutic dose of IV paracetamol.11, 13 The known developmental change from mainly sulfation to glucuronidation was confirmed, and data were added on intestinal and hepatic metabolism of paracetamol in a large paediatric age range. Together with our study, these studies pave the way for microdose studies to be incorporated into paediatric drug development plans to explore PK in this vulnerable population.
This study is limited by the lack of information on the severity of disease and inflammation in these patients and by the wide age range in which extensive development in drug metabolism and transport occurs. The effect of age and disease on CYP3A activity increased the variability in PK of midazolam, possibly obscuring a difference between the PK of a microtracer and a microdose. However, we showed that the age range was comparable in both treatment groups, and we assumed the disease severity was similar in the 2 groups. Another limitation is that the sample size is relatively small. Nevertheless, PK parameters between a microdose and a microtracer were similar and compared with literature values. Moreover, in adults, low sample sizes were used to show dose linearity of midazolam.14
A future perspective more specific to this particular study is that the results indicate that a [14C]midazolam microdose can be used as an alternative to a midazolam therapeutic dose to study CYP3A activity in children. In the case of taking that approach, an attempt can be made in extrapolating the results to other CYP3A‐substrates and predict their disposition using a physiologically based pharmacokinetic modelling approach. Importantly, whether this may be possible will depend on the characteristics of these substrates, as described by Calvier et al.45 As a substantial number of clinically relevant drugs used in children are metabolized by CYP3A,16 this has the potential to impact the efficacy and safety of drug dosing in paediatrics through more informed adaptations of dosing regimens to this population.
We conclude that the PK parameters of [14C]midazolam administered as a microdose did not differ significantly in young infants from that of a microtracer. This supports the dose linearity of an IV [14C]midazolam microdose in children, thus a [14C]midazolam microdosing approach as an alternative to a therapeutic midazolam dose can be used to study developmental changes in hepatic CYP3A activity.
CONTRIBUTORS
B.D.G., W.H.S., B.K.P., E.H.J.K., E.D., L.T.K., W.M., G.G., R.C.G., C.A.J.K., D.T., S.N.W., and M.A.T. wrote the manuscript. B.D.G., W.H.S., B.K.P., E.H.J.K., E.D., L.T.K., W.M., G.G., R.C.G., C.A.J.K., S.N.W., and M.A.T. contributed to the research design. B.D.G., W.H.S., B.K.P., E.H.J.K., E.D., L.T.K., W. M., G.G., R.C.G., S.N.W., and M.A.T. performed the research. B.D.G. and E.H.J.K. analysed the data.
5.
ACKNOWLEDGEMENTS
This project was funded by the ERA‐NET PRIOMEDCHILD, project number 113205022. B.D.G. and S.N.W. were sponsored by a grant from ZonMW, The Netherlands Organization for Health Research and Development, project number 113202007. The other authors have no competing interests to declare.
van Groen BD, Vaes WH, Park BK, et al. Dose‐linearity of the pharmacokinetics of an intravenous [14C]midazolam microdose in children. Br J Clin Pharmacol. 2019;85:2332–2340. 10.1111/bcp.14047
The authors confirm that the PI for this paper is Dr M. A. Turner and that he had direct clinical responsibility for patients.
Data Availability Statement:The data that support the findings of this study are available from the corresponding author upon reasonable request.
Contributor Information
Saskia N. de Wildt, Email: saskia.dewildt@radboudumc.nl.
Mark A. Turner, Email: mark.turner@liverpool.ac.uk.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Hines RN. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol Ther. 2008;118(2):250‐267. [DOI] [PubMed] [Google Scholar]
- 2. Kearns GL, Abdel‐Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology‐‐drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157‐1167. [DOI] [PubMed] [Google Scholar]
- 3. Allegaert K, Rochette A, Veyckemans F. Developmental pharmacology of tramadol during infancy: ontogeny, pharmacogenetics and elimination clearance. Paediatr Anaesth. 2011;21(3):266‐273. [DOI] [PubMed] [Google Scholar]
- 4. de Wildt SN, Kearns GL, Murry DJ, Koren G, van den Anker JN. Ontogeny of midazolam glucuronidation in preterm infants. Eur J Clin Pharmacol. 2010;66(2):165‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Wildt SN, Ito S, Koren G. Challenges for drug studies in children: CYP3A phenotyping as example. Drug Discov Today. 2009;14(1–2):6‐15. [DOI] [PubMed] [Google Scholar]
- 6. Turner MA, Mooij MG, Vaes WH, et al. Pediatric microdose and microtracer studies using 14C in Europe. Clin Pharmacol Ther. 2015;98(3):234‐237. [DOI] [PubMed] [Google Scholar]
- 7. European Medicines Agency . ICH topic M3 (R2) non‐clinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. 2008.
- 8. Food and Drug Administration US Department of Health and Human Services Guidance for Industry Investigators and Reviewers . Exploratory IND studies. 2006.
- 9. Salehpour M, Possnert G, Bryhni H. Subattomole sensitivity in biological accelerator mass spectrometry. Anal Chem. 2008;80(10):3515‐3521. [DOI] [PubMed] [Google Scholar]
- 10. Vuong LT, Blood AB, Vogel JS, Anderson ME, Goldstein B. Applications of accelerator MS in pediatric drug evaluation. Bioanalysis. 2012;4(15):1871‐1882. [DOI] [PubMed] [Google Scholar]
- 11. Mooij MG, van Duijn E, Knibbe CA, et al. Successful use of [14C]paracetamol microdosing to elucidate developmental changes in drug metabolism. Clin Pharmacokinet. 2017;56(10):1185‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garner CR, Park KB, French NS, et al. Observational infant exploratory [(14)C]‐paracetamol pharmacokinetic microdose/therapeutic dose study with accelerator mass spectrometry bioanalysis. Br J Clin Pharmacol. 2015;80(1):157‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mooij MG, van Duijn E, Knibbe CA, et al. Pediatric microdose study of [(14)C]paracetamol to study drug metabolism using accelerated mass spectrometry: proof of concept. Clin Pharmacokinet. 2014;53(11):1045‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lappin G, Kuhnz W, Jochemsen R, et al. Use of microdosing to predict pharmacokinetics at the therapeutic dose: experience with 5 drugs. Clin Pharmacol Ther. 2006;80(3):203‐215. [DOI] [PubMed] [Google Scholar]
- 15. Bosgra S, Vlaming ML, Vaes WH. To apply microdosing or not? Recommendations to single out compounds with non‐linear pharmacokinetics. Clin Pharmacokinet. 2016;55(1):1‐15. [DOI] [PubMed] [Google Scholar]
- 16. Shimada T, Yamazaki H, Mimura M, et al. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270(1):414‐423. [PubMed] [Google Scholar]
- 17. Stevens JC, Hines RN, Gu C, et al. Developmental expression of the major human hepatic CYP3A enzymes. J Pharmacol Exp Ther. 2003;307(2):573‐582. [DOI] [PubMed] [Google Scholar]
- 18. de Wildt SN, Kearns GL, Leeder JS, van den Anker JN. Cytochrome P450 3A: ontogeny and drug disposition. Clin Pharmacokinet. 1999;37(6):485‐505. [DOI] [PubMed] [Google Scholar]
- 19. Stevens JC. New perspectives on the impact of cytochrome P450 3A expression for pediatric pharmacology. Drug Discov Today. 2006;11(9–10):440‐445. [DOI] [PubMed] [Google Scholar]
- 20. Lacroix D, Sonnier M, Moncion A, Cheron G, Cresteil T. Expression of CYP3A in the human liver‐‐evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth. Eur J Biochem. 1997;247(2):625‐634. [DOI] [PubMed] [Google Scholar]
- 21. Leeder JS, Gaedigk R, Marcucci KA, et al. Variability of CYP3A7 expression in human fetal liver. J Pharmacol Exp Ther. 2005;314(2):626‐635. [DOI] [PubMed] [Google Scholar]
- 22. Watkins PB. Noninvasive tests of CYP3A enzymes. Pharmacogenetics. 1994;4(4):171‐184. [DOI] [PubMed] [Google Scholar]
- 23. Streetman DS, Bertino JS Jr, Nafziger AN. Phenotyping of drug‐metabolizing enzymes in adults: a review of in‐vivo cytochrome P450 phenotyping probes. Pharmacogenetics. 2000;10(3):187‐216. [DOI] [PubMed] [Google Scholar]
- 24. Chainuvati S, Nafziger AN, Leeder JS, et al. Combined phenotypic assessment of cytochrome p450 1A2, 2C9, 2C19, 2D6, and 3A, N‐acetyltransferase‐2, and xanthine oxidase activities with the "Cooperstown 5+1 cocktail". Clin Pharmacol Ther. 2003;74(5):437‐447. [DOI] [PubMed] [Google Scholar]
- 25. Fuhr U, Jetter A, Kirchheiner J. Appropriate phenotyping procedures for drug metabolizing enzymes and transporters in humans and their simultaneous use in the "cocktail" approach. Clin Pharmacol Ther. 2007;81(2):270‐283. [DOI] [PubMed] [Google Scholar]
- 26. Hohmann N, Kocheise F, Carls A, Burhenne J, Haefeli WE, Mikus G. Midazolam microdose to determine systemic and pre‐systemic metabolic CYP3A activity in humans. Br J Clin Pharmacol. 2015;79(2):278‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Halama B, Hohmann N, Burhenne J, Weiss J, Mikus G, Haefeli WE. A nanogram dose of the CYP3A probe substrate midazolam to evaluate drug interactions. Clin Pharmacol Ther. 2013;93(6):564‐571. [DOI] [PubMed] [Google Scholar]
- 28. de Wildt SN, de Hoog M, Vinks AA, van der Giesen E, van den Anker JN. Population pharmacokinetics and metabolism of midazolam in pediatric intensive care patients. Crit Care Med. 2003;31(7):1952‐1958. [DOI] [PubMed] [Google Scholar]
- 29. EMA . Guideline on the investigation of medicinal products in the term and preterm neonate. (EMA/PDCO/362462/2016).
- 30. van Duijn E, Sandman H, Grossouw D, Mocking JA, Coulier L, Vaes WH. Automated combustion accelerator mass spectrometry for the analysis of biomedical samples in the low attomole range. Anal Chem. 2014;86(15):7635‐7641. [DOI] [PubMed] [Google Scholar]
- 31. Higton D, Young G, Timmerman P, Abbott R, Knutsson M, Svensson LD. European bioanalysis forum recommendation: scientific validation of quantification by accelerator mass spectrometry. Bioanalysis. 2012;4(22):2669‐2679. [DOI] [PubMed] [Google Scholar]
- 32. Klein MV, Vaes WHJ, Fabriek B, Sandman H, Mous DJW, Gottdang AT. The 1 MV multi‐element AMS system for biomedical applications at the Netherlands Organization for Applied Scientific Research (TNO). Nucl Instrum Methods Phys Res, Sect B. 2013;294:14‐17. [Google Scholar]
- 33. Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add‐in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft excel. Comput Methods Programs Biomed. 2010;99(3):306‐314. [DOI] [PubMed] [Google Scholar]
- 34. Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model‐based drug development‐part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometrics Syst Pharmacol. 2013;2:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model‐based drug development. CPT Pharmacometrics Syst Pharmacol. 2012;1:e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krekels EH, Johnson TN, den Hoedt SM, et al. From pediatric covariate model to Semiphysiological function for maturation: part II‐sensitivity to physiological and physicochemical properties. CPT Pharmacometrics Syst Pharmacol. 2012;1:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2018;46:D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol. 2017;174(Suppl 1):S272‐S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ahn JE, Karlsson MO, Dunne A, Ludden TM. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J Pharmacokinet Pharmacodyn. 2008;35(4):401‐421. [DOI] [PubMed] [Google Scholar]
- 40. de Wildt SN, Kearns GL, Hop WC, Murry DJ, Abdel‐Rahman SM, van den Anker JN. Pharmacokinetics and metabolism of intravenous midazolam in preterm infants. Clin Pharmacol Ther. 2001;70(6):525‐531. [DOI] [PubMed] [Google Scholar]
- 41. Reed MD, Rodarte A, Blumer JL, et al. The single‐dose pharmacokinetics of midazolam and its primary metabolite in pediatric patients after oral and intravenous administration. J Clin Pharmacol. 2001;41(12):1359‐1369. [DOI] [PubMed] [Google Scholar]
- 42. Payne K, Mattheyse FJ, Liebenberg D, Dawes T. The pharmacokinetics of midazolam in paediatric patients. Eur J Clin Pharmacol. 1989;37(3):267‐272. [DOI] [PubMed] [Google Scholar]
- 43. Vet NJ, Brussee JM, de Hoog M, et al. Inflammation and organ failure severely affect midazolam clearance in critically ill children. Am J Respir Crit Care Med. 2016;194(1):58‐66. [DOI] [PubMed] [Google Scholar]
- 44. Roth‐Cline M, Nelson RM. Microdosing studies in children: a US regulatory perspective. Clin Pharmacol Ther. 2015;98(3):232‐233. [DOI] [PubMed] [Google Scholar]
- 45. Calvier EAM, Krekels EHJ, Yu H, et al. Drugs being eliminated via the same pathway will not always require similar pediatric dose adjustments. CPT Pharmacometrics Syst Pharmacol. 2018;7(3):175‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.