

Abstract
In early 2019, the 6th World Symposium on Pulmonary Hypertension (WSPH) released an updated document highlighting the advances in the last five years. During the quinquennial event many experts worked together to suggest new changes in the disease diagnosis and management. Since inception of the WSPH in 1973, this is the first time when the hemodynamic definition of pulmonary hypertension (PH) has been updated. These proceedings have re-defined the different hemodynamic types of PH that occur with the left heart disease along with introduction to the genetic testing as part of pulmonary arterial hypertension (PAH) evaluation. Objective of this review is to highlight the evaluation and diagnosis of PAH based on the proceedings of the 6th WSPH. Accurate early diagnosis and subsequent management of PH is necessary, as despite of treatment advances, survival remains suboptimal.
Keywords: Pulmonary hypertension (PH), 6th World Symposium on Pulmonary Hypertension (6th WSPH), pulmonary arterial hypertension (PAH), chronic thromboembolic pulmonary hypertension (CTEPH), pulmonary veno-occlusive disease (PVOD)
Introduction
Pulmonary hypertension (PH) is a relentless, progressive disease which often leads to premature death. 6th World symposium on pulmonary hypertension (WSPH) created 13 task forces with 124 experts from around the world to review and update the most recent scientific evidence on the evaluation, diagnosis and management of PH (1). With each update every five year, the society of the world experts work together to improve understanding of this disease, as management of this disease is still challenging and life expectancy is still suboptimal.
PH is classified into five groups. The group 1 pulmonary arterial hypertension (PAH), which comprises of diverse diseases that result in similar pathological changes within the pulmonary vasculature. This includes idiopathic, familial, drug and toxin induced PAH and associated forms of PAH like systemic sclerosis, portal hypertension, congenital heart disease and human immunodeficiency virus (HIV). The remaining 4 groups of PH are secondary to other conditions and are usually referred to as secondary PH (2). Treatment of these groups is focused towards correcting the underlying original condition. Group I PAH is the most important of all due to its aggressive nature, poor survival and limited treatment options. With efforts over the last three decades, group 1 PAH survival has improved but is still suboptimal and continues to be an unmet challenge. Registry to Evaluate Early And Long term PAH disease management study (REVEAL) showed five year survival of 57% from the time of diagnostic right heart catheterization (3).
The most controversial recommendations from these proceedings have been the proposal of a new hemodynamic definition of PH. This is the first time, since 1973 (the inception of WSPH), that the definition of PH has been changed (2). These changes are now a topic of active debate within the scientific community. This review will focus on the classification and evaluation of PH based on the 6th WSPH proceedings.
Updated clinical classification of PH
The current classification of PH categorizes clinical conditions associated with PH based on similar pathophysiology, etiologies, clinical presentation, hemodynamic characteristics and therapeutic management. The most updated clinical classification of PH in adults is presented in Table 1. 6th WSPH for the first time included PAH patients who are long term responders to calcium channel blockers (CCB) as a separate entity. Additionally pulmonary veno-occlusive disease (PVOD), pulmonary capillary hemangiomatosis (PCH) and persistent PH of the newborn syndrome is also included in this group (2). The group 2 PH is secondary to the left heart diseases like systolic or diastolic heart failure, left sided valvular diseases and others as shown in Table 1. Group 3 is due to diseases of lung parenchyma or hypoxia related. Group 4 PH is due to chronic thromboembolic pulmonary hypertension (CTEPH) and other pulmonary obstructive processes. Group 5 includes diseases with multifactorial mechanisms or unclear mechanisms.
Table 1. Updated clinical classification of pulmonary hypertension (PH) (adapted from 6th WSPH document).
| PAH |
| Idiopathic PAH |
| Heritable PAH |
| Drug- and toxin-induced PAH (Table 2) |
| PAH associated with |
| Connective tissue disease |
| HIV infection |
| Portal hypertension |
| Congenital heart disease |
| Schistosomiasis |
| PAH long-term responders to calcium channel blockers |
| PAH with overt features of venous/capillaries (PVOD/PCH) involvement |
| Persistent PH of the newborn syndrome |
| PH due to left heart disease |
| PH due to heart failure with preserved LVEF |
| PH due to heart failure with reduced LVEF |
| Valvular heart disease |
| Congenital/acquired cardiovascular conditions leading to post-capillary PH |
| PH due to lung diseases and/or hypoxia |
| Obstructive lung disease |
| Restrictive lung disease |
| Other lung disease with mixed restrictive/obstructive pattern |
| Hypoxia without lung disease |
| Developmental lung disorders |
| PH due to pulmonary artery obstructions |
| Chronic thromboembolic PH |
| Other pulmonary artery obstructions |
| Sarcoma (high or intermediate grade) or angiosarcoma |
| Other malignant tumours |
| Renal carcinoma |
| Uterine carcinoma |
| Germ cell tumours of the testis |
| Other tumours |
| Non-malignant tumours: uterine leiomyoma |
| Arteritis without connective tissue disease |
| Congenital pulmonary artery stenoses |
| Parasites |
| PH with unclear and/or multifactorial mechanisms |
| Haematological disorders: chronic haemolytic anaemia myeloproliferative disorders |
| Systemic and metabolic disorders: pulmonary Langerhans cell histiocytosis, Gaucher disease, glycogen storage disease, neurofibromatosis, sarcoidosis |
| Others: chronic renal failure with or without hemodialysis, fibrosing mediastinitis |
| Complex congenital heart disease |
| Segmental pulmonary hypertension: isolated pulmonary artery of ductal origin, absent pulmonary artery, pulmonary atresia with ventricular septal defect and major aorto-pulmonary collateral arteries, hemitruncus, other |
| Single ventricle: unoperated, operated |
| Scimitar syndrome |
PAH, pulmonary arterial hypertension; PVOD, pulmonary veno-occlusive disease; PCH, pulmonary capillary haemangiomatosis; LVEF, left ventricular ejection fraction.
The 6th WSPH proceedings stressed on the drugs and toxins associated PAH to help treating physicians to identify the culprit drug and perform specific surveillance during evaluation (Table 2). There is clear evidence to suggest that amphetamines/methamphetamines and dasatinib are definitely associated with development of PAH (4,5). Dasatinib is a drug approved for treatment of leukemia and is a type of tyrosine kinase inhibitor. One of the series from France found PAH incidence of 0.45% (5). Dasatinib related PAH generally improves upon cessation of exposure of the drug but in roughly one third of patients it may persist (5,6). In this update, long term responders to CCB were added as a separate subgroup of group1 PAH. Although remodeling of the small pulmonary vasculature is the commonest pathological finding in PAH, pulmonary vasoconstriction also plays an important role in PAH pathophysiology. This is a hallmark of patients with positive vasoreactivity. Acute vasodilator response was observed in 12.5% of a large series of 557 PAH patients, 6.8% of these patients had a long-term clinical and hemodynamic response to CCBs (7). Acute vasodilator response is defined as a drop of more than 10 mmHg in mPAP to go below mPAP ≤40 mmHg associated with an increased or unchanged CO. Many agents have been used for vasoreactivity testing with inhaled nitric oxide at 10–20 ppm as the preferred agent but intravenous epoprostenol, adenosine or inhaled iloprost can be used as well (7,8).
Table 2. Classification of drugs and toxins associated with PAH (adapted from the 6th WSPH).
| Definite | Possible |
|---|---|
| Aminorex | Cocaine |
| Fenfluramine | Phenylpropanolamine |
| Dexfenfluramine | L-tryptophan |
| Benfluorex | St John’s wort |
| Methamphetamines | Amphetamines |
| Dasatinib | Interferon-α and -β |
| Toxic rapeseed oil | Alkylating agents |
| Bosutinib | |
| Direct-acting antiviral agents against hepatitis C virus leflunomide | |
| Indirubin (Chinese herb Qing-Dai) |
PAH, pulmonary arterial hypertension.
Hemodynamic definition of PH
Since 1973, PH has been defined as a mean pulmonary artery (mPAP) of ≥25 mmHg, however, the 6th WSPH, task force recommended that this definition should be changed to mPAP >20 mmHg (2). The original definition of mPAP of ≥25 mmHg was chosen somewhat arbitrarily and does not represent the upper limit of normal mPAP in the general population. All prior hemodynamic studies in healthy individuals have found that normal mPAP is approximately 14±3.3 mmHg and the upper limit (>97.5th percentile) of normal is 20 mmHg (9). This new definition has scientific merit but appears to have less practical value. Major concerns regarding inclusion of individuals with mPAP between 21 and 24 mmHg is the risk of PH diagnosis in these otherwise healthy individuals as very few people are symptomatic at this pressure. Baseline characteristics of REVEAL registry showed mPAP of 50 mmHg at the time of diagnosis (10). This pressure range is way above than the currently accepted range of 25 mmHg. This highlights that patients become clinically symptomatic at a much higher pressure range. Thus, it will be difficult to identify patients at range of 21–24 mmHg. Additionally, there is data lacking at present about the treatment of this population. Although, few recent studies suggest that individuals with mPAP 21–24 tend to progress to “overt PH (mPAP ≥25 mmHg)” more often than patients with mPAP ≤20 mmHg over a 2–3 year follow up (11,12) and these individuals have a high morbidity and mortality over this same time period (13). Based on these data, the 6th WSPH task force recommended that a new mPAP >20 mmHg cut-off for diagnosing PH is both clinically warranted and in the best interest of the patient.
Another major hemodynamic change recommended by the 6th WSPH update is the inclusion of pulmonary vascular resistance (PVR) in all forms of PH. All PH (mPAP >20 mmHg) will be further subclassified as pre-capillary PH (PAH), isolated post-capillary PH (IpcPH) or combined pre- and post-capillary PH (CpcPH) based on pulmonary arterial wedge pressure (PAWP) and PVR. While the threshold and application of significant PAWP has not changed (≤15 mmHg in PAH, >15 mmHg in all post capillary PH), PVR now defines the presence or absence of pre-capillary PH (PVR <3 WU = IpcPH, PVR ≥3 WU in PAH and CpcPH) (2) (Table 3). However, the method of detecting CpcPH remains controversial and that while PVR ≥3 WU has strong evidence to support its diagnostic utility, other hemodynamic markers such as the trans pulmonary gradient (TPG) and pulmonary arterial compliance (PAC) have demonstrated value in two meta-analyses for clarifying the diagnosis in at risk individuals (14,15).
Table 3. Updated hemodynamic definition based on the 6th WSPH, adapted from the original WSPH document (with prior permission).
| Definition | Characteristics | Clinical groups (WHO group) |
|---|---|---|
| Pre-capillary PH | mPAP >20 mmHg, PAWP ≤15 mmHg, PVR ≥3 WU | 1, 3, 4, 5 |
| Isolated post-capillary PH (IpcPH) | mPAP >20 mmHg, PAWP >15 mmHg, PVR <3 WU | 2 and 5 |
| Combined pre- and post-capillary PH (CpcPH) | mPAP >20 mmHg, PAWP >15 mmHg, PVR ≥3 WU | 2 and 5 |
Group 1: PAH; group 2: PH due to left heart disease; group 3: PH due to lung diseases and/or hypoxia; group 4: PH due to pulmonary artery obstructions; group 5: PH with unclear and/or multifactorial mechanisms. mPAP, mean pulmonary arterial pressure; PAWP, pulmonary arterial wedge pressure; PVR, pulmonary vascular resistance; WU, Wood units.
Evaluation of PH
Despite the advances in our understanding of symptomatology, progression and management, there has not been much progress in the early diagnosis of the disease in the last two decades. All guidelines and proceeding documents have a common message of early referral to a PAH expert center but this is yet an unsuccessful goal to achieve. Figure 1 gives an overview of the algorithm which should be used in evaluating patients with suspected PAH. REVEAL registry showed in roughly 21% of patients there was delay of over two years from the symptom onset and the correct diagnosis of PAH (16). A thorough history and examination may give clues for early diagnosis of the disease.
Figure 1.

Algorithm highlighting PH evaluation at a community setting (Tables 4,5). PH, pulmonary hypertension.
Symptoms of PH
There should be a high index of suspicion to diagnose PH. Appropriate screening tests should be performed in patients with suspicion of PH. Symptoms of PH are nonspecific. REVEAL registry data showed dyspnea on exertion to be the most common symptom of PH, seen in over 85% of patients (16). Other common symptoms are chest discomfort, palpitations, leg edema and fatigue. Syncope/presyncope is an important symptom in PAH and indicates severe form of the disease. In REVEAL registry syncope/presyncope was seen in roughly 16% of patients diagnosed within two years of symptom onset and 18% of those diagnosed after two years of symptom onset (16). Presence of edema, ascites, abdominal distension indicates advanced stage or a rapidly progressing disease leading to right-sided heart failure. Rarely, hemoptysis or hoarseness due to unilateral vocal cord paralysis (Ortner’s syndrome) by compression of the left recurrent laryngeal nerve can occur. Wheezing may be caused by large airway compression and angina due to myocardial ischemia caused by compression of the left main coronary artery by enlarged pulmonary artery (17). Dissection and rupture of PA can occur rarely if it is massively dilated or has aneurysmal dilatation and may present as cardiac tamponade.
Physical examination findings
The physical signs of PH include right ventricular heave, an accentuated pulmonary component of the second heart sound, an RV third heart sound, a pansystolic murmur of tricuspid regurgitation and a diastolic murmur of pulmonary regurgitation. Elevated jugular venous pressure, hepatomegaly, ascites, peripheral edema and cool extremities characterize right heart failure. Pulmonary exam is generally devoid of crackles or wheezing (18). In patients with scleroderma or other connective tissue disease, findings of telengectasia, digital ulceration and sclerodactyly may be seen. Presence of inspiratory crackles in these patients may point towards interstitial lung disease. Patients with liver disease may show spider naevi, testicular atrophy and palmer erythema. Digital clubbing is an important finding most frequently encountered in patients with PVOD, cyanotic congenital heart disease, interstitial lung disease or liver disease.
Diagnostic testing
Electrocardiography (EKG)
Suspected patients with PH must undergo a comprehensive evaluation. EKG can provide an early clue to the presence of PH. An abnormal EKG may indicate more severe disease than mild (19,20). EKG may show changes suggestive of right atrial or ventricular dilatation like, right bundle branch block (RBB), right axis deviation (RAD), P pulmonale, RV hypertrophy, RV strain, and QTc prolongation. RV strain is more sensitive finding in comparison to RV hypertrophy.
Hematological testing
Hematological testing does not give a direct diagnostic clue but it is an important testing to explain end-organ compromise. It is also helpful for finding the etiology of PH especially in patients with suspected auto-immue process or connective tissue disease. Routine testing with thyroid function, biochemistry and hematology is essential for evaluation. Evaluation for liver function is essential as liver function abnormalities may be present due to hepatic congestion due to right heart failure, primary liver disease (in cases of porto-PH) or as a result of liver injury due to use of endothelin receptor antagonist. Thyroid disease is common in PAH and may mimic clinical picture of right heart failure. Elevated brain natriuretic peptide (BNP) and N-terminal pro-BNP (NT-proBNP) indicates poor response to treatment or worsening disease. Persistently elevated BNP or NT-proBNP are indicators of worse outcome. All patients must be checked for HIV, hepatitis and CTD. ANA testing should be done routinely and ANA immunofluorescence testing is recommended against ELISA technique and anything more than 1:160 titer should be considered as positive. If there is high index of suspicion, disease specific CTD testing should be done, for example, if scleroderma is suspected, patients should be checked for anticentromere, antitopoisomerase, anti-RNA polymerase III. Hypercoagulable testing like thrombophilia panel including anticardiolipin antibodies, lupus anticoagulant and anti-β2-glycoprotein antibodies should be done in patients with CTEPH.
Pulmonary function tests
A decrease in the diffusion capacity of the lung for carbon monoxide (DLCO) is the earliest change seen in pulmonary function tests in patients with pulmonary vascular diseases. All patients with PAH must undergo routine spirometry measurements with forced expiratory volume, forced vital capacity in addition to total lung capacity measurement. Most patients with PAH may show a mild restrictive defect. In very early disease, diffusion capacity can be normal in PAH. An abnormally lower DLCO, defined as <45% of predicted, is associated with a poor outcome (21,22). Suspect PVOD, scleroderma or concomitant ILD in the setting of marked reduction of DLCO. Patients with PH in the setting of combined pulmonary fibrosis and emphysema (CPFE), marked reduction in DLCO can be seen in association with relatively preserved lung volumes. ILD patients may show reduction of forced vital capacity (FVC) or total lung capacity (TLC) (23). Overnight oximetry or sleep study must be done in patients with suspected sleep disordered breathing. Cardiopulmonary exercise testing (CPET) can also help in evaluating patients with exercise limitation. It can help in pointing towards the etiology of dyspnea on exertion.
Echocardiography
Echocardiography is an excellent screening tool for PH. If echocardiography suggests evidence of PH, it must be confirmed with right heart catheterization (RHC) (18). The pulmonary artery systolic pressure (PASP) is calculated based on the tricuspid regurgitation velocity (TRV). It is calculated based on the simplified Bernoulli equation taking into account right atrial pressure (RAP). Respiratory variation in diameter of the inferior vena cava (IVC) helps in estimating the RAP. An IVC diameter <21 mm with >50% collapsibility with respiration suggests a normal RAP i.e., between 0–5 mmHg, whereas an IVC diameter >21 mm with <50% collapsibility indicates a higher RAP between 10–20 mmHg. In scenarios in which the IVC diameter and collapse do not fit this model, an intermediate value of 8 mmHg may be used (18,24,25). Considering the inaccuracies in calculating RAP, the 6th WSPH recommends (17) using tricuspid regurgitant jet velocity (TRV) to assess the presence/absence or grade the severity of PH (Table 4) in the presence or absence of other pre-specified echocardiographic variables to suggest PH. Table 5 summarizes several additional criteria to indicate probability of PH based on the RV, PA, RA and IVC imaging findings. A contrast echocardiography must be done if obtained images are of poor quality and peak TRV is difficult to estimate. Severe TRV leads to underestimation of tricuspid regurgitant jet and cannot exclude PH.
Table 4. Echocardiographic probability of pulmonary hypertension (PH) in symptomatic patients with a suspicion of PH, adapted from the original WSPH document (with prior permission).
| Peak tricuspid regurgitation velocity, m/s | Presence of other echocardiographic “PH signs” | Echocardiographic probability of PH |
|---|---|---|
| ≤2.8 or not measurable | No | Low |
| >2.8 or not measurable, 2.9–3.4 | Yes, no | Intermediate |
| 2.9–3.4, >3.4 | Yes, not required | High |
Table 5. Chocardiographic signs suggesting pulmonary hypertension (PH), adapted from the original WSPH document (with prior permission).
| (A) The ventricles | (B) Pulmonary artery | (C) Inferior vena cava and right atrium |
|---|---|---|
| Right ventricle/left ventricle basal diameter ratio >1.0 | Right ventricular outflow Doppler acceleration time <105 ms and/or mid-systolic notching | Inferior cava diameter >21 mm with decreased inspiratory collapse (<50% with a sniff or <20% with quiet inspiration) |
| Flattening of the interventricular septum (left ventricular eccentricity index >1.1 in systole and/or diastole) | Early diastolic pulmonary regurgitation velocity >2.2 m·s–1 | Right atrial area (end-systole) >18 cm2 |
| Pulmonary artery diameter >25 mm |
Echocardiographic signs from at least two different categories (A/B/C) from the list should be present to alter the level of echocardiographic probability of PH.
Ventilation-perfusion scan of the lungs
All patients undergoing evaluation for PH must undergo a ventilation/perfusion (V/Q) lung scan testing to rule out CTEPH. It is essential to correctly diagnose CTEPH as this is one of those forms of PH where a potentially curative surgical treatment option is available. Multiple studies have confirmed the superiority of the V/Q scan as being more sensitive and specific to diagnose CTEPH in comparison to CT pulmonary angiogram (CTPA) (26). A V/Q scan has a sensitivity of 90–100% and a specificity of 94–100% to diagnose CTEPH (26). Newer techniques such as three-dimensional magnetic resonance (MR) perfusion mapping have been shown to be as sensitive as traditional perfusion scintigraphy in screening for CTEPH. MR is a radiation-free modality to assess both ventilation and perfusion in CTEPH (27).
Computed tomography: high resolution, contrast-enhanced and pulmonary angiography
Computed tomography (CT) of the chest is an important investigation in the evaluation of PH. It is widely available, easily done and reproducible. It provides important information about lung parenchyma, vascular, cardiac and mediastinal abnormalities. Enlargement of PA or RV or RA on CT imaging may suggest PH. Concomitant presence of lung parenchymal disease may indicate PH due to lung disease. Experienced centers may use a CT angiography for evaluation of CTEPH as well. Presence of esophageal dilation on CT may suggest SSc, congenital cardiac defects such as anomalous pulmonary venous drainage can be detected on contrast CTA and may provide prognostic information (28).
Incidentally detected enlarged PA diameter of more than ≥29 mm or an increased ratio of PA diameter to aortic diameter of more than 1.0 may indicate presence of PH. Presence of increased ratio of segmental artery to bronchus >1:1 in three or four lobes is associated with a high specificity for PH (29,30). High-resolution CT is standard of care to diagnose ILD and emphysema. It may additionally provide information regarding PVOD with the presence of interstitial edema with diffuse central ground-glass opacity and thickened interlobular septa with lymphadenopathy (31). CT angiography of the PA is a very helpful tool in evaluation of PH when CTEPH is suspected. It can help in identifying the lesions which are surgically resect-able with pulmonary endarterectomy (PEA) or benefit from BPA (32,33). Commonly identified lesions are webs, complete obstruction, bands and intimal irregularities (Figure 2). At an experienced center, a CTA can identify these lesions as accurately and reliably as digital subtraction angiography (34,35). Conventional pulmonary angiography is still the standard of care and is mandatory in the evaluation of CTEPH to identify the surgical candidates.
Figure 2.
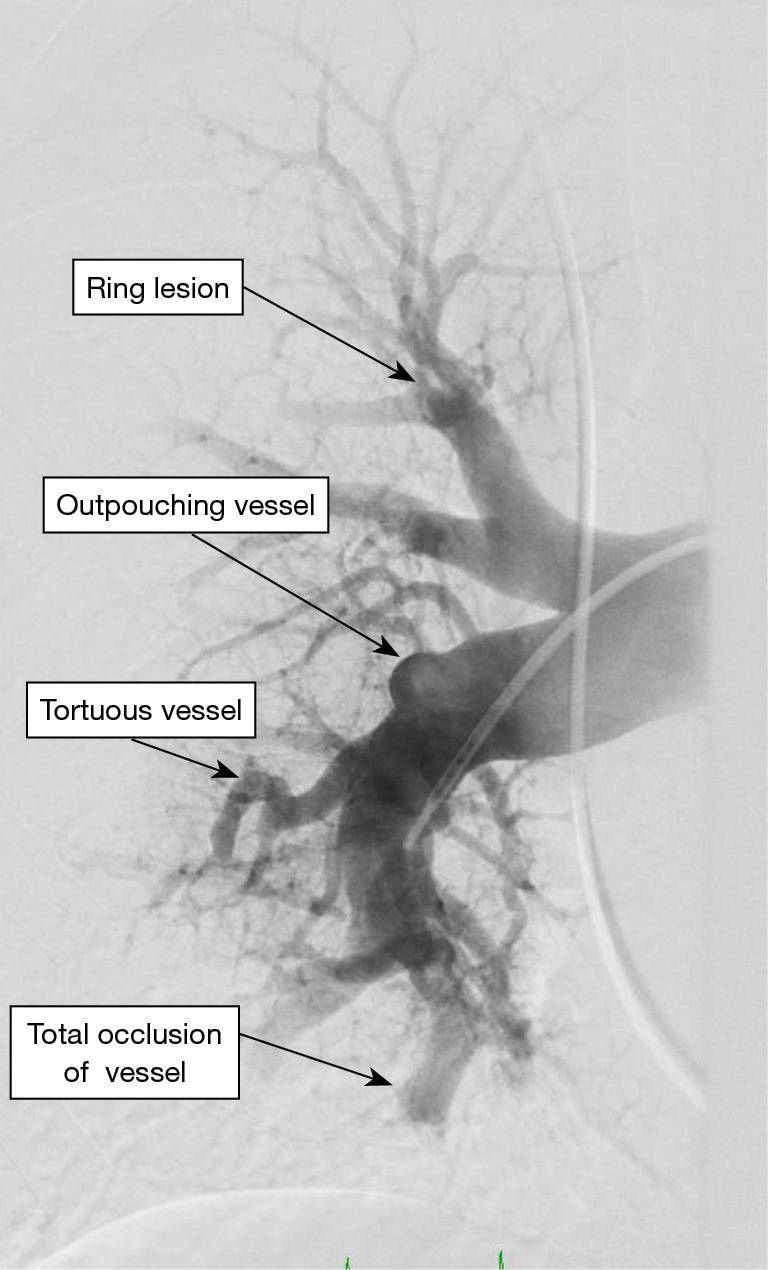
Pulmonary angiogram of a patient with CTEPH showing classical lesions seen in CTEPH. CTEPH, chronic thromboembolic pulmonary hypertension.
Right heart catheterization
RHC is an essential testing needed to confirm the diagnosis of PH. In an experienced center, RHC has low morbidity (1.1%) and mortality of (0.055%) (36). RHC should be done at an experienced center as it requires meticulous attention to details in obtaining the correct hemodynamic information with minimal risk for complications. During a RHC, all pressure measurements are done in end expiration during normal tidal respiration, without breath holding. Pressure measurements should be done in the pulmonary artery (PA), PA wedge position, right ventricle (RV) and right atrium (RA). Oxygen saturation should be done at the minimum from superior vena cava (SVC), RA and PA. If PA saturation is more than 75% then oxygen saturation should be done in each cardiac chamber to evaluate for left to right shunting (18). Thermodilution or the direct Fick methods are used to measure cardiac output. Thermodilution cardiac output is measured in set of three measurements and is considered reliable even in low flow states or severe TR (37). Thermodilution cardiac output may be less reliable in patients with intracardiac shunting due to early recirculation of the injectate volume. Direct Fick method is preferred method but it is not widely available, indirect Fick is acceptable but not a reliable method. Hemodynamic definition and vasoreactivity testing is discussed earlier in this review. Other derived parameters which must be obtained in a RHC for PH evaluation are PVR, transpulmonary pressure and diastolic pressure gradient (DPG). A PVR >3 WU is essential to establish the diagnosis of PAH (2). The DPG is defined as difference between the mean PAWP and diastolic PAP and is not much affected by the filling pressures or flow variations (38) but may not have prognostic value (39). Studies have shown validity of DPG in patients with suspected PH due to left heart disease (40).
Genetic testing
BMPR2 mutations remain the most common genetic cause of PAH, accounting for ~80% of hereditary (HPAH) and ~20% idiopathic PAH (IPAH) (41). Besides BMPR2, other TGF-β superfamily genes including ALK1/ACVRL1 (a heterodimeric partner of BMPR2), BMP9 (a BMPR2 ligand), ENG (a coreceptor for BMPR2 signaling), and SMAD1, 4, and 9 (downstream BMP signaling molecules) and have been linked to both HPAH and IPAH (42). In the last five years whole exome sequencing (WES) in BMPR2 negative HPAH patients led to the discovery of two other genes: CAV1 (involved in BMPR2 membrane localization and signaling) (43) and KCNK3 (a potassium channel that regulates resting membrane potential) (44). In pediatric population, WES recently identified TBX4, a gene linked to small patella syndrome (45) in patients with HPAH.
EIF2AK4 (a kinase involved in amino acid metabolism) is associated with both PVOD and PCH (46,47). In contrast to BMPR2, EIF2AK4 mutations are autosomal recessive and completely penetrant. Presence of EIF2AK4 in a PAH patient can establish the diagnosis of PVOD/PCH in without necessitating lung biopsy (47).
Genetic evaluation of PAH should be done under the guidance of a genetic counselor. Inheritance of these mutations is complex and it adds nuance to the difficult task of genetic counseling to patients with a family or personal history of HPAH looking to conceive. This comes with immense psychological concerns of genetic screening for a disease with no prevention or cure. The genetic screening offers potential benefit with early detection of family members and earlier initiation of therapy when indicated. The 6th WSPH Task Force recommended that genetic screening should be done under the guidance of a genetic counselor or clinical geneticist (48). At this point, a pedigree can be generated to identify relatives at risk, though gene testing or screening should be initiated with affected patients. It should be done be done at experienced centers by the trained and experienced geneticists.
Advanced imaging techniques
Recently many advances have been made in imaging techniques with the goal of early and accurate detection of the disease. VQ single photon emission CT (SPECT) (49-51), dual energy CT (52), three dimensional dynamic contrast enhanced magnetic resonance for assessment of lung perfusion (27,53), functional magnetic resonance imaging for ventilation (54), cardiac magnetic resonance imaging for RV function and strain have been suggested to aid in the diagnosis or prognosis in PH and CTEPH in smaller studies (55-58) but their role in routine clinical practice is yet to be defined.
Conclusions
PH is a complex disease with suboptimal survival. Its diagnosis, assessment and management warrants a comprehensive, detail oriented approach at an expert center. Almost all guidelines, consensus statements and proceeding documents have strongly recommended early referral to an expert center to have a meaningful improvement in the outcome of the patients living with this disease.
Acknowledgments
None.
Ethical Statement: The author is accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Conflicts of Interest: The author serves on the speaker panel for Actelion pharmaceuticals, Bayer and United therapeutics; has received honoraria from these entities for consultancy and speaking engagements. Member of a data safety and monitoring board for an industry sponsored FDA approved study. The author is PI or Sub I on multiple clinical trials sponsored by Actelion and United therapeutics.
References
- 1.Galiè N, McLaughlin VV, Rubin LJ, et al. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur Respir J 2019;53:1802148. 10.1183/13993003.02148-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019;53:1801913. 10.1183/13993003.01913-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benza RL, Miller DP, Barst RJ, et al. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL registry. Chest 2012;142:448-56. 10.1378/chest.11-1460 [DOI] [PubMed] [Google Scholar]
- 4.Zamanian RT, Hedlin H, Greuenwald P, et al. Features and outcomes of methamphetamine-associated pulmonary arterial hypertension. Am J Respir Crit Care Med 2018;197:788-800. 10.1164/rccm.201705-0943OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montani D, Bergot E, Günther S, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 2012;125:2128-37. 10.1161/CIRCULATIONAHA.111.079921 [DOI] [PubMed] [Google Scholar]
- 6.Weatherald J, Chaumais MC, Savale L, et al. Long-term outcomes of dasatinib-induced pulmonary arterial hypertension: a population-based study. Eur Respir J 2017;50:1700217. 10.1183/13993003.00217-2017 [DOI] [PubMed] [Google Scholar]
- 7.Sitbon O, Humbert M, Jaïs X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005;111:3105-11. 10.1161/CIRCULATIONAHA.104.488486 [DOI] [PubMed] [Google Scholar]
- 8.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992;327:76-81. 10.1056/NEJM199207093270203 [DOI] [PubMed] [Google Scholar]
- 9.Kovacs G, Berghold A, Scheidl S, Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J 2009;34:888-94. 10.1183/09031936.00145608 [DOI] [PubMed] [Google Scholar]
- 10.Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest 2010;137:376-87. 10.1378/chest.09-1140 [DOI] [PubMed] [Google Scholar]
- 11.Valerio CJ, Schreiber BE, Handler CE, Borderline mean pulmonary artery pressure in patients with systemic sclerosis: transpulmonary gradient predicts risk of developing pulmonary hypertension. Arthritis Rheum 2013;65:1074-84. 10.1002/art.37838 [DOI] [PubMed] [Google Scholar]
- 12.Coghlan JG, Wolf M, Distler O, Incidence of pulmonary hypertension and determining factors in patients with systemic sclerosis. Eur Respir J 2018;51. doi: . 10.1183/13993003.01197-2017 [DOI] [PubMed] [Google Scholar]
- 13.Douschan P, Kovacs G, Avian A, Mild Elevation of Pulmonary Arterial Pressure as a Predictor of Mortality. Am J Respir Crit Care Med 2018;197:509-16. 10.1164/rccm.201706-1215OC [DOI] [PubMed] [Google Scholar]
- 14.Vanderpool RR, Saul M, Nouraie M, Association Between Hemodynamic Markers of Pulmonary Hypertension and Outcomes in Heart Failure With Preserved Ejection Fraction JAMA Cardiol 2018;3:298-306. 10.1001/jamacardio.2018.0128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caravita S, Dewachter C, Soranna D, Haemodynamics to predict outcome in pulmonary hypertension due to left heart disease: a meta-analysis. Eur Respir J 2018;51. doi: . 10.1183/13993003.02427-2017 [DOI] [PubMed] [Google Scholar]
- 16.Brown LM, Chen H, Halpern S, et al. Delay in recognition of pulmonary arterial hypertension: factors identified from the REVEAL Registry. Chest 2011;140:19-26. 10.1378/chest.10-1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frost A, Badesch D, Gibbs JSR, et al. Diagnosis of pulmonary hypertension. Eur Respir J 2019;53:1801904. 10.1183/13993003.01904-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2015;46:903-75. 10.1183/13993003.01032-2015 [DOI] [PubMed] [Google Scholar]
- 19.Bossone E, Paciocco G, Iarussi D, et al. The prognostic role of the ECG in primary pulmonary hypertension. Chest 2002;121:513-8. 10.1378/chest.121.2.513 [DOI] [PubMed] [Google Scholar]
- 20.Henkens IR, Gan CT, van Wolferen SA, et al. ECG monitoring of treatment response in pulmonary arterial hypertension patients. Chest 2008;134:1250-7. 10.1378/chest.08-0461 [DOI] [PubMed] [Google Scholar]
- 21.Trip P, Nossent EJ, de Man FS, et al. Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: patient characteristics and treatment responses. Eur Respir J 2013;42:1575-85. 10.1183/09031936.00184412 [DOI] [PubMed] [Google Scholar]
- 22.Sun XG, Hansen JE, Oudiz RJ, et al. Pulmonary function in primary pulmonary hypertension. J Am Coll Cardiol 2003;41:1028-35. 10.1016/S0735-1097(02)02964-9 [DOI] [PubMed] [Google Scholar]
- 23.Nathan SD, Barbera JA, Gaine SP, et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J 2019;53:1801914. 10.1183/13993003.01914-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rudski LG, Lai WW, Afilalo J, et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr 2010;23:685-713. 10.1016/j.echo.2010.05.010 [DOI] [PubMed] [Google Scholar]
- 25.Lang RM, Badano LP, Mor-Avi V, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging 2015;16:233-70. 10.1093/ehjci/jev014 [DOI] [PubMed] [Google Scholar]
- 26.Tunariu N, Gibbs SJR, Win Z, et al. Ventilation-perfusion scintigraphy Is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. J Nucl Med 2007;48:680-4. 10.2967/jnumed.106.039438 [DOI] [PubMed] [Google Scholar]
- 27.Rajaram S, Swift AJ, Telfer A, et al. 3D contrast-enhanced lung perfusion MRI is an effective screening tool for chronic thromboembolic pulmonary hypertension: results from the ASPIRE Registry. Thorax 2013;68:677-8. 10.1136/thoraxjnl-2012-203020 [DOI] [PubMed] [Google Scholar]
- 28.Rajaram S, Swift AJ, Condliffe R, et al. CT features of pulmonary arterial hypertension and its major subtypes: a systematic CT evaluation of 292 patients from the ASPIRE Registry. Thorax 2015;70:382-7. 10.1136/thoraxjnl-2014-206088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen Y, Wan C, Tian P, et al. CT-base pulmonary artery measurement in the detection of pulmonary hypertension: a meta-analysis and systematic review. Medicine (Baltimore) 2014;93:e256. 10.1097/MD.0000000000000256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan RT, Kuzo R, Goodman LR, et al. Utility of CT scan evaluation for predicting pulmonary hypertension in patients with parenchymal lung disease. Chest 1998;113:1250-6. 10.1378/chest.113.5.1250 [DOI] [PubMed] [Google Scholar]
- 31.Resten A, Maitre S, Humbert M, et al. Pulmonary hypertension: CT of the chest in pulmonary venoocclusive disease. AJR Am J Roentgenol 2004;183:65-70. 10.2214/ajr.183.1.1830065 [DOI] [PubMed] [Google Scholar]
- 32.Fedullo PF, Auger WR, Kerr KM, et al. Chronic thromboembolic pulmonary hypertension. N Engl J Med 2001;345:1465-72. 10.1056/NEJMra010902 [DOI] [PubMed] [Google Scholar]
- 33.Fukui S, Ogo T, Morita Y, et al. Right ventricular reverse remodelling after balloon pulmonary angioplasty. Eur Respir J 2014;43:1394-402. 10.1183/09031936.00012914 [DOI] [PubMed] [Google Scholar]
- 34.Dartevelle P, Fadel E, Mussot S, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J 2004;23:637-48. 10.1183/09031936.04.00079704 [DOI] [PubMed] [Google Scholar]
- 35.Reichelt A, Hoeper MM, Galanski M, et al. Chronic thromboembolic pulmonary hypertension: evaluation with 64-detector row CT versus digital substraction angiography. Eur J Radiol 2009;71:49-54. 10.1016/j.ejrad.2008.03.016 [DOI] [PubMed] [Google Scholar]
- 36.Hoeper MM, Lee SH, Voswinckel R, et al. Complications of right heart catheterization procedures in patients with pulmonary hypertension in experienced centers. J Am Coll Cardiol 2006;48:2546-52. 10.1016/j.jacc.2006.07.061 [DOI] [PubMed] [Google Scholar]
- 37.Hoeper MM, Maier R, Tongers J, et al. Determination of cardiac output by the Fick method, thermodilution, and acetylene rebreathing in pulmonary hypertension. Am J Respir Crit Care Med 1999;160:535-41. 10.1164/ajrccm.160.2.9811062 [DOI] [PubMed] [Google Scholar]
- 38.Naeije R, Vachiery JL, Yerly P, et al. The transpulmonary pressure gradient for the diagnosis of pulmonary vascular disease. Eur Respir J 2013;41:217-23. 10.1183/09031936.00074312 [DOI] [PubMed] [Google Scholar]
- 39.Tedford RJ, Beaty CA, Mathai SC, et al. Prognostic value of the pre-transplant diastolic pulmonary artery pressure-to-pulmonary capillary wedge pressure gradient in cardiac transplant recipients with pulmonary hypertension. J Heart Lung Transplant 2014;33:289-97. 10.1016/j.healun.2013.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vachiéry JL, Adir Y, Barbera JA, et al. Pulmonary hypertension due to left heart disease. J Am Coll Cardiol 2013;62:D100-8. 10.1016/j.jacc.2013.10.033 [DOI] [PubMed] [Google Scholar]
- 41.Tielemans B, Delcroix M, Belge C, TGFbeta and BMPRII signalling pathways in the pathogenesis of pulmonary arterial hypertension. Drug Discov Today 2019;24:703-16. 10.1016/j.drudis.2018.12.001 [DOI] [PubMed] [Google Scholar]
- 42.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res 2014;115:189-202. 10.1161/CIRCRESAHA.115.303404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Austin ED, Ma L, LeDuc C, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 2012;5:336-43. 10.1161/CIRCGENETICS.111.961888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma L, Roman-Campos D, Austin ED, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 2013;369:351-61. 10.1056/NEJMoa1211097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu N, Gonzaga-Jauregui C, Welch CL, et al. Exome Sequencing in Children With Pulmonary Arterial Hypertension Demonstrates Differences Compared With Adults. Circ Genom Precis Med 2018;11:e001887. 10.1161/CIRCGEN.117.001887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Best DH, Sumner KL, Smith BP, et al. EIF2AK4 Mutations in Patients Diagnosed With Pulmonary Arterial Hypertension. Chest 2017;151:821-8. 10.1016/j.chest.2016.11.014 [DOI] [PubMed] [Google Scholar]
- 47.Eyries M, Montani D, Girerd B, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet 2014;46:65-9. 10.1038/ng.2844 [DOI] [PubMed] [Google Scholar]
- 48.Morrell NW, Aldred MA, Chung WK, et al. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J 2019;53. doi: . 10.1183/13993003.01899-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leblanc M, Leveillée F, Turcotte E. Prospective evaluation of the negative predictive value of V/Q SPECT using 99mTc-Technegas. Nucl Med Commun 2007;28:667-72. 10.1097/MNM.0b013e32827a8e99 [DOI] [PubMed] [Google Scholar]
- 50.Grüning T, Drake BE, Farrell SL, et al. Three-year clinical experience with VQ SPECT for diagnosing pulmonary embolism: diagnostic performance. Clin Imaging 2014;38:831-5. 10.1016/j.clinimag.2014.04.003 [DOI] [PubMed] [Google Scholar]
- 51.Gutte H, Mortensen J, Jensen CV, et al. Detection of pulmonary embolism with combined ventilation-perfusion SPECT and low-dose CT: head-to-head comparison with multidetector CT angiography. J Nucl Med 2009;50:1987-92. 10.2967/jnumed.108.061606 [DOI] [PubMed] [Google Scholar]
- 52.Giordano J, Khung S, Duhamel A, et al. Lung perfusion characteristics in pulmonary arterial hypertension (PAH) and peripheral forms of chronic thromboembolic pulmonary hypertension (pCTEPH): dual-energy CT experience in 31 patients. Eur Radiol 2017;27:1631-9. 10.1007/s00330-016-4500-6 [DOI] [PubMed] [Google Scholar]
- 53.Johns CS, Swift AJ, Rajaram S, et al. Lung perfusion: MRI vs. SPECT for screening in suspected chronic thromboembolic pulmonary hypertension. J Magn Reson Imaging 2017;46:1693-7. 10.1002/jmri.25714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakagawa T, Sakuma H, Murashima S, et al. Pulmonary ventilation-perfusion MR imaging in clinical patients. J Magn Reson Imaging 2001;14:419-24. 10.1002/jmri.1202 [DOI] [PubMed] [Google Scholar]
- 55.Freed BH, Gomberg-Maitland M, Chandra S, et al. Late gadolinium enhancement cardiovascular magnetic resonance predicts clinical worsening in patients with pulmonary hypertension. J Cardiovasc Magn Reson 2012;14:11. 10.1186/1532-429X-14-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.El Abouelnour A, Doyle M, Thompson DV, et al. Does late gadolinium enhancement still have value? Right ventricular internal mechanical work, Ea/Emax and late gadolinium enhancement as prognostic markers in patients with advanced pulmonary hypertension via cardiac MRI. Cardiol Res Cardiovasc Med 2017;2017:CRCM-111. [PMC free article] [PubMed]
- 57.Mehta BB, Auger DA, Gonzalez JA, et al. Detection of elevated right ventricular extracellular volume in pulmonary hypertension using Accelerated and Navigator-Gated Look-Locker Imaging for Cardiac T1 Estimation (ANGIE) cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2015;17:110. 10.1186/s12968-015-0209-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Siqueira ME, Pozo E, Fernandes VR, et al. Characterization and clinical significance of right ventricular mechanics in pulmonary hypertension evaluated with cardiovascular magnetic resonance feature tracking. J Cardiovasc Magn Reson 2016;18:39. 10.1186/s12968-016-0258-x [DOI] [PMC free article] [PubMed] [Google Scholar]