

Abstract
Pulmonary arterial hypertension (PAH) is a condition associated with substantial morbidity and mortality. Over the last 25 years there has been a significant evolution in the therapies to treat PAH. These therapies are effective for patients with group I PAH and group IV PH [chronic thromboembolic pulmonary hypertension (CTEPH)]. PAH is characterized by an imbalance of nitric oxide, prostacyclin and endothelin levels, and current pharmacotherapy involves these three pathways. Earlier clinical trials involving PAH-specific therapies evaluated improvements in 6-minute walk time as a primary improvement whereas contemporary trials have been larger and focused on morbidity and mortality reductions. While there may be a role for monotherapy in disease management, most patients should be considered for dual or triple therapy.
Keywords: Pulmonary arterial hypertension (PAH), nitric oxide, endothelin, prostacyclin, sildenafil, tadalafil, riociguat, bosentan, ambrisentan, macitentan, epoprostenol, treprostinil, selexipag
Introduction
Pulmonary arterial hypertension (PAH) continues to be a condition associated with a high morbidity and mortality despite advances in treatment strategies over the past 25 years. The 6th World Symposium on Pulmonary Hypertension defines pulmonary hypertension as a mean pulmonary arterial pressure >20 mmHg (1). Currently approved therapies for PAH target three primary pathways that are key in the pathogenesis of the disease: the nitric oxide, prostacyclin and endothelin pathways (see Figure 1). The imbalance of vasoactive mediators plays a primary role in the development and progression in the proliferative pathological changes in PAH. PAH-specific therapies have been available since the mid-1990s. Further refinement in appropriate utilization of these medications as well as combination therapy has been critical in the last decade. In this articles, current therapies for the PAH are discussed as well as defining patients appropriate for therapy.
Figure 1.

Pharmacotherapy in PAH. Adapted from (2). PAH, pulmonary arterial hypertension; PDE5, phosphodiesterase-5.
Patients eligible for therapy
PAH is defined by a mean pulmonary arterial pressure >20 mmHg and a wedge pressure (or left atrial or left ventricular end diastolic pressure) <15 mmHg with a pulmonary vascular resistance (PVR) >3 Woods units (see Table 1). A detailed review on the diagnosis of PAH is beyond on the scope of this review and covered in other articles (3-5). Pulmonary hypertension can be classified into 5 groups and current PAH-specific therapy is indicated for group 1 and group 4 patients (see Table 2). A multiparametric risk stratification approach is necessary to define initial therapy and to follow response to therapy in order to maximize clinical outcomes. Several registries and trials have addressed risk stratification and are summarized in Table 3. The 2015 European Society of Cardiology (ESC) guidelines use a multiparametric risk stratification method to define patients at low, medium and high risk for mortality, and these variables can be used for periodic assessment of therapeutic response to medications (see Table 4) (4).
Table 1. Hemodynamic definitions of pulmonary hypertension.
| Definitions | Characteristics | Clinical groups |
|---|---|---|
| Pre-capillary PH | mPAP >20 mmHg, PCWP <15 mmHg, PVR >3 WU | 1, 3, 4, and 5 |
| Isolated post-capillary PH | mPAP >20 mmHg, PCWP >15 mmHg, PVR<3 WU | 2 and 5 |
| Combined pre- and post-capillary PH | mPAP >20 mmHg, PCWP >15 mmHg, PVR >3 WU | 2 and 5 |
PH, pulmonary arterial hypertension; mPAP, mean pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance.
Table 2. Pulmonary hypertension classification.
| Group 1: pulmonary arterial hypertension |
| Idiopathic (IPAH) |
| Heritable (HPAH) |
| Drug and toxin induce PAH |
| Associated with (APAH) |
| Connective tissue disorder |
| HIV infection |
| Portal hypertension |
| Congenital heart disease |
| Schistosomiasis |
| PAH long-term responders to CCB |
| PAH with venous/capillary involvement (PVOD/PCH) |
| Persistent pulmonary hypertension of the newborn syndrome |
| Group 2: left heart disease |
| PH due to HFPEF |
| PH with HFREF |
| Valvular heart disease |
| CV (acquired or congenital) leading to post-capillary PH |
| Group 3: hypoxia or lung disease |
| Obstructive lung disease |
| Restrictive lung disease |
| Other lung disease with mixed restrictive/obstructive pattern |
| Hypoxia without lung disease |
| Developmental lung disorders |
| Group 4: PH due to pulmonary artery obstruction |
| Chronic thromboembolic PH (CTEPH) |
| Other pulmonary artery obstructions |
| Group 5: miscellaneous |
| Hematological disorders |
| Systemic and metabolic disorders |
| Others |
| Complex congenital heart disease |
CCB, calcium-channel blocker; PH, pulmonary arterial hypertension; PAH, pulmonary arterial hypertension.
Table 3. Summary of risk score in PAH.
| Variable | REVEAL | Swedish PAH register | COMPERA | French pulmonary hypertension network |
|---|---|---|---|---|
| Required variable | 12–14 | 8 | 8 | 4 |
| Patients at baseline | 2,716 | 530 | 1,588 | 1,017 |
| Patients at follow-up | 2,529 | 383 | 1,094 | 1,017 |
| Associated PAH included | Yes | Yes | Yes | No |
| Definition of low risk | <6 REVEAL score | <1.5 average score | <1.5 average score | 3–4 out of 4 low-risk criteria |
| 1-year mortality by risk (low/intermediate /high)% | <2.6/7.0/>10.7 | 1.0/7.0/26.0 | 2.8/9.9/21.2 | 1.0/NA/13.0–30.0 |
Adapted from (6). PAH, pulmonary arterial hypertension.
Table 4. Risk stratification in pulmonary arterial hypertension.
| Determinants of prognosis | Low risk <5% | Intermediate risk 5–10% | High risk >10% |
|---|---|---|---|
| Clinical signs of right heart failure | Absent | Absent | Present |
| Progression of Symptoms | No | Slow | Rapid |
| Syncope | No | Isolated syncope | Repeated syncope |
| WHO functional class | I, II | III | IV |
| 6 minute walk (m) | >440 | 165–440 | <165 |
| CPET | Peak VO2 >15 mL/min/kg, >65% predicted, VE/VCO2 slope <36 | Peak VO2 11–15 mL/min/kg, 35–65% predicted, VE/VCO2 slope 36–44.9 | Peak VO2 <11 mL/min/kg, <35% predicted, VE/VCO2 slop >45 |
| NT-proBNP levels | BNP <50 ng/L, NT-proBNP <300 ng/L | BNP 50–300 ng/L, NT-proBNP 300–1,400 ng/L | BNP >300 ng/L, NT-proBNP >1,400 ng/L |
| Imaging | RA area <18 cm2, no pericardial effusion | RA area 18–26 cm2, no/minimal pericardial effusion | RA area >26 cm2, pericardial effusion |
| Hemodynamics | RAP <8 mmHg, CI 2.5 L/min/m2, SvO2 >65% | RAP 8–14 mmHg, CI 2.0–2.4 L/min/m2, SvO2 60–65% | RAP >14 mmHg, CI <2.0 L/min/m2, SvO2 <60% |
Adapted from (4).
An imbalance of nitric oxide, prostacyclin and endothelin pathways are central to the development and propagation of vascular disease in PAH patients. There have been over 40 randomized-control trials evaluating the benefits of these therapies. Although initial trials used improvements in 6-minute walk distance (6MWD) as primary end-points, more contemporary trials have focused on disease progression and clinical deterioration as primary outcomes and have included larger patient numbers.
Nitric oxide pathway
Nitric oxide binds to soluble guanylate cyclase (sGC), which leads to the production of cyclic guanosine monophosphate (cGMP), which leads to arteriole vasodilation, suppresses cell proliferation and promotes vascular remodeling. Phosphodiesterase-5 (PDE5) inhibition prevents the degradation of cGMP (7). Sildenafil and tadalafil are the two PDE5 inhibitors that have demonstrated short-term and long-term clinical benefit in PAH (see Table 5).
Table 5. Nitric oxide pathway in PAH—summary of clinical studies.
| Drug | Study | Background | Primary endpoint | Secondary endpoints | Duration (weeks) | Patients |
|---|---|---|---|---|---|---|
| Sildenafil (Revatio) | SUPER-1 | None | 6MWD | TTCW | 12 | 227 |
| PACES | Epoprostenol | 6MWD | TTCW | 16 | 264 | |
| Iversen, et al. | Bosentan | 6MWD | – | 12 | 20 | |
| Sastry, et al. | None | TT | – | 12 | 22 | |
| Singh, et al. | None | 6MWD | – | 6 | 20 | |
| Tadalafil (Adcirca) | PHIRST | None | 6MWD | TTCW | 16 | 405 |
| Soluble guanylate cyclase stimulators | ||||||
| Riociguat (Adempas) | PATENT | None, bosentan, prostanoids | 6MWD | TTCW | 12 | 443 |
| CHEST | None, bosentan, prostanoids | 6MWD | TTCW | 12 | – | |
PAH, pulmonary arterial hypertension; 6MWD, 6-minute walk distance.
Sildenafil
Sildenafil was evaluated for the treatment of PAH in the SUPER (sildenafil use in pulmonary arterial hypertension) trial. A 12-week, randomized, double blinded, multi-centered trial evaluated the role of sildenafil in patients with symptomatic PAH (idiopathic, congenital, or connective tissue disease related) and to assess whether there was improvement in exercise capacity compared to placebo. The patients received either placebo or sildenafil (20, 40, or 80 mg in a 1:1:1:1 ratio) orally three times daily for 12 weeks with primary end point as change from baseline to week 12 in six minute walk test. The results demonstrated an increase in distance walked in all groups who received sildenafil throughout the end of 12 weeks. Those who finished one year of sildenafil therapy improved by 51 meters in six minutes at the end of the year (8). There was also hemodynamic improvement in all patients who received sildenafil, which included decrease in pulmonary artery pressure, peripheral vascular resistance, and improvement in cardiac index. There was also an improvement in WHO functional class. There was no statistically significant difference between placebo vs. sildenafil group in terms of clinical worsening, although this may have been secondary to a lower risk population (WHO functional class II, III). Of interest, there was no additional improvement in exercise capacity with the patients receiving the higher doses of sildenafil. The majority of side effects associated with the sildenafil group were flushing, dyspepsia, and diarrhea, which were not dose related. This study successfully illustrated the improvement in exercise capacity, hemodynamics, and WHO functional class, without significant adverse events in PAH patients who received sildenafil.
Tadalafil
The PHIRST (pulmonary arterial hypertension and response to tadalafil) trial assessed the utility of tadalafil, a PDE-5 inhibitor, for the treatment for PAH patients (idiopathic, congenital, connective tissue disease, drug-induced). This was a 16-week, randomized, double blinded, multi-centered trial that examined whether tadalafil improved exercise capacity compared to placebo. Patients received either tadalafil 2.5, 10, 20, or 40 mg orally once daily. Fifty-three percent of the patients were on background bosentan therapy. Similar to the SUPER trial, tadalafil improved exercise capacity based on improvement in distance in six-minute walk at the end of 16 weeks, though not to the same degree in the background bosentan group (9). This improvement in exercise capacity carried out to ten months in the extension study. However, unlike the prior sildenafil trial, the response was dose dependent as only the 40 mg dose reached the pre-specified value of significance. Also, unlike the sildenafil trial, there was an improvement in time to clinical worsening, incidence of clinical worsening, and health related quality of life in the tadalafil group. There was also improvement in hemodynamics. There was no improvement in functional class in the tadalafil group, though this was driven by the group on bosentan background therapy based on the post hoc analysis. Similar to sildenafil, tadalafil was well tolerated with mild to moderate adverse events that were not dose dependent. This trial demonstrated the safety and benefit of tadalafil 40 mg daily in PAH patients.
Riociguat
Riociguat is a sGC stimulator and acts independent of nitric oxide to promote positive vascular remodeling and pulmonary vasodilation (see Figure 2). Riociguat has demonstrated benefits in both PAH and CTEPH to increase exercise tolerance and decrease time to clinical worsening (10).
Figure 2.

Riociguat-mechanism of action. sGC, soluble guanylate cyclase; cGMP, cyclic guanosine monophosphate.
PATENT-1 was a large, randomized, double blinded, study that evaluated the role of a sGC stimulator, riociguat, in the PAH population (idiopathic, familial, connective tissue disease, congenital, medication-induced, portal hypertension due to liver cirrhosis). This phase 3 study compared placebo vs. riociguat (2.5 mg three times daily maximum group) vs. riociguat (1.5 mg three times daily maximum group-for exploratory purposes only). At the end of 12 weeks, there was improvement in exercise capacity based on 6MWD in the 2.5 mg max group, especially in patients with WHO class III or IV. There was mean increase by 30 meters in the 6-minute walk (11). This primary endpoint was reflected both in the treatment naive group and also in subjects on background therapy with either ERA or prostanoid. This finding contrasted to the tadalafil study, which did not show the same level of improvement in exercise capacity in patients on background ERA therapy. There was also improvement in hemodynamics, NT-proBNP, WHO functional class, time to clinical worsening, and Borg dyspnea score in the 2.5 mg max group. The most common adverse event in the 2.5 mg riociguat group was syncope (1%). This study validated the use of riociguat in PAH patients based on improvement in the primary and secondary endpoints with an acceptable safety profile.
Riociguat was further studied to see if its efficacy would apply to the chronic thromboembolic pulmonary hypertension (CTEPH) in the CHEST-1 trial. This 16-week, randomized, double-blinded, multi-center trial tested the role of riociguat in patients with inoperable CTEPH or residual or recurrent pulmonary hypertension after pulmonary endarterectomy compared to placebo. There was improvement in exercise capacity in the riociguat group at the end of the 16 weeks with an increase of 39 meters in the six-minute walk from baseline (12). The group who had inoperable CTEPH had a greater improvement in exercise capacity compared to the group that had already received pulmonary endarterectomy. There were also improvements in the secondary endpoints, illustrated by the decrease in NT-proBNP levels, improvement in hemodynamics specifically peripheral vascular resistance, and improvement in WHO functional class. The most common serious drug-related adverse effect was syncope in 2% of the patients. This trial established riociguat as an initial medication for CTEPH patients who were not considered candidates for surgery or continued to have PAH after surgery.
Endothelin pathway
Endothelin-1 (ET1) binds to endothelin (ET) receptors A and B. Endothelin A (ETA) activation leads to pulmonary vasoconstriction and smooth muscles cell proliferation while endothelin B (ETB) acts to clear ET1 and mediate endothelial cell vasodilation and nitric oxide and prostacyclin release. ET1 levels are elevated in PAH and endothelin receptor antagonists (ERA) have demonstrated clear effectiveness in the treatment of PAH (see Table 6).
Table 6. Endothelin pathway in PAH—summary of clinical studies.
| ERA | Study | Background | Primary endpoint | Secondary endpoints | Duration (weeks) | Patients |
|---|---|---|---|---|---|---|
| Bosentan (Tracleer) | Study 351 | None | 6MWT | TTCW | 12 | 32 |
| BREATHE-1 | None | 44 m (P<0.001) | TTCW | 16 | 213 | |
| BREATHE-2 | None | PVR | – | 12 | 33 | |
| EARLY | PDE5i (16%) | PVR, 6MWD | TTCW | 24 | 185 | |
| BREATHE-5 | None | PVR, SaO2 | – | 12 | 54 | |
| Ambrisentan (Letairis) | ARIES-1 | None | 31 m (5 mg) and 51 m (10 mg) | TTCW (ns) | 12 | 202 |
| ARIES-2 | None | 32 m (2.5 mg) and 59 m (5 mg) | TTCW | 12 | 192 | |
| Macitentan (Opsumit) | SERAPHIN | None/PDE5i/iloprost | TTCW: 46.4% placebo, 38.0% 3 mg, 31.4% 10 mg | Safety | 100 | 742 |
PVR, pulmonary vascular resistance; PDE5, phosphodiesterase-5; PAH, pulmonary arterial hypertension; 6MWD, 6-minute walk distance.
Bosentan
Bosentan is non-selective ERA. It requires regular liver function test (LFT) due to elevation in transaminases observed in roughly 11% to 12% of patients. In the BREATHE-1 trial, 213 patients with WHO functional class III and IV PAH were assigned to two doses of bosentan versus placebo and followed for 16 weeks. The primary endpoint was the degree in change in exercise tolerance, and at 16 weeks there was a 44 meter difference in the mean walking distance between the placebo group and combined bosentan groups (95 percent confidence interval, 21 to 67; P<0.001) (13). There were improvements in the Borg dyspnea index, WHO functional class and increased the time to clinical worsening in the bosentan groups. Bosentan also demonstrated improvement walking distance in patients with WHO functional class II PAH (14). BREATHE-5 evaluated the efficacy of bosentan in patients with Eisenmenger’s syndrome, which is associated with increased endothelin expression. Over a 16-week study period, bosentan did not worsen oxygen saturation and it reduced PVR index and mean pulmonary arterial pressures (−5.5 mmHg; P=0.0363) when compared to placebo (15). There was an increase in exercise capacity (53.1 m; P=0.0079) and no increase in adverse events compared to placebo.
Ambrisentan
Ambrisentan and sitaxsentan are selective ETA inhibitors. Due to the potential benefits of selective ETA inhibition while allowing for the salutatory effects of ETB activation, these therapies had potential benefits over non-selective receptors antagonists. Sitaxsentan is an ETa selective ERA that demonstrated benefits in maximum oxygen consumption and improvements in functional status amongst PAH patients (16-18). However, there was a 6% risk of hepatotoxicity and the manufacturer has since withdrawn the drug.
Ambrisentan has ETA selectivity and does not require LFT monitoring. The ARIES-1 and ARIES-2 were concurrent, phase 3, randomized, double-blind, placebo-controlled studies that were identical in design except for the investigative sites and the doses of ambrisentan studied. ARIES-1 randomized patients to placebo or ambrisentan 5 or 10 mg oral daily whereas ARIES-2 randomized patients to placebo or ambrisentan 2.5 or 5 mg oral daily. The initial 12-week study was completed by 183 patients in ARIES-1 and 170 patients in ARIES-2; 280 combined patients completed the 48 weeks of treatment with ambrisentan monotherapy. The primary end point for each study was change in 6MWD from baseline to week 12, and this increased in all ambrisentan groups. Mean placebo-corrected treatment effects were 31 m (P=0.008) and 51 m (P<0.001) in ARIES-1 for 5 and 10 mg ambrisentan, respectively, and 32 m (P=0.022) and 59 m (P<0.001) in ARIES-2 for 2.5 and 5 mg ambrisentan, respectively (19). Improvements in time to clinical worsening, functional status and B-type natriuretic peptide were observed in both studies. In the 280 patients completing 48 weeks of ambrisentan monotherapy, the improvement from baseline in 6-minute walk was 39 m.
Macitentan
Macitentan is a dual endothelin-receptor antagonist that developed by structural modification of bosentan to increase efficacy and safety. It has no significant hepatotoxicity and is a once-daily tablet. Macitentan was studied in a large prospective trial that included 742 PAH patients and demonstrated a significant reduction in morbidity and disease progression. The SERAPHIN trial (Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome) represented a shift in primary end-points towards morbidity and mortality from 6MWD in PAH trials. The primary end point was the time from initiation of treatment to the first occurrence of a composite end point of death, atrial septostomy, lung transplantation, escalation to intravenous or subcutaneous prostanoid or worsening PAH. Background inhaled or oral non-ERA PAH therapy was allowed. Patients with symptomatic PAH were randomly assigned to placebo versus macitentan 3 mg daily or macitentan 10 mg daily. The primary end point occurred in 46.4%, 38.0%, and 31.4% of the patients in these groups, respectively (20). There was significant improvement with both the 3 mg (hazard ratio 0.70, 97.5% CI: 0.52 to 0.96; P=0.01) and 10 mg doses (hazard ratio 0.55, 97.5% CI: 0.39 to 0.76; P<0.001). Worsening of PAH was the most frequent primary end-point event, and the benefit of macitentan was observed regardless of whether the patient was receiving background PAH therapy at baseline. PDE5 inhibitors were used by 61.4% of patients and oral or inhaled prostanoids were used by 5.4% of patients. Adverse events associated with macitentan included headache, nasopharyngitis, and anemia.
MAESTRO (macitentan in Eisenmenger syndrome to restore exercise capacity) study investigated macitentan in patients with Eisenmenger syndrome. Unlike the BREATH-5 trial, there was no significant impact on the primary end point of change from baseline to week 16 in exercise capacity nor were there significant trends in the secondary endpoint of WHO functional class improvement. There were, however, reductions in NT-proBNP levels and PVRi in exploratory endpoint. Conflicting results from the two Eisenmenger trials may be explained by the fact that MAESTRO enrolled more heterogeneous congenital patients including those with simple or complex cardiac defects, and it did not restrict patients due to background therapy or the presence of Down syndrome (21).
Macitentan has also demonstrated benefits in reducing PVR in patients with portopulmonary hypertension without impacting hepatic function in a trial of 85 patients (22). PVR was also reduced in patients with inoperable CTEPH who were treated with macitentan although additional studies are required to assess full clinical benefit (23).
Prostacyclin therapy
Prostacyclins are released by endothelial cells and promote pulmonary vasodilation and have antithrombotic and antiproliferative properties. Prostacyclins can be administered in oral, inhaled, subcutaneous and intravenous forms (see Table 7). Despite significant side effects, prostacyclins are possibly the most aggressive therapies for PAH.
Table 7. Prostacyclin pathway in PAH—summary of clinical studies.
| Prostanoids | Study | Background | Primary endpoint | Secondary endpoints | Duration (weeks) | Patients | |
|---|---|---|---|---|---|---|---|
| Epoprostenol (Flolan) | Rubin, et al. | None | 6MWD | – | 12 | 23 | |
| Barst, et al. | None | 6MWD | Survival | 12 | 81 | ||
| Badesch, et al. | None | 6MWD | – | 12 | 111 | ||
| Treprostinil (Remodulin) | SC | None | 6MWD | – | 12 | 470 | |
| Tyvaso | TRIUMPH (Inhaled) | Bosentan or sildenafil | 6MWD | – | 12 | 235 | |
| Orenitram | FREEDOM-M (po) | None | 6MWD | – | 16 | 185 | |
| FREEDOM-C1 (po) | Bosentan and/or sildenafil | 6MWD (ns) | – | 16 | 354 | ||
| FREEDOM-C2 (po) | Bosentan and/or sildenafil | 6MWD (ns) | – | 16 | 310 | ||
| Iloprost (Ventavis) | AIR | None | 6MWD and FC | – | 12 | 203 | |
| STEP | Bosentan | 6MWD | TTCW | 12 | 67 | ||
| COMBI | Bosentan | 6MWD (ns) | – | 12 | 40 | ||
| Beraprost | ALPHABET | None | 6MWD | – | 12 | 130 | |
| Barst, et al. | None | CW (ns) | – | 52 | 116 | ||
| Prostacyclin IP-receptor agonists | |||||||
| Selexipag (Uptravi) | GRIPHON | Bosentan and/or sildenafil | Death from any cause, PAH complication | 6MWD (ns) | 36 months | 1,156 | |
6MWD, 6-minute walk distance; PAH, pulmonary arterial hypertension.
Epoprostenol
Epoprostenol is available as continuous intravenous therapy and can be used as inhaled therapy. It was one of the initial PAH-specific therapies evaluated for the treatment of advanced PAH. Barst and colleagues demonstrated symptomatic and hemodynamic improvement, in addition to improved survival in patients with severe PAH (24). Among 81 patients with severe PAH and NYHA class III/IV symptoms, exercise capacity and hemodynamics were improved in the 41 patients assigned to epoprostenol. Moreover, there were 8 deaths in the conventional therapy arm and no deaths in the treatment arm over the 12-week study period. Echocardiographic parameters of right ventricular function were also improved over the study follow-up period (25). The hemodynamics effects of epoprostenol are persistent over several months with up to a 53% decline in PVR with doses of 40±15 ng/kg/min (26), and the benefits were demonstrated in high risk populations such as scleroderma-associated PAH (27). Among 162 PAH patients treated with epoprostenol and followed over a mean 363 months, the observed survival with epoprostenol at 1, 2, and 3 years was 87.8%, 76.3%, and 62.8% and was significantly greater than the expected survival of 58.9%, 46.3%, and 35.4% based on historical data (28). Epoprostenol is also available in an inhaled form for acute treatment of PAH in the intensive care unit. Further modifications of the intravenous medication have led to a thermostable and photostable form of the drug that is marketed as Veletri.
Treprostinil-subcutaneous
Treprostinil is a prostacyclin analogue with a longer half-life than epoprostenol, and it has demonstrated clinical improvement as intravenous, subcutaneous, inhaled, and oral therapy. Subcutaneous treprostinil was initially evaluated for PAH and was an alternative to intravenous epoprostenol that avoided the risks of sepsis and thromboembolism associated with a long-term indwelling catheter. In a 12-week double-blind, placebo-controlled multicenter trial of 470 patients with PAH, there was a median 16 meters (P=0.006) improvement in 6-minute walk over a 12-week period (29). Treprostinil therapy improved dyspnea scores and hemodynamics. The PAH patients included those with idiopathic PAH, associated PAH and congenital systemic-to-pulmonary shunts. Infusion site pain occurred in 85% of patients leading discontinuation of therapy in 8% of patients. While the improvement in 6-minute walk was limited compared to other PAH therapies, the mean dose of treprostinil was 9.3 ng/kg/min at 12-week in this trial. In comparison, in a trial of intravenous treprostinil among 44 PAH patients, the improvement in 6-minute walk was a median 83 meters at an average dose of 72 ng/kg/min (30).
Treprostinil and iloprost-inhaled
Inhaled treprostinil therapy was evaluated in the TRIUMPH-1 trial. The study included 235 PAH patients with NYHA class III or IV symptoms who were on a background therapy of bosentan (70%) or sildenafil, and randomized patients to inhaled treprostinil or placebo. The primary end-point was peak 6MWD at 12 weeks and there was a 19 m improvement at 6 weeks (P=0.001) and 20 m at 12 weeks (P=0.0004) (31). There was no improvement in time to clinical worsening or PAH signs or symptoms despite improvements in quality of life measures and NT-proBNP.
Iloprost is a prostacyclin that is administered via inhalation and has demonstrable benefits on clinical function. In the AIR (Aerosolized Iloprost Randomized) study, 203 patients with severe PAH and CTEPH with NYHA class III and IV symptoms were randomized to iloprost 2.5 or 5.0 mcg inhalations (6–9 times per day). The primary end point was improvement in 6MWD by at least 10%. At 12 weeks, iloprost therapy demonstrated an improvement over placebo with greater patients achieving the primary endpoint (16.8% with iloprost compared to 4.9% placebo) (32). The overall improvement in 6MWD was 36.4 and 58.8 m in those patients with idiopathic PAH.
Treprostinil-oral
Oral prostacyclin therapies have been evaluated since 2003. Beraprost was an oral prostacyclin studied in the United States, but the trial was unsuccessful, as improvements in 6MWD seen at 12 weeks were not sustained at one year (33). Oral treprostinil has been evaluated in several clinical trials. The FREEDOM-C trial was a multicenter clinical trial that evaluated the benefits of oral treprostinil on background therapy with PDE-5 inhibitor or ERA. The primary efficacy endpoint was change in 6MWD at 16 weeks relative to baseline. The trial failed to achieve its primary endpoint as the placebo-corrected median 6MWD changes was 11 meters (P=0.0072) (34). However, there was difficulty titrating the drug and those who achieved a dose of 3.25 mg twice daily had median improvements of 34 meters. The FREEDOM-M trial evaluated the utility of oral treprostinil versus placebo in treatment-naive PAH patients. A lower dose 0.25 mg tablet was included in this study to facilitate medication uptitration. The study enrolled 349 patients although only the 228 patients who were provided the 0.25 mg tablet were evaluated in the final analysis. There was median improvement in 6MWD of 23 meters compared to placebo (P=0.0125) (35). There was no difference in WHO functional class, time to clinical worsening or Borg Dyspnea Score compared to placebo. The primary side effects included headache, flushing, nausea, and diarrhea. Despite the improvements with monotherapy, the FREEDOM-C2 trial failed to demonstrate an improvement in exercise capacity over a 16-week period with the addition of oral treprostinil to background ERA and PDE5 inhibitor therapy (36). This trial enrolled 310 patients and the primary end point was a change in 6MWD. There were no improvements in secondary endpoints that included WHO functional class, Borg Dyspnea Scale and time to clinical worsening.
The Food and Drug Administration (FDA) approved oral treprostinil in 2013. The results of the FREEDOM-EV study were presented in 2019. The trial randomized 690 PAH patients who were on one oral PAH medication to oral treprostinil versus placebo and was an event-driven study. The primary end-point was the effect of oral treprostinil on time to first adjudicated clinical worsening event (death, hospitalization due to worsening PAH, initiation of inhaled/infused prostacyclins, disease progression) over 48 weeks. There was a statistically significant improvement in time to disease progression and functional and symptomatic improvements were greatest for patients achieving >3 mg TID of treprostinil (37).
Selexipag
Selexipag is an oral selective prostacyclin receptor (IP receptor) agonist. The IP receptor is a G-protein coupled receptor on vascular smooth muscles and on the surface of platelets, which when activated leads to cyclic adenosine monophosphate and induces vasodilation (38). Selexipag was demonstrated to be safe and beneficial in a phase 2 trial of 43 patients with PAH (39). The GRIPHON study was a phase 3 trial that enrolled 1,156 patients with PAH to selexipag or placebo. The trial included patients naive to therapy as well as patients on background therapy with PDE5 inhibitor, ERA or both. The primary end point was a composite of death from any cause or a complication related to PAH. There was a significant reduction in the primary end-point among the selexipag group compared to placebo (27.0% versus 41.6%, P<0.001) (40). The benefit of selexipag was similar in patients naive to treatment as well as those on monotherapy and dual oral therapy (see Figure 3). The most common adverse events in the selexipag group included headache, diarrhea, nausea, and jaw pain. The benefit of selexipag was significant at low, moderate and high doses and may have reflected the variability of prostacyclin receptor density amongst different patients.
Figure 3.
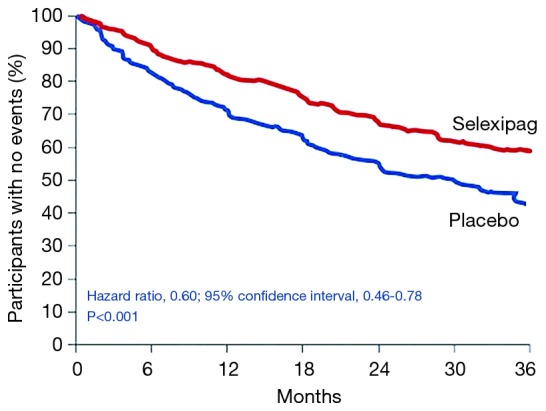
Selexipag as monotherapy of sequential therapy to PDE5 inhibitors and/or ERA. Adapted from (40). PDE5, phosphodiesterase-5.
Combination therapy
Combination therapy was evaluated in several smaller trials that eventually resulted in a large trial evaluating the safety and efficacy of combination oral therapy in the AMBITION trial.
In the BREATH-2 study evaluated the benefits of bosentan in addition to epoprostenol in 33 patients with PAH. There was a trend towards clinical or hemodynamic improvement with the combined therapy of bosentan and epoprostenol but this did not achieve statistical significance. Furthermore, there were several complications including one adverse event and one clinical worsening (41). Although likely underpowered, this study did not support the initial use of combination bosentan and epoprostenol therapy among PAH patients.
In the COMPASS-2 trial, the addition of bosentan to stable sildenafil therapy was not superior to sildenafil monotherapy in the time to first morbidity or mortality event as defined as all-cause death, hospitalization for worsening PAH, atrial septostomy or lung transplantation (42). Among the 334 patients randomized, there was no difference in the mean 60-minute walk distance at 16 weeks although there were improvements in NT-proBNP levels. The addition of bosentan to sildenafil may lead to a reduction in sildenafil levels, and the use of a non-selective ERA may reduce the efficacy of sildenafil through ETB antagonism.
The combination of sildenafil and epoprostenol, however, has demonstrated synergistic effects. The PACES trial evaluated the benefit of sildenafil in patients on background epoprostenol therapy. This study included a total of 267 patients and randomized patients to sildenafil or placebo. Sildenafil improved 6MWD by 28.8 meters (95% CI, 13.9 to 43.8 meters), and there were improvement in cardiac index and reductions mean PA pressures (43). Combined therapy yielded improvement in quality of life and time to clinical worsening although there were increased rates of headaches and dyspepsia.
Ambition trial
The AMBITION trial was a randomized, double blinded, multicenter trial that evaluated the long-term outcomes in patients with PAH (WHO functional class II or III) receiving combination therapy of an ERA and a PDE-5 inhibitor. Patients received either initial combination therapy of 10 mg of ambrisentan plus 40 mg of tadalafil, 10 mg ambrisentan monotherapy, or 40 mg tadalafil monotherapy. The combination therapy group had the lowest first event of clinical failure (death, hospitalization for worsening PAH, disease progression, or unsatisfactory clinical response) compared to the pooled monotherapy groups (18% vs. 31%). At six months, the combination therapy group compared to the pooled monotherapy groups had greater decrease in NT-proBNP, higher percentage of acceptable clinical response, and increased exercise capacity (increase of 49 vs. 24 meters from baseline in six-minute walk) (see Figure 4). Combination therapy had more frequent adverse events related to peripheral edema, headache, nasal congestion, and anemia compared to either monotherapy group. This study elucidated that initial combination therapy with tadalafil and ambrisentan in patients with WHO functional class II or III may be a preferred treatment modality as opposed to sequential addition of therapy or monotherapy with either ambrisentan or tadalafil (44).
Figure 4.
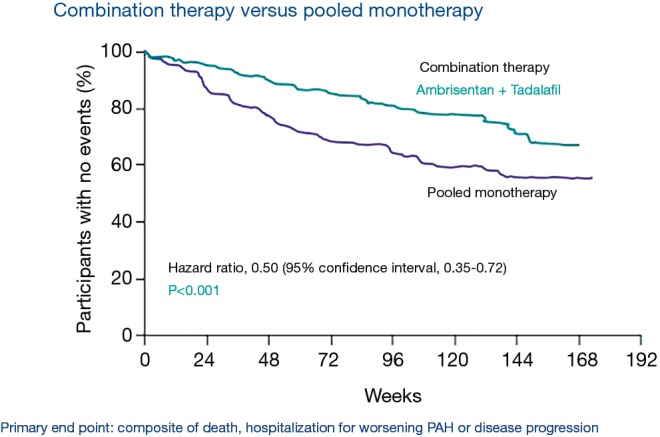
AMBITION trial. Combination therapy (ambrisentan and tadalafil) vs. Pooled monotherapy. Adapted from (44).
Therapeutic approach
Over the last three decades there has been a remarkable increase in the number of therapeutic agents available for PAH. Early initiation of therapy is indicated in PAH patients and careful assessment followed by reassessment of risk is necessary (see Figure 5) (6). In patients who are WHO class IV or are at high risk for clinical deterioration, intravenous epoprostenol should be considered as a first-line agent. In patients with WHO class II and III symptoms, combination therapy should be strongly considered unless the patient is at low risk where monotherapy may be an initial approach. Adjunctive therapies may include anticoagulation, digoxin, aldosterone inhibition and oxygen therapy. Warfarin was initially thought to benefit PAH patients in older retrospective studies although more contemporary studies have questioned the use of warfarin in all PAH patients and its use may be limited to select patients with idiopathic PAH (45,46). The ESC 2015 guidelines outline initial and step-wise approach to PAH therapies, which serves as a general guide to treatment (see Tables 8 and 9). Future therapies focus on targeting growth factors, metabolism, inflammation and immunity, and histamine regulation. In addition, refinement in trial design should help establish the true benefit of such therapies as well as combination therapies among the PAH population. A full discussion of novel therapies is beyond the scope of this review but has been discussed in other publications (47).
Figure 5.

Treatment algorithm in PAH. Adapted from (6). PAH, pulmonary arterial hypertension.
Table 8. European Society of Cardiology Recommendations for initial drug combinations in PAH.
| Measurement/treatment | Class-level | ||
|---|---|---|---|
| WHO-FC II | WHO-FC III | WHO-FCIV | |
| Ambrisentan + tadalafil | I-B | I-B | IIb-C |
| Other ERA + PDE5i | IIa-C | IIa-C | IIb-C |
| Bosentan + sildenafil + iv epoprostenol | – | IIa-C | IIa-C |
| Bosentan + iv epoprostenol | – | IIa-C | IIa-C |
| Other ERA or PDE5i + sc treprostinil | – | IIb-C | IIb-C |
| Other ERA or PDE5i + other iv prostacyclin analogues | – | IIb-C | IIb-C |
Adapted from (4). PAH, pulmonary arterial hypertension; PDE5, phosphodiesterase-5.
Table 9. European Society of Cardiology Recommendations for sequential drug combinations in PAH.
| Measurement/treatment | Class-level | ||
|---|---|---|---|
| WHO-FC II | WHO-FC III | WHO-FCIV | |
| Macitentan added to sildenafil | I-B | I-B | IIa-C |
| Riociugat added to bosentan | I-B | I-B | IIa-C |
| Selexipag added to ERA and/or PDE5I | I-B | I-B | IIa-C |
| Sildenafil added to epoprostenol | I-B | IIa-B | |
| Treatment escalation on background PDE5i + ERA | |||
| Selexipag | I-B | I-B | IIa-C |
| Other triple therapy combinations | IIb-C | IIb-C | IIb-C |
Adapted from (4). PAH, pulmonary arterial hypertension; PDE5, phosphodiesterase-5.
Conclusions
Over the last 15 years, there has been a wide expansion of pharmacotherapy for PAH. While initial therapies focused on exercise capacity, more contemporary studies have focus on combined morbidity and mortality in large study populations. Combination therapy has also demonstrated proven benefit and future studies should establish the role of early upfront triple combination therapy for PAH. Early recognition of the disease may also help abrogate its progression with appropriate medical management. Despite these advances, PAH remains a disease characterized by significant morbidity and mortality and ongoing efforts at halting disease progression through novel therapeutics may allow for additional hope for PAH patients.
Acknowledgments
None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019. doi: . 10.1183/13993003.01913-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Humbert M, Ghofrani HA. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax 2016;71:73-83. 10.1136/thoraxjnl-2015-207170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McLaughlin VV, Shah SJ, Souza R, et al. Management of pulmonary arterial hypertension. J Am Coll Cardiol 2015;65:1976-97. 10.1016/j.jacc.2015.03.540 [DOI] [PubMed] [Google Scholar]
- 4.Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 2015;46:903-75. 10.1183/13993003.01032-2015 [DOI] [PubMed] [Google Scholar]
- 5.Frost A, Badesch D, Gibbs JSR, et al. Diagnosis of pulmonary hypertension. Eur Respir J 2019. doi: . 10.1183/13993003.01904-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galiè N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J 2019;53:180-9. 10.1183/13993003.01889-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Archer SL, Michelakis ED. Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N Engl J Med 2009;361:1864-71. 10.1056/NEJMct0904473 [DOI] [PubMed] [Google Scholar]
- 8.Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353:2148-57. 10.1056/NEJMoa050010 [DOI] [PubMed] [Google Scholar]
- 9.Galiè N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009;119:2894-903. 10.1161/CIRCULATIONAHA.108.839274 [DOI] [PubMed] [Google Scholar]
- 10.Archer SL. Riociguat for pulmonary hypertension--a glass half full. N Engl J Med 2013;369:386-8. 10.1056/NEJMe1306684 [DOI] [PubMed] [Google Scholar]
- 11.Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369:330-40. 10.1056/NEJMoa1209655 [DOI] [PubMed] [Google Scholar]
- 12.Ghofrani HA, D'Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013;369:319-29. 10.1056/NEJMoa1209657 [DOI] [PubMed] [Google Scholar]
- 13.Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896-903. 10.1056/NEJMoa012212 [DOI] [PubMed] [Google Scholar]
- 14.Galiè N, Rubin L, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 2008;371:2093-100. 10.1016/S0140-6736(08)60919-8 [DOI] [PubMed] [Google Scholar]
- 15.Galiè N, Beghetti M, Gatzoulis MA, et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation 2006;114:48-54. 10.1161/CIRCULATIONAHA.106.630715 [DOI] [PubMed] [Google Scholar]
- 16.Barst RJ, Langleben D, Frost A, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med 2004;169:441-7. 10.1164/rccm.200307-957OC [DOI] [PubMed] [Google Scholar]
- 17.Girgis RE, Frost AE, Hill NS, et al. Selective endothelin A receptor antagonism with sitaxsentan for pulmonary arterial hypertension associated with connective tissue disease. Ann Rheum Dis 2007;66:1467-72. 10.1136/ard.2007.069609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benza RL, Barst RJ, Galie N, et al. Sitaxsentan for the treatment of pulmonary arterial hypertension: a 1-year, prospective, open-label observation of outcome and survival. Chest 2008;134:775-82. 10.1378/chest.07-0767 [DOI] [PubMed] [Google Scholar]
- 19.Galiè N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117:3010-9. 10.1161/CIRCULATIONAHA.107.742510 [DOI] [PubMed] [Google Scholar]
- 20.Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809-18. 10.1056/NEJMoa1213917 [DOI] [PubMed] [Google Scholar]
- 21.Gatzoulis MA, Landzberg M, Beghetti M, et al. Evaluation of Macitentan in Patients With Eisenmenger Syndrome. Circulation 2019;139:51-63. 10.1161/CIRCULATIONAHA.118.033575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sitbon O, Bosch J, Cottreel E, et al. Macitentan for the treatment of portopulmonary hypertension (PORTICO): a multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. Lancet Respir Med 2019;7:594-604. 10.1016/S2213-2600(19)30091-8 [DOI] [PubMed] [Google Scholar]
- 23.Ghofrani HA, Simonneau G, D'Armini AM, et al. Macitentan for the treatment of inoperable chronic thromboembolic pulmonary hypertension (MERIT-1): results from the multicentre, phase 2, randomised, double-blind, placebo-controlled study. Lancet Respir Med 2017;5:785-94. 10.1016/S2213-2600(17)30305-3 [DOI] [PubMed] [Google Scholar]
- 24.Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334:296-301. 10.1056/NEJM199602013340504 [DOI] [PubMed] [Google Scholar]
- 25.Hinderliter AL, Willis PW, 4th, Barst RJ, et al. Effects of long-term infusion of prostacyclin (epoprostenol) on echocardiographic measures of right ventricular structure and function in primary pulmonary hypertension. Primary Pulmonary Hypertension Study Group. Circulation 1997;95:1479-86. 10.1161/01.CIR.95.6.1479 [DOI] [PubMed] [Google Scholar]
- 26.McLaughlin VV, Genthner DE, Panella MM, et al. Reduction in pulmonary vascular resistance with long-term epoprostenol (prostacyclin) therapy in primary pulmonary hypertension. N Engl J Med 1998;338:273-7. 10.1056/NEJM199801293380501 [DOI] [PubMed] [Google Scholar]
- 27.Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med 2000;132:425-34. 10.7326/0003-4819-132-6-200003210-00002 [DOI] [PubMed] [Google Scholar]
- 28.McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 2002;106:1477-82. 10.1161/01.CIR.0000029100.82385.58 [DOI] [PubMed] [Google Scholar]
- 29.Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002;165:800-4. 10.1164/ajrccm.165.6.2106079 [DOI] [PubMed] [Google Scholar]
- 30.Hiremath J, Thanikachalam S, Parikh K, et al. Exercise improvement and plasma biomarker changes with intravenous treprostinil therapy for pulmonary arterial hypertension: a placebo-controlled trial. J Heart Lung Transplant 2010;29:137-49. 10.1016/j.healun.2009.09.005 [DOI] [PubMed] [Google Scholar]
- 31.McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol 2010;55:1915-22. 10.1016/j.jacc.2010.01.027 [DOI] [PubMed] [Google Scholar]
- 32.Olschewski H, Simonneau G, Galie N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002;347:322-9. 10.1056/NEJMoa020204 [DOI] [PubMed] [Google Scholar]
- 33.Barst RJ, McGoon M, McLaughlin V, et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2003;41:2119-25. 10.1016/S0735-1097(03)00463-7 [DOI] [PubMed] [Google Scholar]
- 34.Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest 2012;142:1383-90. 10.1378/chest.11-2212 [DOI] [PubMed] [Google Scholar]
- 35.Jing ZC, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation 2013;127:624-33. 10.1161/CIRCULATIONAHA.112.124388 [DOI] [PubMed] [Google Scholar]
- 36.Tapson VF, Jing ZC, Xu KF, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest 2013;144:952-8. 10.1378/chest.12-2875 [DOI] [PubMed] [Google Scholar]
- 37.Tapson V, Sanchez Diaz CJ, Bohns Meter G, et al., editors. Treatment with Oral Treprostinil Delays Time to Clinical Worsening in Patients with Pulmonary Arterial Hypertension - Results from FREEDOM-EV. International Society of Heart Lung Transplantation; 2019; Orlando, FL. [Google Scholar]
- 38.Kuwano K, Hashino A, Asaki T, et al. 2-[4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy]-N-(methylsulfonyl)acetam ide (NS-304), an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther 2007;322:1181-8. 10.1124/jpet.107.124248 [DOI] [PubMed] [Google Scholar]
- 39.Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J 2012;40:874-80. 10.1183/09031936.00137511 [DOI] [PubMed] [Google Scholar]
- 40.Sitbon O, Channick R, Chin KM, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 2015;373:2522-33. 10.1056/NEJMoa1503184 [DOI] [PubMed] [Google Scholar]
- 41.Humbert M, Barst RJ, Robbins IM, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J 2004;24:353-9. 10.1183/09031936.04.00028404 [DOI] [PubMed] [Google Scholar]
- 42.McLaughlin V, Channick RN, Ghofrani HA, et al. Bosentan added to sildenafil therapy in patients with pulmonary arterial hypertension. Eur Respir J 2015;46:405-13. 10.1183/13993003.02044-2014 [DOI] [PubMed] [Google Scholar]
- 43.Simonneau G, Rubin LJ, Galie N, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med 2008;149:521-30. 10.7326/0003-4819-149-8-200810210-00004 [DOI] [PubMed] [Google Scholar]
- 44.Galiè N, Barbera JA, Frost AE, et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N Engl J Med 2015;373:834-44. 10.1056/NEJMoa1413687 [DOI] [PubMed] [Google Scholar]
- 45.Preston IR, Roberts KE, Miller DP, et al. Effect of Warfarin Treatment on Survival of Patients With Pulmonary Arterial Hypertension (PAH) in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL). Circulation 2015;132:2403-11. 10.1161/CIRCULATIONAHA.115.018435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olsson KM, Delcroix M, Ghofrani HA, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 2014;129:57-65. 10.1161/CIRCULATIONAHA.113.004526 [DOI] [PubMed] [Google Scholar]
- 47.Sitbon O, Gomberg-Maitland M, Granton J, et al. Clinical trial design and new therapies for pulmonary arterial hypertension. Eur Respir J 2019. doi: . 10.1183/13993003.01908-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]