

Abstract
Background
One of the clinical hallmarks of low-grade gliomas (LGGs) arising in children with the neurofibromatosis type 1 (NF1) cancer predisposition syndrome is significant clinical variability with respect to tumor growth, associated neurologic deficits, and response to therapy. Numerous factors could contribute to this clinical heterogeneity, including the tumor cell of origin, the specific germline NF1 gene mutation, and the coexistence of additional genomic alterations. Since human specimens are rarely acquired, and have proven difficult to maintain in vitro or as xenografts in vivo, we have developed a series of Nf1 mutant optic glioma mouse strains representing each of these contributing factors.
Methods
Optic glioma stem cells (o-GSCs) were generated from this collection of Nf1 genetically engineered mice, and analyzed for their intrinsic growth properties, as well as the production of chemokines that could differentially attract T cells and microglia.
Results
The observed differences in Nf1 optic glioma growth are not the result of cell autonomous growth properties of o-GSCs, but rather the unique patterns of o-GSC chemokine expression, which differentially attract T cells and microglia. This immune profile collectively dictates the levels of chemokine C-C ligand 5 (Ccl5) expression, the key stromal factor that drives murine Nf1 optic glioma growth.
Conclusions
These findings reveal that genetic and genomic alterations create murine LGG biological heterogeneity through the differential recruitment of T cells and microglia by o-GSC–produced chemokines, which ultimately determine the expression of stromal factors that drive tumor growth.
Keywords: precision oncology, glioma stem cells, tumor microenvironment, chemokine
Key Points.
1. Differences in LGG proliferation are not dictated by differential o-GSC growth.
2. Optic GSC–specific chemokine expression differentially attracts T cells and microglia.
3. T cells and microglia collectively specify tumor Ccl5 levels and LGG growth.
Importance of the Study.
The factors that contribute to the biological variability observed in pediatric LGGs are currently unknown. Defining the etiologies for this clinical heterogeneity is critical for the future implementation of precision oncology approaches to risk assessment and treatment for children with these brain tumors. In an effort to elucidate the cellular and molecular mechanisms that underlie this variability, we employed a series of clinically relevant and authenticated mouse models of Nf1 optic glioma with different germline Nf1 gene mutations and secondary genomic alterations. Using these mice, we demonstrate that the observed differences in overall tumor proliferation do not reflect the intrinsic cell autonomous growth properties of the cancer (optic glioma) stem cells (o-GSCs), but rather the differential recruitment of T cells and microglia by o-GSCs. These findings support a model in which the biological heterogeneity of pediatric LGG is primarily dictated by stromal cell establishment of a supportive microenvironment.
One of the hallmarks of many brain tumors is clinical heterogeneity, such that histologically similar pediatric low-grade gliomas (eg, pilocytic astrocytomas [PAs]) can exhibit strikingly different growth patterns and responses to therapy.1 Some of this heterogeneity could result from the causative genetic mutation (eg, KIAA1549-BRAF alteration versus FGFR1 mutation), tumor location within the neuroaxis (eg, cerebellum versus brainstem), and/or the presence of secondary coexisting genomic changes (eg, PTEN or FGFR1 mutation).2–4 Even in children with the low-grade glioma predisposition syndrome, neurofibromatosis type 1 (NF1), the clinical behavior of the tumors can be dramatically heterogeneous. In this regard, while only 20% of children with NF1 develop PAs of the optic pathway (optic pathway gliomas),5 there are considerable differences between patients with tumors of the same histological subtype.6 This variation includes the age of tumor development, the glioma growth rate, and the response to therapy.
Defining the individual contributions of these causative factors to overall glioma biology has proven challenging in humans, since genomic variation, cell of origin, and tumor microenvironment effects each contribute. Moreover, in the context of NF1, tumors are rarely biopsied or removed, and the few gliomas obtained have proven difficult to maintain in culture or grow as patient-derived xenografts in rodents.7 To define the potential molecular and cellular etiologies for pediatric low-grade glioma heterogeneity, we have leveraged Nf1-mutant mouse strains engineered to develop optic gliomas.8–11 Optic gliomas in the setting of NF1 are caused by a germline mutation in the NF1 tumor suppressor gene coupled with somatic NF1 loss, leading to biallelic NF1 inactivation.12 Similar to children with NF1, optic glioma formation in mice with a germline Nf1 gene mutation occurs following conditional somatic Nf1 loss in neuroglial progenitors during embryogenesis.8
The availability of this experimental platform allows for the introduction of different germline Nf1 gene mutations,11 the addition of other genomic changes,9 and somatic Nf1 loss in different progenitor cell populations.10 Using this strategy, the penetrance of optic gliomas, the latency to tumor formation, and the level of optic glioma growth can be varied, thus creating a population of genetically engineered mouse strains that more fully capture the clinical heterogeneity seen in children with NF1–optic glioma.
In the current study, a series of low-grade glioma stem cell preparations (optic glioma stem cells [o-GSCs]), generated from both previously reported and novel Nf1-mutant mouse strains with optic glioma, were used to demonstrate that mouse optic glioma biological variability is not accounted for by the intrinsic growth properties of the cancer cells. Instead, optic glioma proliferation in situ reflects the differential ability of o-GSCs from mice with different germline and genomic alterations to produce specific chemokines that recruit T cells and microglia. The collective action of T cells and microglia then dictates the levels of the key stromal factor (chemokine C-C ligand 5 [Ccl5]) that drives murine Nf1 optic glioma growth. Taken together, the deployment of these unique strains provided an experimental system to define differences in the immunologic landscape of pediatric low-grade glioma relevant to their biological variability.
Materials and Methods
Mice
All animals were maintained on a C57BL/6 background and used in accordance with an approved Animal Studies Committee protocol at Washington University. As previously described,8–11 we generated Nf1flox/neo; GFAP-Cre (FMC) mice (Nf1+/− mice with Nf1 gene inactivation in neuroglial progenitors), Nf1flox/neo; Ptenflox/wt; GFAP-Cre (FMPC) mice (additional Pten heterozygous deletion in FMC mice), and Nf1flox/neo; Olig2-Cre (FMOC) mice (Nf1+/− mice with Nf1 gene inactivation in Olig2+ precursor cells). Two new strains of mice were generated with one allele containing a nonsense mutation either in exon 10 (c.1149C>A; p.Cys383X) or in exon 28 (c.3827G>C; p.Arg1278Pro; generously provided by Dr Jonathan Epstein, University of Pennsylvania).13Nf1flox/1149; GFAP-Cre (FM1149C) and Nf1flox/1278; GFAP-Cre (FM1278C) mice were produced by intercrossing Nf1+/Cys383X or Nf1+/Arg1278Pro mice with Nf1flox/flox GFAP-Cre mice, respectively. Nf1flox/neoCcr2+/RFP; GFAP-Cre (FMC Ccr2+/RFP) mice were generated by intercrossing Nf1flox/neoCcr2+/RFP and Nf1flox/flox; GFAP-Cre mice.14
Optic Glioma Stem Cells
Optic nerves/chiasms were dissected from 3-month-old FMC, FM1149C, FM1278C, and FMPC mice, as well as from 6-month-old FMOC mice, and used to generate o-GSCs.15 Briefly, single cell suspensions were obtained by digesting the optic nerve/chiasm in Trypsin Digest Medium, followed by maintenance in neural stem cell (NSC) medium (61% Dulbecco’s modified Eagle’s medium [DMEM] low glucose, 35% neurobasal medium, 2 mM L-glutamine, 1% penicillin/streptomycin) supplemented with epidermal growth factor (EGF) (20 ng/mL), fibroblast growth factor (FGF) (20 ng/mL), 1% N2, and 2% B-27. For stem cell marker analysis, o-GSCs were fixed in 4% paraformaldehyde after 24 hours in culture, and stained with nestin, sex determining region Y–box 2 (sox2), brain lipid binding protein (BLBP), cluster of differentiation (CD)133, or Ki67 antibodies (Supplementary Table 1). To measure proliferation, o-GSCs were incubated with 5-ethynyl-2'-deoxyuridine (EdU) and EdU+ cells using the Click-iT EdU assay (Fisher Scientific). To measure apoptosis, 105 o-GSCs were plated into 24-well plates coated with poly-D-lysine (50 μg/mL) and fibronectin (10 μg/mL) in complete NSC medium. After 3 days, cells were grown in NSC medium without N2, B27, FGF, and EGF for 48 hours, fixed in 4% paraformaldehyde, and apoptosis quantitated using an in situ cell death detection kit (Sigma-Aldrich). Self-renewal was assessed using single o-GSC neurospheres from each mouse strain grown in complete NSC. After 7 days, the number of resulting secondary neurospheres was counted, using at least 10 neurospheres per strain. For conditioned medium (CM) experiments, 106 cells were seeded into 6-well plates with 2 mL of NSC medium, which was collected 3 days later. Mouse anti-Ccl2 (3 μg/mL, R&D Systems) and anti-Ccl12 (2.5 μg/mL, R&D Systems) antibodies were used for Ccl2/Ccl12 neutralizing assays. At least 3 independently isolated o-GSC preparations per mouse strain were employed for all experiments. Ras activity in the o-GSC isolates was measured using the RAS activation enzyme-linked immunosorbent assay (ELISA) (Cell Biolabs, STA-440), according to the manufacturer’s instructions.
T Cells
Mouse spleens were homogenized in phosphate buffered saline containing 0.1% bovine serum albumin and 0.6% Na-citrate, incubated with 120 units of DNase I for 15 min at room temperature, and filtered through a 30-µm strainer. Single cell suspensions were obtained after red blood cell lysis (eBioscience, 00433357) and negative selection using the pan-T-cell isolation kit II (Miltenyi Biotec, 130-095-130). T cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% fetal bovine serum (FBS). To assay migration, 105 cells were seeded into the upper chamber of each 24-well Transwell filter (5 µm; Corning) with 0.1 mL RPMI-1640 without FBS. Added to the lower chamber was 0.5 mL of o-GSC CM. After 4 hours, the medium in the lower chamber was collected, and lymphocytes at the lower chamber side of the membrane were counted. Where noted, FM1278C or FMPC CM was neutralized with mouse anti-Ccl2 antibody (0.04 µg/mL; R&D Systems), mouse anti-Ccl12 antibody (0.2 µg/mL; R&D Systems), or both, for 1 h at 37°C. Activated T cells were obtained by 48 hours CD3 (1 μg/mL) and CD28 (5 μg/mL) stimulation in vitro. CM from activated T cells (following CD3/CD28 activation)16 were collected for microglia Ccl5 induction experiments.
Microglia
Mouse brains were homogenized in the gentleMACS Dissociator (Miltenyi) for microglia isolation,16 and cells grown in Minimal Essential Medium Earle’s medium supplemented with 1 mM L-glutamine, 1 mM sodium pyruvate, 0.6% D-(+)-glucose, 100 μg/mL penicillin/streptomycin, 4% FBS, and 6% horse serum. After 2 weeks, microglia/macrophages were separated from astrocytes by shaking (200 g, 5 h, 37°C). Grown in T cell CM were 5 × 105 microglia/macrophages for 24 hours,16 and Ccl5 measured by ELISA (R&D Systems). To assay migration, 6000 microglia were seeded into the upper chamber of the 96-well Transwell filter (5 µm; Corning) with 50 µL F12-DMEM without FBS. Added to the lower chamber was 0.2 mL of o-GSC CM. Microglia on the lower chamber side of the membrane were fixed with 4% paraformaldehyde 4 hours later, and stained with 4′,6′-diamidino-2-phenylindole for quantitation.
Western Blotting
Western blotting was performed as previously reported15 using the antibodies in Supplementary Table 1.
Immunohistochemistry
Mice were euthanized and perfused with Ringer’s solution and then 4% paraformaldehyde. Optic nerves were paraffin embedded, and immunohistochemistry and cell counting performed using primary and secondary antibodies (Supplementary Table 1).16
Chemokine Arrays
For 3 days, 106 o-GSCs were seeded in plates containing 2 mL of complete NSC. Chemokines in the CM were detected using the Proteome Profiler Mouse Chemokine Array Kit (R&D Systems).
RNA Extraction and Real-Time Quantitative PCR
RNA from mouse optic nerves was isolated using Trizol reagent (Life Technologies). RNA from o-GSCs was isolated using the RNeasy kit (Qiagen). Real-time quantitative reverse transcription (qRT)–PCR was performed16 using specific primers (Supplementary Table 2). Values of ΔΔCT were calculated using H3 as an internal control.
Optic Nerve Measurements
Mouse optic nerves were microdissected and photographed, and the optic nerve diameters measured at the chiasm to calculate optic nerve volumes.17
Statistical Analyses
All data were analyzed using GraphPad Prism 6 software. Data between 2 groups were analyzed by unpaired 2-tailed Student’s t-tests. Data for multiple comparisons were analyzed with one-way ANOVA. Each experiment was repeated at least 3 times. Statistical significance was set at P < 0.05.
Results
In order to develop a platform to explore optic glioma biological heterogeneity, we generated a collection of Nf1 optic glioma mouse strains that included the original Nf1 optic glioma strain (FMC), in which a neomycin sequence was inserted into exon 31 of the Nf1 gene,8 as well as mice that differ with respect to the germline Nf1 gene mutation (c.1149C>A mutation [this report], FM1149C; c.3827G>C mutation, FM1278C13), the cell of origin (oligodendrocyte transcription factor 2 [Olig2+] progenitors, FMOC mice10), and the presence of an additional genomic alteration (Pten mutation in neuroglial progenitors, FMPC mice9) (Fig. 1A). At 3 months of age, FMC, FM1278C, and FMPC mice developed optic gliomas with increased proliferation (%Ki67+ cells; Fig. 1B). Strikingly, FMPC mice exhibit the most proliferative tumors, as previously reported.9 In contrast, FMOC mice do not develop tumors until 6 months of age.10 Lastly, only 20–25% of FM1149C mice (3/12 mice) develop optic gliomas (Supplementary Fig. 1).
Fig. 1.

Isolation of o-GSCs from the Nf1 optic glioma mouse strains. (A) Summary of the Nf1 GEM strains used in this study, which differ by the germline Nf1 gene mutation, cell of origin, or additional genetic alteration. (B) Nf1 optic glioma strains exhibit differences in overall tumor proliferation (%Ki67+ cells). (C) Schematic of the workflow used to isolate o-GSCs from the various Nf1 murine optic glioma strains. Asterisks indicate the location of tumor. (D) Stem cell markers (nestin, sox2, BLBP, and CD133) were expressed in the o-GSCs isolated from all of the Nf1 murine optic glioma strains examined. Data are presented as mean ± SEM. *P < 0.05, ∗∗∗P < 0.001; N.T. denotes the low penetrance of optic gliomas in the FM1149C strain. Scale bar, 40 μm.
Using this series of Nf1 genetically engineered mice, we first tested the hypothesis that the differences in tumor proliferation reflected the intrinsic growth properties of the neoplastic cells in these optic gliomas, which we term optic glioma stem cells (o-GSCs).15 For these studies, we generated at least 3 independent o-GSC isolates from 3-month-old FMC, FMPC, FM1149C, and FM1278C mice and 6-month-old FMOC mice, which could be stably propagated after 2 weeks in vitro (Fig. 1C). Each of these o-GSC populations expressed nestin, sox2, BLBP, and CD133, characteristic of NSCs (Fig. 1D).15 Surprisingly, FMC o-GSCs exhibited the highest level of proliferation, as determined by neurosphere diameter, EdU incorporation, Ki67 immunodetection, and direct cell counting (Fig. 2A–D). In addition, FMC o-GSCs showed the highest level of self-renewal at limiting dilution (Fig. 2E). This is in striking contrast to the overall tumor proliferation patterns observed in vivo, where FMPC tumors had the highest level of glioma proliferation.
Fig. 2.

Optic GSCs exhibit different levels of proliferation, self-renewal, and apoptosis. Optic GSC neurosphere diameters (A), %EdU+ cells (B), %Ki67+ cells (C), and direct cell counting (D) demonstrate that FMC o-GSCs exhibit the highest level of proliferation, as well as the highest levels of self-renewal. (E, F) Immunoblotting revealed more cleaved caspase-3 in FMPC o-GSCs after 48 hours starvation relative to FMC o-GSCs. (G) TUNEL assay revealed that FMPC o-GSCs have a higher percentage of TUNEL+ cells relative to FMC o-GSCs after 48 hours starvation. Data are presented as mean ± SEM. *P < 0.05, ∗∗∗P < 0.001. Scale bar: 40 μm.
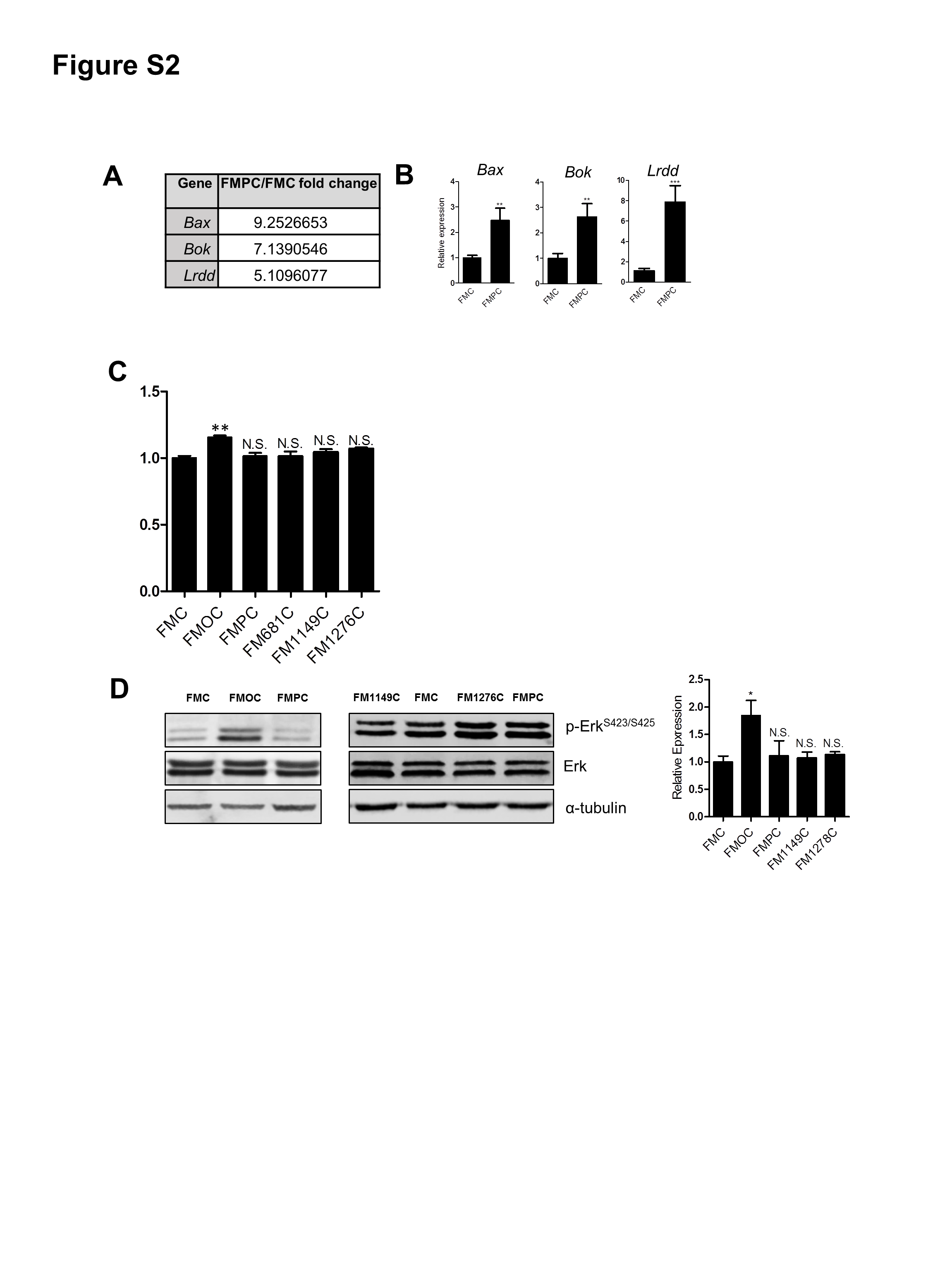
Since FMPC optic gliomas had much higher levels of tumor proliferation relative to their FMC counterparts in situ, we examined o-GSC survival in vitro. Leveraging prior RNA sequencing data from FMC and FMPC o-GSCs (Gene Expression Omnibus accession number, GSE102345),18 increased Bax, Bok, and Lrdd expression was detected in FMPC o-GSCs, which was validated by real-time qRT-PCR on freshly isolated o-GSCs (Supplementary Fig. 2A, B). Consistent with increased Bax, Bok, and Lrdd expression, FMPC o-GSCs had more cleaved caspase-3 expression (Fig. 2F) and percentage of cells positive for terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling (TUNEL) (Fig. 2G) relative to FMC o-GSCs following 48 h of growth factor starvation.
Lastly, the NF1 protein (neurofibromin) is a negative Ras regulator, such that o-GSCs harboring biallelic Nf1 gene inactivation should have higher levels of Ras activation. However, comparisons of the levels of Ras or Ras effector (extracellular signal-regulated kinase) activation did not reveal differences between the various o-GSC populations that could account for the observed differences in o-GSC proliferation or apoptosis (Supplementary Fig. 2C, D). Taken together, these unexpected results indicate that the cell autonomous growth properties of the neoplastic cells did not explain the differences in tumor proliferation seen in situ.
Next, we evaluated the hypothesis that the biological heterogeneity observed in the intact tumors reflected differential recruitment of immune system–like cells previously shown to regulate optic glioma formation and growth. In this regard, microglia (Iba1+ cells)19–21 and T cells (CD3+ cells)16 are required for tumor development and continued growth. As such, we quantified microglia (%Iba1+ cells) and T cell (number of CD3+ cells) content in each of the murine Nf1 optic gliomas. While FM1149C and FM1278C mice did not harbor more microglia than non-neoplastic (FF) optic nerves, FMPC optic nerves had 2-fold more microglia than either FMOC or FMC mice (Fig. 3A). While previous studies from our laboratory have shown that these optic glioma–associated monocytes were CD11b+/CD45low/CX3CR1+ microglia by fluorescence activated cell sorting, we used the microglia-specific marker, Tmem119,22 by immunohistochemistry to confirm their identity. Consistent with their designation as microglia, we observed similar patterns and percentages of Tmem119+ cells in the Nf1 optic gliomas (Fig. 3B). Moreover, there were more ramified Tmem119+ microglia in the FF and FM1149C optic nerves (no tumors), whereas more amoeboid microglia were detected in the FMC, FMC, FM1278C, and FMPC optic nerves bearing gliomas.
Fig. 3.

Differential infiltration of microglia and T cells in the Nf1 optic glioma strains. (A, B) FMC, FMOC, and FMPC mice exhibit more Iba1+ and Tmem119+ cells (microglia; %Iba1+ or %Tmem119+ cells) relative to FF mice, where FMPC optic gliomas have the highest levels of microglia infiltration. (C) FMC, FM1278C, and FMPC mouse optic gliomas have greater CD3+ T cell infiltration relative to FF mice, where FM1278C and FMPC murine optic gliomas have the highest level of T cell infiltration. (D) The majority of the CD3+ cells are CD8+ T cells. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar: 40 µm.
In contrast to microglia infiltration pattern, FMOC mice did not harbor more T cells in their optic gliomas, whereas both FM1278C and FMPC optic gliomas had ~2.5-fold more T cells than FMC mice (Fig. 3C). The differences between these strains are striking, specifically in the FM1278C strain, which exhibits a dissociation between the numbers of microglia and T cells. To characterize these infiltrating T cells, we performed immunohistochemistry using CD4 and CD8 antibodies. While no CD4+ T cells were found in the optic gliomas (data not shown), the majority of the T cells were CD8+ cells (Fig. 3D). When examined at the level of the individual tumor, nearly all CD3+ cells were also CD8+, with only a minor population of CD3+/CD8neg cells.
Given our recent finding that T cells can “educate” microglia to produce Ccl5, a key growth factor required for optic glioma formation and growth,21 we sought to determine whether Ccl5 levels correlated with proliferation in these tumors. RNA was extracted from the optic nerves of 3-month-old FF, FMC, FMPC, and FM1278C mice (n = 4). We specifically focused on these strains, in light of their differences in T cell and microglia content. While the optic gliomas from all three strains (FMC, FM1278C, and FMPC) exhibited higher Ccl5 expression relative to FF mice (Fig. 4A, B), the differences in Ccl5 gene expression correlated with the levels of optic nerve proliferation (%Ki67+ cells, R2 = 0.97; Fig. 4C). For these studies, we coupled Ccl5 levels by qRT-PCR analysis (Fig. 4A) with immunohistochemistry (Fig. 4B; %Ccl5+ cells), with identical results. Since Ccl5 levels are controlled by T cell interactions with microglia, we analyzed microglia (Fig. 4D) and T cell (Fig. 4E) content in each strain, and generated an “immune cell index,” representing the sum of the fold increases in T cell and microglia content relative to FF control mice (Fig. 4F). While neither T cell nor microglia content alone explained the Ccl5 levels, the combination of both (immune index) correlated with both optic nerve Ccl5 levels (Fig. 4G) and proliferation (Fig. 4H). FMOC optic gliomas also exhibited higher Ccl5 levels relative to normal FF optic nerves, whereas the optic nerves from FM1149C mice, which do not form gliomas with significant penetrance, did not exhibit higher Ccl5 expression (Supplementary Fig. 3).
Fig. 4.

Microglia and T cell content correlates with Ccl5 levels and Nf1 optic glioma proliferation. (A) Quantitative RT-PCR revealed increased optic glioma Ccl5 gene expression in FMPC (7-fold) and FM1278C (2.1-fold) mice relative to FMC mice. No Ccl5 expression was detected in FF optic nerves. (B) FMC, FM1278C, and FMPC mice exhibited a greater percentage of Ccl5+ cells relative to FF mice by immunohistochemistry. Scale bar: 40 μm. (C) Optic nerve Ccl5 expression correlates (R2 = 0.97) with relative tumor proliferation (relative %Ki67+ cells). Graphs demonstrating the %Iba1+ cells (D), number of CD3+ cells (E), and the sums of the relative increases of microglia and T cells in the FMC, FM1278C, and FMPC optic gliomas relative to normal (FF) optic nerve (F). The sum of the relative increases of microglia and T cells combined correlates (R2 = 0.97) with optic nerve (G) Ccl5 expression and (H) proliferation (relative %Ki67+ cells). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01.
To determine the etiologic basis for these differences in glioma T cell and microglia recruitment, we hypothesized that the neoplastic cells themselves might produce chemokines, which recruit T cells and microglia. Conditioned medium collected from FMC, FMPC, and FM1278C o-GSCs was assayed using a commercially available chemokine array. Increased Cx3cl1, Cxcl6, Cxcl10, Ccl2, and Ccl12 were detected in the CM from FMPC relative to FMC o-GSCs (Fig. 5A, Supplementary Fig. 4A–C). In addition, Ccl12 was increased, but Cx3cl1, Cxcl1, and Ccl2 levels were reduced in FM1278C relative to FMC o-GSC CM. Since these array experiments did not provide quantitative data, we measured the expression of these chemokines at the mRNA level using qRT-PCR (Fig. 5B, Supplementary Fig. 4D, E). Because only differential expression of Cx3cl1, Ccl2, and Ccl12 were validated (Fig. 5B, Supplementary Fig. 4D), these 3 chemokines were chosen for further study.
Fig. 5.

Differential o-GSC chemokine production dictates microglia and T cell infiltration. (A) FMC and FMPC o-GSC CM have the highest levels of Cx3cl1 expression, FMPC the highest level of Ccl2 expression, and FM1278C and FMPC the highest level of Ccl12 expression. (B) Differential Cx3cl1, Ccl2, and Ccl12 RNA expression was confirmed by qRT-PCR. (C) Greater microglia migration was observed with FMPC o-GSC CM relative to FMC or FM1278C o-GSC CM. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
First, we evaluated the expression of the receptors for these chemokines (Cxcl1-Cxcr2, Cx3cl1-Cx3cr1, Ccl2-Ccr2/Ccr4, and Ccl12-Ccr2) in microglia using online RNA expression atlases (Supplementary Figures 5, 6).23,24 Consistent with earlier reports,25 only Cx3cr1 was highly expressed in microglia. The expression of Cx3cr1 on microglia is consistent with our finding that the CM from 2 o-GSCs with the highest Cx3cl1 (Cx3cr1 ligand) expression (FMC and FMPC) attracted more microglia than CM from FM1278C o-GSCs (Fig. 5C). This observation also correlates to the microglia infiltration pattern in FMC and FMPC optic gliomas, relative to their FM1278C counterparts (Fig. 3A, B). The importance of Cx3cr1 to Nf1 optic glioma formation is further underscored by the observation that FMC mice with reduced Cx3cr1 expression have delayed optic glioma formation.20
Second, to determine whether Ccl2 and Ccl12 were responsible for T cell attraction, we examined the expression of their cognate receptors using the Immunological Genome Project online database RNA expression atlas23 (Supplementary Fig. 6). As expected, Ccr2 and Ccr4 are highly expressed in T cells. Consistent with a role for Ccr2 in Nf1 optic glioma T cell infiltration, the number of CD3+ cells was reduced in FMC mice heterozygous for a knockout Ccr2 allele (Ccr2RFP/+ mice; Fig. 6A). Consistent with a critical role for Ccr2 engagement in the pathogenesis of Nf1 optic glioma, the percentages of Ki67+ cells and Ccl5+ cells were reduced in FMC-Ccr2RFP/+ mice relative to their FMC counterparts (Fig. 6B, C). Since FM1278C o-GSCs produce mainly Ccl12, we sought to determine whether Ccl12 inhibition alone would attenuate T cell migration using a Transwell assay. Following exposure to Ccl12 neutralizing antibodies, T cell migration was reduced (Fig. 6D).
Fig. 6.

Optic glioma T cell infiltration in Nf1 GEM is mediated by Ccl2 and Ccl12. (A–C) FMC-Ccr2+/− mice have fewer T lymphocytes, Ccl5+ cells, and Ki67+ cells in their optic nerves relative to those from FMC mice. Inset depicts a representative CD3+, Ccl5+, and Ki67+ cell. (D) Greater T cell migration was observed with FM1278C relative to FMC CM, which was blocked with Ccl12 neutralizing antibodies. (E) Greater T cell migration was observed with FMPC compared with FMC o-GSC CM, which was inhibited by the addition of neutralizing Ccl2 and Ccl12 antibodies. (F) Proposed model for the differential stromal cell recruitment by o-GSCs. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01. Scale bar: 40 μm.
Third, FMPC o-GSCs produce both Ccl2 and Ccl12, which each could attract T cells. To determine whether these chemokines performed redundant functions, FMPC o-GSC CM was added to T cells in a Transwell assay in the presence of Ccl2 and Ccl12 neutralizing antibodies. While Ccl2 and Ccl12 neutralizing antibodies each reduced T cell migration in response to FMPC CM, the addition of Ccl2 and Ccl12 neutralizing antibodies together exhibited an additive effect, suggesting that both chemokines are involved in T cell attraction by FMPC o-GSCs (Fig. 6E).
Discussion
One of the barriers to the implementation of precision oncology is an incomplete elucidation of the molecular and cellular etiologies that underlie tumor heterogeneity.26 This is challenging using human tumor specimens, as the variables that underlie differential tumor variability cannot be easily controlled. Moreover, the experimental platforms that currently exist exclude critical components of tumor biology, notably the tumor microenvironment, which provides key paracrine support to the growing tumor.27,28 To circumvent some of these obstacles, we have previously deployed murine strains genetically engineered to differentially harbor distinct determinants that each could contribute to glioma growth. In the current study, we leveraged a small series of novel Nf1 mutant mouse strains, incorporating some of these critical variables, to reveal that cancer stem cells create differences in tumor growth through the elaboration of specific chemokines that attract T cells and microglia and create a supportive microenvironment. These findings raise several important points germane to our understanding of low-grade brain tumors.
First, the composite behavior of the tumor reflects synergistic relationships between cancer and noncancer cells. These critical contributions of the tumor microenvironment to glioma growth have been previously revealed using both low-grade and high-grade astrocytoma mouse models.27,28 As such, the processes that control monocyte (microglia and macrophage) tumor infiltration and activation include the requirement for colony stimulating factor 1 receptor,29 CX3CL1 receptor (CX3CR1),20,30 and CCL5 receptor function.16,21 To further demonstrate that o-GSC–produced chemokines determine the expression of stromal factors that drive immune cell infiltration and optic glioma growth, future in vivo experiments, such as those employing the inhibition of key chemokines (eg, Cx3cl1, Ccl12) as previously performed using Ccl2 neutralizing antibodies,16 will be necessary. Coupling these experiments with genetic strategies and tumor explant modeling will help to firmly establish the requirements for this immune circuitry.
Moreover, other tumor–stroma circuit interactions involve versican activation of microglia31 and toll-like receptor engagement.32 In this regard, human PA tumor cell lines frequently undergo senescence in culture, which may reflect the need for trophic factors produced by non-neoplastic cells, which are not currently incorporated into these in vitro model systems.7,33 The established network of supportive signals from the tumor microenvironment, especially in the case of low-grade gliomas, highlights the need to consider therapies that target these co-dependencies. This is underscored by the findings in this report, which demonstrate that cell-intrinsic differences in o-GSC growth in vitro do not account for the observed heterogeneity in optic glioma growth in vivo. Instead, it is the combined effects of T cells and microglia infiltration by o-GSCs, leading to the differential Ccl5 production, which are ultimately responsible for dictating the growth pattern of the tumor as a whole.
Second, the impact of specific germline NF1 gene mutation on the intrinsic tumor cell properties (apoptosis, proliferation) appears to be limited, since all patient derived Nf1 gene mutations were similar with respect to o-GSC growth. The notable exception was o-GSCs from genetically engineered mice (GEM) strains harboring the artificial Nf1 knockout allele. It is not clear why o-GSCs from mice with this non-naturally occurring mutation exhibit higher levels of proliferation and self-renewal. While this mutation affects the Ras GTPase activating protein domain (similar to the Arg1278 mutation), this proliferative advantage is not due to higher levels of Ras or mitogen-activated protein kinase kinase (MEK) activation. Preliminary experiments suggest that these differences might result from variations in levels of cyclic adenosine monophosphate (cAMP); however, the mechanism(s) governing cAMP homeostasis in o-GSCs are currently not known. Studies are currently in progress to expand these analyses to o-GSCs from additional Nf1 optic glioma mouse strains with different germline Nf1 gene mutations, as well as to explore other signaling pathways activated in these neoplastic cells. Nonetheless, these findings underscore the importance of the tumor microenvironment to tumor proliferation involving non–cell autonomous (stromal) drivers of glioma growth.
Third, the impact of coexisting genomic alterations on the non–cell autonomous properties of tumor growth provides an additional mechanism to control stromal production of growth factors and influence glioma biology. In this regard, studies using experimental models of high-grade glioma have demonstrated that AXL regulates the glioblastoma microenvironment,34 enhancer of zeste homolog 2 controls monocyte M1 phenotypes,35 and isocitrate dehydrogenase modulates immune system infiltration.36 While the number of additional genomic alterations in PA is small, mutations in ATRX, ALT, KIAA1549:BRAF, PTEN, FGFR1, and H3-K27M have been reported.4,37–40 As observed in o-GSCs from murine Nf1 optic glioma harboring a heterozygous Pten mutation, a different chemokine module accompanies Pten mutation, leading to both increased T cell and microglia infiltration, as well as higher levels of tumoral Ccl5 expression. Collectively, these findings support the idea that additional mutations confer unique properties on cancer stem cells to facilitate the establishment of a supportive microenvironment through differential chemokine production and immune cell recruitment.
Fourth, the cooperativity between T cells and microglia is highlighted by the finding that Ccl5 levels reflect the abundance of T cells and microglia. Recently, we showed that T cells produce little Ccl5, even following CD28/CD3 activation, but are capable of stimulating microglia to produce Ccl5 through the elaboration of paracrine signals.16 In this manner, we envision that similar levels of resident microglia Ccl5 expression in the Arg1278 optic glioma strain relative to the Nf1 knockout strain results from greater T cell infiltration and microglia Ccl5 induction by the larger number of coexisting T cells. Along those lines, the highest levels of tumor Ccl5 expression were observed in the Nf1 optic glioma strain harboring a heterozygous Pten deletion. These o-GSCs had the highest levels of chemokine expression, especially Cx3cl1, Ccl2, and Ccl12, which are likely responsible for attracting T cells and microglia and facilitating maximal microglia education and Ccl5 production. The importance of T cells to microglia function and the establishment of a supportive tumor microenvironment is highlighted by studies in human tumors, where CD4+ T cell content positively correlated with tumor grade.41–43 Studies are currently under way to define the immunologic circuitry that attracts T cells and microglia, physiologically activates T cells, and culminates in T cell–mediated microglia reprogramming, Ccl5 production, and enhanced glioma growth (Fig. 6F).
Taken together, the deployment of a series of Nf1 GEM optic glioma strains with different germline Nf1 gene mutations, secondary genomic alterations, and cell of origin provides an unprecedented opportunity to explore the cellular and molecular etiologies for low-grade glioma clinical diversity. The finding that these etiologies converge on cancer stem cell chemokine attraction of microglia and T cells establishes the foundation for co-dependent glioma circuitry. In this regard, low-grade gliomas likely harbor multiple vulnerabilities that reflect these stromal dependencies, which could be exploited to design more effective therapies. Finally, our ability to dissect the factors that create heterogeneity provides additional opportunities to understand the molecular and cellular pathogenesis of these tumors, as well as to potentially develop more personalized treatments for these common childhood gliomas.
Funding
This work was funded by grants from the National Cancer Institute (1-R01-CA214146-01) and the National Institute of Neurological Disorders and Stroke (1-R35-NS07211-01) to D.H.G, as well as UL1-TR000448 (Hope Center Viral Vectors Core and the Genome Technology Access Center). Y.P. was supported by a fellowship grant from the James S. McDonnell Foundation.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We appreciate the technical assistance of Ms Yu Ma.
Conflict of interest statement.
None declared.
Authorship statement
XG and DHG conceived the study. XG and YP performed the experiments. XG and DHG wrote the manuscript. DHG supervised the study.
References
- 1. Sadighi Z, Slopis J. Pilocytic astrocytoma: a disease with evolving molecular heterogeneity. J Child Neurol. 2013;28(5):625–632. [DOI] [PubMed] [Google Scholar]
- 2. Rodriguez FJ, Ligon AH, Horkayne-Szakaly I, et al. BRAF duplications and MAPK pathway activation are frequent in gliomas of the optic nerve proper. J Neuropathol Exp Neurol. 2012;71(9):789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68(21):8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jones DT, Hutter B, Jäger N, et al. ; International Cancer Genome Consortium PedBrain Tumor Project Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45(8):927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63–66. [DOI] [PubMed] [Google Scholar]
- 6. Campen CJ, Gutmann DH. Optic pathway gliomas in neurofibromatosis type 1. J Child Neurol. 2018;33(1):73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raabe EH, Lim KS, Kim JM, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17(11):3590–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bajenaru ML, Hernandez MR, Perry A, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63(24):8573–8577. [PubMed] [Google Scholar]
- 9. Kaul A, Toonen JA, Gianino SM, Gutmann DH. The impact of coexisting genetic mutations on murine optic glioma biology. Neuro Oncol. 2015;17(5):670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Solga AC, Toonen JA, Pan Y, et al. The cell of origin dictates the temporal course of neurofibromatosis-1 (Nf1) low-grade glioma formation. Oncotarget. 2017;8(29):47206–47215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Toonen JA, Anastasaki C, Smithson LJ, et al. NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum Mol Genet. 2016;25(9):1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gutmann DH, Donahoe J, Brown T, James CD, Perry A. Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol Appl Neurobiol. 2000;26(4):361–367. [DOI] [PubMed] [Google Scholar]
- 13. Yzaguirre AD, Padmanabhan A, de Groh ED, et al. Loss of neurofibromin Ras-GAP activity enhances the formation of cardiac blood islands in murine embryos. Elife. 2015;4:e07780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saederup N, Cardona AE, Croft K, et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One. 2010;5(10):e13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen YH, McGowan LD, Cimino PJ, et al. Mouse low-grade gliomas contain cancer stem cells with unique molecular and functional properties. Cell Rep. 2015;10(11):1899–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pan Y, Xiong M, Chen R, et al. Athymic mice reveal a requirement for T-cell-microglia interactions in establishing a microenvironment supportive of Nf1 low-grade glioma growth. Genes Dev. 2018;32(7-8):491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hegedus B, Banerjee D, Yeh TH, et al. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res. 2008;68(5):1520–1528. [DOI] [PubMed] [Google Scholar]
- 18. Pan Y, Bush EC, Toonen JA, et al. Whole tumor RNA-sequencing and deconvolution reveal a clinically-prognostic PTEN/PI3K-regulated glioma transcriptional signature. Oncotarget. 2017;8(32):52474–52487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet. 2007;16(9):1098–1112. [DOI] [PubMed] [Google Scholar]
- 20. Pong WW, Higer SB, Gianino SM, Emnett RJ, Gutmann DH. Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Ann Neurol. 2013;73(2):303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Solga AC, Pong WW, Kim KY, et al. RNA sequencing of tumor-associated microglia reveals Ccl5 as a stromal chemokine critical for neurofibromatosis-1 glioma growth. Neoplasia. 2015;17(10):776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bennett ML, Bennett FC, Liddelow SA, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113(12):E1738–E1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Y, Chen K, Sloan SA, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shay T, Kang J. Immunological genome project and systems immunology. Trends Immunol. 2013;34(12):602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liang KJ, Lee JE, Wang YD, et al. Regulation of dynamic behavior of retinal microglia by CX3CR1 signaling. Invest Ophthalmol Vis Sci. 2009;50(9):4444–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seoane J, De Mattos-Arruda L. The challenge of intratumour heterogeneity in precision medicine. J Intern Med. 2014;276(1):41–51. [DOI] [PubMed] [Google Scholar]
- 27. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31(3):326–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feng X, Szulzewsky F, Yerevanian A, et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget. 2015;6(17):15077–15094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hu F, Dzaye O, Hahn A, et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro Oncol. 2015;17(2):200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vinnakota K, Hu F, Ku MC, et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro Oncol. 2013;15(11):1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jacob K, Quang-Khuong DA, Jones DT, et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res. 2011;17(14):4650–4660. [DOI] [PubMed] [Google Scholar]
- 34. Sadahiro H, Kang KD, Gibson JT, et al. Activation of the receptor tyrosine kinase AXL regulates the immune microenvironment in glioblastoma. Cancer Res. 2018;78(11):3002–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yin Y, Qiu S, Li X, Huang B, Xu Y, Peng Y. EZH2 suppression in glioblastoma shifts microglia toward M1 phenotype in tumor microenvironment. J Neuroinflammation. 2017;14(1):220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amankulor NM, Kim Y, Arora S, et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017;31(8):774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rodriguez FJ, Brosnan-Cashman JA, Allen SJ, et al. Alternative lengthening of telomeres, ATRX loss and H3-K27M mutations in histologically defined pilocytic astrocytoma with anaplasia. Brain Pathol. 2019;29(1):126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Orillac C, Thomas C, Dastagirzada Y, et al. Pilocytic astrocytoma and glioneuronal tumor with histone H3 K27M mutation. Acta Neuropathol Commun. 2016;4(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reinhardt A, Stichel D, Schrimpf D, et al. Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol. 2018;136(2):273–291. [DOI] [PubMed] [Google Scholar]
- 40. Becker AP, Scapulatempo-Neto C, Carloni AC, et al. KIAA1549: BRAF gene fusion and FGFR1 hotspot mutations are prognostic factors in pilocytic astrocytomas. J Neuropathol Exp Neurol. 2015;74(7):743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Han S, Zhang C, Li Q, et al. Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Br J Cancer. 2014;110(10):2560–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang W, Wu S, Guo K, Hu Z, Peng J, Li J. Correlation and clinical significance of LC3, CD68+ microglia, CD4+ T lymphocytes, and CD8+ T lymphocytes in gliomas. Clin Neurol Neurosurg. 2018;168:167–174. [DOI] [PubMed] [Google Scholar]
- 43. Yang I, Han SJ, Sughrue ME, Tihan T, Parsa AT. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: evidence of distinct immunological microenvironments that reflect tumor biology. J Neurosurg. 2011;115(3):505–511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.