

Abstract
Dichloroacetate (DCA) is an investigational drug targeting the glycolytic hallmark of cancer by inhibiting pyruvate dehydrogenase kinases (PDK). It is metabolized by GSTZ1, which has common polymorphisms altering enzyme or promoter activity. GSTZ1 is also irreversibly inactivated by DCA. In the first clinical trial of DCA in a hematological malignancy, DiCAM (DiChloroAcetate in Myeloma), we have examined the relationship between DCA concentrations, GSTZ1 genotype, side effects, and patient response. DiCAM recruited seven myeloma patients in partial remission. DCA was administered orally for 3 months with a loading dose. Pharmacokinetics were performed on day 1 and 8. Trough and peak concentrations of DCA were measured monthly. GSTZ1 genotypes were correlated with drug concentrations, tolerability, and disease outcomes. One patient responded and two patients showed a partial response after one month of DCA treatment, which included the loading dose. The initial half‐life of DCA was shorter in two patients, correlating with heterozygosity for GSTZ1*A genotype, a high enzyme activity variant. Over 3 months, one patient maintained DCA trough concentrations approximately threefold higher than other patients, which correlated with a low activity promoter genotype (−1002A, rs7160195) for GSTZ1. This patient displayed the strongest response, but also the strongest neuropathy. Overall, serum concentrations of DCA were sufficient to inhibit the constitutive target PDK2, but unlikely to inhibit targets induced in cancer. Promoter GSTZ1 polymorphisms may be important determinants of DCA concentrations and neuropathy during chronic treatment. Novel dosing regimens may be necessary to achieve effective DCA concentrations in most cancer patients while avoiding neuropathy.
Keywords: Chemotherapy, clinical pharmacology, drug metabolism, genetic polymorphism, toxicology
Abbreviations
- AUC
area under the plasma concentration curve
- CyBorD
cyclophosphamide-bortezomib-dexamethasone
- DCA
dichloroacetate
- DEX
dexamethasone
- DiCAM
DiChloroAcetate in Myeloma
- IMWG
International Myeloma Working Group
- LEN
lenalidomide
- MM
multiple myeloma
- PDH
pyruvate dehydrogenase
- PDK
pyruvate dehydrogenase kinase
- PN
peripheral neuropathy
- SNP
single nucleotide polymorphism
- TNSc
Total Neuropathy Score
1. INTRODUCTION
Deregulated energetics is an established hallmark of cancer that includes the glycolytic phenotype (elevated glucose uptake and glycolysis, with conversion of pyruvate to lactate in the presence of adequate oxygen).1, 2 The glycolytic phenotype is widespread amongst many cancer types, and is exploited clinically in imaging of cancers with [18F]‐fluorodeoxyglucose positron emission tomography (18F‐FDG PET).2, 3 Dichloroacetate (DCA) is an established investigational drug that can target the deregulated energetics of cancer. DCA reverses the glycolytic phenotype, redirecting the metabolism of pyruvate away from lactate production and into mitochondrial oxidation.4, 5 It does this through the inhibition of pyruvate dehydrogenase (PDH) kinases (PDKs) http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=608, a family of four kinases (PDK1‐4) that phosphorylate and inactivate PDH.5 Antitumor activity in vivo was first demonstrated in lung and breast cancer models,6, 7 and has subsequently been shown in a range of solid tumor types (reviewed in 4). The actions of DCA include inhibiting proliferation, inducing apoptosis, enhancing apoptosis of other cytotoxic agents (particularly those that target mitochondrial metabolism 8, 9, 10, 11), and inhibiting angiogenesis, through mechanisms including decreasing the mitochondrial membrane potential, increasing p53 activity, decreasing hypoxia inducible factor‐1α expression, and decreased VEGF expression.4
With decades of clinical use in adults and children with congenital lactic acidosis,12, 13 DCA has progressed rapidly into clinical trials against cancer, resulting in four published reports on phase I/II trials in solid tumors.14, 15, 16, 17 Chu et al14 concluded the recommended phase 2 dose (RP2D) for oral DCA is 6.25 mg/kg b.i.d., based on the maximum tolerated dose, with peripheral neuropathy (PN) arising after one or more months of treatment being one of the most common concerns, however, high variability in pharmacokinetics and the occurrence of neuropathy was observed among patients.14 The previous DCA trials in cancer included limited drug serum concentration monitoring, thus it remains unclear whether the RP2D is adequate for efficacy in the cancer setting. Further, it was noted in two of the trials16, 17 that it took several weeks for DCA to become detectable in serum, suggesting a loading dose may be of value. Thus, an effective dosing regimen of DCA for the treatment of cancer patients has not been established. An expected effective concentration of DCA can be proposed based on its ability to inhibit the PDKs in vitro. DCA is a pan‐PDK inhibitor, with the constitutive isoform, PDK2, having the lowest IC50 (0.2 mmol/L), whereas PDK1 and PDK3 require considerably more DCA for inhibition (1.0 mmol/L and 8.0 mmol/L, respectively).5, 18 PDK1 and PDK3 are both target genes of the transcription factor hypoxia inducible factor (HIF), and so may be expressed at elevated levels in hypoxic regions of solid cancers.5
DCA is metabolized primarily in the liver by glutathione transferase Zeta (GSTZ1). The GSTZ1 gene has three common nonsynonymous single nucleotide polymorphisms (SNPs) resulting in four common isoforms of the protein19, 20 and one rare isoform.21 The four main isoforms have been characterized in vitro for activity with haloacids, with GSTZ1*A» GSTZ1*B ~ GSTZ1*C > GSTZ1*D for activity toward DCA.19, 20 GSTZ1 also has polymorphisms in the promoter, including SNP rs7160195 at nt −1002, which impact on the level of expression of luciferase reporter constructs,22 and may alter the level of enzyme expression in the liver.23 A luciferase reporter promoter construct containing the −1002G vs −1002A allele resulted in approximately sixfold higher promoter activity when tested in transiently transfected HepG2 cells (figure 5B in22). Consistent with this, expression of GSTZ1 in frozen human liver samples, estimated by both protein and mRNA‐based methods, was found to be approximately threefold higher in Caucasians who did not carry the −1002A allele (figures 1 and 3 in 23). It should be noted that these associations are not necessarily true in African‐Americans, where different haplotypes are present.22, 23 The impact of the genetic variants of GSTZ1 on DCA kinetics and dynamics in patients has been reported in only a small number of individuals, and in only one of the cancer clinical trials,15 but has led to the development of a test to enable genetic‐based dosing for DCA.24
In addition to genetic polymorphisms influencing DCA pharmacokinetics, DCA is an inhibitor of its own metabolism, irreversibly inhibiting GSTZ1 activity and leading to degradation of the protein,25, 26 explaining why subsequent doses of DCA have a longer half‐life compared to the initial dose.27 This adds complexity in understanding when DCA might achieve steady‐state concentrations during multiple dosing, requiring pharmacokinetic investigations on multiple days. Furthermore, giving a loading dose to achieve steady‐state concentrations more rapidly may be a useful strategy.
Multiple myeloma (MM) is a plasma cell malignancy affecting bone marrow and lytic lesions in the bones that remains incurable despite improvements in therapy including cyclophosphamide‐bortezomib‐dexamethasone (CyBorD or CVD), lenalidomide/ thalidomide, melphalan, and autologous stem cell transplantation.28, 29 Poor prognostic variants are often related to clonal genetic aberrations including 1p‐ and t(4,14) amongst others.30, 31 MM is associated with expression of a clonal immunoglobulin protein (paraprotein or M‐band) that is used routinely to monitor disease and assess response to therapy. The importance of metabolism changes has only recently been appreciated in hematological cancers.32 The metabolic phenotype of myeloma includes the glycolytic phenotype, present in approximately 50% of MM cell lines examined,33, 34 with high lactate dehydrogenase (LDH) and PDK1 expression associated with poorer patient outcomes33 and drug resistance, which may be overcome by reversing the glycolytic phenotype.35, 36 DCA combined with bortezomib showed additive cytotoxic effects in vitro and improved outcomes in a mouse model.34 Most recently, lactate has been identified as a survival factor,37 and inhibition of lactate transporters (MCTs) could reduce cell viability.37, 38 Extrapolating from these observations of the glycolytic phenotype, MM patients may be good candidates to benefit from DCA treatment.
In this study, we designed the DiCAM (DiChloroAcetate in Myeloma) clinical trial to improve upon previous trials. Pharmacogenetics were examined in both the acute and chronic treatment settings. The spectrum of cancer types treated with DCA was expanded into hematological malignancies, and MM patients were chosen allowing easy monitoring of disease via blood paraprotein levels. The MM patients were in plateau phase and in relatively good health, increasing the likelihood of them completing the treatment regimen and giving reliable pharmacokinetic information. Existing PN was not an exclusion criteria so that the significance of this potential side effect could be examined in a real patient setting. A loading dose of DCA was included to take advantage of the inhibition of GSTZ1, which was predicted to lead to more rapid achievement of therapeutic concentrations. While conducted in a limited number of patients, this study has been able to provide valuable insights for improved future trial design for testing efficacy of DCA in cancer patients.
2. MATERIALS AND METHODS
This study was approved by the ACT Health Human Research Ethics Committee and the Australian National University Human Research Ethics Committee. The study was registered with the Australian New Zealand Clinical Trials Registry (ANZCTR registration number: ACTRN12615000226505).
2.1. Patient Eligibility
Patients were eligible for inclusion if they had: plasma cell myeloma in plateau phase (defined as neither progression nor response for at least 28 days); no change in myeloma therapy for at least 16 weeks (except bisphosphonates), excluding reinduction therapy, but enabling recruitment if stable on maintenance therapy; aged over 18 years; an Eastern Cooperative Oncology Group performance score of ≤2 and; a life expectancy of at least 6 months and; measurable disease (defined as quantifiable serum paraprotein on electrophoresis of at least 2 g/L, or elevated free kappa (>21 mg/L) or lambda light chains (>30 mg/L) with an abnormal serum free light chain ratio (normal κ:λ = 0.26‐1.26). Progression was defined as per International Myeloma Working Group (IMWG) to include an increase in the paraprotein by ≥25% and at least 5g/L, or >25% increase in difference between involved and uninvolved light chain level, with an absolute increase of >0.1g/L. Response was defined as at least an IMWG minimal response, including a reduction in the paraprotein by ≥25%.39 In the case of light chain only myeloma, a modified criteria of a 25% decrease in the difference between the involved and uninvolved light chains and an absolute reduction of at least 100 mg/L was chosen as a minimal response criteria is not available for light chain only myeloma.
2.2. Study Design
This was a prospective, open label, nonrandomized phase 2 study (Simon's Mini‐max 2‐stage design) of the efficacy of DCA in plateau phase myeloma. The recruitment aim was for 15 patients in the first stage, and 10 patients in the second stage. The primary efficacy endpoint was the overall response rate during 12 weeks of treatment. The secondary objectives were to (a) establish the drug concentrations of DCA in vivo with the dosing schedule, (b) confirm the tolerability and safety of DCA at these doses, and (c) genotype patients for glutathione transferase Zeta (GSTZ1) and correlate genotypes with DCA concentrations and tolerability.
The final patient in this cohort (P007) was enrolled in an updated protocol which is ongoing. The dosing on d1 in the new protocol is identical to the DiCAM protocol, thus the pharmacokinetic and pharmacogenetic results of P007 on d1 are reported in this study, as P007 shares an uncommon genotype with P001. The updated protocol deviates from this protocol after d3, thus no other results from P007 are consistent with the patient cohort and so are not reported here.
2.3. Drug formulation and dosing
Clinical grade crystalline sodium DCA (TCI America, Portland, OR) was purchased and compounded into 500 mg, 125 mg, and 25 mg gelatine capsules with inert filler by Capital Compounding Pharmacists (Woden, ACT, Australia). Capsules were prepared as patients were recruited, stored by The Canberra Hospital Oncology Pharmacy, and dispensed within 6 months of compounding. Patients received five doses of 25 mg/kg over days 1‐3: dose 1 in the morning of d1, then 25 mg/kg b.i.d. on d2 and 3. From d4 onwards, the dose was 6.25 mg/kg b.i.d., with the exception of d8, when a single dose of 25 mg/kg was given in the morning, to replicate the pharmacokinetic measurements of d1. DCA treatment was in addition to other maintenance treatments and was continued for 12 weeks, at which time DCA was discontinued.
2.4. Monitoring of patient residual disease and DCA toxicity
A physical examination including Total Neuropathy Score (TNSc),40 blood counts, serum biochemistry tests were undertaken at baseline and repeated on day 8, 28, 56, 84, 112 (4 months), 168 (6 months), and 252 (9 months) of the trial. Paraprotein level and/or free light chain levels were measured at baseline and days 28, 56, 84, 112, 168, and 252. Patients were withdrawn for new onset or progression to Grade‐3 peripheral neuropathy (defined as any of the seven aspects of TNSc having a score > 3), or any other emergent grade 3 or 4 toxicity if the symptoms were not alleviated after supportive treatments.
2.5. Patient serum pharmacokinetic sampling
Measurements of serum DCA concentrations were made for determination of pharmacokinetics of DCA on d1 and d8 of the study. Blood samples were collected at time 0, 1, 2, 3, 4, 5, 6, and 24 hours after administration of DCA. For ongoing monitoring of DCA concentrations, the original protocol onto which P001 was recruited collected through samples only on d28, d56, and d84. This was revised for the subsequent patients to include collection of trough and peak concentrations as follows: On d15 and d22, blood samples were taken before DCA administration to measure maintenance dose trough concentration. On d28, d56, and d84, blood samples were taken before and 2 hours after DCA administration to monitor the maintenance dose trough concentration and peak concentration.
2.6. DNA isolation, genotyping, and haplotype analysis
DNA was extracted from peripheral blood (QIAGEN blood kit, Cat.No.51106) and GSTZ1 genotyping was performed by published methods for SNPs in the promoter (nt −1002, rs7160195) and in protein‐coding exons (nt 94, rs3177427; nt 124, rs7972; and nt 245, rs1046428). Haplotypes were inferred from those present in the Australian European population studied previously.19, 20 The expected proteins are described with the standard nomenclature for glutathione transferases.41
2.7. Analysis of serum DCA concentrations
Serum DCA concentrations were analyzed in duplicate by liquid chromatography‐mass spectrometry (LC‐MS) for patients P001‐P004, and by gas chromatography‐mass spectrometry (GC‐MS) for patients P005‐P007. Prism 7 was used to calculate the area under the plasma concentration curve (AUC) on d1 from 0‐6 hours and the half‐life of DCA (using a nonlinear regression, one phase decay curve fit) from drug concentrations on d1 from peak (at 1 or 2 hours) to 6 hours (See Appendix S1 for details.)
2.8. In vitro drug combination studies
The MM cell lines RPMI 8226, U266, NCI‐H929, MM.1S, and MM.1R were treated with DCA (D54702, Sigma Chemical Co St. Louis, MO) and chemotherapy agents for 72 or 96 hours, and the neutral red uptake assay was used to determine total viable cell number, expressed as a percentage of untreated/vehicle control cells. (See Appendix S1 for full details.) At least three independent experiments were performed for each treatment.
3. RESULTS
3.1. Patient characteristics
Six Caucasian patients were enrolled in this study between May 2015 and June 2016. Patient demographics, baseline disease, prior treatments, and comorbidities are listed in Table 1. Four of these six patients had myeloma with intact paraproteins, whereas two patients had free light chain myeloma (P001, free kappa; P002 free lambda). The mean age at enrollment was 65.6 years (range 52‐77). They had 2‐7 prior therapies. Five of six patients were on comaintenance treatments during the study (Table 1). Maintenance treatment in P002 (CyBorD) was stopped at study d30 due to steroid myopathy and insomnia.
Table 1.
Patient characteristics
| Patient ID | P001 | P002 | P003 | P004 | P005 | P006 |
|---|---|---|---|---|---|---|
| Age of enrollment | 70 | 72 | 77 | 61 | 62 | 52 |
| Gender | Male | Male | Female | Male | Male | Male |
| Age of diagnosis | 65 | 62 | 75 | 60 | 60 | 49 |
| Diagnosis (year) | 2010 | 2005 | 2013 | 2014 | 2013 | 2012 |
| Ethnicity | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian |
| Cytogenetics | Normal | Unknown | Normal | Complex b | Normal | Normal |
| Light chain only myeloma (κ/λ) | No | λ | No | No | No | No |
|
Intact paraprotein (HC/LC Isotypes) |
a IgA/κ | No | IgG/κ | IgG/ λ | IgG/ λ | IgG/κ |
| Baseline disease |
FLC (λ‐κ) 45 mg/L |
FLC (λ‐κ) 239 mg/L |
Paraprotein 9 g/L |
Paraprotein 4 g/L |
Paraprotein 4 g/L |
Paraprotein 10 g/L |
| Comaintenance treatment | None | CyBorD(until study day 30) | Thalidomide | Thalidomide, DEX | Thalidomide | Lenalidomide, DEX |
| Best response received after 1st therapy | VGPR | VGPR | SD | PR | PR | PR |
| Lines of prior therapy | 3 | 7 | 2 | 3 | 3 | 3 |
| Comorbidities |
Deep vein thrombosis/ pulmonary embolism; kidney stones |
Unknown | Dyslipidemia | No | Gastro‐esophageal reflux |
Deep vein thrombosis/ pulmonary embolism |
Abbreviations: CyBorD, Cyclophosphamide + Bortezomib +Dexamethasone; DEX: dexamethasone; CR, complete response; FLC, free light chain; HC, heavy chain; LC, light chain; sCR, stringent complete response; PR, partial response; SD, stable disease; VGPR, very good partial response.
P001: P001 showed an IgA paraprotein at diagnosis in 2010, but the IgA paraprotein was not detected after 5 years on the screening day for the trial in 2015, thus his disease readout was presented as free light chain myeloma.
P004 cytogenetics results: (43,X, Y, add (1)(q21), add (2)(p23), add (2)(q33), add (4)(p15), der (8) t (8; 17)(p21; q11.2), del (11)(p12), add (12)(q13), −13, t (14; 22)(q32; q12), −16, −16, −17, −20, +mar1‐3[12]/sdl, del (6)(q25)[5]/46, XX [36].
Four patients had normal cytogenetics and one patient was untested (Table 1). Patient P004 had confirmed complex cytogenetics including chromosome 1 abnormalities, 14q32 rearrangements and monosomy 13 which are associated with poor prognosis.30, 31 P004 was withdrawn from the study after 1 month due to rapid disease progression.
3.2. Patient response
One of the six patients (P002) showed a response to DCA treatment at d28 maintained to d84 (Figure 1A, Figure S1). Two patients had transient falls in measurable disease at d28 (P001 and P003), but did not meet response criteria. After DCA treatment was completed, P002 steadily progressed. P004 was taken off the trial at d28 due to disease progression. P005 had a 25% (1g) increase in paraprotein on d56, which did not meet criteria for progression and returned to the baseline level (4 g/L) and remained at that level throughout the rest of the trial. P006 had a gradual increase in paraprotein during the trial, reaching the criteria for disease progression on d84, the last day of DCA treatment (Figure 1A, Figure S1).
Figure 1.
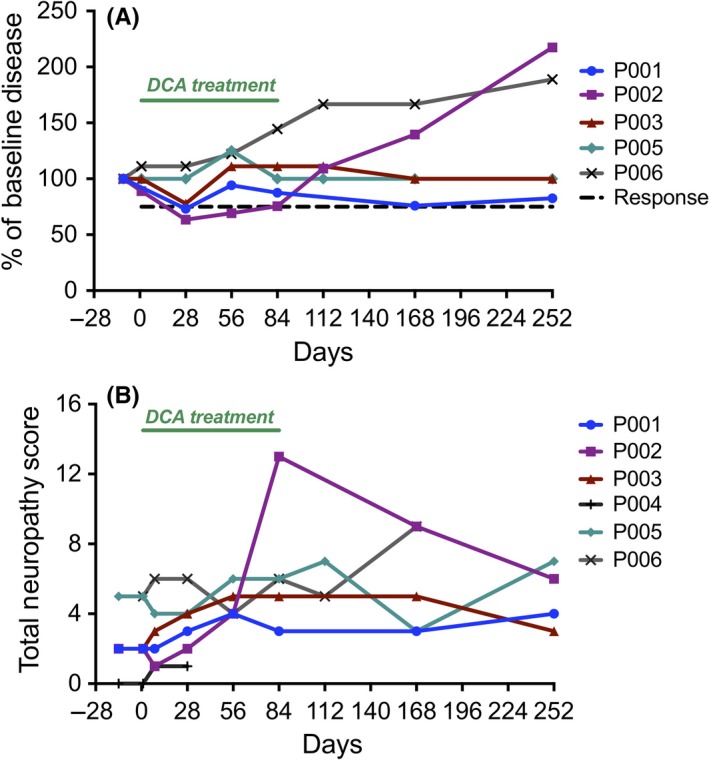
(A) Patient disease outcomes. Paraprotein level or FLC levels were measured at screening, and then on 28, 56, 84, 112, 168, and 252 of study. Patient disease readout was normalized to the baseline disease level on screening day (100%). DCA treatment period is indicated by the green bar. Graphs of raw readouts for each patient are included in Figure S1. (B) Patient Total Neuropathy Scores (TNSc). The neuropathy of each patient was assessed through seven components, as described in the methods. Graphs detailing the seven component scores for each patient are included in Figure S2. DCA treatment period is indicated by the green bar
3.3. Toxicity
No patient withdrew from the trial due to DCA induced toxicity, and none required dose reduction during the 3‐month DCA treatment period. Blood counts, liver, and kidney function were not adversely affected, consistent with previous findings from DCA trials in cancer patients.14, 15, 17 There were no problems with compliance as indicated by interviewing the patients and return of unused medication.
Five of six patients started the trial with some PN from previous myeloma treatments, and five of six patients experienced some small increase in TNSc while on DCA, but the TNSc decreased after completing DCA treatment, indicating the PN induced by DCA was reversible (Figure 1B). On the last day of treatment (d84), P002 presented with a significant increase in TNSc, with both the strength and motor symptoms having a score of 3 (Figure S2). This made his TNSc much higher than the other patients (Figure 1B), but as with the milder changes in the other patients, the symptoms gradually abated over the 6 months of monitoring after the DCA treatment was completed.
3.4. Pharmacokinetics
Serum DCA concentrations were determined on d1 and d8 of the trial after a single oral dose of 25 mg/kg taken at approximately 9 am. Patients were not given special breakfast instructions, which may contribute to variation in absorption rates and peak concentrations. The effect of food on DCA absorption has not been studied; published pharmacokinetic studies have always been done in initially fasting subjects. On d1, serum DCA concentrations peaked rapidly in all patients (median peak time 1 hour, mean peak concentration 0.33 mmol/L, range 0.15‐0.68 mmol/L, Figure 2A). DCA was then cleared with a mean half‐life of 93 min (Table 2), with 90% of the drug being cleared by 6 hours, and DCA being undetectable at 24 hours (Figure 2A). Notably, P001 and P007 had faster apparent clearance of DCA compared to other patients (CL/F > 65 L/h, Table 2). This was also reflected in a smaller AUC (Table 2) and a sharp drop in DCA concentrations between 1 and 2 hours (Figure 2A). On d8 of the study (Figure 2B), the same oral dose of 25 mg/kg DCA resulted in peak serum concentrations approximately twofold those achieved on d1 (median peak time 2 hours, mean peak concentration 0.59 mmol/L, range 0.50‐0.66 mmol/L on d8). The serum DCA concentration remained high (0.3‐0.6 mmol/L) 6 hours post‐DCA administration, and was readily detected at 24 hours. In contrast to d1, on d8 the drug concentration profile of P001 was not remarkably different from other patients. (P007 d8 could not be compared due to a different dosing regime in the modified protocol.)
Figure 2.
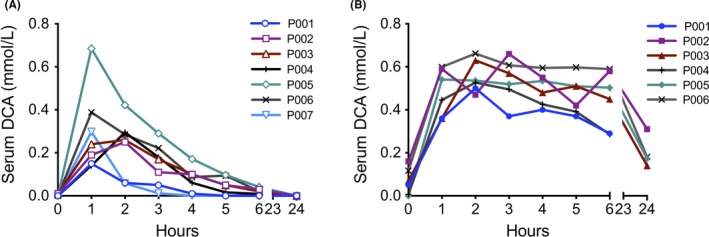
Patient serum DCA pharmacokinetics on (A) day 1, and (B) day 8 of the trial. Serum was collected hourly after a single oral dose of 25 mg/kg (high loading dose), and DCA measured by mass spectrometry
Table 2.
Patient GSTZ1 genotypes and correlation with DCA metabolism
|
aHalf‐life DCA (min) Day 1 |
aAUC (mg/L.h) Day 1 |
bCL/F (L/h) Day 1 |
Haplotypes/ Protein Isoforms |
cAverage trough [DCA] (mmol/L) |
Promoter nt −1002 |
||
|---|---|---|---|---|---|---|---|
| P001 | 61 | 34.8 | 50.3 | Z1*A / Z1*C | (KRT/EGT) | 0.13 (n = 4) | A/G |
| P002 | 53 | 92.8 | 21.5 | Z1*B/ Z1*B | (KGT/KGT) | 0.34 (n = 5) | A/A |
| P003 | 125 | 107.7 | 13.9 | Z1*C/ Z1*D | (EGT/EGM) | 0.10 (n = 5) | G/G |
| P004 | 79 | 89.7 | 23.7 | Z1*C/ Z1*C | (EGT/EGT) | 0.04 (n = 4) | G/G |
| P005 | 105 | 217.4 | 12.6 | Z1*C/ Z1*D | (EGT/EGM) | 0.21 (n = 5) | G/G |
| P006 | 201 | 140.2 | 16.1 | Z1*C/ Z1*C | (EGT/EGT) | 0.17 (n = 6) | G/G |
| P007 | 25 | 47.4 | 34.2 | Z1*A / Z1*C | (KRT/EGT) | N/A | A/G |
| Means by Genotype | |||||||
|
Protein – All genotypes |
93 | 104.3 | 24.6 | ||||
|
P002‐P006 (No Z1*A) |
113 | 129.6 | 17.6 | ||||
|
P001, P007 (One Z1*A) |
43 | 41.1 | 42.3 | ||||
|
Promoter nt −1002 All genotypes |
0.165 | ||||||
| G/G and G/A (n = 5) | 0.130 | ||||||
| A/A (P002) | 0.34 | ||||||
After the initial dose of 25 mg/kg.
Apparent clearance (CL/F).
During the 3 month maintenance treatment period (6.25 mg/kg b.i.d.). Average derived from values presented in Table S1
Bold indicates the unusual values and genotypes discussed in the text.
The serum trough and peak (2 hours post dose) concentrations were monitored during the 3 month DCA maintenance treatment (6.25 mg/kg b.i.d.). The average DCA trough concentration increased with time, from 0.074 mmol/L on d8 to 0.309 mmol/L on d84 (Figure 3A, Table S1). This trough concentration range is very similar to the data reported by Chu et al.14 Strikingly, P002 had DCA trough concentrations two‐ to threefold higher than the average of the other patients on d56 and d84 (Figure 3A, Table S2), correlating with the highest TNSc (Figure 1B). Neither neuropathy score nor trough concentrations were elevated during the first month of DCA treatment. The DCA peak concentrations ranged from 0.04 to 0.69 mmol/L when patients were on maintenance dose (Figure 3B, Table S1).
Figure 3.
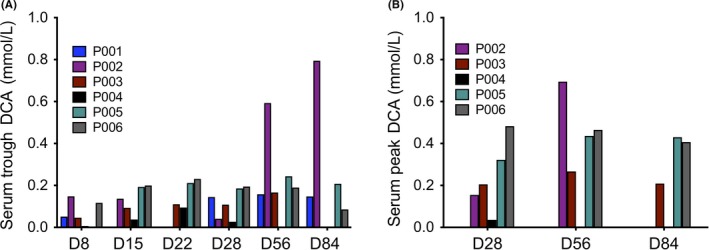
Patient serum DCA (A) trough and (B) peak concentrations over 12 weeks of the trial. Serum was collected before (trough) and 2 h after (peak) the morning oral maintenance dose of 6.25 mg/kg. Full details of values and variations in data points collected for each patient are included in Table S1. (Note: Peak concentration samples were not collected under the original protocol onto which P001 was recruited.)
3.5. Pharmacogenetics
We determined the genotypes of the patients for the common promoter and coding region SNPs of GSTZ1. Overall, the alleles present in our seven patients were consistent with expected frequencies for Caucasians. Notably, P001 and P007 possessed one allele of GSTZ1*A, a minor haplotype with 3.6‐fold enzymatic activity toward DCA in vitro compared to wild‐type protein.20 The presence of GSTZ1*A correlated with the shorter half‐life of DCA and lower AUC on d1 (Figure 2A, Table 2).
All isoforms of GSTZ1 are irreversibly inactivated by DCA requiring expression of new protein to restore enzyme activity. Promoter SNPs have been described that result in expression differences of GSTZ1. An A at nt −1002 is associated with lower promoter activity in vitro22 and lower protein expression in human liver.23 P002 possessed an uncommon genotype at nt −1002, being homozygous A for the low activity promoter. This correlated with the highest average DCA trough concentration (Figure 3, Table 2) and the highest TNSc (Figure 1B).
3.6. Drug combinations in vitro
We examined the effect of DCA in combination with CyBorD on cell viability of myeloma cell lines in vitro. A fixed ratio of the cytotoxic agents (400: 1: 10, based on P002’s CyBorD dosing regimen) for cyclophosphamide: bortezomib: dexamethasone (DEX) was tested against U266 and RPMI 8226 cells, which have previously been shown to have different levels of glycolytic activity.34 Consistent with Sanchez et al,34 5 mmol/L DCA alone significantly reduced the total viable cell number of RPMI 8226 cells (high glycolytic activity), but not U266 cells (low glycolytic activity) (Figure 4A). Regardless of this difference in response to DCA alone, DCA did not increase the effectiveness of cytotoxic doses of the CyBorD drug combination in either cell line (Figure 4A).
Figure 4.
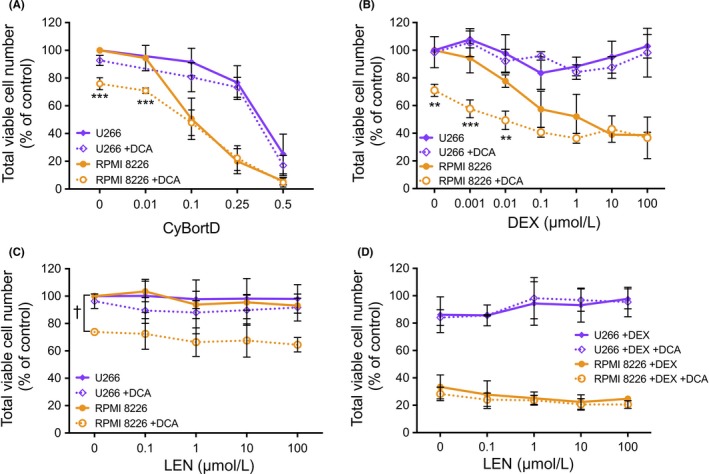
In vitro total viability cell number of cell lines treated with DCA in combination with myeloma maintenance therapies used during the trial. RPMI 8226 and U266 cell lines were treated with 5 mmol/L DCA in combination with (A) CyBorD (72 h), (B) DEX (72 h), (C) LEN (96 h), or (D) LEN + DEX (96 h). CyBorD combination was at a fixed ratio of 400:1:10, where 1x concentrations were 4000 nmol/L cyclophosphamide, 10 nmol/L bortezomib, and 100 nmol/L dexamethasone. All data points shown represent mean ± SD from at least three independent experiments. **P < .01 and ***P < .001 compared to non‐DCA‐treated counterpart. †P < .001 for all points in the series. (Note: x‐axes are not linear/ logarithmic.)
The impact of DCA on cell viability in vitro when used in combination with the standard myeloma therapies DEX and lenalidomide (LEN) was also examined. RPMI 8226 cells were moderately sensitive to DEX, whereas U266 cells were unresponsive (Figure 4B). The addition of DCA to low concentrations of DEX further decreased total viable cell number in RPMI8226 cells, however, the viability of U266 cells did not change. Neither cell line was sensitive to LEN at the concentrations tested (Figure 4C). DCA was effective at reducing total viable cell number in the presence of LEN in RPMI 8226 cells but did not alter total viable cell number of U266 cells treated with LEN (Figure 4C). Similarly, when DCA was added the combination of LEN and DEX, there was no difference in total viable cell number (Figure 4D). A similar lack of synergistic effect of DCA with these agents was observed in the additional myeloma cell lines MM.1S and MM.1R which were sensitive to LEN (Figure S3).
4. DISCUSSION
Targeting the cancer metabolic phenotype may offer a novel cancer treatment strategy not yet in routine clinical use. The glycolytic phenotype has received much interest, as it can be targeted by DCA, a prototypic pan‐PDK inhibitor that has been in clinical use for other indications for decades.4 In this small pilot study we found that, although response rates were low, there were very different pharmacokinetic profiles for DCA which are likely to have impacted both on the response rates and toxicity and suggest alternative approaches to dosing may be required.
Several early phase clinical trials in cancer have demonstrated its low toxicity and safety in patients with solid tumors, with reversible PN being the primary concern.14, 15, 16, 17 In this study, we too found that PN was the only significant side effect, seen only after chronic use and was reversible. Five of six patients entered the trial with some preexisting PN, which was not greatly exacerbated by DCA at 6.25 mg/kg b.i.d., thus DCA can be used in patients with existing peripheral neuropathy.
Significant DCA‐related PN was only experienced in one patient, P002 (Figure 1B) in association with higher trough DCA concentrations (Figure 3A) and an uncommon GSTZ1 genotype – homozygosity for the −1002A promoter SNP (Table 2). The −1002A promoter allele has been demonstrated to have reduced activity in vitro.22 We propose that during chronic DCA treatment, when GSTZ1 is persistently inactivated and degraded, −1002A/A individuals will re‐express GSTZ1 protein at a lower rate or level making them more susceptible to chronic accumulation of DCA and thus PN. This reduced ability to re‐express the protein may be more important in determining DCA kinetics and side effects than the activity (ie, protein haplotype) of the enzyme toward DCA.
In Caucasians, −1002A is in dis‐equilibrium with E32K23 (ie, GSTZ1*A (KRT) or GSTZ1*B (KGT)). These two isoforms are ~3.6‐fold different in activity toward DCA20 and thus this weaker promoter may express protein with high (GSTZ1*A) or intermediate (GSTZ1*B) catalytic activity. P002 was homozygous Z1*B, which would be predicted to be the most susceptible/ lowest activity combination genotype. Heterozygosity for −1002A in P001 did not associate with unusual peak or trough DCA concentrations, suggesting that homozygosity is necessary for the phenotype to be significant. The −1002A SNP has a population frequency of 0.33 in Australian Europeans,22 which would result in homozygotes constituting 11% of the population.
In addition to having the highest total neuropathy score, P002 also demonstrated the strongest reduction in disease in response to DCA treatment, maintaining a response until the end of the treatment period (Figure 1A). This may be attributed to the high drug concentrations experienced by P002 compared to the other patients. The alternative explanation of a combination effect of DCA with CyBortD was explored in vitro, but no synergism was demonstrated. Thus, both the response and increased toxicity are attributable to altered drug concentrations due to underlying genetic polymorphisms.
The pharmacokinetics on d1 after the initial high dose of DCA (acute treatment setting) showed an interesting correlation between the GSTZ1*A isoform and a reduced half‐life/ smaller AUC (Table 2) for DCA. However, this phenotype may be of less importance in the setting of long term cancer treatment, as the difference between P001 and other patients with different GSTZ1 genotypes had disappeared after 1 week of treatment (Figure 2B), presumably due to inactivation of GSTZ1 protein by DCA.
Considering the low frequency of homozygosity for the −1002A, the lack of difference between protein‐coding variants after 1 week of treatment, and that the DCA‐related PN only arose with chronic treatment and was reversible, it remains unclear that genotyping for GSTZ1 before treatment is necessary for safe use of DCA. Should a −1002A/A genotype be detected, additional monitoring for PN and potential dose reduction could be implemented. None‐the‐less, a clinical genotyping assay for the protein‐coding GSTZ1 variants has been developed.24 Note that this test does not consider the promoter SNPs, and so may be overlooking an important component of DCA pharmacogenetics. For the genotyping test to be most informative for predicting a personalized dose, both the promoter and coding SNPs need to be considered. Analysis of additional patients for both DCA pharmacokinetics and pharmacogenetics is warranted to be sure of the impact of GSTZ1 genotypes on DCA concentrations.
Previous trials of DCA in brain and solid cancers showed that DCA therapy was associated with disease stabilization in heavily pretreated patients.14, 15 Similarly, our evidence of response is modest, with only one patient (P002) showing a response (Figure 1A). The initial low disease burden in these patients (Table 1) made robust detection of improvement difficult. The responder had the highest sustained DCA concentrations, but two others had minor reductions in the myeloma‐related proteins at d28, suggesting a trend toward a clinical response in 50% of patients, consistent with expected response rates from in vitro cell line data. We suggest this transient response may be due to the initial high loading dose of DCA, and that the average drug concentration range when on 6.25 mg/kg b.i.d. was not high enough to be effective. Excluding P002, trough concentrations ranged from 0.03 to 0.24 mmol/L, sufficient to inhibit PDK2 (IC50 0.2 mmol/L), whereas peak concentrations ranged from 0.15 to 0.48 mmol/L. While this peak concentration may inhibit both PDK2 and PDK4 (IC50 0.5 mmol/L), it remains below the IC50 for PDK1 (1.0 mmol/L) and PDK3 (8 mmol/L), which are likely to be induced in many cancer types.5 For this reason, patient recruitment to this version of the trial protocol was stopped, and our investigations are continuing with a modified dosing regimen that aims to maintain higher concentrations of DCA as observed on d8, while avoiding the chronic side effects of DCA treatment.
These discussions are based on DCA as a single agent, however, DCA is most likely to be used in combination regimens. In our in vitro studies, we saw no indication of beneficial combination interactions between DCA and commonly used myeloma treatments (Figure 4 and Figure S3), however, DCA did not interfere with their efficacy. Where DCA was effective alone, this effect was often additive with the other agents, particularly at low doses of the other agent. We cannot exclude the possibility of pharmacokinetic interactions between DCA and these drugs, as there are no published studies on this topic. However, based on our in vitro results, DCA may be most beneficial for myeloma patients where the cytotoxicity of the other drug is low (eg, Figure 4C) or when the concentration of the cytotoxic agent is low between doses (eg, Figure 4A and B).
This study concludes that further study of DCA in myeloma is warranted in patients with a higher disease burden and in combination with other therapies. While the small number of patients does not allow firm conclusions to be drawn about associations between GSTZ1 genetics and pharmacokinetics or peripheral neuropathy, future studies should include GSTZ1 genotyping of promoter and protein‐coding polymorphisms to address these questions in additional patients. Regardless of GSTZ1 genotypes, a dosing regime based on drug concentrations and biomarkers rather than maximum tolerated dose, that can achieve higher concentrations without peripheral neuropathy is likely to be needed to achieve maximum benefit from DCA in the treatment of cancer.
ACKNOWLEDGEMENTS
We thank Celgene for providing lenalidomide for the in vitro experiments. We thank Anais Le Gall for her input into the early stages of the trial design. We thank the patients for their willingness to participate.
DISCLOSURE
None of the authors have any competing interests to declare.
AUTHORS’ CONTRIBUTIONS
Dan Dan Tian – managed the patient sample collection, performed the DCA assays (GC‐MS) for patients 001‐004, performed the in vitro experiments, analyzed all the data, drafted the manuscript. Samuel K Bennett – wrote major portion of the protocol, managed ethics submissions, and other regulatory administration. Lucy A Coupland – provided scientific background, contributed to design of protocol, statistical considerations, and patient information sheets. Kathryn Forwood – contributed to overall design, processed minor amendments, and collected patient samples and data. Yadanar Lwin – contributed to overall design, processed minor amendments, and collected patient samples and data. Niloofar Pooryousef – performed and analyzed the GC‐MS DCA assays for patients 005‐007, interpreted the pharmacogenetics. Illa Tea – developed and supervised mass spectrometry (GC‐MS) DCA data acquisition, analysis, and interpretation. Thy Truong – developed and supervised mass spectrometry (LC‐MS) DCA data acquisition, analysis, and interpretation. Teresa Neeman – supervised statistical aspects of protocol design and data analysis. Philip Crispin – contributed to design of protocol, definition of patient criteria, patient recruitment and statistical considerations. James D’Rozario – contributed to design of protocol, definition of patient criteria, patient recruitment, and statistical considerations. Anneke C Blackburn – provided scientific background, dosing, and toxicity information, designed pharmacokinetic and pharmacogenetic aspects of protocol, prepared the manuscript.
Supporting information
Tian DD, Bennett SK, Coupland LA, et al. GSTZ1 genotypes correlate with dichloroacetate pharmacokinetics and chronic side effects in multiple myeloma patients in a pilot phase 2 clinical trial. Pharmacol Res Perspect. 2019;e00526 10.1002/prp2.526
PI statement: The authors confirm that the Principal Investigator for this paper is James D’Rozario and that he had direct clinical responsibility for patients.
Funding information
This work was supported by The Canberra Hospital Private Practice Trust Fund, Cancer Council ACT Project Grant APP1103848, and the Monaro Committee for Cancer Research.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 2. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic‐targeting therapy for cancer. Br J Cancer. 2008;99:989‐994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kankotia S, Stacpoole PW. Dichloroacetate and cancer: new home for an orphan drug? Biochem Biophys Acta. 2014;1846:617‐629. [DOI] [PubMed] [Google Scholar]
- 5. Papandreou I, Goliasova T, Denko NC. Anticancer drugs that target metabolism: is dichloroacetate the new paradigm? Int J Cancer. 2011;128:1001‐1008. [DOI] [PubMed] [Google Scholar]
- 6. Bonnet S, Archer SL, Allalunis‐Turner J, et al. A mitochondria‐K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37‐51. [DOI] [PubMed] [Google Scholar]
- 7. Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat. 2010;120:253‐260. [DOI] [PubMed] [Google Scholar]
- 8. Gang BP, Dilda PJ, Hogg PJ, Blackburn AC. Targeting of two aspects of metabolism in breast cancer treatment. Cancer Biol Ther. 2014;15:1533‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen H, Decollogne S, Dilda PJ, et al. Dual‐targeting of aberrant glucose metabolism in glioblastoma. Journal of Experimental & Clinical Cancer Research : CR. 2015;34:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stockwin LH, Yu SX, Borgel S, et al. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer. 2010;127:2510‐2519. [DOI] [PubMed] [Google Scholar]
- 11. Sun RC, Board PG, Blackburn AC. Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells. Mol Cancer. 2011;10:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaufmann P, Engelstad K, Wei Y, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66:324‐330. [DOI] [PubMed] [Google Scholar]
- 13. Stacpoole PW, Kerr DS, Barnes C, et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519‐1531. [DOI] [PubMed] [Google Scholar]
- 14. Chu QS, Sangha R, Spratlin J, et al. A phase I open‐labeled, single‐arm, dose‐escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest New Drugs. 2015;33(3):603‐610. 10.1007/s10637-015-0221-y. [DOI] [PubMed] [Google Scholar]
- 15. Dunbar EM, Coats BS, Shroads AL, et al. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs. 2014;32:452‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garon EB, Christofk HR, Hosmer W, et al. Dichloroacetate should be considered with platinum‐based chemotherapy in hypoxic tumors rather than as a single agent in advanced non‐small cell lung cancer. J Cancer Res Clin Oncol. 2014;140:443‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra34. [DOI] [PubMed] [Google Scholar]
- 18. Bowker‐Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue‐specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329(Pt 1):191‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blackburn AC, Coggan M, Tzeng HF, et al. GSTZ1d: a new allele of glutathione transferase zeta and maleylacetoacetate isomerase. Pharmacogenetics. 2001;11:671‐678. [DOI] [PubMed] [Google Scholar]
- 20. Blackburn AC, Tzeng HF, Anders MW, Board PG. Discovery of a functional polymorphism in human glutathione transferase zeta by expressed sequence tag database analysis. Pharmacogenetics. 2000;10:49‐57. [DOI] [PubMed] [Google Scholar]
- 21. Shroads AL, Langaee T, Coats BS, et al. Human polymorphisms in the glutathione transferase zeta 1/maleylacetoacetate isomerase gene influence the toxicokinetics of dichloroacetate. J Clin Pharmacol. 2012;52:837‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fang YY, Kashkarov U, Anders MW, Board PG. Polymorphisms in the human glutathione transferase zeta promoter. Pharmacogenet Genomics. 2006;16:307‐313. [DOI] [PubMed] [Google Scholar]
- 23. Langaee TY, Zhong G, Li W, et al. The influence of human GSTZ1 gene haplotype variations on GSTZ1 expression. Pharmacogenet Genomics. 2015;25:239‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Langaee T, Wagner R, Horne LP, et al. Personalized dosing of dichloroacetate using GSTZ1 clinical genotyping assay. Genetic Testing and Molecular Biomarkers. 2018;22:266‐269. [DOI] [PubMed] [Google Scholar]
- 25. Anderson WB, Board PG, Gargano B, Anders MW. Inactivation of glutathione transferase zeta by dichloroacetic acid and other fluorine‐lacking alpha‐haloalkanoic acids. Chem Res Toxicol. 1999;12:1144‐1149. [DOI] [PubMed] [Google Scholar]
- 26. Tzeng HF, Blackburn AC, Board PG, Anders MW. Polymorphism‐ and species‐dependent inactivation of glutathione transferase zeta by dichloroacetate. Chem Res Toxicol. 2000;13:231‐236. [DOI] [PubMed] [Google Scholar]
- 27. Stacpoole PW, Henderson GN, Yan Z, James MO. Clinical pharmacology and toxicology of dichloroacetate. Environ Health Perspect. 1998;106(Suppl 4):989‐994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12:335‐348. [DOI] [PubMed] [Google Scholar]
- 29. Quach H, Joshua D, Ho J, et al. Treatment of patients with multiple myeloma who are eligible for stem cell transplantation: position statement of the Myeloma Foundation of Australia Medical and Scientific Advisory Group. Intern Med J. 2015;45:94‐105. [DOI] [PubMed] [Google Scholar]
- 30. Avet‐Loiseau H, Facon T, Grosbois B, et al. Oncogenesis of multiple myeloma: 14q32 and 13q chromosomal abnormalities are not randomly distributed, but correlate with natural history, immunological features, and clinical presentation. Blood. 2002;99:2185‐2191. [DOI] [PubMed] [Google Scholar]
- 31. Fonseca R, Blood E, Rue M, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101:4569‐4575. [DOI] [PubMed] [Google Scholar]
- 32. Leni Z, Parakkal G, Arcaro A. Emerging metabolic targets in the therapy of hematological malignancies. Biomed Res Int. 2013;2013:946206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fujiwara S, Kawano Y, Yuki H, et al. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br J Cancer. 2013;108:170‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sanchez WY, McGee SL, Connor T, et al. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br J Cancer. 2013;108:1624‐1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maiso P, Huynh D, Moschetta M, et al. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Can Res. 2015;75:2071‐2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zub KA, Sousa MM, Sarno A, et al. Modulation of cell metabolic pathways and oxidative stress signaling contribute to acquired melphalan resistance in multiple myeloma cells. PLoS ONE. 2015;10:e0119857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fujiwara S, Wada N, Kawano Y, et al. Lactate, a putative survival factor for myeloma cells, is incorporated by myeloma cells through monocarboxylate transporters 1. Experimental Hematology & Oncology. 2015;4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hanson DJ, Nakamura S, Amachi R, et al. Effective impairment of myeloma cells and their progenitors by blockade of monocarboxylate transportation. Oncotarget. 2015;6:33568‐33586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Landgren O, Rajkumar SV. New developments in diagnosis, prognosis, and assessment of response in multiple myeloma. Clin Cancer Res. 2016;22:5428‐5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cavaletti G, Jann S, Pace A, et al. Multi‐center assessment of the Total Neuropathy Score for chemotherapy‐induced peripheral neurotoxicity. J Peripher Nerv Syst. 2006;11:135‐141. [DOI] [PubMed] [Google Scholar]
- 41. Mannervik B, Board PG, Hayes JD, Listowsky I, Pearson WR. Nomenclature for mammalian soluble glutathione transferases. Methods Enzymol. 2005;401:1‐8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.