

Abstract
Among mammalian phylogenies, those characterized by rapid radiations are particularly problematic. The New World monkeys (NWMs, Platyrrhini) comprise 3 families and 7 subfamilies, which radiated within a relatively short time period. Accordingly, their phylogenetic relationships are still largely disputed. In the present study, 56 nuclear non-coding loci, including 33 introns (INs) and 23 intergenic regions (IGs), from 20 NWM individuals representing 18 species were used to investigate phylogenetic relationships among families and subfamilies. Of the 56 loci, 43 have not been used in previous NWM phylogenetics. We applied concatenation and coalescence tree-inference methods, and a recently proposed question-specific approach to address NWM phylogeny. Our results indicate incongruence between concatenation and coalescence methods for the IN and IG datasets. However, a consensus was reached with a single tree topology from all analyses of combined INs and IGs as well as all analyses of question-specific loci using both concatenation and coalescence methods, albeit with varying degrees of statistical support. In detail, our results indicated the sister-group relationships between the families Atelidae and Pitheciidae, and between the subfamilies Aotinae and Callithrichinae among Cebidae. Our study provides insights into the disputed phylogenetic relationships among NWM families and subfamilies from the perspective of multiple non-coding loci and various tree-inference approaches. However, the present phylogenetic framework needs further evaluation by adding more independent sequence data and a deeper taxonomic sampling. Overall, our work has important implications for phylogenetic studies dealing with rapid radiations.
Keywords: coalescence, concatenation, non-coding nuclear genes, primates, phylogenetics
The New World monkeys (NWMs, Platyrrhini) are a group of arboreal primates distributed in South and Central America, ranging from southern Mexico to northern Argentina (Kinzey 1997; Mittermeier et al. 2013). The evolutionary history of NWMs is characterized by rapid bursts of diversification at the family levels that occurred within a 10 million year window (Kay et al. 2008; Hodgson et al. 2009; Fleagle 2013). Taxonomically, NWMs are divided into the 3 families Pitheciidae with the 2 subfamilies Pitheciinae (uakaris and sakis) and Callicebinae (titi monkeys), Atelidae with the 2 subfamilies Atelinae (spider and woolly monkeys, and muriquis) and Alouattinae (howler monkeys), and Cebidae with the 3 subfamilies Cebinae (capuchin and squirrel monkeys), Aotinae (night monkeys) and Callithrichinae (marmosets and tamarins) (Fleagle 2013). Aotinae and Callithrichinae are sometimes elevated to the family level (Mittermeier et al. 2013), but we follow here a NWM classification with 3 families and 7 subfamilies.
Over the last decades, a cascade of molecular phylogenetic studies of NWMs using mitochondrial and nuclear DNA sequences has been conducted (Schneider et al. 1993, 1996; Poux and Douzery et al. 2004; Opazo et al. 2006; Schrago 2007; Fabre et al. 2009; Osterholz et al. 2009; Wildman et al. 2009; Chatterjee et al. 2009; Perelman et al. 2011; Springer et al. 2012; Finstermeier et al. 2013; Jameson-Kiesling et al. 2015; Aristide et al. 2015). Owing to this rapid radiation and recent speciation events, the phylogeny of NWMs is still incompletely resolved as earlier studies revealed contradictory branching patterns among and within the 3 NWM families (Opazo et al. 2006; Fabre et al. 2009; Osterholz et al. 2009; Wildman et al. 2009; Chatterjee et al. 2009; Perelman et al. 2011; Perez et al. 2012; Ting and Sterner 2013; Finstermeier et al. 2013; Jameson-Kiesling et al. 2015; Aristide et al. 2015; Delgado et al. 2016). All 3 alternative sister group relationships among Pitheciidae, Atelidae, and Cebidae have been proposed (Figure 1Aa–Ac) (Schneider et al. 1993, 1996; Poux and Douzery et al. 2004; Opazo et al. 2006; Fabre et al. 2009; Osterholz et al. 2009; Wildman et al. 2009; Chatterjee et al. 2009; Perelman et al. 2011; Springer et al. 2012; Finstermeier et al. 2013; Schrago et al. 2014; Jameson-Kiesling et al. 2015; Aristide et al. 2015). Similar to the branching patterns among families, the phylogenetic relationships among the 3 subfamilies of the Cebidae (Aotinae, Callithrichinae, and Cebinae) are also not well resolved, with earlier studies providing support for each of the 3 possible relationships (Figure 1Ba–Bc) (Poux and Douzery et al. 2004; Opazo et al. 2006; Schrago 2007; Fabre et al. 2009; Wildman et al. 2009; Chatterjee et al. 2009; Springer et al. 2012; Finstermeier et al. 2013; Jameson-Kiesling et al. 2015; Aristide et al. 2015). The poor resolution and discordances among gene trees coupled with short internal branches are consistent with a recent and rapid radiation of NWMs (Kay et al. 2008; Hodgson et al. 2009; Fleagle 2013).
Figure 1.

Alternative phylogenetic relationships that have been proposed among (A) NWM families and (B) subfamilies of the Cebidae family.
Here, we used 56 nuclear non-coding loci, including 33 introns (INs) and 23 intergenic (IGs) regions, from representatives of all NWM families and subfamilies to test previous phylogenetic hypotheses. The majority of loci (43) have not been used in previous NWM phylogenetic studies, and most are from INs, a class of non-coding DNA less commonly employed (Schneider et al. 1996) compared with the widely used IGs regions (Wildman et al. 2009; Jameson-Kiesling et al. 2015). In addition to traditional concatenation tree reconstruction methods, we applied 2 coalescent-based species-tree estimation methods for the resolution of higher-level relationships in NWMs. The coalescence methods have been thought to suffer less from analytical biases relative to concatenation methods in the case of rapid radiations by accounting for differences between gene and species trees (Swenson and EI-Mabrouk 2012; Roch and Warnow 2015). In addition, we utilized a recently developed question-specific approach for reducing incongruence associated with large data sets and tree-inference methods in phylogenomics (Chen et al. 2015).
Materials and Methods
Material
Blood and tissue samples of 20 NWMs from 18 species representing all 3 NWM families and 7 subfamilies were obtained from the zoos in Cologne, Gettorf, Kunming, Landau, Romagne, Rostock, Stockholm, and Toronto (Table 1). Blood samples were taken during routine health checks, whereas muscle samples were obtained from deceased specimens. Blood samples were immediately subjected to DNA extraction after arrival in the laboratory, whereas tissue was stored frozen in 96% ethanol before further processing.
Table 1.
Information about investigated species, their origin, and genbank accession numbers.
| Family | Subfamily | Species | Common name | Genbank | Origin |
|---|---|---|---|---|---|
| Cebidae | Callithrichinae | Callithrix jacchus | Common Marmoset | KY458990-KY459995 | Toronto zoo |
| Cebuella pygmaea | Pygmy Marmoset | KY458990-KY459995 | Cologne zoo | ||
| Cebuella pygmaea | Pygmy Marmoset | KY458990-KY459995 | Stockholm zoo | ||
| Leontopithecus rosalia | Golden Lion Tamarin | KY458990-KY459995 | Cologne zoo | ||
| Saguinus bicolor | Pied Tamarin | KY458990-KY459995 | Magdeburg zoo | ||
| Callimico goeldii | Goeldi’s Monkey | KY458990-KY459995 | Cologne zoo | ||
| Aotinae | Aotus azarae | Azara’s Night Monkey | KY458990-KY459995 | Gettorf zoo | |
| Cebinae | Cebus capucinus | Colombian White-faced Capuchin | KY458990-KY459995 | Romagne zoo | |
| Sapajus apella | Guianan Brown Capuchin | KY458990-KY459995 | Kunming zoo | ||
| Sapajus apella | Guianan Brown Capuchin | KY458990-KY459995 | Rostock zoo | ||
| Saimiri sciureus | Guianan Squirrel Monkey | KY458990-KY459995 | Kunming zoo | ||
| Saimiri boliviensis | Black-capped Squirrel Monkey | KY458990-KY459995 | Romagne zoo | ||
| Atelidae | Atelinae | Ateles paniscus | Red-faced Back Spider Monkey | KY458990-KY459995 | wild deceased species |
| Ateles fusciceps | Black-headed Spider Monkey | KY458990-KY459995 | Landau zoo | ||
| Lagothrix lagotricha | Humboldt’s Woolly Monkey | KY458990-KY459995 | Romagne zoo | ||
| Alouattinae | Alouatta caraya | Paraguayan Howler Monkey | KY458990-KY459995 | Cologne zoo | |
| Pitheciidae | Pitheciinae | Pithecia pithecia | White-faced Saki | KY458990-KY459995 | wild deceased species |
| Chiropotes albinasus | Red-nosed Bearded Saki | KY458990-KY459995 | Cologne zoo | ||
| Cacajao calvus | Bald Uakari | KY458990-KY459995 | Cologne zoo | ||
| Callicebinae | Plecturocebus cupreus | Coppery Titi Monkey | KY458990-KY459995 | Romagne zoo | |
| Cercopithecidae | Macaca mulatta | Rhesus Macaque | rheMac2 | UCSC Genome Browser | |
| Hominidae | Pongo abelii | Sumatra Orangutan | ponAbe2 | UCSC Genome Browser | |
| Pan troglodytes | Chimpanzee | panTro2 | UCSC Genome Browser | ||
| Homo sapiens | Human | hg18 | UCSC Genome Browser |
Data sets and laboratory work
Total genomic DNA from blood or tissue was isolated using standard proteinase K or phenol/chloroform extraction (Sambrook et al. 1989). We amplified and sequenced a total of 56 nuclear non-coding loci (Table 2). Forty-three of these loci were taken from a study investigating the phylogenetic relationships among colobine genera (leaf-eating Old World monkeys) (Wang et al. 2012), whereas the other 13 loci derived from a study on NWMs (Wildman et al. 2009). PCR conditions and primer sequences are shown in Supplementary Table S1. The amplified DNA fragments were purified and sequenced in both directions with an ABI PRISM™ 3700 DNA or 3130xL sequencer following the manufacturer’s protocol. In the case of poor performance of direct sequencing resulting from complex DNA structures or tandem repeats, PCR products were cloned into the PMD18-T Vector and transformed into ultracompetent E. coli cells (TaKaRa Biotechnology Co., Ltd. Dalian, China). Five positive clones per ligation reaction were sequenced. All sequences were checked and queried in BLAST to assess homology. For some species, PCR attempts failed to produce sequence data. These sequences were excluded from the corresponding independent gene analyses and treated as missing data in the combined analyses. In total, 1006 newly determined non-coding sequences have been generated in this study (GenBank Accession Numbers KY458990-KY459009; Table 1). To expand our dataset, orthologous sequences from 4 non-NWM primates, that is, human (Homo sapiens, hg18), common chimpanzee (Pan troglodytes, panTro2), orangutan (Pongo abelii, ponAbe2), and rhesus macaque (Macaca mulatta, rheMac2), were downloaded from GenBank and used as outgroups.
Table 2.
Characterization of 56 nuclear non-coding genes examined in the present study
| Fragment Name | Chromosome Location | Data Type | Aligned Length | Variable Sites | Parsimony- Informative sites | Best- fitModel | Nucleotide CompositionA-T | Pairwise Distance | Grouping in question-specific dataset |
|---|---|---|---|---|---|---|---|---|---|
| chr1-4 | chr1 | IN | 362 | 103 | 56 | SYM+G | 0.5 | 7.30E-02 | non-matching |
| chr3-2 | chr3 | IN | 461 | 111 | 55 | GTR+G | 0.59 | 5.50E-02 | matchA |
| chr3-5 | chr3 | IN | 234 | 60 | 31 | HKY | 0.68 | 6.50E-02 | matchA |
| chr4-7 | chr4 | IN | 335 | 59 | 28 | HKY | 0.58 | 4.80E-02 | non-matching |
| chr5-8 | chr5 | IN | 453 | 71 | 36 | GTR+G | 0.64 | 3.60E-02 | non-matching |
| chr6-5 | chr6 | IN | 325 | 75 | 40 | GTR+G | 0.64 | 5.60E-02 | matchA |
| chr7-6 | chr7 | IN | 367 | 85 | 35 | TVMef+G | 0.5 | 5.30E-02 | non-matching |
| chr8-1 | chr8 | IN | 466 | 145 | 69 | GTR+G | 0.56 | 8.00E-02 | shared matchA and matchB |
| chr8-2 | chr8 | IN | 454 | 121 | 65 | TVM+G | 0.69 | 6.70E-02 | shared matchA and matchB |
| chr10-5 | chr10 | IN | 426 | 56 | 31 | TVMef+I+G | 0.5 | 3.10E-02 | non-matching |
| chr11-2 | chr11 | IN | 263 | 86 | 54 | K80+G | 0.5 | 9.10E-02 | matchB |
| chr12-1 | chr12 | IN | 464 | 105 | 47 | GTR+G | 0.53 | 4.60E-02 | shared matchA and matchB |
| chr12-2 | chr12 | IN | 355 | 105 | 61 | K80+G | 0.5 | 8.00E-02 | non-matching |
| chr13-3 | chr13 | IN | 316 | 89 | 33 | TVM+G | 0.72 | 6.30E-02 | matchB |
| chr13-6 | chr13 | IN | 337 | 88 | 38 | TVM+G | 0.7 | 6.60E-02 | shared matchA and matchB |
| chr15-1 | chr15 | IN | 586 | 162 | 86 | TVM+G | 0.55 | 6.50E-02 | shared matchA and matchB |
| chr17-8 | chr17 | IN | 336 | 95 | 55 | TrN+G | 0.45 | 8.10E-02 | non-matching |
| chr18-4 | chr18 | IN | 362 | 98 | 48 | HKY | 0.61 | 6.20E-02 | matchA |
| chr19-1 | chr19 | IN | 462 | 114 | 53 | TIM1+G | 0.43 | 5.30E-02 | matchA |
| chr19-5 | chr19 | IN | 349 | 60 | 22 | HKY | 0.42 | 4.00E-02 | non-matching |
| chr20-4 | chr20 | IN | 464 | 115 | 46 | TrN+G | 0.53 | 6.00E-02 | matchA |
| chr20-5 | chr20 | IN | 407 | 119 | 47 | K80+G | 0.5 | 6.10E-02 | non-matching |
| ENC2 | chr22 | IN | 372 | 82 | 42 | HKY+G | 0.48 | 5.60E-02 | non-matching |
| ENC5 | chr7 | IN | 607 | 139 | 71 | TrN+G | 0.57 | 5.50E-02 | shared matchA and matchB |
| ENC14 | chr14 | IN | 466 | 118 | 54 | GTR+G | 0.57 | 5.30E-02 | shared matchA and matchB |
| ENC35 | chr21 | IN | 460 | 108 | 55 | TVM+I | 0.69 | 5.00E-02 | matchA |
| X45 | chrX | IN | 476 | 122 | 61 | GTR | 0.65 | 5.90E-02 | shared matchA and matchB |
| X61 | chrX | IN | 544 | 126 | 56 | TVM+G | 0.61 | 4.60E-02 | non-matching |
| 6p22.3 | chr6 | IN | 625 | 110 | 47 | GTR+G | 0.56 | 4.20E-02 | matchA |
| 8q23.1 | chr8 | IN | 568 | 80 | 33 | TVM+G | 0.68 | 2.90E-02 | non-matching |
| 10p12.33 | chr10 | IN | 429 | 95 | 36 | TPM1uf | 0.62 | 4.40E-02 | non-matching |
| 2p21 | chr2 | IN | 679 | 158 | 75 | TVM+G | 0.51 | 5.00E-02 | matchA |
| 14q32.13 | chr14 | IN | 710 | 50 | 18 | HKY+I+G | 0.57 | 1.20E-02 | non-matching |
| chr1-6 | chr1 | IG | 454 | 112 | 59 | TVM+G | 0.65 | 5.50E-02 | non-matching |
| chr2-1 | chr2 | IG | 272 | 70 | 28 | K80+G | 0.5 | 5.20E-02 | shared matchA and matchB |
| chr2-8 | chr2 | IG | 375 | 75 | 26 | HKY | 0.6 | 4.40E-02 | shared matchA and matchB |
| chr4-2 | chr4 | IG | 295 | 26 | 0 | TIM1 | 0.42 | 1.90E-02 | shared matchA and matchB |
| chr5-6 | chr5 | IG | 400 | 127 | 43 | TVM | 0.55 | 8.50E-02 | matchA |
| chr6-6 | chr6 | IG | 351 | 70 | 36 | TVM+G | 0.58 | 4.30E-02 | shared matchA and matchB |
| chr9-5 | chr9 | IG | 390 | 93 | 50 | HKY+I | 0.52 | 5.60E-02 | non-matching |
| chr11-3 | chr11 | IG | 374 | 122 | 56 | K80+G | 0.5 | 7.30E-02 | matchA |
| chr18-3 | chr18 | IG | 388 | 164 | 96 | TrN+G | 0.41 | 1.17E-01 | non-matching |
| ENC15 | chr14 | IG | 773 | 175 | 85 | HKY+G | 0.53 | 5.80E-02 | matchB |
| ENC19 | chr16 | IG | 398 | 100 | 42 | HKY | 0.65 | 5.70E-02 | non-matching |
| ENC25 | chr21 | IG | 390 | 120 | 64 | K80+G | 0.5 | 7.70E-02 | shared matchA and matchB |
| X5 | chrX | IG | 357 | 89 | 46 | TVM | 0.61 | 5.80E-02 | matchB |
| X37 | chrX | IG | 531 | 120 | 49 | TVM+G | 0.64 | 4.70E-02 | non-matching |
| X65 | chrX | IG | 495 | 137 | 75 | TVM+G | 0.57 | 6.60E-02 | non-matching |
| 1p31.1 | chr1 | IG | 534 | 128 | 56 | HKY | 0.65 | 4.50E-02 | non-matching |
| 1q31.3 | chr1 | IG | 569 | 142 | 81 | TVM+G | 0.62 | 6.40E-02 | non-matching |
| 2p22.3 | chr2 | IG | 593 | 119 | 56 | TrN+G | 0.59 | 4.30E-02 | non-matching |
| 3p13 | chr3 | IG | 695 | 68 | 32 | TVM+G | 0.56 | 2.00E-02 | shared matchA and matchB |
| 3q22.2 | chr3 | IG | 499 | 124 | 52 | HKY+G | 0.56 | 5.20E-02 | matchB |
| 5p15.33 | chr5 | IG | 421 | 124 | 63 | TVM | 0.45 | 7.80E-02 | matchA |
| 10q23.1 | chr10 | IG | 528 | 137 | 53 | K80+G | 0.5 | 5.50E-02 | matchA |
| Xq22.1 | chrX | IG | 602 | 64 | 14 | TVM | 0.58 | 2.30E-02 | shared matchA and matchB |
| INs | IN | 14520 | 3310 | 1584 | GTR+G | 0.57 | 4.20E-02 | ||
| IGs | IG | 10684 | 2509 | 1157 | TVM+G | 0.56 | 5.20E-02 | ||
| matchA | 13796 | 3477 | 1711 | TVM+G | 0.58 | 5.90E-02 | |||
| matchB | 11108 | 2840 | 1386 | TVM+G | 0.59 | 5.90E-02 | |||
| 56NWM | 25204 | 5819 | 2741 | TVM+G | 0.57 | 4.80E-02 |
matchA and matchB, loci that match branching patterns presented in figure 4, respectively
Alignments and sequence characterization
Sequences were aligned using Muscle 3.8.31 (Edgar 2004) under default settings. All 56 genes were analyzed separately and concatenated. The concatenated alignment was divided into 3 datasets: 1) 33 INs combined, 2) 23 IGs regions combined, and 3) INs and IGs combined. All alignments were visually corrected, and poorly aligned positions and indels were removed with Gblocks 0.91 b (Castresana 2000) using default settings. Statistical attributes of the nucleotide sequence data were estimated with MEGA 7 (Kumar et al. 2016) and DAMBE 7.0.5 (Xia 2017) was used to check for substitution saturation.
Phylogenetic analyses based on consensus and coalescent methods
Phylogenetic trees for individual and concatenated loci were reconstructed with maximum-likelihood (ML) and Bayesian methods in RAxML 8.0.12 (Stamatakis 2006; Silvestro and Michalak 2010) and Mr Bayes 3.2.2 (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003), respectively. The best-fit models of sequence evolution for each locus were selected under the Akaike Information Criterion (AIC) (Akaike 1974; Posada and Buckley 2004) with jModeltest 1.1.0 (Posada 2008, 2009). The chosen models and their parameters were applied to infer both ML and Bayesian trees. For tree reconstructions based on concatenated datasets, each locus was treated as a separate partition with its own substitution model. ML trees were calculated with the heuristic algorithm, 10 random-addition sequence replicates and TBR branch swapping. Tree reliability was assessed using a bootstrap (BS) analysis with 100 replicates (Felsenstein 1985). For Bayesian analyses, we used 3-heated chains and a single cold chain in all Metropolis-coupled Markov chain Monte Carlo (MCMC) runs. We performed 3 independent runs for each dataset, each for 2 million generations with parameter sampling every 100 generations. The average standard deviation of split frequencies was close to 0.001 when the runs were finished. The first 25% of the trees were discarded as burn-in. A 50% majority-rule consensus of post burn-in trees was constructed to summarize the posterior probability (PP) for each split.
In addition to the traditional concatenation methods, we applied 2 coalescent-based species-tree estimation methods, that is, Accurate Species TRee ALgorithm (ASTRAL-II) (Mirarab et al. 2014) and Species Tree estimation using Average Ranks of coalescence (STAR) (Liu et al. 2009). The ASTRAL analyses used the unrooted gene trees as the input file and the Maximum Quartet Support Species Tree (MQSST) was searched. The STAR analyses, conducted in STRAW (Shaw et al. 2013), were performed with multilocus BSs (Seo 2008) to estimate statistical support. For both analyses, individual gene trees for each of the non-coding sequences were estimated using RAxML 8.0.12 under the GTR+G model with 1000 BS replicates.
Phylogenetic analyses based on question-specific approach
Increasing the number of investigated loci does not always allow for better resolution of phylogenetic relationships, particularly when single locus analyses reveal contrasting results. In such cases, building and investigating question-specific datasets may be a more powerful approach to resolving questionable branching patterns (Chen et al. 2015). Chen et al. (2015) proposed 2 question-specific strategies to resolve such problematic nodes. In the “hypothesis-control approach” loci whose gene trees do not support any of the hypotheses for given question are removed, whereas in the “node-control strategy” only loci are selected whose gene trees recover a control node. The second approach is more relaxed than the first (Chen et al. 2015), and hence we selected the more strict “hypothesis-control approach” to address the branching patterns among NWM families and cebid subfamilies. Accordingly, loci whose phylogenetic trees do not support any of the 3 hypotheses for the relationships among platyrrhine families (Figure 1Aa–Ac) and cebid subfamilies (Figure 1Ba–Bc) were removed. The resulting 2 question-specific datasets were used to conduct phylogenetic analyses as described above.
Divergence time estimation
Divergence times for the NWM radiation were estimated using a relaxed lognormal molecular clock in BEAST 2.4.7 (Drummond and Rambaut 2007). We assumed a GTR+I + G model of sequence evolution with 4 rate categories. Uniform priors were employed for GTR substitution parameters (0, 100), the gamma shape parameter (0, 100) and the proportion of invariant sites parameter (0, 1). The uncorrelated lognormal relaxed molecular clock model was used to estimate substitution rates for all nodes in the tree, with uniform priors on the mean (0, 100) and standard deviation (0, 10) of this clock model. We employed the Yule (pure-birth process) of speciation as the tree prior and a UPGMA tree to construct a starting tree.
We applied 6 calibration points that were used in earlier studies (Springer et al. 2012; Benton et al. 2015; Byrne et al. 2016) and derived from the fossil record. For all 6 nodes, a uniform distribution prior was selected. We used 1) the origin of Anthropoidea: minimum = 33.9 million years ago (Ma), maximum = 66.0 Ma (Benton et al. 2015), 2) Homo–Pan split: minimum = 5.11 Ma (Springer et al. 2012), maximum = 10.0 Ma (Benton et al. 2015), 3) the origin of Hominidae: minimum = 11.6 Ma, maximum = 28.5 Ma (Springer et al. 2012), 4) the origin of Catarrhini: minimum = 24.44 Ma, maximum = 34 Ma (Benton et al. 2015), 5) the origin of Pitheciidae: minimum = 15.7 Ma, maximum = 26.0 Ma (Byrne et al. 2016) and 6) the origin of Cebinae: minimum = 12.5 Ma, maximum = 26.0 Ma (Byrne et al. 2016). Posterior distributions of parameters were approximated by sampling from 2 independent MCMC analyses. Each analysis ran for 100 million generations with parameters logged every 1000 generations. Convergence was assessed in Tracer 1.5 (Rambaut and Drummond 2009) after excluding the first 25% as burn-in. A consensus chronogram with node height distribution was generated with TreeAnnotator 2.4.7 and visualized with FigTree 1.4.3 (Rambaut 2012).
For studies with animals
All applicable institutional and/or national guidelines for the care and use of animals were followed. No animals were sacrificed for this study. Blood samples were taken during routine health checks by experienced veterinarians and not specifically for this study. Tissue samples were obtained from deceased animals. All research adhered to the legal requirements of the countries in which research was conducted. The study was carried out in compliance with the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES) and the principles of the American Society of Primatologists for the ethical treatment of nonhuman primates.
Results
Characteristics of the nuclear non-coding data
General information about the 56 nuclear non-coding regions from 18 NWM species is summarized in Table 2. Alignments for individual loci varied from 422 bp (chr2-8) to 1251 bp (2p22.3). After the removal of poorly aligned positions and indels alignment lengths varied from 234 bp (chr3-5) to 773 bp (ENC15). Alignments differed also in the number of parsimony-informative sites, ranging from 0 (chr4-2) to 96 (chr18-3). An A-T bias (average ratio = 56.4%) was apparent in most loci, as typically observed in non-coding regions (Yu and Zhang 2005; Matthee et al. 2007; Wang et al. 2012). The optimal model of sequence evolution varied by locus, suggesting different evolutionary histories of individual loci. Pairwise K2P distances among all NWM species ranged from 1.2% (14q32.13) to 11.7% (chr18-3), with an average of 5.6%.
The concatenation of all non-coding regions (alignment 3) recovered a total of 25, 204 sites, of which 2741 (10.9%) were parsimony-informative. The average K2P distance is 4.8%. The 33 concatenated INs (alignment 1) resulted in 14, 520 sites, comprising 1584 (10.9%) parsimony-informative sites and showing an average K2P distance of 4.2%. In comparison, the concatenated 23 IGs (alignment 2) resulted in 10, 684 sites, of which 1157 (10.8%) were parsimony-informative and the average K2P distance was 5.2%. Therefore, a slightly slower evolutionary rate for INs relative to IGs was observed, consistent with the fact that there are selective constraints on INs but not on IGs due to the presence of pre-mRNA secondary structures (Ometto et al. 2006). We found that there was no evidence of substitution saturation across our INs and IGs dataset based on the Iss statistic in DAMBE, as evidenced by the significantly lower values of Iss (index of substitution saturation) than Iss.c (critical value for symmetrical tree topology) (IN: 0.2584 < 0.8558, P = 0, P < 0.001; IG: 0.2898 < 0.8473, P = 0, P < 0.001).
Phylogenetic relationships based on the consensus and coalescent methods
To test different hypotheses about the branching pattern among NWM families and Cebidae subfamilies, different datasets (INs, IGs, and INs and IGs combined) were investigated with concatenation (ML and BA) and coalescent-based tree-inference methods (ASTRAL and STAR) (Figures 2 and 3). On family level, a consistent tree topology was obtained from all 3 datasets and the 4 applied methods, although the sister grouping of Atelidae and Pitheciidae was weakly supported. For relationships among subfamilies within the Cebidae, weakly supported and inconsistent results were obtained for IN and IG datasets using the concatenation and coalescence tree-inference methods. Even the 2 coalescent-based tree reconstruction methods resulted in inconsistent tree topologies for the same dataset (ASTRAL: Figure 2a, b; STAR: Figure 2a, c). However, statistical support for the nodes of interest, that is, the relationships among families and cebid subfamilies, were generally low, except for the tree topology derived from the IG dataset where strong support was found for a sister grouping of Atelidae and Pitheciidae (hypothesis in Figure 1Ac), and Aotinae and Callithrichinae within Cebidae (hypothesis in Figure 1Ba), at least in ML and Bayesian reconstructions (Figure 2c: ML BS: 78% and 95%, Bayesian PP: 0.95 and 0.98, respectively).
Figure 2.

Phylogenetic tree reconstructions based on the analyses of 33 INs and 23 IGs regions. Tree topologies revealed by ML/Bayesian/STAR analyses (INs) and ASTRAL analysis (IGs) (A), ASTRAL analysis (INs) (B), and ML/Bayesian/STAR analyses (IGs) (C). Numbers at nodes indicate statistical support values.
Figure 3.

Ultrametric tree as obtained from the analyses of all non-coding loci combined and the matchA and matchB datasets. The support values for the 2 nodes of interest (branching pattern among NWM families and subfamilies within the Cebidae family) are shown (ML BS/Bayesian PP/STAR BS/ASTRAL BS). For both nodes, the top values are those from the combined non-coding loci analyses, and those from matchA and matchB are shown in the middle and the bottom, respectively. The divergence time estimation is based on the dataset including all non-coding loci.
Phylogenetic analyses inferred from combining all non-coding regions using both concatenation (ML and BA) and coalescence (ASTRAL and STAR) methods resulted in an identical tree topology (Figure 3). All analyses suggested a division of NWMs into the 3 monophyletic families Cebidae, Atelidae and Pitheciidae, and consistently supported the sister grouping of Atelidae and Pitheciidae to the exclusion of Cebidae, corresponding to hypothesis Ac (ML BS: 89%, Bayesian PP: 1.00, STAR BS: 68%, ASTRAL BS: 71%). Within Cebidae, the subfamilies Aotinae and Callithrichinae form a clade to the exclusion of Cebinae in all analyses, corresponding to hypothesis Ba (ML BS: 70%, Bayesian PP: 0.97, STAR BS: 51%, ASTRAL BS: <50%).
Phylogenetic relationships based on question-specific approach
The individual analyses of 56 non-coding regions produced a variety of tree topologies with low levels of nodal support, probably owing to the limited phylogenetic information harbored in a single region (Supplementary Figure S1). For interfamilial and inter-subfamilial relationships of NWMs, the analyses of 22 loci (Table 2 and Figure 4) do not support any of the 6 hypotheses presented in Figure 1. In accordance with the question-specific strategy (Chen et al. 2015), these “non-matching” loci were excluded, with the aim of improving the signal strength of the data for the questions of interest. The resulting data sets comprise 30 loci for the interfamilial relationships (13,796 bp in total; referred to as matchA dataset hereafter) and 24 for the inter-subfamilial relationships (11,108 bp in total; referred to as matchB dataset hereafter) (Table 1). MatchA and matchB contain 1,711 bp (12.4%) and 1386 bp (12.5%) parsimony-informative sites, respectively. The average K2P distance for both matchA and matchB was 5.9%, which is higher than for the IN, IG and combined datasets. Interestingly, all concatenation and coalescence analyses of the matchA and matchB datasets produced an identical tree topology to that inferred from the analyses of all non-coding regions combined (Figure 3), thus supporting hypothesis Ac (ML BS: 97% and 70%, Bayesian PP: 1.00 and <0.90, STAR BS: 95% and 50%, ASTRAL BS: 92% and <50%) and hypothesis Ba (ML BS: both <50% %, Bayesian PP: both <0.90, STAR BS: both BS < 50%, ASTRAL BS: both BS < 50%).
Figure 4.
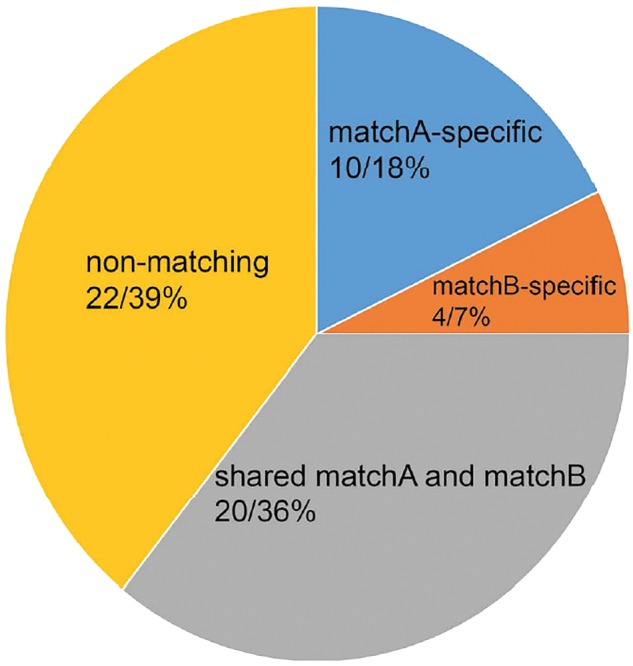
The matchA and matchB datasets comprise 30 (44%) and 24 (43%) genes that support any of the 3 hypotheses about NWM interfamilial (Figure 1A) and inter-subfamilial (Figure 1B) relationships, respectively. Both datasets shared 20 (36%) loci. A total of 22 (39%) loci do not match any of the 6 hypotheses.
Divergence time estimation
Divergence time calculations for the origin and diversification among NWM families and subfamilies based on the tree topology inferred from combining all non-coding regions revealed extremely short branches suggesting diversification within relative short time periods. According to our time estimates, the family Cebidae diverged from the common ancestor of Pitheciidae and Atelidae at 25.7 (95% HPD: 20.64–31.12) Ma, while latter 2 split shortly afterwards 24.73 (19.86–29.98) Ma. Within Cebidae, the subfamily Cebinae separated from the ancestor of Aotinae and Callithrichinae at 22.27 (17.92–27.14) Ma, whereas Aotinae and Callithrichinae diverged 21.7 (17.4–26.42) Ma. Within Pitheciidae and Atelidae, subfamilies split 19.7 (15.7–23.79) Ma (Pitheciinae and Callicebinae) and 16.24 (12.54–20.4) Ma (Atelinae and Alouattinae), respectively. NWM genera appeared between 19.84 (15.78–24.3) Ma (Cebus/Sapajus and Saimiri within Cebinae) and 4.98 (3.66–6.44) Ma (Callithrix and Cebuella within Callithrichinae). The split within Cebinae is relatively old and Cebus/Sapajus and Saimiri are sometimes classified as distinct subfamilies Cebinae and Saimirinae (Hershkovitz 1972, 1977; Mittermeier et al. 2013).
Discussion
Among mammalian phylogenies, those characterized by rapid species radiations have long been a challenging problem. Our study based on a set of nuclear non-coding loci, including INs and IGs regions, using both concatenation and coalescence tree-inference methods as well as a question-specific approach, provides insights into the phylogenetic relationships among NWM families and subfamilies.
In our study, we obtained consistent branching patterns among NWM families from IGs and INs and different tree-inference methods, but different relationships were recovered for the 3 Cebidae subfamilies (Figure 2). However, a consensus tree supporting hypotheses Ac and Ba was consistently recovered from all the analyses of the combined IG and IN datasets (Figure 3), albeit with varying degrees of statistical support. Among the 3 NWM families, a closer affinity between Pitheciidae and Atelidae than either is to Cebidae was obtained, that is, hypothesis Ac. This result is in agreement with studies using nuclear protein-coding loci and combined nuclear and mitochondrial loci (Schneider et al. 1993, 1996; Harada et al. 1995; Opazo et al. 2006; Schrago 2007), but disagrees with some other studies relying on nuclear protein-coding genes (Poux and Douzery 2004; Perelman et al. 2011) and genomic segments (Schrago et al. 2014), mitochondrial genome data (Finstermeier et al. 2013), non-coding genes (Wildman et al. 2009; Jameson-Kiesling et al. 2015), transposable elements analyses (Osterholz et al. 2009) and the combined datasets of different classes of genes (Fabre et al. 2009; Chatterjee et al. 2009; Springer et al. 2012; Aristide et al. 2015), in which 2 alternative hypotheses Aa or Ab were suggested instead. In this study, the proposals of hypotheses Aa and Ab were not found in any of the datasets (Figures 2 and 3). In contrast, the results from the concatenation and coalescence analyses all supported hypothesis Ac. Notably, the nodal supports for hypothesis Ac for the combined IG dataset are relatively high in ML and Bayesian analyses (BS: 78%, PP: 0.95).
Depending on the taxa examined and analytical methods used, previous studies have supported each of the 3 hypotheses of the relationships among the 3 subfamilies of Cebidae (Figure 1b). In the present study, a sister grouping of Aotinae and Callithrichinae to the exclusion of Cebinae, that is, hypothesis Ba, was recovered from the concatenation analyses and STAR analyses based on the IG dataset (Figure 2c) as well as those from all the analyses of the combined INs and IGs (Figure 3) with high nodal supports in the concatenation analyses. This result is in agreement with previous nuclear and mitochondrial analyses (Perelman et al. 2011; Perez et al. 2012; Springer et al. 2012; Finstermeier et al. 2013; Jameson-Kiesling et al. 2015; Aristide et al. 2015). The alternative hypothesis Bc, that is, the sister grouping of Callithrichinae and Cebinae, was found in concatenation and STAR analyses of INs and ASTRAL analyses of IGs (Figure 2a), but with low nodal support in all cases. The hypothesis Bb for the grouping of Aotinae and Cebinae is only recovered here in ASTRAL analyses of INs (Figure 2b).
Previous phylogenetic studies of NWMs have been based mainly on concatenation tree-inference methods (Schneider et al. 1993, 1996; Poux and Douzery et al. 2004; Opazo et al. 2006; Schrago 2007; Fabre et al. 2009; Osterholz et al. 2009; Wildman et al. 2009; Chatterjee et al. 2009; Perelman et al. 2011; Springer et al. 2012; Finstermeier et al. 2013; Jameson-Kiesling et al. 2015; Aristide et al. 2015). A coalescence-based method (*BEAST) has been applied by Perez et al. (2012) using data from published studies (Opazo et al. 2006; Wildman et al. 2009; Perelman et al. 2011), whereas Schrago et al. (2014) analyzed 92Mbp of genomic segments of a limited number of samples using STAR and MPEST, which resulted in an unresolved tree topology. In our study, analyses of the independent nuclear non-coding datasets from previous studies using both traditional concatenation (ML and Bayesian) and 2 recently-developed summary coalescence methods (ASTRAL and STAR) provide an opportunity to examine their application in addressing the phylogenetic resolution among NWMs. Intriguingly, our analyses suggest a consensus tree on interfamilial relationships and an incongruence concerning inter-subfamilial relationships between concatenation and coalescence phylogenetic results in the case of IN and IG analyses, respectively (Figure 2). Phylogenetic inconsistent between different studies may be caused by different markers, incomplete lineage sorting and different tree-building methods. Phylogenetic incongruence between concatenation and coalescence trees has been reported in mammalian orders (McCormack et al. 2012; Song et al. 2012; Tsagkogeorga et al. 2013; Kumar et al. 2013; Giarla and Esselstyn 2015), snakes (Pyron et al. 2014; Ruane et al. 2014), birds (Haddrath and Baker 2012; Fuchs et al. 2013; Jarvis et al. 2014) and plants (Springer and Gatesy 2012; Zhao et al. 2013; Zhong et al. 2013, 2014; Xi et al. 2013, 2014; Wickett et al. 2014; Tang et al. 2015; Simmons and Gatesy 2015; Nater et al. 2015). It has been thought that the probability of the occurrence of such conflicting signals would increase when splitting times between taxa are short (McCormack et al. 2012; Song et al. 2012; Tsagkogeorga et al. 2013; Kumar et al. 2013; Xi et al. 2013, 2014; Pyron et al. 2014; Tang et al. 2015; Nater et al. 2015), as typical for platyrrhines. It is noted that when all non-coding regions (INs and IGs) are combined, a congruence supporting hypotheses Ac for interfamilial and Ba for inter-subfamilial relationships from both tree-inference methods was retrieved (Figure 3). Perez et al. (2012); Perez and Rosenberger (2014) mentioned that the discrepancy between coalescence and concatenation methods in resolving the rapid radiation events among NWMs is not unexpected most likely as a result of incomplete lineage sorting. However, it should be also mentioned that the support values from the coalescence analyses in the present study are low (Figures 2 and 3), in contrast to the generally high support from the concatenation analyses. In fact, the coalescent-based analyses which use summary methods that estimate the species tree by combining individual gene trees have been thought to suffer from insufficient phylogenetic signal in the case of short gene regions for which the alignments will increase gene tree estimation error (Pollock et al., 2002; Roch and Warnow 2015). For our dataset, 56 individual loci were taken from previous studies (Wildman et al. 2009; Wang et al. 2012) and not all sequence length of these individual loci are longer than 1 kb. So we assume that the coalescent-based analyses of our dataset are more likely to suffer from insufficient phylogenetic signal in the case of very short gene regions, which may lead to the low nodal supports observed in coalescent-based analyses. An increasing number of informative loci used may likely increase the power of coalescence methods for our dataset to further phylogenetic resolution. We suggested that the dataset used maybe a more important factor for the phylogenetic studies dealing with the family and subfamily-level relationships of NWMs given that the combined dataset and the question-specific loci dataset retrieve the consistent relationships regardless of the tree-building methods.
Chen et al. (2015) developed a question-specific approach which operates by selecting those gene sequences that yield support for one of several predefined hypotheses, with the aim of concentrating the phylogenetic signal for a specific question and not allowing it to be swamped by individual gene history. By alleviating the incongruences associated with data size and the tree inference method, the authors demonstrated the enhanced performance of their method for resolving problematic relationships within jawed vertebrates. Interestingly, using 2 question-specific datasets in our study recovered a single tree in favor of the combined non-coding regions trees, regardless of the tree-inference methods used (Figure 3), providing further support for hypotheses Ac and Ba. Hence, the application of this approach showed its resolving power at the family and subfamilial level among platyrrhines. The comparison of different datasets examined here found that the 2 question-specific datasets matchA and matchB demonstrated slightly higher variable sites (25.2% and 25.6%, respectively), and parsimony-informative sites (12.4% and 12.5%, respectively), than the INs (22.8% and 10.9%), IGs (23.5% and 10.8%) and all combined dataset (23.1% and 10.9%). Thus, it seems that question-specific methods may collect more phylogenetic signals to reconstruct the evolutionary history of NWMs.
In conclusion, our study provides support for some previously suggested relationships among families (a sister-group relationship between Pitheciidae and Atelidae) and subfamilies (a sister group relationship between Aotinae and Callithrichinae) within NWMs from the perspective of multiple non-coding loci and various tree-inference methods (STAR and ASTRAL) as well as a question-specific approach. Nonetheless, to clarify the NWMs phylogenetic framework still needs future evaluation by the addition of independent sequence data and a deeper taxonomic sampling.
Supplementary Material
Acknowledgments
We are grateful to the staff of the zoos in Cologne, Gettorf, Kunming, Landau, Magdeburg, Romagne, Rostock, Stockholm, and Toronto for providing valuable primate samples. This work was supported by the Animal Branch of the Germplasm Bank of Wild Species, Chinese Academy of Sciences (Large Research Infrastructure Funding). We would also like to thank Nicole Otto and Christiane Schwarz for help in the laboratory.
Funding
Financial support for this project was obtained from the Yunnan Provincial Education Fund project in Scientific Research to XPW (No. 2015Y003), the National Youth Talent Support Program to LY, and the German Primate Center to CR.
Author contributions
L.Y. and C.R. designed and managed the project. X.P.W., B.K.L., and C.R. helped with sample collection. X.P.W., C.R., and Y.P.L. performed the experiments. X.P.W. and J.Y.H. performed the data analyses. X.P.W., L.Y., C.R., B.K.L., and N.T. wrote the paper. All authors read and approved the final manuscript.
References
- Akaike H, 1974. A new look at the statistical model identification. IEEE Trans Automat Contr 19:716–723. [Google Scholar]
- Aristide L, Rosenberger AL, Tejedor MF, Perez SI, 2015. Modeling lineage and phenotypic diversification in the New World monkey (Platyrrhini, Primates) radiation. Mol Phylogenet Evol 82:375–385. [DOI] [PubMed] [Google Scholar]
- Benton MJ, Donoghue PCJ, Asher RJ, Friedman M, Near TJ. et al. , 2015. Constraints on the timescale of animal evolutionary history. Palaeontol Electron 18:1–107. [Google Scholar]
- Byrne H, Rylands AB, Carneiro JC, Lynch-Alfaro JW, Bertuol F. et al. , 2016. Phylogenetic relationships of the New World titi monkeys (Callicebus): first appraisal of taxonomy based on molecular evidence. 13:10 BMC Evol Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J, 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552. [DOI] [PubMed] [Google Scholar]
- Chen MY, Liang D, Zhang P, 2015. Selecting question-specific genes to reduce incongruence in phylogenomics: a case study of jawed vertebrate backbone phylogeny. Syst Biol 64:1104–1120. [DOI] [PubMed] [Google Scholar]
- Chatterjee HJ, Ho SY, Barnes I, Groves C, 2009. Estimating the phylogeny and divergence times of primates using a supermatrix approach. BMC Evol Biol 9:259.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado NM, Galbany J, Pérez-Pérez A, 2016. Molar shape variability in platyrrhine primates. J Hum Evol 99:79–92. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A, 2007. BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre PH, Rodrigues A, Douzery EJ, 2009. Patterns of macroevolution among Primates inferred from a supermatrix of mitochondrial and nuclear DNA. Mol Phylogenet Evol 53:808–825. [DOI] [PubMed] [Google Scholar]
- Felsenstein J, 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. [DOI] [PubMed] [Google Scholar]
- Fleagle JG, 2013. Primate Adaptation and Evolution, 3rd edn. New York: Academic Press. [Google Scholar]
- Finstermeier K, Zinner D, Brameier M, Meyer M, Kreuz E. et al. , 2013. A mitogenomic phylogeny of living primates. PLoS ONE 8:e69504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs J, Pons JM, Liu L, Ericson PGP, Couloux A. et al. , 2013. A multi-locus phylogeny suggests an ancient hybridization event between Campephilus and melanerpine woodpeckers (Aves: picidae). Mol Phylogenet Evol 67:578–588. [DOI] [PubMed] [Google Scholar]
- Giarla TC, Esselstyn JA, 2015. The challenges of resolving a rapid, recent radiation: empirical and simulated phylogenomics of Philippine shrews. Syst Biol 64:727–740. [DOI] [PubMed] [Google Scholar]
- Haddrath O, Baker AJ, 2012. Multiple nuclear genes and retroposons support vicariance and dispersal of the palaeognaths, and an Early Cretaceous origin of modern birds. Proc Biol Sci 279:4617–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada ML, Schneider H, Schneider MPC, Sampaio I, Czelusniak J. et al. , 1995. DNA evidence on the phylogenetic systematics of New World monkeys: support for the sister-grouping of Cebus and Saimiri from two unlinked nuclear genes. Mol Phylogenet Evol 4:331–349. [DOI] [PubMed] [Google Scholar]
- Hershkovitz P, 1972. Notes on New World monkeys. Int Zoo Yearb 12:3–12. [Google Scholar]
- Hershkovitz P, 1977. Living New World Monkeys (Platyrrhini). With an Introduction to Primates. Chicago (IL: ): University of Chicago Press. [Google Scholar]
- Hodgson JA, Sterner KN, Matthews LJ, Burrell AS, Jani RA. et al. , 2009. Successive radiations, not stasis, in the South American primate fauna. Proc Natl Acad Sci USA 106:5534–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F, 2001. MrBayes: bayesian inference of phylogenetic trees. Bioinformatics 17:754–755. [DOI] [PubMed] [Google Scholar]
- Jameson-Kiesling NM, Yi SV, Xu K, Gianluca Sperone F, Wildman DE, 2015. The tempo and mode of New World monkey evolution and biogeography in the context of phylogenomic analysis. Mol Phylogenet Evol 82:386–399. [DOI] [PubMed] [Google Scholar]
- Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P. et al. , 2014. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 346:1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay RF, Fleagle JG, Mitchell TRT, Colbert M, Bown T. et al. , 2008. The anatomy of Dolichocebus gaimanensis, a stem platyrrhine monkey from Argentina. J Hum Evol 54:323–382. [DOI] [PubMed] [Google Scholar]
- Kinzey WG, 1997. New World Monkeys: Ecology, Evolution and Behavior. New York: de Gruyter. [Google Scholar]
- Kumar V, Hallström BM, Janke A, 2013. Coalescent-based genome analyses resolve the early branches of the Euarchontoglires. PLoS ONE 8:e60019.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K, 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yu L, Pearl DK, Edwards SV, 2009. Estimating species phylogenies using coalescence times among sequences. Syst Biol 58:468–477. [DOI] [PubMed] [Google Scholar]
- Matthee CA, Eick G, Willows-Munro S, Montgelard C, Pardini AT. et al. , 2007. Indel evolution of mammalian introns and the utility of non-coding nuclear markers in eutherian phylogenetics. Mol Phylogenet Evol 42:827–837. [DOI] [PubMed] [Google Scholar]
- McCormack JE, Faircloth BC, Crawford NG, Gowaty PA, Brumfiled RT. et al. , 2012. Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species-tree analysis. Genome Res 10:746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S, Reaz R, Bayzid MS, Zimmermann T, Swenson MS. et al. , 2014. ASTRAL: genome-scale coalescent-based species tree. Bioinformatics 30:i541–i548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittermeier RA, Rylands AB, Wilson DE, 2013. Handbook of the Mammals of the World, Vol. 3, Primates. Barcelona: Lynx Edicions. [Google Scholar]
- Nater A, Burri R, Kawakami T, Smeds L, Ellegren H, 2015. Resolving evolutionary relationships in closely related species with whole-genome sequencing data. Syst Biol 64:1000–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ometto L, De Lorenzo D, Stephan W, 2006. Contrasting patterns of sequence divergence and base composition between Drosophila introns and intergenic regions. Biol Lett 2:604–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo JC, Wildman DE, Prychitko T, Johnson RM, Goodman M, 2006. Phylogenetic relationships and divergence times among New World monkeys (Platyrrhini, Primates). Mol Phylogenet Evol 40:274–280. [DOI] [PubMed] [Google Scholar]
- Osterholz M, Walter L, Roos C, 2009. Retropositional events consolidate the branching order among New World monkey genera. Mol Phylogenet Evol 50:507–513. [DOI] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE. et al. , 2011. A molecular phylogeny of living primates. PLoS Genet 7:e1001342.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SI, Klaczko J, dos Reis SF, 2012. Species tree estimation for a deep phylogenetic divergence in the New World monkeys (Primates: platyrrhini). Mol Phylogenet Evol 65:621–630. [DOI] [PubMed] [Google Scholar]
- Perez SI, Rosenberger AL, 2014. The status of platyrrhine phylogeny: a meta-analysis and quantitative appraisal of topological hypotheses. J Hum Evol 76:177–187. [DOI] [PubMed] [Google Scholar]
- Pollock DD, Zwickl DJ, McGuire JA, Hillis DM, 2002. Increased taxon sampling is advantageous for phylogenetic inference. Syst Biol 51:664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D, Buckley TR, 2004. Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst Biol 53:793–808. [DOI] [PubMed] [Google Scholar]
- Posada D, 2008. jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256. [DOI] [PubMed] [Google Scholar]
- Posada D, 2009. Selection of models of DNA evolution with jModelTest. Methods Mol Biol 537:93–112. [DOI] [PubMed] [Google Scholar]
- Poux C, Douzery EJP, 2004. Primate phylogeny, evolutionary rate variations, and divergence times: a contribution from the nuclear gene IRBP. Am J Phys Anthropol 124:1–16. [DOI] [PubMed] [Google Scholar]
- Pyron RA, Hendry CR, Chou VM, Lemmon EM, 2014. Effectiveness of phylogenomic data and coalescent species-tree methods for resolving difficult nodes in the phylogeny of advanced snakes (Serpentes: caenophidia). Mol Phylogenet Evol 81:221–231. [DOI] [PubMed] [Google Scholar]
- Rambaut A, Drummond AJ, 2009. Tracer v15. Available from: http://beast.bio.ed.ac.uk/software/tracer (accessed 1 December 2009).
- Rambaut A, 2012. FigTree v1. 4.0. A graphical viewer of phylogenetic trees. Available from: http://tree.bio.ed.ac.uk/software/figtree/ (accessed 21 January 2012).
- Roch S, Warnow T, 2015. On the robustness to gene tree estimation error (or lack thereof) of coalescent-based species tree methods. Syst Biol 64:663–676. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP, 2003. MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. [DOI] [PubMed] [Google Scholar]
- Ruane S, Bryson RW Jr, Pyron RA, Burbrink FT, 2014. Coalescent species delimitation in milksnakes (genus Lampropeltis) and impacts on phylogenetic comparative analyses. Syst Biol 63:231–250. [DOI] [PubMed] [Google Scholar]
- Sambrook E, Fritsch EF, Maniatis T, 1989. Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Press. [Google Scholar]
- Schneider H, Schneider MP, Sampaio I, Harada ML, Stanhope M. et al. , 1993. Molecular phylogeny of the New World monkeys (Platyrrhini, Primates). Mol Phylogenet Evol 2:225–242. [DOI] [PubMed] [Google Scholar]
- Schneider H, Sampaio MIC, Harada ML, Barroso CML, Stanhope MPC. et al. , 1996. Molecular phylogeny of the New World monkeys (Platyrrhini, Primates) based on two unlinked nuclear genes: iRBP intron 1 and e-globin sequences. Am J Phys Anthropol 100:153–179. [DOI] [PubMed] [Google Scholar]
- Schrago GC, 2007. On the time scale of New World primate diversification. Am J Phys Anthropol 132:344–354. [DOI] [PubMed] [Google Scholar]
- Schrago GC, Menezes NA, Furtado C, Bonvicino RC, Seuanez NH, 2014. Multispecies coalescent analysis of the early diversification of neotropical Primates: phylogenetic inference under strong gene trees/species tree conflict. Genome Biol Evol 6:3105–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo TK, 2008. Calculating bootstrap probabilities of phylogeny using multilocus sequence data. Mol Biol Evol 25:960–971. [DOI] [PubMed] [Google Scholar]
- Shaw TI, Ruan Z, Glenn TC, Liu L, 2013. STRAW: species TRee Analysis Web server. Nucleic Acids Res 41:238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestro D, Michalak I, 2010. raxmlGUI: A Graphical Front-End for RAxML Available from: http://sourceforge.net/projects/raxmlgui/.
- Simmons MP, Gatesy J, 2015. Coalescence vs. concatenation: sophisticated analyses vs. first principles applied to rooting the angiosperms. Mol Phylogenet Evol 91:98–122. [DOI] [PubMed] [Google Scholar]
- Song S, Liu L, Edwards SV, Wu S, 2012. Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model. Proc Natl Acad Sci USA 109:14942–14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer MS, Meredith RW, Gatesy J, Emerling CA, Park J. et al. , 2012. Macroevolutionary dynamics and historical biogeography of primate diversification inferred from a species supermatrix. PLoS ONE 7: e49521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer MS, Gatesy J, 2012. Land plant origins and coalescence confusion. Trends Plant Sci 19:267–269. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Swenson KM, EI-Mabrouk N, 2012. Gene trees and species trees: irreconcilable differences. BMC Bioinformatics 13:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Zou XX, Zhang LB, Ge S, 2015. Multilocus species tree analyses resolve the ancient radiation of the subtribe Zizaniinae (Poaceae). Mol Phylogenet Evol 84:232–239. [DOI] [PubMed] [Google Scholar]
- Ting N, Sterner KN, 2013. Primate molecular phylogenetics in a genomic era. Mol Phylogenet Evol 66:565–568. [DOI] [PubMed] [Google Scholar]
- Tsagkogeorga G, Parker J, Stupka E, Cotton JA, Rossiter SJ, 2013. Phylogenomic analyses elucidate the evolutionary relationships of bats. Curr Biol 23:2262–2267. [DOI] [PubMed] [Google Scholar]
- Wang XP, Yu L, Roos C, Ting N, Chen CP. et al. , 2012. Phylogenetic relationships among the colobine monkeys revisited: new insight from analyses of complete mt genomes and 44 nuclear non-coding markers. PLoS One 7: e36274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickett NJ, Mirarab S, Nguyen N, Warnow T, Carpenter E. et al. , 2014. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc Natl Acad Sci USA 111: E4859–E4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildman DE, Jameson NM, Opazo JC, Yi SV, 2009. A fully resolved genus level phylogeny of Neotropical primates (Platyrrhini). Mol Phylogenet Evol 53:694–702. [DOI] [PubMed] [Google Scholar]
- Xia X, 2017. DAMBE6: new tools for microbial genomics, phylogenetics and molecular evolution. J Hered 108:431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi ZX, Rest JR, Davis CC, 2013. Phylogenomics and coalescent analyses resolve extant seed plant relationships. PLoS ONE 8: e80870.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi ZX, Liu L, Rest JS, Davis CC, 2014. Coalescent versus concatenation methods and the placement of Amborella as sister to water lilies. Syst Biol 63:919–932. [DOI] [PubMed] [Google Scholar]
- Yu L, Zhang YP, 2005. Evolutionary implications of multiple SINE insertions unusually occurred in an intronic region from diverse mammals. Mamm Genome 16:651–660. [DOI] [PubMed] [Google Scholar]
- Zhao L, Zhang N, Ma PF, Liu Q, Li DZ. et al. , 2013. Analyses of nuclear genes reveal the evolutionary relationships within the BEP clade and the evidence of positive selection in Poaceae. PLoS ONE 8:e64642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Liu L, Yan Z, Penny D, 2013. Origin of land plants using the multispecies coalescent model. Trends Plant Sci 18:492–495. [DOI] [PubMed] [Google Scholar]
- Zhong B, Liu L, Yan Z, Penny D, 2014. The multispecies coalescent model and land plant origins: a reply to Springer and Gatesy. Trends Plant Sci 19:270–272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.