

Abstract
Although heterogeneity in the observed outcomes in clinical trials is often assumed to reflect a true heterogeneous response, it could actually be due to random variability. This retrospective analysis of 4 randomized, double-blind, placebo-controlled multi-period (i.e., episode) cross-over trials of fentanyl for breakthrough cancer pain illustrates the use of multi-period cross-over trials to examine heterogeneity of treatment response. A mixed effects model including fixed effects for treatment and episode and random effects for patient and treatment-by-patient interaction was used to assess the heterogeneity in patients’ responses to treatment during each episode. A significant treatment-by-patient interaction was found for 3 of 4 trials (p < 0.05), suggesting heterogeneity of the effect of fentanyl among different patients in each trial. Similar analyses in other therapeutic areas could identify conditions and therapies that should be investigated further for predictors of treatment response in efforts to maximize the efficiency of developing precision medicine strategies.
Keywords: Precision or personalized medicine, heterogeneity, multi-period cross-over trials
Introduction
It is often assumed both in medical practice and in published interpretations of randomized clinical trials (RCTs) that patients respond differently to the same medical intervention; that is, some patients respond very well, while others respond poorly or not at all.1,2 In response to these assumptions, which are largely unproven, substantial investments have been made to advance “precision medicine,” that is, strategies to identify patients who would have a greater response to (or less harm from) treatment using either genotyping or phenotyping. There are many highly beneficial implications of such precision. Patients and study participants would not be unnecessarily exposed to treatments from which they are not likely to receive benefit and the funds spent on ineffective or harmful treatments would decrease. In fact, this approach has been successful for treating tumors with specific oncogenic mutations.3 However, differential responses to treatment based on baseline participant (rather than tumor) phenotype or genotype have rarely been identified in RCTs.4,5 RCTs have an advantage in testing these hypotheses because, unlike observational studies, they can minimize unmeasured confounding through randomization. However, these types of RCTs require specific hypotheses regarding phenotypic or genotypic predictors of treatment response in order to define the relevant subgroups. Before committing major investments of effort and funding to discover possible phenotypes or genotypes that predict differential treatment responses, it would be prudent to rigorously test whether patients truly respond differently to particular treatments.6
In RCTs, substantial variability in observed outcomes is often present and it is intuitive to assume that this variability occurs because patients are inherently different in terms of how they respond to treatment. However, there are 4 possible sources of variability in outcomes that are observed in clinical trials: (1) variability from the treatment effect (i.e., the difference in mean outcome between the treatment groups), (2) between-patient variability (i.e., variability in the outcome for different patients, irrespective of the treatment), (3) within-patient variability (i.e., variability in the outcome over time, for a given treatment, for individual patients); later, we refer to the latter as “residual” error, and (4) treatment-by-patient interaction (i.e., variability in how patients respond to the treatment).6–10 Variability from (2) and (3) above can erroneously be interpreted as patient variability in treatment response in parallel group trials or clinical observation.
In order to investigate whether the effect of a treatment (vs. placebo) truly varies between patients (i.e., treatment-by-patient interaction exists), patients must be exposed to at least one of, and ideally both of, the treatment and placebo on at least 2 occasions using a multi-period cross-over design.9 To our knowledge, few, if any, previous studies have utilized data from multi-period cross-over trials that evaluate each treatment at least twice in order to directly test whether patients’ seemingly variable treatment response is truly due to a treatment-by-patient interaction.9 We analyzed data from 4 multi-period cross-over trials conducted in patients with breakthrough cancer pain (i.e., relatively brief episodes of severe pain occurring in patients with ongoing cancer pain) to examine whether responses to treatment with fentanyl were truly heterogeneous among patients. The goal of this article is to provide an illustrative example of how data from multi-period cross-over trials can be used to examine the degree of variability in patient response to specific treatments. Such studies do not themselves identify predictors of treatment response, but they could represent an efficient strategy to help to inform the decision regarding whether or not to allocate resources for investigations of phenotypic and genotypic predictors of treatment response.
Results
Table 1 summarizes the following characteristics of each trial: (1) the number of participants enrolled in the dose titration and double-blind randomized phases, (2) the number of participants who provided at least 1 post-treatment pain assessment for (a) at least 1 episode of placebo and active treatment and (b) for all planned episodes, and (3) the number of active and placebo episodes that contributed to the analyses for each trial.
Table 1.
Trial characteristics
| Trial | 099–14 | CP-043 | FEN-201 | INS-05–001 |
|---|---|---|---|---|
| Primary publication | Portenoy, 2006 | Portenoy, 2010 | Rauck, 2010 | Rauck 2012 |
| DESIGN FEATURES | ||||
| Response definition for enrollment | At least 2 consecutive episodes at a particular dose yielded satisfactory pain relief without unacceptable AEs | At least 2 consecutive episodes at a particular dose yielded acceptable pain relief without unacceptable AEs | At least 2 episodes at a particular dose yielded satisfactory pain relief | At least 2 consecutive episodes at a particular dose yielded acceptable pain relief without unacceptable AEs |
| Episodes | 7 active, 3 placebo | 7 active, 3 placebo | 6 active, 3 placebo | 7 active, 3 placebo |
| SPID calculation | (0–15)*15+(0–30)*15 | (0–5)*5+(0–10)*5+ (0–15)*5+(0–30)*15 | (0–5)*5+(0–10)*5+ (0–15)*5+(0–30)*15 | (0–5)*5+(0–10)*5+ (0–15)*5+(0–30)*15 |
| Restrictions on sequence order | 1 of 18 possible sequences; the first episode could not be placebo and 2 consecutive episodes could not be placebo | Not indicated in paper or study report synopsis | 2 consecutive episodes could not be placebo | Not indicated in paper or study report synopsis |
| RESULTS | ||||
| Number of participants enrolled in the dose-titration phase | 123 | 114 | 151 | 130 |
| Number of participants randomized | 77 | 83 | 82 | 98 |
| Number of participants who provided data for all 10 (9) episodes | 61 | 48 | 46 | 81 |
| Number of participants who provided post-treatment data for at least 1 active and 1 placebo episode (included in the analyses) | 72 | 73 | 75 | 93 |
| Number of active episodes with data from at least 1 post-treatment time point | 490 | 459 | 392 | 635 |
| Number of placebo episodes with data from at least 1 post-treatment time point | 206 | 200 | 196 | 269 |
AE: adverse event; SPID: summed pain intensity difference
Numbers within parentheses for the SPIC calculation refer to the pain score at that time in minutes post dose. Thus, for example, (0–15) represents the pain score at the time of the dose of medication minus the pain score 15 minutes after the dose of medication.
A significant treatment-by-patient interaction was observed for the individual analyses of 3 of the 4 trials. Although the variance component related to treatment-by-patient interaction was statistically significant in 3 trials, the magnitude of this variance component was small in all cases relative to the magnitude of the variance component associated with residual random error (Table 2). Figure 1 presents the predictions (i.e., shrunken estimates) from the statistical models of the mean difference in SPID30 between placebo and active episodes for each patient in each of the 4 trials, with associated 95% confidence intervals, sorted by the magnitude of the estimated mean difference in SPID30 between active and placebo treatments. Figure 2 presents plots of the naïve vs. the shrunken estimates of the treatment effects for each patient in each of the 4 trials. Heterogeneity of the treatment effects among different patients (i.e., the treatment-by-patient interaction) is apparent from Figure 1; however, the degree of shrinkage in the estimated treatment effects for individual patients is substantial in Figure 2, suggesting that the between-patient heterogeneity in treatment effect is modest relative to the within-patient variability in the treatment effect (see Methods section for more guidance on interpretation of these analyses).
Table 2.
Estimates of variance components from the mixed effects model
| Estimates of variance by trial | Estimate | P-value |
|---|---|---|
| 099–14 | ||
| Patient | 988.7 | <0.0001 |
| Treatment-by-patient | 166.0 | 0.0202 |
| Residual | 1244.5 | |
| CP-043 | ||
| Patient | 1163.7 | <0.0001 |
| Treatment-by-patient | 249.2 | 0.0009 |
| Residual | 1142.3 | |
| FEN-201 | ||
| Patient | 823.7 | <0.0001 |
| Treatment-by-patient | 133.8 | 0.0878 |
| Residual | 1194.8 | |
| INS-05–001 | ||
| Patient | 1110.4 | <0.0001 |
| Treatment-by-patient | 287.6 | 0.0001 |
| Residual | 1473.7 | |
Figure 1.
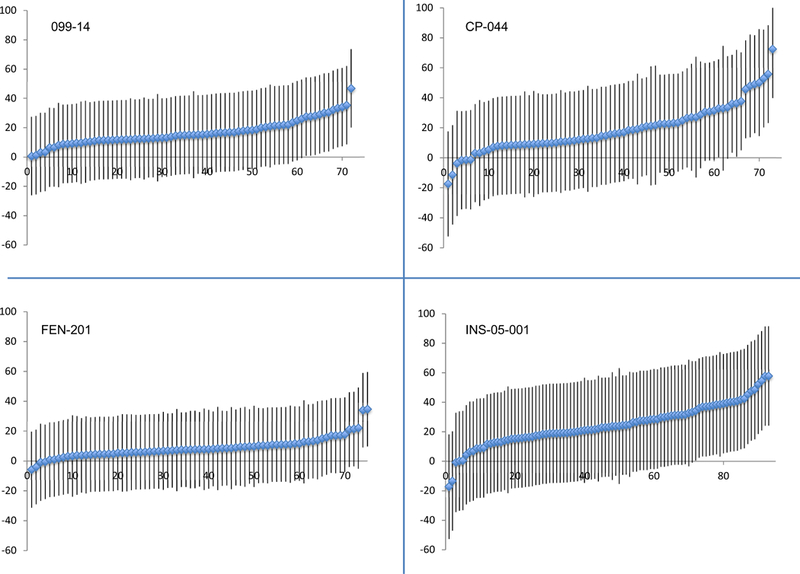
Shrunken estimates of treatment effects (active episodes – placebo episodes) for individual patients in each of the 4 trials, with associated 95% confidence intervals.
Figure 2.
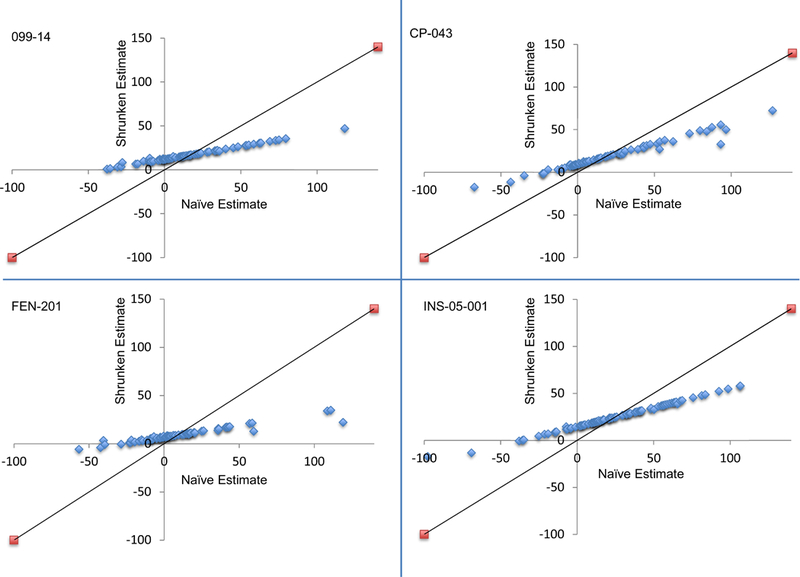
Plots of the naively estimated treatment effects (active episodes – placebo episodes) (X-axis) vs. the predicted treatment effects (shrunken estimates) (Y-axis) for individual patients in each of the 4 trials. The solid line indicates the expected pattern in the absence of shrinkage (i.e., if there were no within-patient variability in the estimated treatment effect). The substantial shrinkage indicates that the observed between-patient heterogeneity in the treatment effect, while statistically significant in the 099–14, CP-043, and INS-05–001 trials, is modest relative to the within-patient variability.
Discussion
The analyses performed in the illustrative example included in this article demonstrate that some of the variability in response to treatment with fentanyl for breakthrough cancer pain is indeed due to treatment-by-patient interaction, that is, that there are differences among patients in how they respond to fentanyl treatment (Figure 1). Nevertheless, the magnitude of this component of variation was modest. Our analyses also demonstrate that it is feasible to identify treatment-by-patient interaction using data from multi-period crossover trials. If a meaningful treatment-by-patient interaction is not identified in a multi-period crossover trial, the implication is that future efforts to identify phenotypic or genotypic predictors of treatment effect are not likely to be successful.9 Examples of similar ongoing efforts include a clinical trial to evaluate the effects of melatonin on sleep in a stimulant-treated ADHD population, which plans to enroll 300 participants with 3 active and 3 placebo periods.19 Another published protocol outlines a planned trial with 200 patients to evaluate statin-induced muscle symptoms with 3 active and 3 placebo periods.20 Analyses of these data similar to the ones performed here may help to determine whether it is worthwhile to invest resources to identify predictors of response to drugs with known efficacy in particular therapeutic areas. Subsequent research to identify predictors of treatment response would be highly useful to improve the efficacy, safety, and efficiency of clinical care.
An alternative to using multi-period cross-over trials to identify potential heterogeneity in treatment response is to perform exploratory subgroup analyses in trials that were aimed at determining the main effect of a treatment. This is commonly done in Phase 3 trials, but it is usually the case that several subgroups are examined and the power to detect anything other than very large subgroup differences in response to treatment is relatively low.21 Reproducibility of such findings that could be due to chance would be challenging and would require large scale trials for confirmation, that is, trials that could be avoided if the absence of substantial heterogeneity were determined in an efficiently designed trial.
Although conducting parallel group trials with placebo arms after a drug has been approved by regulatory agencies may be challenging, performing multi-period cross-over trials that could also function as N-of-1 trials22–25 to help individual patients determine whether a drug is actually providing them benefit over placebo is certainly feasible. For example, series of N-of-1 trials that evaluate the usefulness of interventions for chronic pain have been published.26–28
This study has strengths and limitations. The analysis was repeated in 4 data sets from trials of different formulations of the drug and the treatment-by-patient interaction yielded a p-value < 0.05 in 3 of them, with the p-value in the other trial being 0.09, providing consistent support for heterogeneity among patients of the effect of fentanyl for treating breakthrough cancer pain. These results are limited by the fact that they were performed using data from trials of a single class of therapy in a single pain condition; they are therefore not generalizable to other therapies or conditions. The analysis treated “episode” as the treatment period in the statistical analysis; however, unlike most cross-over trials in which the timing and duration of a treatment period are fixed, the breakthrough pain episodes occurred at different times in different patients and the duration of time between episodes varied between patients. In addition, variability in the type of pain experienced during different episodes could have contributed to variability in treatment response between patients. The models used assume a continuous normal mixture and one could postulate other models such as a finite mixture of two distinct groups of, say, “responders” and “non-responders”. Such models may be more difficult to fit and may require larger sample sizes. We did not have access to patient-level adverse event (AE) data. It is possible that participants who had better response to active treatment experienced more AEs on treatment than the patients who responded less consistently, and therefore the observed treatment-by-patient interaction could partially be due to greater unblinding of the treatment assignment in some patients than in others. It is also possible that differential patient response to treatment could be due to differential exposure to the treatment (e.g., variable drug concentrations in people based on weight), which was not measured in these trials. Collection of pharmacokinetic and pharmacodynamic data would be important in any trial designed to examine the issue of whether variation in drug exposure is associated with the heterogeneity in treatment response.
Cross-over trials are not appropriate for all conditions and therapies (e.g., rapidly progressing conditions such as cancer, Alzheimer’s disease, or ALS or curative therapies such as antibiotics). However, they would be applicable in many other conditions such as insomnia, migraines, asthma, chronic pain, depression, anxiety, fatigue. Also, although the approach presented here includes a statistical test for heterogeneity of treatment response and a method to estimate the magnitude of the observed heterogeneity, it does not determine the clinical importance of the magnitude of this heterogeneity. Such judgements regarding clinical importance would depend on the treatment characteristics (including adverse effects and costs) and the disease. For example, clinical implementation of treatments that are associated with moderate to severe side effects, especially those that are potentially irreversible, or treatments that are very expensive, would likely benefit more from having valid predictors of treatment response. In such cases, a relatively low degree of heterogeneity could motivate pursuit of a future study of predictors of treatment response.
Future studies using methodologies similar to the one presented here in other therapeutic areas and with other treatments, where applicable, could help to inform efficient allocation of resources for investigations of phenotypic and genotypic predictors of treatment response. Identification of such predictors could enhance efficiency of therapeutic development and clinical implementation, decreasing monetary costs and patient exposure to potentially harmful therapies with low likelihood of robust response.
Methods
This secondary data analysis included data from 4 multi-period cross-over trials for breakthrough cancer pain. The data were obtained from the Food and Drug Administration (FDA) database through a broad agency agreement between the FDA and the Analgesic, Anesthetic, and Addiction Clinical Trial Translations, Innovations, Opportunities, and Networks (ACTTION) public-private partnership. These data include all of the multi-period crossover trials of fentanyl for breakthrough cancer pain that were available electronically, which were phase 3 trials for bioerodible mucoadhesive fentanyl (FEN-201),11 fentanyl buccal tablet (099–14),12 fentanyl nasal spray (CP-043),13 and fentanyl sublingual spray (INS-05–001).14 The protocol for these retrospective analyses was approved by the University of Rochester Medical Center Research Subjects Review Board. Informed consent was obtained from all patients enrolled in the original trials.
The 4 double-blind RCTs all had similar study designs in which eligible patients had cancer, were being treated with opioid analgesics for cancer pain but were experiencing breakthrough cancer pain, and had a sufficient response to fentanyl treatment for breakthrough cancer pain episodes in an open-label titration period. “Sufficient response” was defined as significant pain relief from 2 consecutive episodes in the titration period. In at least 3 of 4 trials, the 2 episodes were also required to be without unacceptable adverse events (Table 1). The eligible participants then received 3 doses of placebo and 6 or 7 doses of fentanyl in random order and in a double-blind fashion to treat 9 or 10 consecutive breakthrough cancer pain episodes. The primary efficacy endpoint for each trial was the summed pain intensity difference score for 30 minutes post dose (SPID30). For these analyses, we calculated the SPID30 value for each trial using a time weighted formula that utilizes all of the available pain ratings up to 30 minutes after each dose. Table 1 indicates the formula that was used to calculate SPID for each trial. We used this strategy so that the SPID units would be consistent across all trials. All participants who provided at least 1 post-treatment pain assessment from at least 1 active and 1 placebo episode were included in these analyses.
A mixed effects linear model was used to investigate treatment-by-patient interaction for the SPID30. The model included fixed effects for treatment, episode, age, gender, and race, and random effects for patient and treatment-by-patient interaction. Variance components were estimated using restricted maximum likelihood (REML) and a test of the null hypothesis of no treatment-by-patient interaction was performed using a mixture of chi-square distributions.15,16 The predicted treatment effects from this model for individual patients (i.e., the best linear unbiased predictors (BLUPs)) were computed using the metafor package in R,17 as described by Araujo et al.18 These estimates are weighted averages of the estimated overall treatment effect (i.e., what would be expected if there were no heterogeneity in response to treatment) and the individual patient’s estimated treatment effect (i.e., what would be expected if the observed differences in patients’ response to treatment were completely due to true heterogeneity in response between patients), the latter obtained naively from an analysis that treats patient and treatment-by-patient interaction as fixed effects. In the computation of the BLUPs, more weight is given to the estimated overall treatment effect if there is large within-patient variability relative to between-patient variability in the treatment effect. This results in more shrinkage of the patient’s estimated treatment effect toward the overall average. Therefore, if there is substantial true heterogeneity between patients in their response to treatment, the predicted treatment effects should not deviate too much from the observed treatment effects (i.e., they should not be “shrunk” very much toward the overall average). Each trial was analyzed separately, and a p-value < 0.05 was considered significant for the treatment-by-patient interaction.
Study Highlights.
What is the current knowledge on this topic?
Many investigators and consumers of clinical trial data erroneously assume that observed variability in treatment outcomes between patients in clinical trials is certainly due to true heterogeneity in response to treatment. To our knowledge, no previous trials have used multi-period cross-over trials to definitively demonstrate true heterogeneity.
What question did the study address?
Whether heterogeneity in treatment response can be isolated from other sources of variability using data from multi-period cross-over trial data in which each treatment is used at least twice.
What does this study add to our knowledge?
This study provides an illustrative example of how data from multi-period cross-over trials can be used to examine the degree of variability in patient response to specific treatments.
How might this change clinical pharmacology or translational science?
Performance of analyses similar to the ones presented here to measure the degree of true heterogeneity in patient responses to treatment will inform decisions as to whether to pursue future studies to identify predictors of treatment response, avoiding wasted resources pursuing predictors of treatment response where true heterogeneity is lacking.
Acknowledgements
This article was reviewed and approved by the Executive Committee of the Analgesic, Anesthetic, and Addiction Clinical Trial Translations, Innovations, Opportunities, and Networks (ACTTION) public-private partnership with the United States Food and Drug Administration.
Funding: Financial support for this project was provided by the ACTTION public-private partnership, which has received unrestricted research contracts, grants, or other revenue from the FDA, multiple pharmaceutical and device companies, philanthropy, and other sources.
Members of ACTTION’s leadership team (all of whom were authors) were involved in the design and execution of the study and preparation of the manuscript. The funders of ACTTION played no role in this project. The views expressed in this article are those of the authors and no official endorsement by the Food and Drug Administration (FDA) or the pharmaceutical and device companies that provided unrestricted grants to support the activities of the ACTTION public-private partnership should be inferred. The authors declare no financial interests related to this work.
Footnotes
Conflict of Interest:
References
- 1.Moore RA, Cai N, Skljarevski V, et al. Duloxetine use in chronic painful conditions--individual patient data responder analysis. Eur J Pain 2014;18(1):67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore RA, Derry S, McQuay HJ, et al. Clinical effectiveness: an approach to clinical trial design more relevant to clinical practice, acknowledging the importance of individual differences. Pain 2010;149(2):173–6. [DOI] [PubMed] [Google Scholar]
- 3.National Cancer Institute. Precision Medicine in Cancer Treatment [Available from: https://www.cancer.gov/about-cancer/treatment/types/precision-medicine accessed 3/19/2018].
- 4.Demant DT, Lund K, Vollert J, et al. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double-blind, placebo-controlled phenotype-stratified study. Pain 2014;155(11):2263–73. [DOI] [PubMed] [Google Scholar]
- 5.Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 screening for hypersensitivity to abacavir. The NEJM 2008;358(6):568–79. [DOI] [PubMed] [Google Scholar]
- 6.Senn S. Individual response to treatment: is it a valid assumption? BMJ 2004;329(7472):966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dworkin RH, McDermott MP, Farrar JT, et al. Interpreting patient treatment response in analgesic clinical trials: Implications for genotyping, phenotyping, and personalized pain treatment. Pain 2014;155:457–60. [DOI] [PubMed] [Google Scholar]
- 8.Lonergan M, Senn SJ, McNamee C, et al. Defining drug response for stratified medicine. Drug Discov Today 2017;22(1):173–79. [DOI] [PubMed] [Google Scholar]
- 9.Senn S, Rolfe K, Julious SA . Investigating variability in patient response to treatment--a case study from a replicate cross-over study. Statistical Methods in Medical Research 2011;20(6):657–66. [DOI] [PubMed] [Google Scholar]
- 10.Senn S. Mastering variation: variance components and personalised medicine. Statistics in Medicine 2016;35(7):966–77. doi: 10.1002/sim.6739 [published Online First: 2015/09/30] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rauck R, North J, Gever LN, et al. Fentanyl buccal soluble film (FBSF) for breakthrough pain in patients with cancer: a randomized, double-blind, placebo-controlled study. Annals of Oncology 2010;21(6):1308–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Portenoy RK, Taylor D, Messina J, et al. A randomized, placebo-controlled study of fentanyl buccal tablet for breakthrough pain in opioid-treated patients with cancer. The Clinical Journal of Pain 2006;22(9):805–11. [DOI] [PubMed] [Google Scholar]
- 13.Portenoy RK, Burton AW, Gabrail N, et al. A multicenter, placebo-controlled, double-blind, multiple-crossover study of Fentanyl Pectin Nasal Spray (FPNS) in the treatment of breakthrough cancer pain. Pain 2010;151(3):617–24. [DOI] [PubMed] [Google Scholar]
- 14.Rauck R, Reynolds L, Geach J, et al. Efficacy and safety of fentanyl sublingual spray for the treatment of breakthrough cancer pain: a randomized, double-blind, placebo-controlled study. Current Medical Research and Opinion 2012;28(5):859–70. [DOI] [PubMed] [Google Scholar]
- 15.Stram DO, Lee JW. Variance components testing in the longitudinal mixed effects model. Biometrics 1994;50(4):1171–7. [PubMed] [Google Scholar]
- 16.Morrell CH. Likelihood ratio testing of variance components in the linear mixed-effects model using restricted maximum likelihood. Biometrics 1998;54(4):1560–8. [PubMed] [Google Scholar]
- 17.Viechtbauer W. Conducting meta-analysis in R with metafor package. J Statist Software 2010;36:1–48. [Google Scholar]
- 18.Araujo A, Julious S, Senn S. Understanding Variation in Sets of N-of-1 Trials. PloS One 2016;11(12):e0167167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Punja S, Nikles CJ, Senior H, et al. Melatonin in Youth: N-of-1 trials in a stimulant-treated ADHD Population (MYNAP): study protocol for a randomized controlled trial. Trials 2016;17:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herrett E, Williamson E, Beaumont D, et al. Study protocol for statin web-based investigation of side effects (StatinWISE): a series of randomised controlled N-of-1 trials comparing atorvastatin and placebo in UK primary care. BMJ Open 2017;7(12):e016604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.European Medicines Agency. Guideline on the investigation of subgroups in confirmatory clinical trials. 2014; Accessed 9/25/2018 [http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/02/WC500160523.pdf].
- 22.Guyatt GH, Keller JL, Jaeschke R, Rosenbloom D, Adachi JD, Newhouse MT. The n-of-1 randomized controlled trial: clinical usefulness. Our three-year experience. Annals of Internal Medicine 1990;112:293–299. [DOI] [PubMed] [Google Scholar]
- 23.Guyatt GH, Heyting A, Jaeschke R, Keller J, Adachi JD, Roberts RS. N of 1 randomized trials for investigating new drugs. Controlled Clinical Trials 1990;11:88–100. [DOI] [PubMed] [Google Scholar]
- 24.Mahon J, Laupacis A, Donner A, Wood T. Randomised study of n of 1 trials versus standard practice. BMJ 1996;312:1069–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lillie EO, Patay B, Diamant J, Issell B, Topol EJ, Schork NJ. The n-of-1 clinical trial: the ultimate strategy for individualizing medicine? Personalized Medicine 2011;8:161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yelland MJ, Poulos CJ, Pillans PI, et al. N-of-1 randomized trials to assess the efficacy of gabapentin for chronic neuropathic pain. Pain Med 2009;10:754–761. [DOI] [PubMed] [Google Scholar]
- 27.Yelland MJ, Nikles CJ, McNairn N, Del Mar CB, Schluter PJ, Brown RM. Celecoxib compared with sustained-release paracetamol for osteoarthritis: a series of n-of-1 trials. Rheumatology 2007;46:135–140. [DOI] [PubMed] [Google Scholar]
- 28.Zucker DR, Ruthazer R, Schmid CH, et al. Lessons learned combining N-of-1 trials to assess fibromyalgia therapies. The Journal of Rheumatology 2006;33:2069–2077. [PubMed] [Google Scholar]