

Supplemental Digital Content is available in the text.
Keywords: arrhythmogenic right ventricular, dysplasia, calcineurin, GSK3β, therapy, TMEM43
Background:
Arrhythmogenic cardiomyopathy/arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiac disease characterized by fibrofatty replacement of the myocardium, resulting in heart failure and sudden cardiac death. The most aggressive arrhythmogenic cardiomyopathy/ARVC subtype is ARVC type 5 (ARVC5), caused by a p.S358L mutation in TMEM43 (transmembrane protein 43). The function and localization of TMEM43 are unknown, as is the mechanism by which the p.S358L mutation causes the disease. Here, we report the characterization of the first transgenic mouse model of ARVC5.
Methods:
We generated transgenic mice overexpressing TMEM43 in either its wild-type or p.S358L mutant (TMEM43-S358L) form in postnatal cardiomyocytes under the control of the α-myosin heavy chain promoter.
Results:
We found that mice expressing TMEM43-S358L recapitulate the human disease and die at a young age. Mutant TMEM43 causes cardiomyocyte death and severe fibrofatty replacement. We also demonstrate that TMEM43 localizes at the nuclear membrane and interacts with emerin and β-actin. TMEM43-S358L shows partial delocalization to the cytoplasm, reduced interaction with emerin and β-actin, and activation of glycogen synthase kinase-3β (GSK3β). Furthermore, we show that targeting cardiac fibrosis has no beneficial effect, whereas overexpression of the calcineurin splice variant calcineurin Aβ1 results in GSK3β inhibition and improved cardiac function and survival. Similarly, treatment of TMEM43 mutant mice with a GSK3β inhibitor improves cardiac function. Finally, human induced pluripotent stem cells bearing the p.S358L mutation also showed contractile dysfunction that was partially restored after GSK3β inhibition.
Conclusions:
Our data provide evidence that TMEM43-S358L leads to sustained cardiomyocyte death and fibrofatty replacement. Overexpression of calcineurin Aβ1 in TMEM43 mutant mice or chemical GSK3β inhibition improves cardiac function and increases mice life span. Our results pave the way toward new therapeutic approaches for ARVC5.
Clinical Perspective.
What Is New?
This is the first animal model to reproduce human arrhythmogenic right ventricular cardiomyopathy (ARVC) type 5, the most aggressive subtype of arrhythmogenic cardiomyopathy.
Transgenic mice expressing TMEM43 (transmembrane protein 43)–S358L show fibrofatty replacement of the myocardium and die at a young age.
This model confirms that TMEM43 is localized mostly at the nuclear membrane and provides new information on the pathophysiological mechanism of ARVC5.
As in other forms of ARVC, the glycogen synthase kinase-3β signaling pathway plays an important role in this disease.
What Are the Clinical Implications?
Using this animal model, our work tests 2 new therapeutic approaches for ARVC5, for which there are currently no effective therapies to prevent disease progression in humans.
Although the antifibrotic drug GM-CT-01 did not show a beneficial effect on transgenic mice expressing TMEM43-S358L, inhibition of glycogen synthase kinase-3β improved cardiac function and survival, opening the way to a new therapeutic approach focused on glycogen synthase kinase-3β inhibition that could be used in humans with ARVC5 in the future.
Arrhythmogenic cardiomyopathy (ACM)/arrhythmogenic right ventricular cardiomyopathy (ARVC) is an incurable genetically inherited disease that causes heart failure and sudden cardiac death.1,2 ACM/ARVC is an autosomal dominant disease with rare recessive forms. To date, 15 independent loci and 13 dominant ACM/ARVC–causing genes have been linked to the condition. Up to 60% of ACM/ARVC cases have been attributed to mutations in genes encoding proteins of the desmosomal complex, with a significant minority caused by mutations in nondesmosomal genes.3–5 The most aggressive ACM/ARVC subtype is ACM/ARVC type 5 (ARVC5), which is caused by a point substitution (p.S358L) in a highly conserved TMEM43/LUMA (transmembrane region of transmembrane protein 43). The causal gene for ARVC5 was first described in families from Newfoundland (Canada) in 2008 as the cause of a fully penetrant aggressive disease with a high incidence of malignant ventricular arrhythmias.6 Since then, several other patients have been diagnosed around the globe. Prognosis is very poor, with more than half of affected men dying by the age of 50 years.7 The pathogenic mechanism of ARVC5 and the role of TMEM43 in the genesis of the disease are poorly understood, precluding the development of effective therapies.
Here, we report the characterization of the first transgenic mouse model of ARVC5. We analyzed disease development and progression in depth and determined the localization, function, and mechanism of action of TMEM43. Finally, we show that glycogen synthase kinase-3β (GSK3β) inhibition with a chemical inhibitor or by calcineurin Aβ1 (CnAβ1) overexpression improves cardiac function and survival of mice with ARVC5.
Methods
Data Availability
The data that support the findings of this study are available from the corresponding authors on reasonable request. RNA sequencing data are available at the GEO repository (GSE101301). Proteomic data are available at the PeptideAtlas repository (http://www.peptideatlas.org/PASS/PASS01063). Full Methods can be found in the online-only Data Supplement.
Mice
TMEM43 wild-type (TMEM43WT) and TMEM43 mutant (TMEM43mut) mice, in the C57BL/6JCrl (Charles River Laboratories) background, express human WT TMEM43 and human TMEM43-S358L, respectively, specifically in cardiomyocytes under the control of the myosin heavy chain (MHC) promoter. WT C57BL/6JCrl mice were used as controls. TMEM43mut male mice were crossed with α-myosin heavy chain–CnAβ1 mice8 to generate the double-transgenic mouse line TMEM43mut-CnAβ1. Male and female mice were used throughout the study. Mice were housed in an air-conditioned room with a 12-hour light/dark cycle with free access to water and chow. For the inhibition of galectin 3, TMEM43mut mice were randomized to the saline-treated control group (n=8) or the GM-CT-01–treated group (n=8). Mice were treated twice a week with intravenous injections of GM-CT-01 (120 mg/kg) or placebo (normal saline) in the tail vein from 5 weeks to 4 months of age. For GSK3β inhibition, mice were injected intraperitoneally daily with SB216763 (2.5 mg/kg per day). All procedures were approved by the Centro Nacional de Investigaciones Cardiovasculares Carlos III Ethics Committee and the Regional Government of Madrid (PA-27/13, PROEX-177/17). All animal experiments conformed to EU Directive 2010/63EU and Recommendation 2007/526/EC, enforced in Spanish law under Real Decreto 53/2013.
Echocardiography
Cardiac function, chamber dilatation, and wall thickness were analyzed in neonatal mice and in mice 3 and 5 weeks and 2 and 4 months of age by transthoracic 2D and M-mode echocardiography. Measurements were carried out by a blinded operator using a high-frequency ultrasound system with a 50-MHz linear probe for neonates and a 30-MHz probe for older mice (Vevo 2100, Visualsonics Inc). For ultrasound scans, mice were placed on a heating pad; neonates were not anesthetized, whereas older mice were kept under light anesthesia with isoflurane adjusted to obtain a target heart rate of 500±50 bpm. Left ventricular (LV) ejection fraction (EF) and LV end-diastolic volume were obtained from the long-axis view, and LV posterior wall in diastole was obtained from the short-axis view. Right ventricular systolic function was assessed indirectly from the tricuspid annular plane systolic excursion, estimated from maximum lateral tricuspid annulus movement obtained from a 2D 4-chamber apical view. Images were analyzed offline by an expert using the Vevo 2100 Workstation software. Animals were euthanized by gradually filling the chamber with carbon dioxide. Mice, hearts, and lungs were weighed after death.
Electrocardiograms
ECGs were obtained in unanesthetized neonates and in 3- and 5-week-old and 2- and 4-month-old anesthetized mice with bipolar limb leads (leads I, II, and III) and unipolar limb leads (leads aVR, aVL, and aVF) for 60 to 90 seconds. Measurements were taken by a blinded operator with mice (except for neonates) placed under light anesthesia with isoflurane (MP36R, BIOPAC Systems, Inc). ECGs were analyzed by an expert using Acqknowledge 4.1.1. for MP36R (BIOPAC Systems, Inc). Mean values were calculated from 10 consecutive standard ECG time intervals and waves.
Cell Culture, Transfection, and Immunofluorescence
P19 cells were transfected with the following HA-tagged expression vectors for TMEM43: TMEM43WT-Ct-HA, Nt-HA-TMEM43WT, TMEM43-S358L-Ct-HA, and Nt-HA-TMEM43-S358L, where Nt and Ct indicate attachment of the HA tag to the N-terminus or the C-terminus. Cells were fixed in 4% paraformaldehyde in PBS for 10 minutes at 4°C, permeabilized for 10 minutes with 0.1% Triton X-100/PBS, and incubated in 10% goat serum/PBS for 30 minutes at room temperature. Cells were incubated overnight in 1% goat serum/PBS with anti-HA (11583816001, Roche). After primary antibody incubation, cells were washed with PBS, incubated for 1 hour at room temperature with Alexa Fluor 488 goat anti-mouse immunoglobulin G (A-11029; Thermo Fisher Scientific) and DAPI in 1% goat serum/PBS, and mounted in Vectashield mounting medium. Images were acquired with a Nikon A1R multiline inverted confocal microscope, a Plan Apo VC 60×/1,4 Oil DIC N2 Oil objective, and Nikon NIS reprocessing software. Brightness and contrast were linearly adjusted with Adobe Photoshop CS5.1.
Immunoprecipitation
TMEM43 immunoprecipitation experiments were performed in the P19 cell line. Briefly, cells were transfected as described above. Cells were lysed with TBS buffer (150 mmol/L NaCl, 20 mmol/L Tris-HCl, pH 7.4) supplemented with 1% Nonidet P-40, 5 mmol/L EDTA, 5 mmol/L MgCl2, and 1× complete protease, phosphatase, and acetylase inhibitors. Protein extracts were incubated with anti-HA–conjugated Dynabeads (Life Technologies) for 1 hour at 4°C. Beads were washed 3 times with lysis buffer containing 0.05% Nonidet P-40 and 5 times with lysis buffer without added detergent. Bound proteins were released from beads by boiling in 4× Laemmli sample buffer. Immunoprecipitates and input samples were resolved by SDS-PAGE or subjected to protein digestion followed by nanoliquid chromatography coupled to mass spectrometry for protein identification and quantification by peptide counting.9 The proteomics data set (raw and msf files and protein database) is available in the PeptideAtlas repository (http://www.peptideatlas.org/PASS/PASS01063, which can be downloaded via ftp.peptideatlas.org: username, PASS01063; password, FZ6532b).
Statistical Analysis
All data are presented as mean±SEM. All data sets were analyzed for statistical significance by regular or repeated-measures 1-way ANOVA followed by Bonferroni or Dunnett posttest for multiple comparisons or 2-way ANOVA followed by Bonferroni posttest (GraphPad Prism), as indicated in the figure legends. Survival curves were compared by the log-rank (Mantel-Cox) test. Differences were considered statistically significant at P<0.05.
Results
Cardiac-Restricted Expression of Human TMEM43-S358L Induces Biventricular Dysfunction and Reduces Survival
We generated transgenic mice expressing human TMEM43-S358L under the control of the α-myosin heavy chain promoter (TMEM43mut mice) to provide cardiac-specific expression of the mutant protein. As controls, we used mice overexpressing human WT TMEM43 (TMEM43WT mice) and WT littermates (Figure IA–IC in the online-only Data Supplement). TMEM43mut mice were dead by 6 months of age, with a median life span of 23 weeks, whereas TMEM43WT and WT mice showed no notable mortality at this age (Figure 1A).
Figure 1.
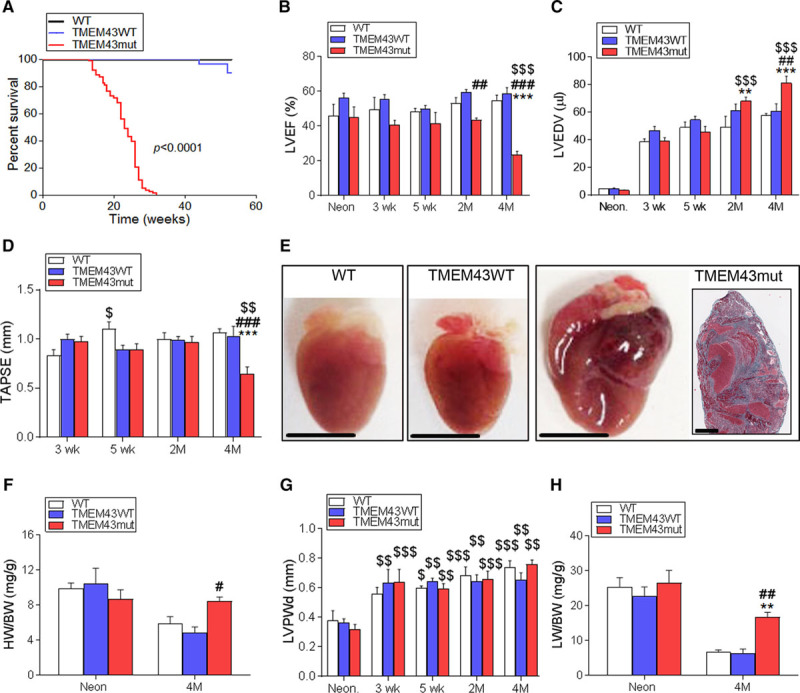
TMEM43 (transmembrane protein 43)–S358L expression in cardiomyocytes causes severe cardiac dysfunction. A, Wild-type mice (WT; n=27) and mice overexpressing either WT TMEM43 (TMEM43WT; n=29) or TMEM43-S358L (TMEM43mut; n=84) under the control of the α-myosin heavy chain (αMHC) promoter were monitored for 60 weeks, and their survival rate was determined from a Kaplan-Meier curve. The indicated P value was obtained with a log-rank test. B through D, Left ventricular ejection fraction (LVEF), end-diastolic volume (LVEDV), and tricuspid annular plane systolic excursion (TAPSE) measured by echocardiography at birth (Neon) and at 3 and 5 weeks and 2 and 4 months of age. E, Gross morphology of representative hearts from 4-month-old WT, TMEM43WT, and TMEM43mut mice; bar, 500 μm. Inset, A thrombus in the left atrium of a TMEM43mut mouse; bar, 50 μm. F, Ratio of heart weight to body weight (HW/BW) determined at birth and at 4 months. G, LV posterior wall thickness in diastole (LVPWd) analyzed by echocardiography. H, Ratio of lung weight to body weight (LW/BW) determined at birth and at 4 months. Graphs show mean±SEM. **P<0.01, ***P<0.001, TMEM43mut vs WT. #P<0.05, ##P<0.01, ###P<0.001, TMEM43mut vs TMEM43WT. $P<0.05, $$P<0.01, $$$P<0.001 for different time points vs neonates (B, C, and G) or 3 weeks (D) for each mouse line; 2-way regular (B, C, F, and H) or repeated-measures (D and G) ANOVA was used, followed by Bonferroni post-test; n=5 to 6 per group.
Echocardiography showed a decline in LVEF that was already evident in the TMEM43mut group at 2 months of age, indicating a loss of LV contractility (Figure 1B and Table I and Video I in the online-only Data Supplement). Accordingly, TMEM43mut mice also developed progressive LV dilatation with a high LV end-diastolic volume (Figure 1C). Right ventricular contractility was also reduced in TMEM43mut mice at 4 months of age (Figure 1D). Although death was slightly delayed in females, no significant differences in survival and cardiac function were observed between males and females (Figure ID–IF in the online-only Data Supplement).
Gross morphology at 4 months revealed a clear enlargement of TMEM43mut hearts compared with TMEM43WT and WT hearts (Figure 1E). TMEM43mut mice also had an elevated ratio of heart weight to body weight at 4 months (Figure 1F). However, no changes were observed in ventricular wall thickness (Figure 1G). Decreased cardiac function in TMEM43mut mice was accompanied by pulmonary congestion at 4 months (Figure 1H).
Electrocardiographic analysis showed progressive P-wave prolongation and defective depolarization of the atria in TMEM43mut mice, some of which developed atrial fibrillation and atrial paralysis (Figure 2A and 2D and Table II in the online-only Data Supplement). Similarly, TMEM43mut mice had ventricular disease, as evidenced by a progressive widening of the QRS complex (Figure 2B and 2E) and a progressive decrease in QRS amplitude (Figure 2C and 2F). No differences were observed between males and females (Figure II in the online-only Data Supplement). Together, the results of the ECG suggest that TMEM43mut mice develop electric defects already noticeable at 5 weeks of age.
Figure 2.
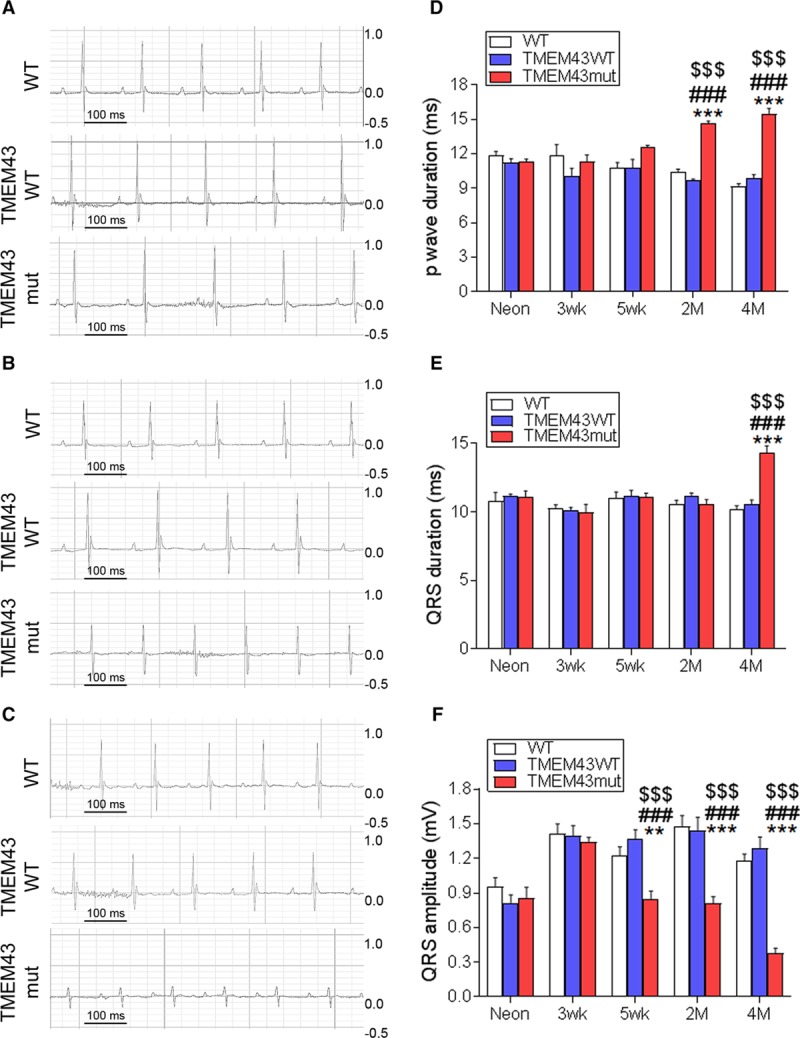
TMEM43 (transmembrane protein 43) mutant (mut) mice show cardiac conduction defects. A through C, Surface electrocardiographic analysis of cardiac conduction in wild-type (WT), TMEM43WT, and TMEM43mut mice 1 day (A), 5 weeks (B), and 4 months (C) of age. Shown are 0.6-second traces from the V3 lead. D through F, The p-wave duration (D) and QRS duration and amplitude (E and F) assessed at birth (Neon) and at 3 and 5 weeks and 2 and 4 months of age. Graphs show mean±SEM. **P<0.01, ***P<0.001, TMEM43mut vs WT. ###P<0.001, TMEM43mut vs TMEM43WT. $$$P<0.001 for different time points vs neonates for each mouse line; 2-way repeated-measures ANOVA followed by Bonferroni post-test; n=6.
TMEM43-S358L Expression in the Heart Induces Cardiomyocyte Death and Fibrofatty Replacement
An important hallmark of human ACM/ARVC is the progressive fibrofatty replacement of the ventricular myocardium10; however, whether fibrofatty replacement is preceded by cardiomyocyte death has not been clearly demonstrated. We found a significant increase in circulating cardiac troponin I levels in TMEM43mut mice as early as 5 weeks of age (Figure 3A), indicating cardiomyocyte necrosis. We also investigated the activation of apoptosis by assessing the presence of procaspase 3 and cleaved caspase 3 in the hearts of these mice. At 2 months of age, TMEM43mut mice showed increased levels of cleaved caspase 3, which were maintained at 4 months, although the differences did not reach statistical significance (Figure 3B and Figure IIIA in the online-only Data Supplement). In addition, a similar trend was found for Beclin1, a key protein involved in autophagy11 (Figure 3C and Figure IIIB in the online-only Data Supplement). These results provide the first clear evidence implicating cell-death pathways in the early and sustained cardiomyocyte loss in ARVC5.
Figure 3.
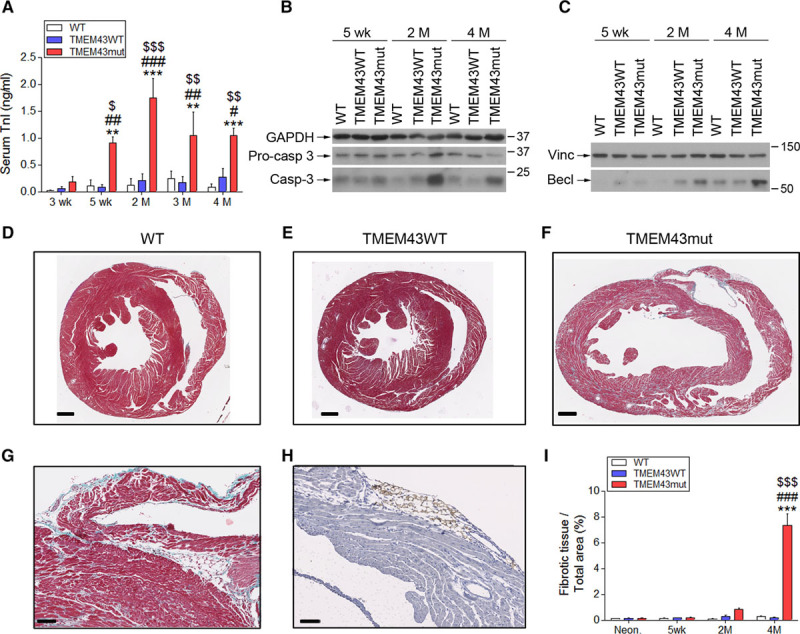
TMEM43 (transmembrane protein 43)–S358L causes cardiomyocyte death and replacement by fibrofatty tissue. A, Serum troponin I (TnI) determined by ELISA in mice 3 and 5 weeks and 2, 3, and 4 months of age. B and C, Western blot analysis of the presence in myocardium of activated and total caspase 3 (casp-3; B) and beclin1 (Becl; C) at the indicated time points. D through I, Masson trichrome staining in myocardial sections from wild-type (WT; D), TMEM43WT (E), and TMEM43 mutant (mut) mice (F and G). Subepicardial adipocytes were stained with perilipin (H), and the percentage of fibrotic tissue was quantified (I). Bar, 500 μm (D–F), 50 μm (G), and 100 μm (H). Graphs show data points for individual mice and mean±SEM. Vinc indicates vinculin. A and I, **P<0.01, ***P<0.001, TMEM43mut vs WT; #P<0.05, ##P<0.01, ###P<0.001, TMEM43mut vs TMEM43WT; $P<0.05, $$P<0.01, $$$P<0.001 for different time points vs 3 weeks (A) or neonates (I) for each mouse line; 2-way regular ANOVA followed by Bonferroni post-test; n=6 to 18.
To determine whether dead cardiomyocytes were substituted by fibrotic tissue, we analyzed heart sections by Masson trichrome staining. TMEM43mut mice showed massive fibrosis in both ventricles at 4 months of age (Figure 3D–3G). Mutant mice also showed accumulation of adipose tissue in the subepicardial layer and the myocardium, as revealed by perilipin staining (Figure 3H). Quantification of the fibrotic area confirmed the strong fibrotic response as disease progressed (Figure 3I). In line with these results, TMEM43mut mice showed an increase in collagen 1α1 mRNA at 2 and 4 months, together with strong induction of lysyl oxidase, which crosslinks collagen and elastin (Figure IIIC and IIID in the online-only Data Supplement). These changes were accompanied by increased expression of the myocardial remodeling markers skeletal muscle actin α1 and brain natriuretic peptide (Figure IIIE and IIIF in the online-only Data Supplement). Together, these results demonstrate that TMEM43-S358L induces cardiomyocyte death followed by fibrotic replacement.
Epicardium-Derived Cells Contribute to the Fibrotic Replacement of Cardiomyocytes
Using the Cre-LoxP technology and cell type–specific promoters, we generated 4 sets of lineage-tracing mice to determine the contribution made to the fibrotic replacement by endothelial cells (Tie2-Cre), macrophages (LysM-Cre), cardiomyocytes (cTnT-Cre), and epicardium-derived cells (Wt1-Cre). In addition to the TMEM43mut transgene, each line carried a floxed reporter allele (EYFP, RFP, or GFP) preceded by a STOP codon that was removed on recombination by the Cre recombinase. In these mice, Cre-expressing cells and their progeny were permanently labeled by expression of the reporter (Figure IVA–IVQ in the online-only Data Supplement). To locate collagen fibers in myocardial tissue, we used second-harmonic-generation microscopy (Figure IVA in the online-only Data Supplement), which highlights organized mature collagen fibers, and confocal reflection microscopy (Figure IVB in the online-only Data Supplement), which reveals general tissue fibrosis. Labeled macrophages were found in the tissue but did not colocalize with the collagen fibers (Figure IVC–IVE in the online-only Data Supplement), and neither did the cardiomyocytes or endothelial cells (Figure IVF–IVH and IVO–IVQ in the online-only Data Supplement). In contrast, both second-harmonic-generation and reflection microscopy revealed colocalization of epicardium-derived cells and collagen fibers in TMEM43mut hearts (Figure IVI–IVN in the online-only Data Supplement). These results indicate that epicardium-derived cells (which are likely resident fibroblasts derived from the epicardium during development) are the main cell type contributing to the massive interstitial fibrosis observed in ARVC5.
TMEM43 Is Located in the Nuclear Membrane and Interacts With Cytoskeleton-Binding Proteins
There is currently controversy about whether TMEM43 is located within the nuclear rim,12 the endoplasmic reticulum,13 or the intercalated disk.14 To investigate the cellular localization of WT and S358L human TMEM43 and their interacting partners, we first modeled TMEM43-S358L and WT TMEM43 proteins in silico. The p.S358L mutation in transmembrane domain 3 (red in Figure 4A and 4B) predicted disruption of 3D protein structure and lower structural stability for TMEM43-S358L compared with WT TMEM43. TMEM43-S358L also showed higher hydrophobicity than the WT form, reducing the availability of the transmembrane domain 4 (dark blue in Figure 4A and 4B), which is predicted to interact with a partner protein. The p.S358L mutation also affected the structure of the transmembrane domain 1 (orange in Figure 4A and 4B), which is necessary for TMEM43 dimerization. Because a previous report suggested that TMEM43 function requires dimerization through transmembrane domain 1,15 we also modeled homodimers and heterodimers of TMEM43WT and TMEM43mut. In silico modeling of the heterodimer WT TMEM43-TMEM43-S358L revealed a distorted structure compared with the WT TMEM43 homodimer (Figure 4C and 4D).
Figure 4.
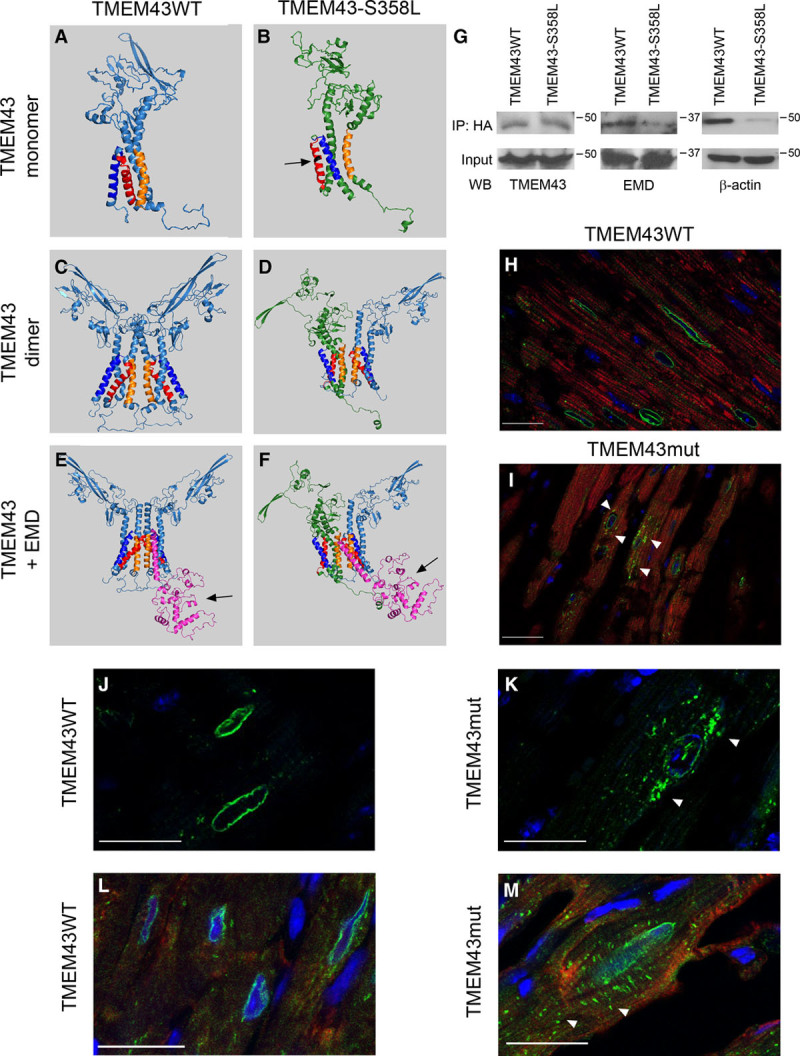
The S358L mutation alters TMEM43 (transmembrane protein 43) conformation and protein interactions. A through F, In silico modeling of the tertiary structure of wild-type (WT; A, C, and E, light blue) and mutant (mut) TMEM43 (B, D, and F, green) as monomers (A and B), dimers (C and D), or in complexes with emerin (EMD; purple molecule; E and F). Orange indicates transmembrane domain 1; red, transmembrane domain 3; dark blue, transmembrane domain 4; and black residue, S358L mutation. G, HA-tagged TMEM43WT and TMEM43mut were expressed in P19 cells and immunoprecipitated (IP) with anti-HA. The presence of TMEM43, EMD, and β-actin in the input and immunoprecipitate was analyzed by Western blot. H through M, Immunofluorescence analysis of TMEM43 localization in myocardial sections from 4-month-old TMEM43WT (H, J, and L) and TMEM43mut mice (I, K, and M) with 3 different anti-TMEM43 antibodies (H and I, Abcam; J and K, Santa Cruz; L and M, antibodies generated by Franke et al14). Green indicates TMEM43; red, troponin I (only for H and I); and blue, DAPI. White arrowheads indicate partial TMEM43 localization in the cytoplasm. Bar, 20 μm.
TMEM43 protein sequence is highly conserved along evolution.8 Nevertheless, the in silico model of mouse TMEM43 shows a different tertiary structure compared with the human protein, with a nonstructured loop arranging transmembrane domains 3 and 4 differently (Figure VA in the online-only Data Supplement). Most important, the p.S358L mutation has virtually no effect on the mouse TMEM43 structure, and mouse TMEM43-S358L shows only minimal differences with the WT protein (Figure VB and VC in the online-only Data Supplement). These results probably explain why TMEM43-S358L knock-in mice have no disease phenotype,16 whereas transgenic mice expressing the human protein develop ARVC5.
TMEM43 has been suggested to interact with emerin (Emery-Dreifuss muscular dystrophy [EMD]) and SUN2 (SUN domain-containing protein).15 We therefore modeled the interaction of these proteins with human TMEM43 dimers. The WT-mutant TMEM43 heterodimer showed an interaction with EMD (purple in Figure 4E); however, this interaction was predicted to abnormally embed EMD in the nuclear membrane (Figure 4F). This would have the effect of blocking EMD function because it would prevent its interaction with lamin A (the pocket in EMD that interacts with lamin A is indicated by an arrow in Figure 4E and 4F). This in silico prediction was validated by immunoprecipitation in P19 cells transfected with expression plasmids for HA-tagged WT TMEM43 and TMEM43-S358L, which revealed weaker interaction of EMD with TMEM43-S358L than with the WT form (Figure 4G). These results suggest that the p.S358L point mutation in human TMEM43 results in a dominant-negative form of TMEM43, disrupting the activity of the WT form.
Histological analysis of hearts from 4-month-old mice using 3 different anti-TMEM43 antibodies showed perinuclear localization of TMEM43 in TMEM43WT mice, whereas the TMEM43-S358L protein was partially delocalized in the cytoplasm in TMEM43mut mice (Figure 4H–4M). As in desmoplakin ACM/ARVC and other ARVC models,17,18 TMEM43mut mouse hearts showed mislocalization of connexin 43 (Figure VI in the online-only Data Supplement).
To validate those results, we analyzed TMEM43 localization in P19 cells transfected with plasmids expressing HA-tagged TMEM43 proteins. We found partial cytoplasmic localization of TMEM43-S358L, regardless of whether the HA tag was placed at the N- or the C-terminus, whereas the WT protein showed a mainly perinuclear staining (Figure VII in the online-only Data Supplement). The perinuclear localization of WT TMEM43 in the nuclear membrane is in agreement with several previous reports.13,15,16,19–21 It should be acknowledged, however, that TMEM43 has also been reported in desmosomes.14 Although we could not detect TMEM43 in these structures by immunofluorescence, we did see an interaction with desmosomal proteins in immunoprecipitation experiments (Table III in the online-only Data Supplement), suggesting that a fraction of TMEM43 may be localized in desmosomes.
To gain insight into the molecular partners of TMEM43, we immunoprecipitated the WT and the mutant protein from transfected P19 cells using an anti-HA antibody. Quantitative proteomics of the coprecipitating proteins identified several cytoskeleton and cytoskeleton-interacting proteins among the binding partners for WT TMEM43, including actin, actinin, spectrin, shroom 3, myosin, and formin-like 2 (Table III in the online-only Data Supplement). Many of these interactions were reduced in cells expressing TMEM43-S358L. Immunoprecipitation and Western blot analysis confirmed the reduced interaction of TMEM43-S358L with α-actin compared with the WT TMEM43 form (Figure 4G). Interestingly, the mutant TMEM43 protein showed increased interaction with the AKT modulator Pa2g4 (Table III in the online-only Data Supplement), which might affect this signaling pathway.
Antifibrotic Treatment Does Not Improve Cardiac Function in ARVC5 Mice
Increased interstitial fibrosis is a hallmark of human ARVC5 and was evident in TMEM43mut mice. Interstitial fibrosis increases passive stiffness, causes electric remodeling, and enhances arrhythmogenicity, further contributing to cardiac remodeling and dysfunction.1,22 To investigate whether reducing interstitial fibrosis could improve heart function in ARVC5, we treated mice with the β-galactoside–binding lectin Gal-3 (galectin-3) inhibitor GM-CT-01, a known antifibrotic drug23.
Gal-3 is highly expressed in fibrotic tissues23 and mediates extracellular matrix remodeling in heart failure.24 Gal-3–null mice are resistant to fibrotic disease of several organs, including liver, kidney, and lung.25 RNA sequencing revealed increased expression of the Gal-3-encoding gene (Lgals3) in the hearts of 2-month-old TMEM43mut mice, together with other genes involved in the immune response and fibrosis (Table IV and Figure VIII in the online-only Data Supplement). No major changes in transcript isoforms were detected (Table V in the online-only Data Supplement). Validation by quantitative reverse-transcribed polymerase chain reaction showed a strong induction of Lgals3 in TMEM43mut mice at 2 and 4 months of age (Figure 5A), which was confirmed by Western blot (Figure 5B and 5C). To investigate whether inhibition of Gal-3–regulated profibrotic pathways could improve heart function in ARVC5, we conducted blinded controlled in vivo experiments in TMEM43mut mice. The mice were treated with either saline or GM-CT-01, a galactomannan with antifibrotic properties26 that inhibits Gal-3 by binding to its carbohydrate-binding domain, which is necessary for the formation of Gal-3 pentamers.23 We then analyzed heart sections for collagen content by Masson trichrome staining and type by staining with Picrosirius red. Picrosirius red binds specifically to collagen fibrils of varying diameters and distinguishes between collagen type I, which confers stiffness to the tissue, and collagen type III, which is more elastic. Treatment with GM-CT-01 triggered a significant switch in the collagen type in TMEM43mut hearts from collagen type I to collagen type III (Figure 5D). Although the sum of the collagen fibers seemed to decrease with GM-CT-01, the total fibrotic area was not reduced (Figure 5E). LVEF was comparable in GM-CT-01–treated and saline-treated TMEM43mut mice (Figure 5F). These results suggest that although collagen in the heart changes to a more elastic type, this is not sufficient to restore cardiac function.
Figure 5.
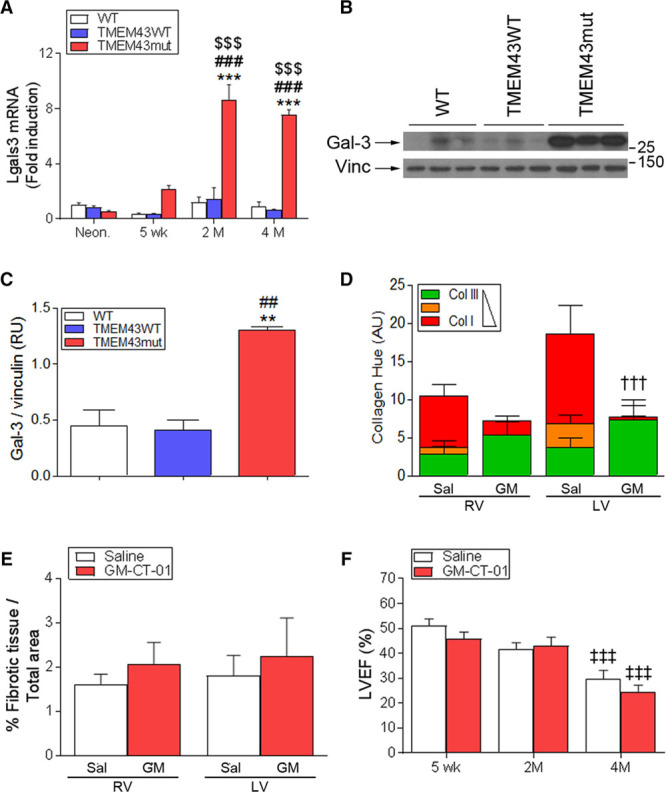
Inhibition of fibrosis does not improve cardiac function in TMEM43 (transmembrane protein 43) mutant (mut) mice. A, Quantitative reverse-transcribed polymerase chain reaction analysis of myocardial galectin-3 (Lgals3) expression in the mouse lines at the indicated ages. B and C, Western blot analysis (B) and quantification (C) of Gal-3 (galectin-3) protein levels at different time points in the 3 mouse lines at 4 months of age. D, TMEM43mut mice were treated with GM-CT-01 (GM; 120 mg/kg) or saline (Sal), and myocardial sections were stained with Picrosirius red to determine the maturity of the collagen fibers in each ventricle. E, The percentage of fibrotic tissue in the hearts of 4-month-old mice was quantified by Masson trichrome staining. F, Echocardiography-determined left ventricular (LV) ejection fraction (LVEF) in 5-week-, 2-month-, and 4-month-old TMEM43mut mice treated with GM-CT-01 or saline. Graphs show data points for individual mice and mean±SEM. AU indicates arbitrary units; and RV, right ventricle. A, ***P<0.001, TMEM43mut vs WT; ###P<0.001, TMEM43mut vs TMEM43WT; $$$P<0.001 for different ages vs neonates for each mouse line; n=3 to 8; 2-way regular ANOVA followed by Bonferroni post-test. C, **P<0.01, TMEM43mut vs WT; ##P<0.01, TMEM43mut vs TMEM43WT; 1-way ANOVA with Bonferroni correction. D, †††P<0.001, GM-CT-01 vs saline. E and F, ‡‡‡P<0.001 vs 5 weeks; 2-way regular ANOVA followed by Bonferroni post-test; n=7 to 8.
TMEM43-S358L Causes Cell Death by Activating GSK3β
GSK3β is mislocalized and activated in other ARVCs, both in patients and in mouse models.18,27 In addition, GSK3β inactivation is beneficial in several pathological settings, including myocardial infarction28,29 and pressure overload.30 To determine whether this signaling pathway was disturbed in ARVC5, we first investigated whether GSK3β activation was altered by TMEM43-S358L. We observed that GSK3β phosphorylation is decreased in TMEM43mut mice compared with their WT littermates (Figure 6A and 6B), indicating increased GSK3β activation. This was accompanied by reduced phosphorylation (indicating decreased activation) of its upstream regulatory kinase AKT (Figure 6A and 6C). To determine whether TMEM43-S358L had any effect on β-catenin transcriptional activity, which is inhibited by GSK3β we transfected neonatal cardiomyocytes with an expression vector for the different TMEM43 constructs together with a reporter plasmid in which luciferase is controlled by several β-catenin–activated TCF binding sites. We found that human TMEM43-S358L decreased β-catenin activation, whereas mouse TMEM43-S358L did not (Figure 6D). This decrease was prevented by the GSK3β inhibitor CHIR99021 (Figure 6E).
Figure 6.
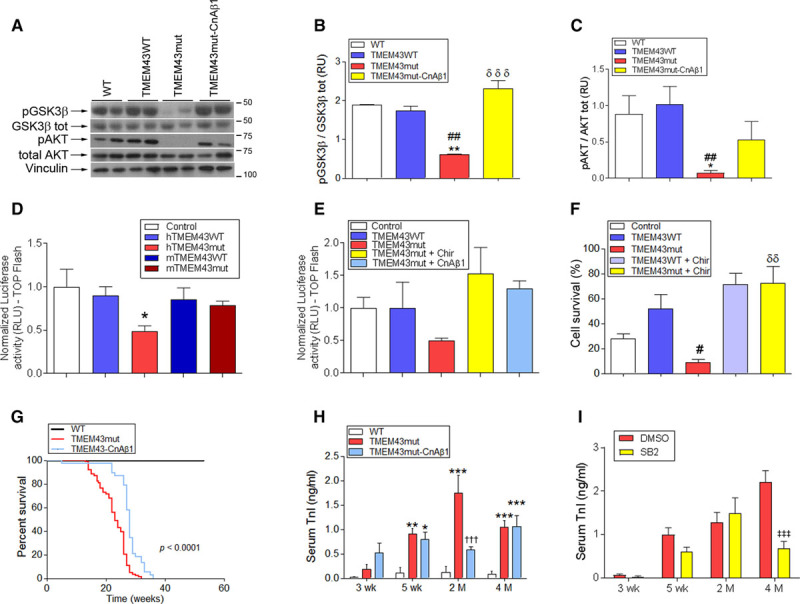
TMEM43 (transmembrane protein 43) p.S358L promotes cell death by activating glycogen synthase kinase-3β (GSK3β). A through C, TMEM43 mutant (mut) mice were crossed with α-myosin heavy chain–calcineurin Aβ1(CnAβ1) mice overexpressing CnAβ1 in cardiomyocytes (TMEM43mut-CnAβ1 mice). The presence of total and phosphorylated (Ser9) GSK3β, as well as total and phosphorylated (Ser473) AKT, was assessed by Western blot in myocardial samples from wild-type (WT), TMEM43WT, TMEM43mut, and TMEM43mut-CnAβ1 mice (A) and quantified (B and C). Increased phosphorylation of GSK3β indicates inactivation; n=3 to 11. D and E, Neonatal mouse cardiomyocytes were transfected with a reporter plasmid in which luciferase expression is controlled by a β-catenin–dependent TCF multimer, together with expression plasmids for human (h; D and E) or mouse (m; only in D) WT TMEM43, TMEM43-S358L, and CnAβ1. Empty pcDNA3.1 was used as a negative control. When indicated, the GSK3β inhibitor CHIR99021 (Chir; 3 μmol/L) was added. F, Percentage of viable P19 cells transfected with expression plasmids for human WT TMEM43 and TMEM43-S358L grown in the absence of serum for 32 hours. The GSK3β inhibitor CHIR99021 (3 μmol/L) was added when indicated. As a positive control of cell death, P19 cells were incubated with 10% ethanol for 5 hours. G, Sixty-week Kaplan-Meier survival curves for WT (n=27), TMEM43mut (n=84), and TMEM43mut-CnAβ1 mice (n=38). The indicated P value was obtained with a log-rank test. H and I, Serum troponin I (TnI) determined by ELISA in the indicated mice 3 and 5 weeks and 2 and 4 months of age. In I, TMEM43mut mice were treated with the GSK3β inhibitor SB-216763 (SB) or dimethyl sulfoxide (DMSO) as a vehicle control; n=4 to 5. Graphs show mean±SEM. B through D and F, *P<0.05, **P<0.01, ***P<0.001 vs WT animals or control vector; ##P<0.01, ###P<0.001 vs TMEM43WT. B, δδδP<0.001 vs TMEM43mut; δδP<0.01 CHIR99021-treated cells vs untreated. H, **P<0.01, ***P<0.001 vs WT; †††P<0.001, TMEM43mut vs TMEM43mut-CnAβ1. I, ‡‡‡P<0.001, SB vs DMSO. B through F, One-way ANOVA followed by Dunnett posttest; n=3 to 11. H and I, Two-way ANOVA followed by Bonferroni posttest; n=10. Note that WT and TMEM43mut mice data for G and H are those shown in Figures 1A and 3A, respectively, and are repeated here for comparative purposes.
Because the AKT pathway is strongly involved in cell survival, we investigated the effect of TMEM43-S358L on cell viability. Expression of TMEM43-S358L in P19 cells resulted in increased cell death in the absence of growth factors. This effect was precluded by the GSK3β inhibitor (Figure 6F). Together, these results indicate that TMEM43-S358L expression interferes with the AKT signaling pathway, resulting in GSK3β activation and increased cell death.
Treatment of TMEM43mut Mice With CnAβ1 Prolongs Survival and Improves Cardiac Function
CnAβ1, a naturally occurring splice variant of calcineurin Aβ, has a beneficial effect on the heart after injury.8,31 The distinct properties of CnAβ1 are conferred by a unique C-terminal domain, not present in any other known protein.32 More important, CnAβ1 is required for the activation of the AKT/GSK3β/β-catenin signaling pathway, which results in inhibition of GSK3β and activation of β-catenin.33
To determine whether CnAβ1 overexpression could revert the activation of GSK3β induced by TMEM43-S358L, we crossed TMEM43mut mice with mice overexpressing CnAβ1 in a cardiomyocyte-specific manner.8 We found that AKT activity was preserved in the hearts of double-transgenic mice and that, accordingly, GSK3β was not activated (Figure 6A–6C). In addition, CnAβ1 restored β-catenin activity (Figure 6E).
Given that CnAβ1 activates AKT and inhibits GSK3β, we next investigated whether CnAβ1 overexpression improves cardiac function in the double-transgenic animals. Most notably, CnAβ1 significantly expanded the life span of the mutant mice to a median of 28 weeks (Figure 6G). Circulating cardiac troponin I levels were significantly reduced at 2 months of age in TMEM43mut-CnAβ1 mice (Figure 6H), suggesting a partial reduction of cardiomyocyte necrosis in the double-transgenic mice. Similarly, chemical inhibition of GSK3β with SB-216763 in TMEM43mut mice resulted in reduced serum cardiac troponin I, reinforcing the idea that GSK3β activation has a causal role in cardiomyocyte death induced by TMEM43-S358L.
Echocardiography assessment showed significantly superior LV function in TMEM43mut-CnAβ1 mice compared with TMEM43mut mice at 4 months of age (Figure 7A and Table VI and Video II in the online-only Data Supplement). LV dilatation was also reduced in double-transgenic animals, although the differences with TMEM43mut mice did not reach significance (Figure 7B). TMEM43mut-CnAβ1 mice also showed a small improvement in right ventricular function (tricuspid annular plane systolic excursion; Figure 7C) and improved electrocardiographic parameters compared with TMEM43mut mice (Figure 7D–7F and Table VII in the online-only Data Supplement).
Figure 7.
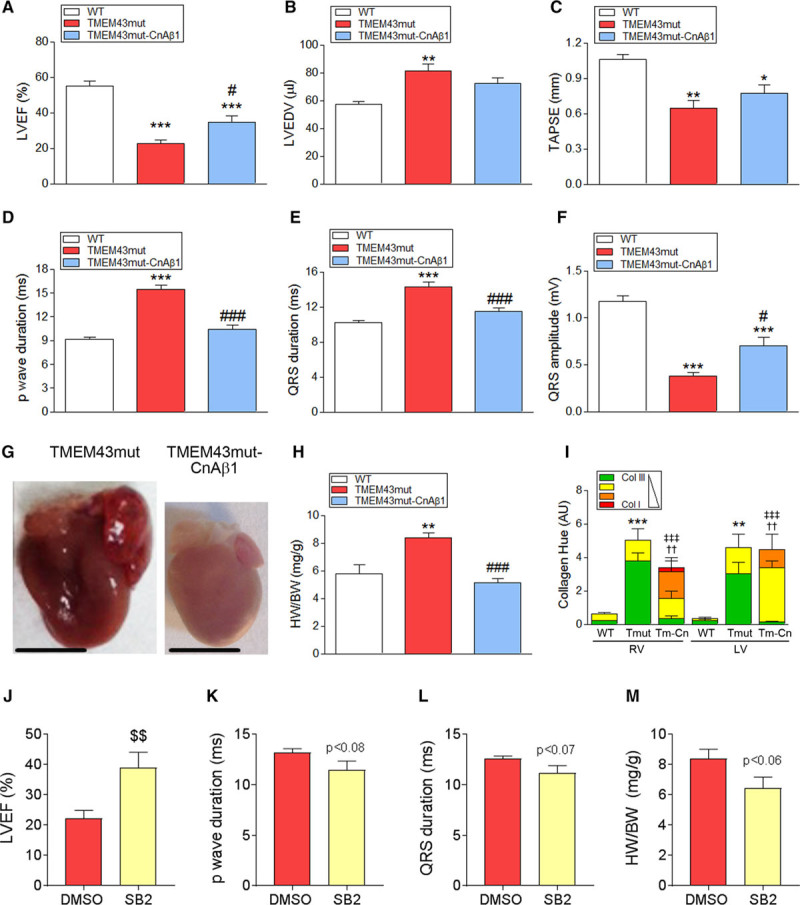
Calcineurin Aβ1 (CnAβ1) overexpression and chemical inhibition of glycogen synthase kinase-3β (GSK3β) improve cardiac function in TMEM43 (transmembrane protein 43) mutant (mut) mice. A through C, Left ventricular (LV) ejection fraction (EF; A), LV end-diastolic volume (LVEDV; B), and tricuspid annular plane systolic excursion (TAPSE; C) were determined by echocardiography in 4-month-old mice. D through F, Surface electrocardiographic determination of p-wave duration (D) and QRS duration and amplitude (E and F). G and H, Gross heart morphology (G) and ratio of heart weight to body weight (HW/BW; H) in 4-month-old mice; bar, 500 μm. I, Picrosirius red staining analysis of collagen fibers in myocardial sections. J, LVEF was determined by echocardiography in 4-month-old TMEM43mut mice treated with the GSK3β inhibitor SB-216763 (SB) or dimethyl sulfoxide (DMSO) as a control. K and L, The p-wave (K) and QRS (L) durations were analyzed by ECG in TMEM43mut (Tmut) mice treated as in J. M, Ratio of heart weight to body weight (HW/BW; H) in 4-month-old TMEM43mut mice treated as in J. Graphs in A to F, H and J-M show data with mean±SEM. A through F and H, *P<0.05, **P<0.01, ***P<0.001, TMEM43mut or TMEM43-CnAβ1 vs WT; #P<0.05, ###P<0.001, TMEM43mut-CnAβ1 (Tm-Cn) vs TMEM43mut; 1-way ANOVA followed by Bonferroni posttest; n=6 to 8. I, **P<0.01, ***P<0.001, collagen (Col) type III TMEM43mut vs WT; ††P<0.01 Col I TMEM43mut-CnAβ1 vs TMEM43mut; ‡‡‡P<0.001, Col III TMEM43-CnAβ1 vs TMEM43mut; 2-way ANOVA followed by Bonferroni posttest; n=3 to 6. Note that WT and TMEM43mut mice data for A through F and H are those shown in Figures 1B through 1D, 2D through 2F, and 1F and are repeated here for comparative purposes. J through M, $$P<0.01, SB2 vs DMSO, 2-sample ttest; n=7 to 9.
Evaluation of gross heart morphology at 4 months showed that TMEM43-CnAβ1 hearts were considerably smaller than those of the TMEM43mut mice (Figure 7G). In addition, TMEM43-CnAβ1 mice had a significantly lower ratio of heart weight to body weight than TMEM43mut mice (Figure 7H) and showed no evidence of pulmonary congestion (Figure IXA in the online-only Data Supplement).
Examination of fibrotic tissue by Masson trichrome staining revealed no differences in total collagen content between TMEM43-CnAβ1 and TMEM43mut hearts (Figure IXB and IXC in the online-only Data Supplement). However, analysis of fibers by Picrosirius red staining indicated that TMEM43-CnAβ1 mice accumulated stiffer fibers (richer in collagen I) than TMEM43mut mice (Figure 8I).
Figure 8.
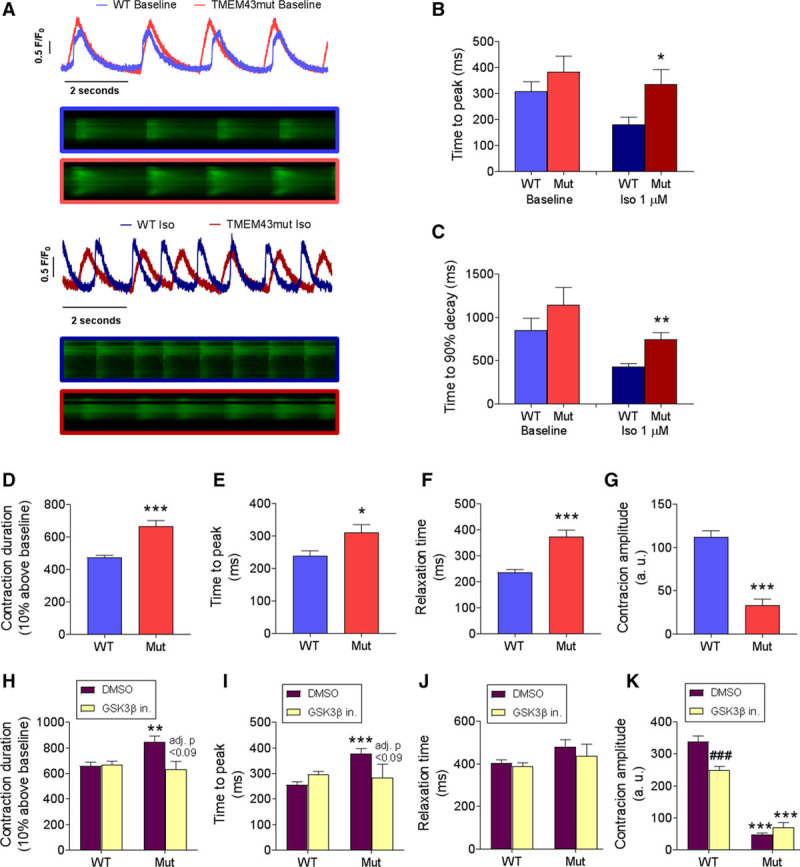
Human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) bearing the p.TMEM43 (transmembrane protein 43)–S358L mutation develop contractile dysfunction. A through C, hiPSC-CMs bearing the wild-type (WT) alleles or a heterozygous p.TMEM43-S358L mutation (Mut) were loaded with 5 µmol/L Fluo 4-AM, and Ca2+ transients were imaged in individual cells under a confocal microscope in the presence or absence of 1 µmol/L isoproterenol (Iso). D through G, WT and mutant hiPSC-CM beating was video recorded, and contraction duration (D), time to peak (E), relaxation time (F), and contraction amplitude (G) were measured. H through K, WT and mutant hiPSC-CMs were treated with the glycogen synthase kinase-3β (GSK3β) inhibitor CHIR99021 (GSK3β in.) or dimethyl sulfoxide (DMSO) as a control and different contraction parameters were measured. *P<0.05, **P<0.01, ***P<0.001, mutant vs WT cells. ###P<0.001 and adjusted P values in H and I refer to GSK3β inhibitor vs DMSO. D through G, Two-sample t test; H through K, regular 2-way ANOVA with Bonferroni posttest; n=10 to 20; (B and C) 46 to 49; and (D–G); n=12-69.
In agreement with the beneficial effect of CnAβ1 on cardiac function, treatment of TMEM43mut mice with the GSK3β inhibitor SB-216763 resulted in improved EF and a partial normalization of electric abnormalities (Figure 7J–7M). Together, these results demonstrate that GSK3β inhibition, either chemically or by CnAβ1 overexpression, improves cardiac function in mice with ARVC5.
Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes Bearing the p.S358L Mutation Show Contraction Abnormalities That Are Normalized by GSK3β Inhibition
To determine whether the p.S358L mutation induces a disease-associated phenotype also in human cells, we developed mutant human induced pluripotent stem cell (hiPSC)–derived cardiomyocytes (hiPSC-CMs) and compared their contraction behavior with that of WT cells. No significant differences in Ca2+ transient parameters were found between phenotypes at baseline (Figure 8A–8C). However, in the presence of 1 µmol/L isoproterenol, both the rising and decay phases of the Ca2+ transient were slower in the mutant phenotype. These results were confirmed by measuring hiPSC-CM contraction with a recently reported algorithm.34 We found significantly increased contraction duration, time to peak, and relaxation time in mutant hiPSC-CMs, accompanied by decreased contraction amplitude (Figure 8D–8G). GSK3β inhibition partially reduced contraction time and improved contraction overall (Figure 8H–8K), reinforcing the idea that GSK3β plays a relevant role in ARVC5.
Discussion
ARVC5 is a devastating disease that causes sudden cardiac death and heart failure.6 It is caused by a point mutation in TMEM43, a transmembrane protein that, as shown here, is located in the nuclear membrane in its WT form. Despite efforts, this disease remains incurable and has no specific therapy. To develop therapies to delay the onset or to slow the progression of ARVC5, it is necessary to define the initial molecular events and pathophysiological mechanisms. We therefore performed a step-by-step characterization of the first ARVC5 transgenic mouse line (TMEM43mut) from the early stages of the disease to the latest manifestations of the ACM/ARVC phenotype. TMEM43mut mice show biventricular systolic dysfunction as the disease progresses and considerable accumulation of fibrofatty tissue, thus reproducing the human condition, which also shows biventricular affectation in 43% of the cases.7 Unlike other ACM/ARVC mouse models,17,27,35–37 TMEM43mut hearts accumulate fat in the right ventricle, although to a lower extent than observed in human patients, likely as a result of the absence of subepicardial fat in the mouse heart.
The first disease manifestations in TMEM43mut mice are electric abnormalities, observed as early as 5 weeks of age in ECG. Interestingly, no sudden cardiac death or appropriate implantable cardioverter-defibrillator discharge has been reported so far in ARVC5 patients <19 years of age.38 This corresponds to the onset of the disease at 5 weeks of age observed in our ARVC5 mouse model. Electric features preceded anatomic defects, as also described in other ARVC mouse models.39 Although the molecular mechanisms that underlie cellular uncoupling and consequent conduction abnormalities are poorly understood,40 mislocalization of connexin 43 that results in gap-junction remodeling would contribute to defects in impulse propagation and might explain the early phenotype.17
We show that mutant TMEM43 causes progressive cardiomyocyte loss through activation of cell-death pathways. Necrosis started early in life, as shown by cardiac troponin I levels, whereas apoptosis and autophagy appeared only in the later stages of the disease, suggesting that necrosis is the main pathway leading to early cardiomyocyte death. Sustained cardiomyocyte loss in TMEM43mut hearts progressively led to the replacement of dead cells by fibrofatty tissue, eventually leading to massive interstitial fibrosis in all 4 chambers. We show that epicardium-derived cells contribute strongly to cardiac fibrosis in this model, as has also been shown for other heart diseases, including myocardial infarction.41 Given that many resident fibroblasts in the adult heart are derived from Wt1+ epicardial cells during embryonic development, it is likely that the epicardium-derived cells contributing to cardiac fibrosis in TMEM43mut mice are resident cardiac fibroblasts. We found no contribution from macrophages, endothelial cells, or cardiomyocytes, although we cannot rule out contributions from other cell types not tested here.
The role of fibrosis in ACM/ARVC is unclear; it could represent either a pathological mechanism or a compensatory response. Treating our mouse lines with a Gal-3 inhibitor that has antifibrotic properties altered the collagen type III/I ratio to a more elastic fiber conformation. This, however, had no beneficial effect on cardiac function. These results suggest that the fibrotic response developed in ARVC5 tries to compensate for the loss of cardiomyocytes but would not be an appropriate therapeutic target.
The Wnt/GSK3β/β-catenin signaling pathway seems to play a central role in the pathogenesis of other forms of ARVC and is a key regulator of myogenesis.7.GSK3β modulates this signaling pathway by phosphorylating β-catenin, promoting its rapid turnover by the proteasome. We show here that TMEM43-S358L inhibits AKT, which results in GSK3β activation and inhibition of β-catenin–dependent transcription. We also show that expression of mutant TMEM43 in cell culture causes significant cell death in a GSK3-dependent manner, demonstrating the functional involvement of GSK3β in the deleterious effects of TMEM43-S358L. Although it is unclear how it inhibits AKT, we found that TMEM43-S358L interacts with the AKT modulator Pa2g4 (EBP1 [Erbb3 interacting protein]). Interaction of Pa2g4 with AKT inhibits apoptosis and promotes cell survival.42,43 Therefore, it is tempting to speculate that by binding Pa2g4, TMEM43-S358L interferes with the Pa2g4-AKT interaction and thereby reduces the antiapoptotic effect of AKT. Overexpression of the calcineurin variant CnAβ1, which activates the AKT pathway and inhibits GSK3β, significantly expands the life span and improves cardiac function in TMEM43mut mice. TMEM43mut-CnAβ1 mice showed a partial reduction in cardiomyocyte death and less severe electric abnormalities, suggesting that CnAβ1 overexpression preserves cardiomyocyte function in the TMEM43mut myocardium. Similarly, chemical inhibition of GSK3β reduced cardiomyocyte death and electric abnormalities and improved cardiac contraction in TMEM43mut mice. The role of GSK3β in the pathogenesis of ARVC5 was further confirmed in hiPSC-CMs bearing the p.S358L mutation, which showed improved contraction after GSK3β inhibition.
Our computer models show that the p.S358L point mutation in TMEM43 increases the hydrophobicity of the molecule and would lead to a distorted protein, impairing its function. The dimer formed between TMEM43-S358L and the WT form adopts a deformed structure that reduces its interaction with other nuclear membrane proteins such as EMD, as well as with cytoskeleton-interacting proteins. This defective interaction is illustrated by the partial delocalization of TMEM43-S358L to the cytoplasm. The functional implications of these protein-protein interactions need to be fully clarified; however, it is plausible that a reduced interaction between the nucleus and the cytoskeleton contributes to cardiomyocyte death under biomechanical stress.
Interestingly, our computer models also showed that the third transmembrane domain in mouse TMEM43 has a different conformation and orientation from that of the human protein. More important, the p.S358L mutation has no effect on the structure of mouse TMEM43. The lack of a structural change in the mutant mouse protein is likely the reason why it does not inhibit β-catenin signaling and why knock-in mice bearing the p.S358L mutation have no pathological phenotype.14 In addition, by using a cardiomyocyte-specific promoter to express TMEM43-S358L, we can conclude that ARVC5 is originated in cardiomyocytes and not in other cell types.
We show that human TMEM43-S358L causes cardiomyocyte death and fibrofatty replacement. Although it appears that fibrosis is not an effective therapeutic target, GSK3β inhibition by CnAβ1 overexpression or a chemical inhibitor has a beneficial effect on the mutant hearts. It will be interesting to explore GSK3β inhibitors as potential therapeutic agents for ARVC5 to improve the life of patients with this incurable disease.
Limitations
Although our transgenic mouse is the only model that reproduces human ARVC5, certain characteristics of the human disease were not presented by this model. No significant differences were found between males and females, in contrast to human patients. In addition, mice show fat infiltration in the myocardium but not to the extent found in ARVC5 human hearts.
Acknowledgments
The authors thank F. Montoya for mouse work, E. Arza and V. Labrador from the Centro Nacional de Investigaciones Cardiovasculares Carlos III Microscopy Unit for help with microscopy, R. Doohan for immunostaining advice, J.A. Bernal for the hiPSC line, M.A. Sanguino for assistance with hiPSCs, J. Jalife for the Gal3 inhibitor treatment, and S. Bartlett for English editing. RNA sequencing experiments were performed in the Centro Nacional de Investigaciones Cardiovasculares Carlos III Genomics Unit and analyzed by the Bioinformatics Unit. Anti-TMEM43 was a kind gift of Dr Franke Werner (DKFZ, Heidelberg, Germany). Conceptualization: E.L.-P., P.G.-P., and L.P.-B.; methodology: L.P.-B., M.V.-O., J.G.-S., P.O.-S., C.M.-G., F.M., D.J.S., B.P., G.G., E.B.-K., J.V., and M.V.G.-G.; investigation: L.P.-B., M.V.-O., J.G.-S., M.R., J.L.-A., P.O.-S., C.M.-G., F.M., M.L.-O., E.B.-K., J.V., D.J.S, B.P., G.G., S.P., F.D., and M.V.G.-G.; formal analysis: L.P.-B., M.V.-O., E.B.-K., J.V., F.M., M.V.G.-G., C.M.-G., S.P., D.J.S., P.G.-P., and E.L.-P.; writing, original draft: L.P.-B., E.L.-P., and P.G-P.; writing, review and editing: L.P.-B., E.L.-P., and P.G-P.; funding acquisition: E.L.-P. and P.G.-P.; supervision, E.L.-P. and P.G.-P.; and project administration, E.L.-P. and P.G.-P.
Sources of Funding
This work was supported by grants from the European Union (CardioNeT-ITN-289600 and CardioNext-608027 to Dr Lara-Pezzi), the Spanish Ministry of Economy and Competitiveness (RTI2018-096961-B-I00, SAF2015-65722-R, and SAF2012-31451 to Dr Lara-Pezzi; SAF2015-71863-REDT to Dr Garcia-Pavia), the Spanish Carlos III Institute of Health (PI14/0967 to Dr Garcia-Pavia, CPII14/00027 to Dr Lara-Pezzi; RD012/0042/0066 to Drs Garcia-Pavia and Lara-Pezzi), the Regional Government of Madrid (2010-BMD-2321 “Fibroteam” to Dr Lara-Pezzi), the Isabel Gemio Foundation (Todos somos Raros grant to Dr Garcia-Pavia), and the Spanish Society of Cardiology (2014 Basic Research Grant to Dr Garcia-Pavia). This work was also supported by the Plan Estatal de I+D+I 2013-2016–European Regional Development Fund (FEDER) “A way of making Europe,” Spain. The Centro Nacional de Investigaciones Cardiovasculares Carlos III is supported by the Ministerio de Ciencia, Innovación y Universidades (MCNU), and Pro Centro Nacional de Investigaciones Cardiovasculares Carlos III Foundation and is a Severo Ochoa Center of Excellence (SEV-2015-0505).
Disclosures
None.
Supplementary Material
Footnotes
Drs Garcia-Pavia and Lara-Pezzi contributed equally as senior authors.
Sources of Funding, see page 1203
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/circulationaha.119.040366.
References
- 1.Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376:61–72. doi: 10.1056/NEJMra1509267. doi: 10.1056/NEJMra1509267. [DOI] [PubMed] [Google Scholar]
- 2.Stadiotti I, Catto V, Casella M, Tondo C, Pompilio G, Sommariva E. Arrhythmogenic cardiomyopathy: the guilty party in adipogenesis. J Cardiovasc Transl Res. 2017;10:446–454. doi: 10.1007/s12265-017-9767-8. doi: 10.1007/s12265-017-9767-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–1297. doi: 10.1016/j.jacc.2013.06.033. doi: 10.1016/j.jacc.2013.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Padrón-Barthe L, Domnguez F, Garcia-Pavia P, Lara-Pezzi E. Animal models of arrhythmogenic right ventricular cardiomyopathy: what have we learned and where do we go? Insight for therapeutics. Basic Res. Cardiol. 2017;112:50–56. doi: 10.1007/s00395-017-0640-3. [DOI] [PubMed] [Google Scholar]
- 5.Ye L, Ni X, Zhao ZA, Lei W, Hu S. The application of induced pluripotent stem cells in cardiac disease modeling and drug testing. J Cardiovasc Transl Res. 2018;11:366–374. doi: 10.1007/s12265-018-9811-3. doi: 10.1007/s12265-018-9811-3. [DOI] [PubMed] [Google Scholar]
- 6.Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–821. doi: 10.1016/j.ajhg.2008.01.010. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodgkinson KA, Connors SP, Merner N, Haywood A, Young TL, McKenna WJ, Gallagher B, Curtis F, Bassett AS, Parfrey PS. The natural history of a genetic subtype of arrhythmogenic right ventricular cardiomyopathy caused by a p.S358L mutation in TMEM43. Clin Genet. 2013;83:321–331. doi: 10.1111/j.1399-0004.2012.01919.x. doi: 10.1111/j.1399-0004.2012.01919.x. [DOI] [PubMed] [Google Scholar]
- 8.Felkin LE, Narita T, Germack R, Shintani Y, Takahashi K, Sarathchandra P, López-Olañeta MM, Gómez-Salinero JM, Suzuki K, Barton PJ, et al. Calcineurin splicing variant calcineurin Aβ1 improves cardiac function after myocardial infarction without inducing hypertrophy. Circulation. 2011;123:2838–2847. doi: 10.1161/CIRCULATIONAHA.110.012211. doi: 10.1161/CIRCULATIONAHA.110.012211. [DOI] [PubMed] [Google Scholar]
- 9.Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, Pérez-Hernández D, Vázquez J, Martin-Cofreces N, Martinez-Herrera DJ, Pascual-Montano A, Mittelbrunn M, Sánchez-Madrid F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980. doi: 10.1038/ncomms3980. doi: 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavandero S, Chiong M, Rothermel BA, Hill JA. Autophagy in cardiovascular biology. J Clin Invest. 2015;125:55–64. doi: 10.1172/JCI73943. doi: 10.1172/JCI73943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dreger M, Bengtsson L, Schöneberg T, Otto H, Hucho F. Nuclear envelope proteomics: novel integral membrane proteins of the inner nuclear membrane. Proc Natl Acad Sci U S A. 2001;98:11943–11948. doi: 10.1073/pnas.211201898. doi: 10.1073/pnas.211201898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bengtsson L, Otto H. LUMA interacts with emerin and influences its distribution at the inner nuclear membrane. J Cell Sci. 2008;121(pt 4):536–548. doi: 10.1242/jcs.019281. doi: 10.1242/jcs.019281. [DOI] [PubMed] [Google Scholar]
- 14.Franke WW, Dörflinger Y, Kuhn C, Zimbelmann R, Winter-Simanowski S, Frey N, Heid H. Protein LUMA is a cytoplasmic plaque constituent of various epithelial adherens junctions and composite junctions of myocardial intercalated disks: a unifying finding for cell biology and cardiology. Cell Tissue Res. 2014;357:159–172. doi: 10.1007/s00441-014-1865-1. doi: 10.1007/s00441-014-1865-1. [DOI] [PubMed] [Google Scholar]
- 15.Liang WC, Mitsuhashi H, Keduka E, Nonaka I, Noguchi S, Nishino I, Hayashi YK. TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy. Ann Neurol. 2011;69:1005–1013. doi: 10.1002/ana.22338. doi: 10.1002/ana.22338. [DOI] [PubMed] [Google Scholar]
- 16.Stroud MJ, Fang X, Zhang J, Guimarães-Camboa N, Veevers J, Dalton ND, Gu Y, Bradford WH, Peterson KL, Evans SM, et al. Luma is not essential for murine cardiac development and function. Cardiovasc Res. 2018;114:378–388. doi: 10.1093/cvr/cvx205. doi: 10.1093/cvr/cvx205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lyon RC, Mezzano V, Wright AT, Pfeiffer E, Chuang J, Banares K, Castaneda A, Ouyang K, Cui L, Contu R, et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum Mol Genet. 2014;23:1134–1150. doi: 10.1093/hmg/ddt508. doi: 10.1093/hmg/ddt508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chelko SP, Asimaki A, Andersen P, Bedja D, Amat-Alarcon N, DeMazumder D, Jasti R, MacRae CA, Leber R, Kleber AG, et al. Central role for GSK3β in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight. 2016;1:e85923. doi: 10.1172/jci.insight.85923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schirmer EC, Florens L, Guan T, Yates JR, 3rd, Gerace L. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science. 2003;301:1380–1382. doi: 10.1126/science.1088176. doi: 10.1126/science.1088176. [DOI] [PubMed] [Google Scholar]
- 20.Dreger M, Bengtsson L, Schöneberg T, Otto H, Hucho F. Nuclear envelope proteomics: novel integral membrane proteins of the inner nuclear membrane. Proc Natl Acad Sci U S A. 2001;98:11943–11948. doi: 10.1073/pnas.211201898. doi: 10.1073/pnas.211201898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajkumar R, Sembrat JC, McDonough B, Seidman CE, Ahmad F. Functional effects of the TMEM43 Ser358Leu mutation in the pathogenesis of arrhythmogenic right ventricular cardiomyopathy. BMC Med Genet. 2012;13:21. doi: 10.1186/1471-2350-13-21. doi: 10.1186/1471-2350-13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Spreeuwel ACC, Bax NAM, van Nierop BJ, Aartsma-Rus A, Goumans MTH, Bouten CVC. Mimicking cardiac fibrosis in a dish: fibroblast density rather than collagen density weakens cardiomyocyte function. J Cardiovasc Transl Res. 2017;10:116–127. doi: 10.1007/s12265-017-9737-1. doi: 10.1007/s12265-017-9737-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Traber PG, Zomer E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS One. 2013;8:e83481. doi: 10.1371/journal.pone.0083481. doi: 10.1371/journal.pone.0083481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu L, Ruifrok WP, Meissner M, Bos EM, van Goor H, Sanjabi B, van der Harst P, Pitt B, Goldstein IJ, Koerts JA, et al. Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ Heart Fail. 2013;6:107–117. doi: 10.1161/CIRCHEARTFAILURE.112.971168. doi: 10.1161/CIRCHEARTFAILURE.112.971168. [DOI] [PubMed] [Google Scholar]
- 25.Takemoto Y, Ramirez RJ, Yokokawa M, Kaur K, Ponce-Balbuena D, Sinno MC, Willis BC, Ghanbari H, Ennis SR, Guerrero-Serna G, et al. Galectin-3 regulates atrial fibrillation remodeling and predicts catheter ablation outcomes. JACC Basic Transl. Sci. 2016;1:143–154. doi: 10.1016/j.jacbts.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmad N, Gabius HJ, André S, Kaltner H, Sabesan S, Roy R, Liu B, Macaluso F, Brewer CF. Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J Biol Chem. 2004;279:10841–10847. doi: 10.1074/jbc.M312834200. doi: 10.1074/jbc.M312834200. [DOI] [PubMed] [Google Scholar]
- 27.Martherus R, Jain R, Takagi K, Mendsaikhan U, Turdi S, Osinska H, James JF, Kramer K, Purevjav E, Towbin JA. Accelerated cardiac remodeling in desmoplakin transgenic mice in response to endurance exercise is associated with perturbed Wnt/β-catenin signaling. Am J Physiol Heart Circ Physiol. 2016;310:H174–H187. doi: 10.1152/ajpheart.00295.2015. doi: 10.1152/ajpheart.00295.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woulfe KC, Gao E, Lal H, Harris D, Fan Q, Vagnozzi R, DeCaul M, Shang X, Patel S, Woodgett JR, et al. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ Res. 2010;106:1635–1645. doi: 10.1161/CIRCRESAHA.109.211482. doi: 10.1161/CIRCRESAHA.109.211482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miura T, Tanno M. Mitochondria and GSK-3beta in cardioprotection against ischemia/reperfusion injury. Cardiovasc Drugs Ther. 2010;24:255–263. doi: 10.1007/s10557-010-6234-z. doi: 10.1007/s10557-010-6234-z. [DOI] [PubMed] [Google Scholar]
- 30.Matsuda T, Zhai P, Maejima Y, Hong C, Gao S, Tian B, Goto K, Takagi H, Tamamori-Adachi M, Kitajima S, et al. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc Natl Acad Sci U S A. 2008;105:20900–20905. doi: 10.1073/pnas.0808315106. doi: 10.1073/pnas.0808315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.López-Olañeta MM, Villalba M, Gómez-Salinero JM, Jiménez-Borreguero LJ, Breckenridge R, Ortiz-Sánchez P, García-Pavía P, Ibáñez B, Lara-Pezzi E. Induction of the calcineurin variant CnAβ1 after myocardial infarction reduces post-infarction ventricular remodelling by promoting infarct vascularization. Cardiovasc Res. 2014;102:396–406. doi: 10.1093/cvr/cvu068. doi: 10.1093/cvr/cvu068. [DOI] [PubMed] [Google Scholar]
- 32.Lara-Pezzi E, Winn N, Paul A, McCullagh K, Slominsky E, Santini MP, Mourkioti F, Sarathchandra P, Fukushima S, Suzuki K, et al. A naturally occurring calcineurin variant inhibits FoxO activity and enhances skeletal muscle regeneration. J Cell Biol. 2007;179:1205–1218. doi: 10.1083/jcb.200704179. doi: 10.1083/jcb.200704179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gómez-Salinero JM, López-Olañeta MM, Ortiz-Sánchez P, Larrasa-Alonso J, Gatto A, Felkin LE, Barton PJR, Navarro-Lérida I, Ángel Del Pozo M, García-Pavía P, et al. The calcineurin variant CnAβ1 controls mouse embryonic stem cell differentiation by directing mTORC2 membrane localization and activation. Cell Chem Biol. 2016;23:1372–1382. doi: 10.1016/j.chembiol.2016.09.010. doi: 10.1016/j.chembiol.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 34.Sala L, van Meer BJ, Tertoolen LGJ, Bakkers J, Bellin M, Davis RP, Denning C, Dieben MAE, Eschenhagen T, Giacomelli E, et al. MUSCLEMOTION: a versatile open software tool to quantify cardiomyocyte and cardiac muscle contraction in vitro and in vivo. Circ Res. 2018;122:e5–e16. doi: 10.1161/CIRCRESAHA.117.312067. doi: 10.1161/CIRCRESAHA.117.312067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pilichou K, Remme CA, Basso C, Campian ME, Rizzo S, Barnett P, Scicluna BP, Bauce B, van den Hoff MJ, de Bakker JM, et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J Exp Med. 2009;206:1787–1802. doi: 10.1084/jem.20090641. doi: 10.1084/jem.20090641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kant S, Krull P, Eisner S, Leube RE, Krusche CA. Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell Tissue Res. 2012;348:249–259. doi: 10.1007/s00441-011-1322-3. doi: 10.1007/s00441-011-1322-3. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hodgkinson KA, Howes AJ, Boland P, Shen XS, Stuckless S, Young T-L, Curtis F, Collier A, Parfrey PS, Connors SP. Long-term clinical outcome of arrhythmogenic right ventricular cardiomyopathy in individuals with a p.S358L mutation in TMEM43 following implantable cardioverter defibrillator therapy. Circ Arrhythm Electrophysiol. 2016;9:e003589. doi: 10.1161/CIRCEP.115.003589. doi: 10.1161/CIRCEP.115.003589. [DOI] [PubMed] [Google Scholar]
- 39.Gomes J, Finlay M, Ahmed AK, Ciaccio EJ, Asimaki A, Saffitz JE, Quarta G, Nobles M, Syrris P, Chaubey S, et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur Heart J. 2012;33:1942–1953. doi: 10.1093/eurheartj/ehr472. doi: 10.1093/eurheartj/ehr472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Patel VV, Kostetskii I, Xiong Y, Chu AF, Jacobson JT, Yu C, Morley GE, Molkentin JD, Radice GL. Cardiac-specific loss of n-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ Res. 2005;97:474–481. doi: 10.1161/01.RES.0000181132.11393.18. [DOI] [PubMed] [Google Scholar]
- 41.Smart N, Bollini S, Dubé KN, Vieira JM, Zhou B, Davidson S, Yellon D, Riegler J, Price AN, Lythgoe MF, et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature. 2011;474:640–644. doi: 10.1038/nature10188. doi: 10.1038/nature10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Ahn JY, Liu X, Ye K. Ebp1 isoforms distinctively regulate cell survival and differentiation. Proc Natl Acad Sci U S A. 2006;103:10917–10922. doi: 10.1073/pnas.0602923103. doi: 10.1073/pnas.0602923103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahn JY, Liu X, Liu Z, Pereira L, Cheng D, Peng J, Wade PA, Hamburger AW, Ye K. Nuclear Akt associates with PKC-phosphorylated Ebp1, preventing DNA fragmentation by inhibition of caspase-activated DNase. EMBO J. 2006;25:2083–2095. doi: 10.1038/sj.emboj.7601111. doi: 10.1038/sj.emboj.7601111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors on reasonable request. RNA sequencing data are available at the GEO repository (GSE101301). Proteomic data are available at the PeptideAtlas repository (http://www.peptideatlas.org/PASS/PASS01063). Full Methods can be found in the online-only Data Supplement.