

Abstract
Several lines of evidence suggest that norepinephrine (NE) can modulate seizure activity. However, the experimental methods used in the past cannot exclude the possible role of other neurotransmitters coreleased with NE from noradrenergic terminals. We have assessed the seizure susceptibility of genetically engineered mice that lack NE. Seizure susceptibility was determined in the dopamine β-hydroxylase null mutant (Dbh −/−) mouse using four different convulsant stimuli: 2,2,2-trifluroethyl ether (flurothyl), pentylenetetrazol (PTZ), kainic acid, and high-decibel sound.Dbh −/− mice demonstrated enhanced susceptibility (i.e., lower threshold) compared with littermate heterozygous (Dbh +/−) controls to flurothyl, PTZ, kainic acid, and audiogenic seizures and enhanced sensitivity (i.e., seizure severity and mortality) to flurothyl, PTZ, and kainic acid. c-Fos mRNA expression in the cortex, hippocampus (CA1 and CA3), and amygdala was increased in Dbh −/− mice in association with flurothyl-induced seizures. Enhanced seizure susceptibility to flurothyl and increased seizure-induced c-fos mRNA expression were reversed by pretreatment withl-threo-3,4-dihydroxyphenylserine, which partially restores the NE content in Dbh −/− mice. These genetically engineered mice confirm unambiguously the potent effects of the noradrenergic system in modulating epileptogenicity and illustrate the unique opportunity offered by Dbh −/− mice for elucidating the pathways through which NE can regulate seizure activity.
Keywords: dopamine β-hydroxylase, c-fos mRNA, norepinephrine, flurothyl, epilepsy, seizure, kainic acid
Chen et al. (1954) first suggested that the noradrenergic system modifies seizure activity. Since then, four major observations have supported an anticonvulsant role for norepinephrine (NE): (1) selective lesioning of noradrenergic neurons (with 6-hydroxydopamine or DSP-4) increases seizure susceptibility to a variety of convulsant stimuli (Arnold et al., 1973; Jerlicz et al., 1978; Mason and Corcoran, 1979; Snead, 1987; Trottier et al., 1988;Sullivan and Osorio, 1991; Mishra et al., 1994); (2) direct stimulation of the locus coeruleus (LC, the major concentration of noradrenergic cell bodies in the CNS) and the subsequent release of NE reduce CNS sensitivity to convulsant stimuli (Libet et al., 1977; Turski et al., 1989); (3) genetically epilepsy-prone rats (GEPRs), a widely used animal model of epilepsy, have deficient presynaptic NE content, NE turnover, tyrosine hydroxylase levels, dopamine β-hydroxylase (DBH) levels, and NE uptake (Jobe et al., 1984; Dailey and Jobe, 1986;Browning et al., 1989; Lauterborn and Ribak, 1989; Dailey et al., 1991); and (4) adrenergic agonists acting at the α-2 adrenoreceptor (α2-AR) have anticonvulsant action (Papanicolaou et al., 1982; Baran et al., 1985; Loscher and Czuczwar, 1987; Fletcher and Forster, 1988;Jackson et al., 1991).
Although there is significant evidence that the NE system is anticonvulsant, there are several considerations that temper one's confidence in the hypothesis that NE, itself, reduces seizure sensitivity. For example, although the lesioning studies (i.e., chemical destruction of noradrenergic terminals) reduce the amount of NE release, this manipulation also reduces the release of other transmitters coreleased with NE. The neuropeptides galanin and neuropeptide Y (NPY) and the neurotransmitter adenosine (i.e., ATP) are released at noradrenergic terminals and have been shown to exert anticonvulsant effects against several convulsant stimuli (Murray et al., 1985; Mazarati et al., 1992, 1998; Dichter, 1994; Erickson et al., 1996; Baraban et al., 1997). A similar argument can be made for the anticonvulsant effect of direct LC stimulation, which results in the release not only of NE but also of these cotransmitters. The enhanced seizure sensitivity of the GEPRs may not be caused solely by their abnormal noradrenergic system, because these animals also have abnormalities in their central serotonergic, GABAergic, and excitatory amino acid systems (Faingold et al., 1986; Dailey et al., 1992;Meyerhoff et al., 1992); moreover, other animal models of epilepsy have a higher than normal central NE content (Noebels, 1986; Hara et al., 1993). Finally, the α2-AR pharmacological studies are difficult to interpret because the effect of clonidine (α2-AR agonist) on seizure-induced activity can be biphasic, nonexistent, or even proconvulsant (King and Burnham, 1982; Tacke and Kolonen, 1984;Lapin and Ryzor, 1990). Such multiple responses to α2-AR agonists may be caused by the localization of the affected α2-AR. Activation of presynaptic α2-AR autoreceptors would reduce transmitter released at NE terminals (L'Heureux et al., 1986), whereas activation of postsynaptic α2-ARs would mimic the effect of released NE. Because it has not been determined whether the anticonvulsant effect of α2-AR agonists is mediated via pre- or postsynaptic receptors, it remains unclear whether increased NE release is anti- or proconvulsant.
Taken together, these studies suggest that changes in noradrenergic functions (terminal NE content or release) can modulate seizure activity, but they do not resolve the issue of whether NE is, itself, anticonvulsant. It is this issue that we have addressed with the DBH null mutant (Dbh −/−) mouse. These animals selectively lack NE and epinephrine (dopamine content tends to be elevated) because DBH is required for the conversion of dopamine to NE (Thomas et al., 1998).
MATERIALS AND METHODS
Animals. Mice were derived from a hybrid line (129/Sv/Ev and C57BL/6J). Dbh −/− and heterozygote (Dbh +/−) mice were bred as described previously (Thomas et al., 1995). Mice were maintained on a 12 hr light/dark cycle in a specific pathogen-free facility at the University of Washington (Seattle, WA). Food and water were available ad libitum, and animals were maintained according to the guidelines outlined in theNIH Guide for Care and Use of Laboratory Animals. All animal procedures were approved by the University of Washington Animal Care Committee. Genotype was deduced from phenotype (Dbh −/− mice exhibit delayed growth during adolescence and ptosis), and a subset of mice was confirmed by PCR (Thomas et al., 1995).Dbh +/− mice are indistinguishable from wild-type (+/+) mice as to NE and epinephrine levels (Thomas et al., 1998). Preliminary studies showed no significant difference in seizure susceptibility [2,2,2-trifluroethyl ether (flurothyl)-induced seizures] between wild-type (+/+) and heterozygote Dbh (+/−) mice; therefore,Dbh +/− mice were used as controls in all experiments. Adult (3–6 months) male and female littermates of each genotype were evenly distributed to experimental and control groups for each convulsant stimulus. A subset of animals will receive a single intraperitoneal injection ofl-threo-3,4-dihydroxphenylserine (DOPS; 1 mg/gm). DOPS is converted to NE by aromaticl-amino acid decarboxylase, which is present in all biogenic amine neurons. Five hours after a single administration of DOPS, NE levels peak in peripheral and central regions; dopamine levels are not affected by DOPS (Thomas et al., 1998).
Flurothyl susceptibility. Flurothyl seizure thresholds were determined for Dbh +/− and Dbh −/− mice, with and without previous administration of DOPS. Mice were placed in an air-tight Plexiglas chamber, and the volatile convulsant flurothyl (Aldrich, Milwaukee, WI) was infused (20 μl/min) onto filter paper from which it vaporized (Prichard et al., 1969). The latencies (seconds) to the first myoclonic jerk (focal seizure) and to generalized (clonic/tonic) seizure served as the measurements of seizure susceptibility. Each mouse was tested individually, removed immediately from the chamber after seizure onset, and received only one exposure to flurothyl. Some animals received DOPS (1 mg/gm, i.p.) 6 hr before seizure-threshold testing. Latency (seconds) data from each group were expressed as the mean ± SEM and were analyzed with Student's t test comparisons; statistical significance was taken at p < 0.05. For each group (Dbh−/− and Dbh +/− mice, with and without DOPS), we also determined the percentage of animals proceeding to tonic extension followed by recovery versus the percentage progressing to death. Surviving animals were killed 1 hr after the seizure to measure c-fos mRNA expression.
Pentylenetetrazol susceptibility. Pentylenetetrazol (PTZ) at two different doses (30 and 40 mg/kg, i.p.) was administered to bothDbh +/− and Dbh −/− mice. After injection, the animals were placed into a clear container and closely monitored for 10 min. The latencies (seconds) to the first myoclonic jerk (focal seizure), to forelimb clonus, and to generalized (clonic/tonic) seizures were measured and analyzed as described above.
Kainic acid susceptibility. Kainic acid (stock solution, 4 mg/ml) was dissolved in neutral-buffered saline and administered to both Dbh +/− and Dbh −/− mice at an intraperitoneal dose of 20 mg/kg. After injection, the animals were placed into a clear container and closely monitored for 40 min. The latency (seconds) to the first generalized (clonic/tonic) seizure was measured and was analyzed as described above. For each group, we also determined the percentage of animals that progressed to death.
Audiogenic seizure susceptibility. Audiogenic seizure sensitivity in Dbh +/− and Dbh −/− mice was determined by exposing animals to a 115 dB sound for 60 sec with an SR Pilot (San Diego Instruments, San Diego, CA). After the sound was started, the mouse was closely monitored for occurrence of a seizure. If no seizure occurred, the sound was terminated after 60 sec. If a seizure was observed during the 60 sec period, the sound was immediately terminated, and the animal was removed. Mice were scored as exhibiting or not exhibiting a seizure.
c-Fos mRNA expression after flurothyl-induced seizures. The mice that survived flurothyl-induced generalized seizures were killed by cervical dislocation 1 hr after the seizure [Dbh +/− (n = 8) and Dbh −/− (n = 6) without DOPS; Dbh +/− (n = 8) andDbh −/− (n = 9) with DOPS]. To determine basal c-fos mRNA expression Dbh +/− (n = 6) and Dbh −/− (n = 6) mice (same age as the flurothyl-tested mice) were also killed. Brains were collected from each animal and immediately frozen on dry ice. Twenty micrometer coronal sections containing neocortex and hippocampus were cut on a cryostat and mounted onto Fisher Superfrost slides (Fisher Scientific, Houston, TX). Slides were stored at −70°C until assayed.
Tissue preparation and labeling of the c-fos oligonucleotide was performed as described previously (Szot et al., 1997). The c-fos oligonucleotide probe was a 51-base probe complementary to nucleotides 270–319 of the c-fos mRNA (Curran et al., 1987). The oligonucleotide probe was 3′-end-labeled with [33P]dATP (New England Nuclear, Boston, MA) using terminal deoxyribonucleotidyl transferase (Life Technologies, Gaithersburg, MD) and then purified on NEN-Sorb columns (New England Nuclear). The c-fos hybridization buffer for the flurothyl-induced seizure assay contained 0.3 × 106 cpm/50 ml. The c-fos hybridization buffer for the basal assay contained 0.4 × 106 cpm/50 ml. Hyperfilm (Amersham, Arlington Heights, IL) was exposed to slides containing tissue hybridized with c-fos [33P]oligonucleotide for 1 d for the flurothyl-induced seizure assay and 5 d for the basal assay. To quantitate c-fos mRNA expression in the specific regions of the CNS, all sections were processed, hybridized, and washed in the same experimental session. Each sheet of Hyperfilm contained sections from all four groups (Dbh +/− and Dbh −/− mice with and without DOPS). To determine basal c-fos mRNA expression, sections from Dbh +/− and Dbh −/− mice were processed, hybridized, and washed in a similar manner. Optical densities were measured from films using the MicroComputer Imaging Device (Imaging Research, Ontario, Canada). Separate optical density measurements were made of the left and right hemispheres over three successive sections, which were anatomically matched across animals according to the atlas of Franklin and Paxinos (1997). Background optical density was subtracted from each image. Each mean ± SEM reported here is the averaged value of six optical density readings (after background subtraction) for each animal. Data were analyzed by Student's t test; statistical significance was taken as p < 0.05.
RESULTS
Dbh −/− mice have increased susceptibility to epileptic stimuli
Flurothyl
Dbh −/− mice without DOPS had significantly reduced latencies to the first myoclonic jerk (MJ) and clonic/tonic (C/T) seizure compared with Dbh +/− controls (Fig.1A,B, without DOPS). The latency to the first MJ was affected to a greater degree (46% reduction) than the latency to C/T convulsion (29% reduction). The percent of Dbh −/− and Dbh +/− mice progressing to tonic extension after a C/T seizure was identical (45%); however, 100% of the Dbh −/− mice died after C/T seizure, whereas only 60% of the Dbh +/− mice died after tonic extension (Table 1). The higher mortality rate of Dbh −/− mice was not a function of the duration of flurothyl exposure, because the average duration of exposure was shorter for the Dbh −/− than for theDbh +/− animals.
Fig. 1.
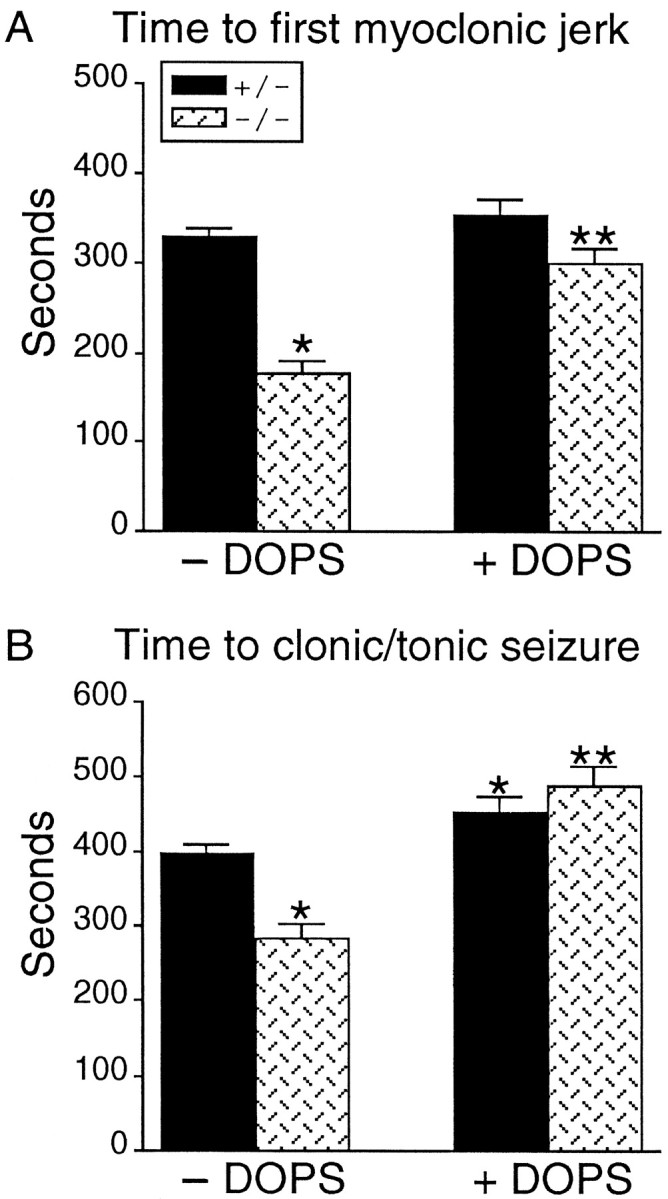
Responsiveness of Dbh +/− andDbh −/− mice to flurothyl-induced seizures. Latencies (seconds) to first MJ (A) and clonic/tonic seizure (B) recorded in Dbh +/− and Dbh −/− mice, with and without the administration of DOPS. Dbh −/− mice without DOPS had significantly shorter latencies to MJ (A) and C/T seizures (B) compared with Dbh +/− mice without DOPS (mean ± SEM; singleasterisks denote p < 0.05). Administration of DOPS (1 mg/gm) 6 hr before flurothyl significantly increased flurothyl latencies in the Dbh −/− mice compared with the Dbh −/− mice without DOPS for both MJ (A) and C/T seizures (B) (mean ± SEM; double asterisks denotep < 0.05). Latencies to both MJ and C/T convulsions in Dbh −/− mice with DOPS were not significantly different compared with those in Dbh +/− mice with DOPS.
Table 1.
Number of animals used for flurothyl-induced seizures with and without the administration of DOPS
| Without DOPS | With DOPS | |||
|---|---|---|---|---|
| +/− | −/− | +/− | −/− | |
| Flurothyl-induced seizures | n = 11 | n = 11 | n = 9 | n = 11 |
| % tonic extension | 45 (5/11) | 45 (5/11) | 44 (4/9) | 54 (6/11) |
| % mortality | 60 (3/5) | 100 (5/5) | 25 (1/4) | 33 (2/6) |
Percentage of animals exhibiting tonic extension and the percent of animals that died (mortality) after tonic extension forDbh +/− and Dbh −/− mice after flurothyl-induced seizure.
NE levels are partially restored in the CNS of Dbh −/− mice by the administration of DOPS (Thomas et al., 1998). Administration of DOPS to Dbh −/− mice significantly lengthened the latency to the first MJ and C/T convulsion (Fig.1A,B with DOPS); latencies to MJ and C/T convulsions in Dbh −/− mice with DOPS were not statistically different from latencies in Dbh +/− mice with DOPS. Administration of DOPS to Dbh +/− mice did not significantly alter the latency to the first MJ but significantly increased the latency time to C/T seizures (Fig. 1). Administration of DOPS to Dbh −/− and Dbh +/− mice did not affect the number of animals progressing to tonic extension but did reduce the number of animals dying after tonic extension in both groups (Table 1).
Pentylenetetrazol
Dbh −/− and Dbh +/− mice were challenged with PTZ at 30 and 40 mg/kg, and the latencies (seconds) to the first MJ, forelimb clonus (FC), and C/T were measured (Fig.2). PTZ (40 mg/kg) induced generalized seizures in all Dbh −/− mice (eight of eight) but in only four of seven Dbh +/− mice. Latencies to MJ, FC, and C/T seizures in Dbh −/− mice were significantly shorter than those in Dbh +/− mice (Fig.2A). The percent of animals exhibiting tonic extension was greater in Dbh −/− mice (100%) than inDbh +/− mice (29%); however, for both genotypes, all animals exhibiting tonic extension died (Fig.2A).
Fig. 2.

Responsiveness of Dbh +/− andDbh −/− mice to PTZ injections at 40 mg/kg (A) and 30 mg/kg (B).Left, Graphs show seizure latencies (seconds) to the first myoclonic jerk, forelimb clonus, and clonic/tonic seizure inDbh −/− and Dbh +/− mice. At both PTZ concentrations Dbh −/− mice had significantly shorter latencies compared with those in Dbh +/− mice (mean ± SEM; singleasteriskdenotes p < 0.01; doubleasterisks denote p < 0.001).Right, Graphs show the percentage of animals progressing to tonic extension and the percentage of animals that died after tonic extension (mortality).
PTZ (30 mg/kg) induced C/T seizures in 8 of 10 Dbh −/− mice and in 2 of 9 Dbh +/− mice. Of these animals exhibiting seizures, the Dbh −/− mice had significantly shorter latencies to the first MJ, FC, and C/T seizures than didDbh +/− mice (Fig. 2B). Again 100% of the Dbh −/− mice that exhibited seizure activity progressed to tonic extension and death; however, only 11% of theDbh +/− mice that exhibited seizure activity had tonic extension, and of those, only 44% died.
Kainic acid
Kainic acid (KA; 20 mg/kg) induced some seizure behavior (i.e., staring, head nodding, and forelimb clonus) in most animals in both groups; however KA induced generalized C/T convulsions in 100% of theDbh −/− mice (eight of eight) but in only 38% of theDbh +/− mice (three of eight). Of the animals showing C/T convulsions, Dbh −/− mice had a significantly shorter latency to generalized seizure (1587 ± 188 sec) than didDbh +/− mice (2243 ± 120 sec). The Dbh−/− mice also exhibited enhanced sensitivity to KA compared withDbh +/− mice; 50% of the Dbh −/− mice died after the KA-induced seizure, whereas none of the Dbh +/− mice died.
Audiogenic seizures
The Dbh −/− mice were more sensitive to the acoustic stimuli than were Dbh +/− mice, in that 50% (5 of 10) of the Dbh −/− mice exhibited a generalized seizure during the sound stimulus, whereas only 11% (1 of 9) of the Dbh+/− exhibited a generalized convulsion. Seizures were initiated shortly after onset of the sound (latencies between 3 and 12 sec) and manifested initially as jumping behavior that progressed quickly to explosive running–bouncing activity and finally to tonic extension and death. Sensitivity to sound-induced seizure was identical between the groups of animals; all animals (i.e., in both Dbh −/− andDbh +/− groups) that exhibited a sound-induced generalized seizure died.
Dbh −/− mice have increased c-fos mRNA associated with flurothyl-induced seizures
The animals that survived flurothyl-induced seizures [Dbh +/− (n = 8) and Dbh −/− (n = 6) without DOPS; Dbh +/− (n = 8) and Dbh −/− (n = 9) with DOPS] were killed 1 hr after C/T seizures to measure c-fos mRNA expression. Seizure-induced c-fos mRNA expression was quantitated in the neocortex, amygdala, and hippocampus [CA1, CA3, and dentate gyrus (DG)] [see Figs. 3 (for representative autoradiograms), 4 (for quantitative comparisons)].
Fig. 3.
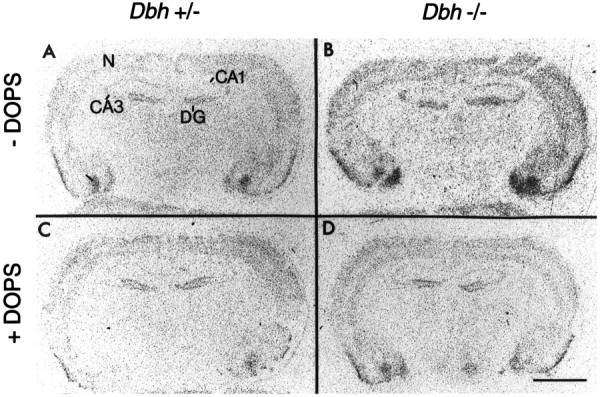
Representative autoradiograms of c-fos mRNA expression after flurothyl-induced seizures. A,C, Flurothyl-induced c-fos mRNA expression inDbh +/− mice without (A) and with (C) DOPS (1 mg/gm). B, D, Flurothyl-induced c-fos mRNA expression in Dbh −/− mice without (B) and with (D) DOPS (1 mg/gm). Note the higher c-fos mRNA expression in Dbh −/− animals without DOPS (B). DOPS (1 mg/gm) administration not only reduces c-fos mRNA expression of Dbh −/− mice (compareB with D) but also normalizes c-fos mRNA expression in Dbh −/− mice relative to that inDbh +/− mice (compare C withD). Scale bar, 2 mm. N, Neocortex.
In the neocortex, Dbh −/− mice had significantly greater seizure-induced c-fos mRNA expression than did the Dbh +/− mice, even though the two different genotypes had similar seizure-induced behavior (generalized seizures). Administration of DOPS to Dbh −/− mice reduced seizure-associated c-fos mRNA expression to a level comparable with that seen in the neocortex ofDbh +/− mice, without or with DOPS (Fig. 4). Administration of DOPS to Dbh +/− mice did not alter seizure-associated c-fos mRNA expression in the neocortex.
Fig. 4.
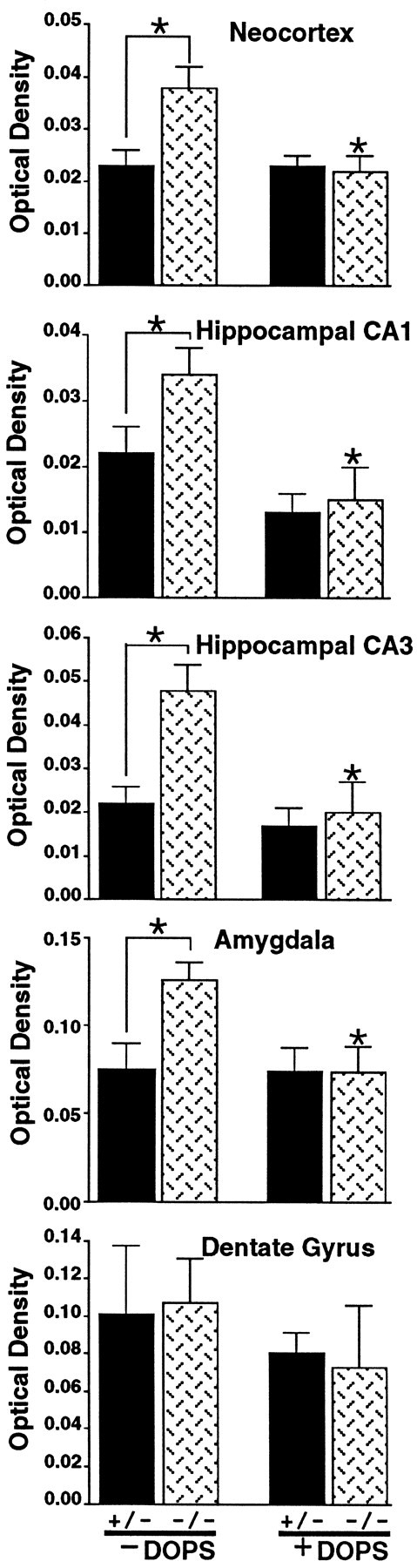
Quantification of flurothyl seizure-associated c-fos mRNA expression in the cortex, hippocampus (CA1, CA3, and DG), and amygdala in Dbh −/− and Dbh +/− mice, with and without DOPS (1 mg/gm). Flurothyl seizure-associated c-fos mRNA expression was significantly higher in Dbh −/− mice without DOPS than in Dbh +/− mice without DOPS in all regions but the dentate gyrus (mean ± SEM; asterisks denotep < 0.01). DOPS (1 mg/gm) administration toDbh −/− mice significantly reduced c-fos mRNA expression in all regions but the dentate gyrus (mean ± SEM;asterisks denote p < 0.05); c-fos mRNA expression in Dbh −/− mice and Dbh+/− mice with DOPS was not significantly different. Basal c-fos mRNA expression in Dbh −/− mice is not significantly different from basal c-fos mRNA expression in Dbh +/− mice (data not shown).
Similar results were obtained in the hippocampal CA1 and CA3 regions and the amygdala. Flurothyl seizure-associated c-fos mRNA expression inDbh −/− mice was significantly higher than that inDbh +/− mice in CA1 and CA3 regions and the amygdala. Administration of DOPS to Dbh −/− mice significantly reduced the flurothyl seizure-associated c-fos mRNA expression (to the level observed in Dbh +/− mice with DOPS). Flurothyl seizure-associated c-fos mRNA expression in Dbh +/− mice was not significantly changed by DOPS pretreatment. The only region where c-fos mRNA expression was not significantly different betweenDbh −/− and Dbh +/− mice was in the DG. Administration of DOPS to both genotypes also had no effect on flurothyl-induced c-fos mRNA expression in the DG. Because theDbh −/− mice had elevated seizure-associated c-fos mRNA expression in the neocortex, hippocampal CA1 and CA3, and amygdala, basal c-fos mRNA was measured in Dbh +/− and Dbh−/− mice. Basal c-fos mRNA expression in Dbh −/− mice was not significantly different from that in Dbh +/− mice (data not shown).
DISCUSSION
These studies provide evidence that endogenous NE exerts a profound inhibitory effect on seizure induction. The enhanced susceptibility of Dbh −/− mice to such a diverse set of seizure-inducing stimuli (convulsant stimuli potentially acting at excitatory or inhibitory receptors, sodium channels, and brainstem activation) (Olney et al., 1974; Schwob et al., 1980; Woodbury, 1980; Browning, 1985; Snead, 1992) suggests a “global” suppressive action of NE. The loss of NE's inhibitory action in Dbh−/− mice is also associated with increased c-fos mRNA expression after flurothyl-induced seizures. LC axons have a high degree of collateralization, and a single neuron can innervate several distant regions (Fallon and Loughlin, 1982; Loughlin et al., 1982). This diffuse noradrenergic innervation pattern would allow NE release from LC terminals to suppress neuronal activity throughout the brain, including regions such as the cortex and hippocampus that are important in regulating seizures. Our studies support the hypothesis that there is an inverse relationship between the release of NE and seizure susceptibility; i.e., reducing NE release increases seizure susceptibility and increasing NE release has a protective effect against seizures.
Although many studies have implicated NE as an endogenous neuromodulator of seizure activity (Chen et al., 1954; Arnold et al., 1973; Libet et al., 1977; Jerlicz et al., 1978; Mason and Corcoran, 1979; Snead, 1987; Trottier et al., 1988; Turski et al., 1989; Sullivan and Osorio, 1991; Mishra et al., 1994), the evidence has not always been consistent. The data presented here demonstrate that a selective loss of NE from noradrenergic terminals is proconvulsant. One could argue that the NE deficiency in a knock-out mouse is not definitive because developmental changes associated with the deletion of theDbh gene might contribute to the seizure-susceptibility phenotype seen in the Dbh −/− mice. However, we have also shown that increasing NE content in the CNS with DOPS administration (Thomas et al., 1998) can normalize seizure susceptibility ofDbh −/− mice. The ability of DOPS to rescue Dbh−/− mice has also been demonstrated with most other behavioral and physiological deficiencies in these mice (Thomas et al., 1995; Thomas and Palmiter, 1997a,b,c). DOPS rescue of noradrenergic function in theDbh −/− mice suggests that there is a normal anatomical development of the “noradrenergic” system during gestation in these animals; this prediction of a normal pattern of noradrenergic terminals in Dbh −/− mice has been confirmed in studies of NE transporter-binding sites (D. Weinshenker, unpublished observation). If one assumes a normal organization of noradrenergic terminals inDbh −/− mice, the conversion of DOPS to NE byl-aromatic amino acid decarboxylase could theoretically restore NE at appropriate terminals.
The absence of NE in Dbh −/− mice resulted in their greater sensitivity (i.e., enhanced seizure severity and higher mortality) to convulsant stimuli than that in Dbh +/− animals. After PTZ- and kainic acid-induced seizures, a higher percentage of the Dbh −/− mice progressed to tonic extension and death. A similar finding was observed with flurothyl-induced seizures; although the percentage of animals progressing to tonic extension was the same for Dbh +/− andDbh −/− mice without DOPS, more Dbh −/− mice than Dbh +/− mice died after tonic extension. Administration of DOPS to both Dbh +/− and Dbh−/− mice had little effect on seizure severity but reduced the number of animals that died after tonic extension, especially inDbh −/− mice. These results reflect the ability of DOPS to reverse the higher sensitivity of Dbh −/− mice to flurothyl-induced seizures. The increased survival of DOPS-treatedDbh +/− mice may be caused by an elevation in NE content above a normal catecholamine content (Thomas et al., 1998).
Associated with the enhanced susceptibility to convulsant stimuli inDbh −/− mice is an elevation in seizure-induced c-fos mRNA expression in the cortex, hippocampus (CA1 and CA3), and amygdala. The immediate early gene c-fos has long been considered a marker of neuronal activity (Dragunow and Robertson, 1987; Morgan et al., 1987; Sonnenberg et al., 1989; Morgan and Curran, 1991). A correlation of seizure severity and c-fos expression has been observed with different convulsant stimuli (White and Price, 1993; Szot et al., 1997;Robbins et al., 1998). The enhanced seizure-induced c-fos mRNA expression in Dbh −/− mice is not a function of an elevated basal c-fos state, because basal c-fos mRNA expression inDbh −/− mice is not different from that in Dbh+/− mice. This enhanced c-fos mRNA expression in the CNS ofDbh −/− mice was measured in animals with a consistent behavioral seizure phenotype, suggesting a relationship between c-fos mRNA expression and seizure threshold. Acute DOPS administration normalized the seizure-associated c-fos mRNA expression inDbh −/− mice. We conclude that the ability of the noradrenergic system to regulate seizure activity is a direct result of NE-mediated suppression of CNS excitability in such regions as the neocortex, hippocampus, and amygdala.
The ability of NE to have an inhibitory effect on seizures seems inconsistent with its general role on the arousal state of an animal. Noradrenergic neurons are active in awake animals but quiescent during sleep (Jouvet, 1969; Hobson et al., 1975; Aston-Jones and Bloom, 1981;Robbins, 1984). Basal c-fos mRNA expression in the cortex corresponds to the arousal state of the rat (Cirelli et al., 1996). When noradrenergic neurons in the LC were destroyed with 6-hydroxydopamine, the amount of basal c-fos mRNA expression in the cortex of the awake animal was reduced to levels comparable with that in an animal during sleep (Cirelli et al., 1996). Although these studies suggest a relationship between basal c-fos mRNA expression and NE, our study failed to find a change in basal c-fos mRNA expression inDbh −/− mice relative to Dbh +/− mice. This difference emphasizes the gross effects of lesioning noradrenergic neurons, which results not only in the loss of NE but also affects the level of all neurotransmitters coreleased with NE. These cotransmitters released with NE may contribute to the basal excitability of the neurons.
The dual action of NE as an inhibitory and excitatory neurotransmitter can be attributed to the large diversity of noradrenergic receptors. Iontophoretic application of NE to neocortex or hippocampus results in both excitatory and inhibitory responses (Szabadi, 1979; Langmoen et al., 1981; Nishi et al., 1981; Segal, 1981; Madison and Nicoll, 1986;Waterhouse, 1986; Stanton, 1992). The excitatory response of NE appears to be mediated via the β-receptors and/or α1-ARs, whereas the inhibitory response is mediated via the α2-ARs (Curet and deMontigny, 1988; Parfitt et al., 1988; Licata et al., 1993). This dual action of NE on neuronal activity is apparent when synaptic NE content is elevated with NE reuptake blockers; these agents do not alter the animal's susceptibility to convulsant stimuli (Kleinrok et al., 1991; Yacobi and Burnham, 1991). We postulate that the anticonvulsant action of NE is mediated via α2-ARs. Indeed, agonists selective for the α2-ARs have been shown to exert anticonvulsant effects against audiogenic seizures in mice, as well as against PTZ-, kainic acid-, and bicuculline-induced seizures; α2-AR antagonists have the reverse effect (Papanicolaou et al., 1982; Baran et al., 1985;Loscher and Czuczwar, 1987; Fletcher and Forster, 1988; Jackson et al., 1991). However, it has not been determined whether the anticonvulsant effect of α2-AR agonists is mediated via the pre- or postsynaptic receptors. A recently developed transgenic mouse with nonfunctional α2A-ARs (MacMillan et al., 1996) responded to a kindling paradigm (a process of repetitively applied stimuli resulting in generalized seizures) similarly to wild-type mice treated with an α2-AR antagonist (Janumpalli et al., 1998). Although the Dbh−/− mice are not the same as the α2A-AR mutant, the combined results provide compelling evidence that NE acting at least partially via inhibitory postsynaptic α2-ARs dampens seizure excitability. The lack of spontaneous seizure activity in Dbh −/− mice suggests that NE release may only become important under conditions of high activity (e.g., seizures) when the LC is sufficiently activated; i.e., NE serves as a potent modulator of excitability.
In conclusion, the data presented here show unambiguously that NE is capable of modulating seizure activity induced by different convulsant stimuli. The pervasive inhibitory action of NE on excitability is reflected in the increased seizure-associated c-fos mRNA expression in the Dbh −/− mice. Because galanin and NPY are also inhibitory neuromodulators that are coreleased from the same terminals as NE, it seems that the noradrenergic projection system may use multiple neurotransmitters to dampen excitability. Because of this complexity, the Dbh −/− mice provide an especially useful and new system to examine the pathways through which NE regulates seizure activity.
Footnotes
This work was supported by the National Alliance for Research on Schizophrenia and Depression (P.S.), the Department of Veterans Affairs (P.S.), National Institutes of Health Grant NS-18895 (P.A.S.), and the Howard Hughes Medical Institute (D.W. and N.C.R). We thank Sumitomo Pharmaceuticals for their generous donation of DOPS.
P.S. and D.W. contributed equally to this work.
Correspondence should be addressed to Dr. Patricia Szot, Geriatric Research, Education, and Clinical Center (182B), Veterans Affairs Medical Center, 1660 South Columbian Way, Seattle, WA 98108. E-mail:szot@u.washington.edu.
REFERENCES
- 1.Arnold PS, Racine RJ, Wise RS. Effects of atropine, reserpine, 6-hydroxydopamine and handling on seizure development in the rat. Exp Neurol. 1973;40:457–470. doi: 10.1016/0014-4886(73)90087-3. [DOI] [PubMed] [Google Scholar]
- 2.Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep–waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baraban SC, Hollopeter G, Erickson JC, Schwartzkroin PA, Palmiter RD. Knock-out mice reveal a critical antiepileptic role for neuropeptide Y. J Neurosci. 1997;17:8927–8936. doi: 10.1523/JNEUROSCI.17-23-08927.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baran H, Sperk G, Hortnagl H, Sapetschnig G, Hornykiewicz O. Alpha 2-adrenoceptors modulate kainic acid-induced limbic seizures. Eur J Pharmacol. 1985;113:263–269. doi: 10.1016/0014-2999(85)90744-7. [DOI] [PubMed] [Google Scholar]
- 5.Browning RA. Role of the brain-stem reticular formation in tonic-clonic seizures: lesions and pharmacological studies. Fed Proc. 1985;44:2425–2431. [PubMed] [Google Scholar]
- 6.Browning RA, Wade DR, Marcinczyk M, Long GL, Jobe PC. Regional brain abnormalities in norepinephrine uptake and dopamine β-hydroxylase activity in the genetically epilepsy-prone rat. J Pharmacol Exp Ther. 1989;249:229–235. [PubMed] [Google Scholar]
- 7.Chen G, Ensor GR, Bohner B. A facilitation action of reserpine on the central nervous system. Proc Soc Exp Biol Med. 1954;86:507–510. doi: 10.3181/00379727-86-21149. [DOI] [PubMed] [Google Scholar]
- 8.Cirelli C, Pompeiano M, Tononi G. Neuronal gene expression in the waking state: a role for the locus coeruleus. Science. 1996;274:1211–1215. doi: 10.1126/science.274.5290.1211. [DOI] [PubMed] [Google Scholar]
- 9.Curet O, deMontigny C. Electrophysiological characterization of adrenoceptors in the rat dorsal hippocampus. I. Receptors mediating the effect of microiontophoretically applied norepinephrine. Brain Res. 1988;475:35–46. doi: 10.1016/0006-8993(88)90196-5. [DOI] [PubMed] [Google Scholar]
- 10.Curran T, Gordon MB, Rubino KL, Sambucetti LC. Isolation and characterization of the c-fos (rat) cDNA and analysis of post-translational modifications in vitro. Oncogene. 1987;2:79–84. [PubMed] [Google Scholar]
- 11.Dailey JW, Jobe PC. Indices of noradrenergic function in the central nervous system of seizure-naive genetically epilepsy-prone rats. Epilepsia. 1986;27:665–670. doi: 10.1111/j.1528-1157.1986.tb03593.x. [DOI] [PubMed] [Google Scholar]
- 12.Dailey JW, Mishra PK, Ko KH, Penny JE, Jobe PC. Noradrenergic abnormalities in the central nervous system of seizure-naive genetically epilepsy-prone rats. Epilepsia. 1991;32:168–173. doi: 10.1111/j.1528-1157.1991.tb05240.x. [DOI] [PubMed] [Google Scholar]
- 13.Dailey JW, Mishra PK, Ko KH, Penny JE, Jobe PC. Serotonergic abnormalities in the central nervous system of seizure-naive genetically epilepsy-prone rats. Life Sci. 1992;50:319–326. doi: 10.1016/0024-3205(92)90340-u. [DOI] [PubMed] [Google Scholar]
- 14.Dichter MA. Emerging insights into mechanisms of epilepsy: implications for new antiepileptic drug development. Epilepsia. 1994;35[Suppl 4]:s51–s57. doi: 10.1111/j.1528-1157.1994.tb05956.x. [DOI] [PubMed] [Google Scholar]
- 15.Dragunow M, Robertson HA. Kindling stimulation induces c-fos protein(s) in granule cells of the rat dentate gyrus. Nature. 1987;329:441–442. doi: 10.1038/329441a0. [DOI] [PubMed] [Google Scholar]
- 16.Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–421. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- 17.Faingold CL, Gehlach B, Caspary DM. Decreased effectiveness of GABA-mediated inhibition in the inferior colliculus of the genetically epilepsy-prone rat. Exp Neurol. 1986;93:145–159. doi: 10.1016/0014-4886(86)90154-8. [DOI] [PubMed] [Google Scholar]
- 18.Fallon JH, Loughlin SE. Monoamine innervation of the forebrain: collateralization. Brain Res Bull. 1982;9:295–307. doi: 10.1016/0361-9230(82)90143-5. [DOI] [PubMed] [Google Scholar]
- 19.Fletcher A, Forster EA. A proconvulsant action of selective α2-adrenoceptor antagonists. Eur J Pharmacol. 1988;151:27–34. doi: 10.1016/0014-2999(88)90688-7. [DOI] [PubMed] [Google Scholar]
- 20.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; New York: 1997. [Google Scholar]
- 21.Hara M, Sasa M, Kawabata A, Serikawa T, Yamada T, Yamada J, Takaori S. Decreased dopamine and increased norepinephrine levels in the spontaneously epileptic rat, a double mutant rat. Epilepsia. 1993;34:433–440. doi: 10.1111/j.1528-1157.1993.tb02583.x. [DOI] [PubMed] [Google Scholar]
- 22.Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science. 1975;189:55–58. doi: 10.1126/science.1094539. [DOI] [PubMed] [Google Scholar]
- 23.Jackson HC, Dickinson SL, Nutt DJ. Exploring the pharmacology of the pro-convulsant effects of alpha 2-adrenoceptor antagonists in mice. Psychopharmacology (Berl) 1991;105:558–562. doi: 10.1007/BF02244380. [DOI] [PubMed] [Google Scholar]
- 24.Janumpalli S, Butler LS, MacMillan LB, Limbird LE, McNamara JO. A point mutation (D79N) of the alpha2a-adrenergic receptor abolishes the antiepileptic action of endogenous norepinephrine. J Neurosci. 1998;18:2004–2008. doi: 10.1523/JNEUROSCI.18-06-02004.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jerlicz M, Kostowski W, Bidzi'nski A, Hauptman M, Dymecki J. Audiogenic seizures in rats: relation to noradrenergic neurons of the locus coeruleus. Acta Physiol Pol. 1978;29:409–412. [PubMed] [Google Scholar]
- 26.Jobe PC, Ko KH, Dailey JW. Abnormalities in norepinephrine turnover rate in the central nervous system of the genetically epilepsy-prone rat. Brain Res. 1984;290:357–360. doi: 10.1016/0006-8993(84)90956-9. [DOI] [PubMed] [Google Scholar]
- 27.Jouvet M. Biogenic amines and the states of sleep. Science. 1969;163:32–41. doi: 10.1126/science.163.3862.32. [DOI] [PubMed] [Google Scholar]
- 28.King GA, Burnham WM. Alpha 2-adrenergic antagonists suppress epileptiform EEG activity in a petit mal seizure model. Life Sci. 1982;30:293–298. doi: 10.1016/0024-3205(82)90511-2. [DOI] [PubMed] [Google Scholar]
- 29.Kleinrok Z, Gustaw J, Czuczwar SJ. Influence of antidepressant drugs on seizure susceptibility and the anticonvulsant activity of valproate in mice. J Neural Transm [Suppl] 1991;34:85–90. doi: 10.1007/978-3-7091-9175-0_11. [DOI] [PubMed] [Google Scholar]
- 30.Langmoen IA, Segal M, Andersen P. Mechanisms of norepinephrine actions on hippocampal pyramidal cells in vitro. Brain Res. 1981;208:349–362. doi: 10.1016/0006-8993(81)90563-1. [DOI] [PubMed] [Google Scholar]
- 31.Lapin IP, Ryzor IV. Effect of catecholaminergic drugs on quinolinate- and kynurenine-induced seizures in mice. J Neurol Transm Gen Sect. 1990;82:55–65. doi: 10.1007/BF01244834. [DOI] [PubMed] [Google Scholar]
- 32.Lauterborn JC, Ribak CR. Differences in dopamine beta-hydroxylase immunoreactivity between the brains of genetically epilepsy-prone and Sprague-Dawley rats. Epilepsy Res. 1989;4:161–176. doi: 10.1016/0920-1211(89)90001-6. [DOI] [PubMed] [Google Scholar]
- 33.L'Heureux R, Dennis T, Curet O, Scatton B. Measurement of endogenous noradrenaline release in the rat cerebral cortex in vivo by transcortical dialysis: effects of drugs affecting noradrenergic transmission. J Neurochem. 1986;46:1794–1801. doi: 10.1111/j.1471-4159.1986.tb08498.x. [DOI] [PubMed] [Google Scholar]
- 34.Libet B, Gleason CA, Wright EW, Feinstein B. Suppression of an epileptiform type of electrocortical activity in the rat by stimulation of the locus coeruleus. Epilepsia. 1977;18:451–462. doi: 10.1111/j.1528-1157.1977.tb04991.x. [DOI] [PubMed] [Google Scholar]
- 35.Licata F, Li-Volsi G, Maugeri G, Ciranna L, Santangelo F. Effects of noradrenaline on the firing rate of vestibular neurons. Neuroscience. 1993;53:149–158. doi: 10.1016/0306-4522(93)90293-o. [DOI] [PubMed] [Google Scholar]
- 36.Loscher W, Czuczwar SJ. Comparison of drugs with different selectivity for central alpha1- and alpha2-adrenoceptors in animal models of epilepsy. Epilepsy Res. 1987;1:165–172. doi: 10.1016/0920-1211(87)90037-4. [DOI] [PubMed] [Google Scholar]
- 37.Loughlin SE, Foote SL, Fallon JH. Locus coeruleus projections to cortex: topography, morphology and collateralizations. Brain Res Bull. 1982;9:287–294. doi: 10.1016/0361-9230(82)90142-3. [DOI] [PubMed] [Google Scholar]
- 38.MacMillan LB, Hein L, Smith MS, Piascik MT, Limbird LE. Central hypotensive effects of the α2a-adrenergic receptor subtype. Science. 1996;273:801–803. doi: 10.1126/science.273.5276.801. [DOI] [PubMed] [Google Scholar]
- 39.Madison DV, Nicoll RA. Actions of noradrenaline recorded intracellularly in rat hippocampal CA1 pyramidal neurons in vitro. J Physiol (Lond) 1986;372:221–244. doi: 10.1113/jphysiol.1986.sp016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mason ST, Corcoran ME. Catecholamines and convulsions. Brain Res. 1979;170:497–507. doi: 10.1016/0006-8993(79)90967-3. [DOI] [PubMed] [Google Scholar]
- 41.Mazarati AM, Hal'aszi E, Telegdy G. Anticonvulsive effects of galanin administered into the central nervous system upon the picrotoxin-kindled seizure syndrome in rats. Brain Res. 1992;589:164–166. doi: 10.1016/0006-8993(92)91179-i. [DOI] [PubMed] [Google Scholar]
- 42.Mazarati AM, Liu H, Soomets U, Sankar R, Shin D, Katsumori H, Langel U, Wasterlain CG. Galanin modulation of seizures and seizure modulation of hippocampal galanin in animal models of status epilepticus. J Neurosci. 1998;18:10070–10077. doi: 10.1523/JNEUROSCI.18-23-10070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyerhoff JL, Carter RE, Yourick DL, Slusher BS, Coyle JT. Genetically epilepsy-prone rats have increased brain regional activity of an enzyme which liberates glutamate from N-acetyl-aspartyl-glutamate. Brain Res. 1992;593:140–143. doi: 10.1016/0006-8993(92)91276-k. [DOI] [PubMed] [Google Scholar]
- 44.Mishra PK, Burger RL, Bettendorf AF, Browning RA, Jobe PC. Role of norepinephrine in forebrain and brainstem seizures: chemical lesioning of locus ceruleus with DSP4. Exp Neurol. 1994;125:58–64. doi: 10.1006/exnr.1994.1006. [DOI] [PubMed] [Google Scholar]
- 45.Morgan JI, Curran T. Proto-oncogene transcription factors and epilepsy. Trends Pharmacol Sci. 1991;12:343–349. doi: 10.1016/0165-6147(91)90594-i. [DOI] [PubMed] [Google Scholar]
- 46.Morgan JI, Cohen DR, Hempstead JL, Curran T. Mapping patterns of c-fos expression in the central nervous system after seizure. Science. 1987;237:192–197. doi: 10.1126/science.3037702. [DOI] [PubMed] [Google Scholar]
- 47.Murray TF, Sylvester D, Schultz CS, Szot P. Purinergic modulation of pentylenetetrazol seizure threshold in the rat. Neuropharmacology. 1985;24:761–766. doi: 10.1016/0028-3908(85)90010-3. [DOI] [PubMed] [Google Scholar]
- 48.Nishi H, Watanabe S, Ueki S. Effects of monoamines injected into the hippocampus on hippocampal seizure discharges in the rabbit. J Pharmacobiodyn. 1981;4:7–14. doi: 10.1248/bpb1978.4.7. [DOI] [PubMed] [Google Scholar]
- 49.Noebels JL. Mutational analysis of inherited epilepsies. Adv Neurol. 1986;44:97–113. [PubMed] [Google Scholar]
- 50.Olney JW, Rhee V, Ho OL. Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res. 1974;77:507–512. doi: 10.1016/0006-8993(74)90640-4. [DOI] [PubMed] [Google Scholar]
- 51.Papanicolaou J, Summers RJ, Vajda FJE, Louis WJ. The relationship between alpha 2-adrenoceptor selectivity and anticonvulsant effect in a series of clonidine-like drugs. Brain Res. 1982;241:393–397. doi: 10.1016/0006-8993(82)91086-1. [DOI] [PubMed] [Google Scholar]
- 52.Parfitt KD, Freedman R, Bickford-Wimer PC. Electrophysiological effects of locally applied noradrenergic agents at cerebellar Purkinje neurons: receptor specificity. Brain Res. 1988;462:242–251. doi: 10.1016/0006-8993(88)90552-5. [DOI] [PubMed] [Google Scholar]
- 53.Prichard JW, Gallagher BB, Glaser GH. Experimental seizure-threshold testing with flurothyl. J Pharmacol Exp Ther. 1969;166:170–178. [PubMed] [Google Scholar]
- 54.Robbins CA, Kim DW, Szot P, Rho JM, White SS, Schwartzkroin PA. c-Fos mRNA expression is increased in mouse brain after seizure induced by flurothyl. Epilepsia. 1998;39[Suppl 6]:26. [Google Scholar]
- 55.Robbins TW. Cortical noradrenaline, attention and arousal. Psychol Med. 1984;14:13–21. doi: 10.1017/s0033291700003032. [DOI] [PubMed] [Google Scholar]
- 56.Schwob JE, Fuller T, Price JL, Olney JW. Widespread patterns of neuronal damage following systemic or intracerebral injections of kainic acid: a histological study. Neuroscience. 1980;5:991–1014. doi: 10.1016/0306-4522(80)90181-5. [DOI] [PubMed] [Google Scholar]
- 57.Segal M. The action of norepinephrine in the rat hippocampus: intracellular studies in the slice preparation. Brain Res. 1981;206:107–128. doi: 10.1016/0006-8993(81)90104-9. [DOI] [PubMed] [Google Scholar]
- 58.Snead OC., III Noradrenergic mechanisms in gamma-hydroxybutyrate-induced seizure activity. Eur J Pharmacol. 1987;136:103–108. doi: 10.1016/0014-2999(87)90785-0. [DOI] [PubMed] [Google Scholar]
- 59.Snead OC., III Pharmacological models of generalized absence seizures in rodents. J Neural Transm Suppl. 1992;35:7–19. doi: 10.1007/978-3-7091-9206-1_2. [DOI] [PubMed] [Google Scholar]
- 60.Sonnenberg JL, Mitchelmore C, McGregor-Leon PF, Hempstead J, Morgan JI, Curran T. Glutamate receptor agonists increase the expression of Fos, Fra and AP-1 DNA binding activity in the mammalian brain. J Neurosci Res. 1989;24:72–80. doi: 10.1002/jnr.490240111. [DOI] [PubMed] [Google Scholar]
- 61.Stanton PK. Noradrenergic modulation of epileptiform bursting and synaptic plasticity in the dentate gyrus. Epilepsy Res Suppl. 1992;7:135–150. [PubMed] [Google Scholar]
- 62.Sullivan HC, Osorio I. Aggravation of penicillin-induced epilepsy in rats with locus ceruleus lesion. Epilepsia. 1991;32:591–596. doi: 10.1111/j.1528-1157.1991.tb04697.x. [DOI] [PubMed] [Google Scholar]
- 63.Szabadi E. Adrenoceptors on central neurons: microelectrophoretic studies. Neuropharmacology. 1979;18:831–843. doi: 10.1016/0028-3908(79)90079-0. [DOI] [PubMed] [Google Scholar]
- 64.Szot P, White SS, Veith RC. Effect of pentylenetetrazol on the expression of tyrosine hydroxylase mRNA and norepinephrine and dopamine transporter mRNA. Mol Brain Res. 1997;44:46–54. doi: 10.1016/s0169-328x(96)00217-3. [DOI] [PubMed] [Google Scholar]
- 65.Tacke U, Kolonen S. The effect of clonidine and yohimbine on audiogenic seizures (AGS) in rats. Pharmacol Res Commun. 1984;16:1019–1030. doi: 10.1016/s0031-6989(84)80066-1. [DOI] [PubMed] [Google Scholar]
- 66.Thomas SA, Palmiter RD. Disruption of the dopamine β-hydroxylase gene in mice suggests roles for norepinephrine in motor function, learning, and memory. Behav Neurosci. 1997a;111:579–589. doi: 10.1037//0735-7044.111.3.579. [DOI] [PubMed] [Google Scholar]
- 67.Thomas SA, Palmiter RD. Thermoregulatory and metabolic phenotypes of mice lacking noradrenaline. Nature. 1997b;387:94–97. doi: 10.1038/387094a0. [DOI] [PubMed] [Google Scholar]
- 68.Thomas SA, Palmiter RD. Impaired maternal behavior in mice lacking norepinephrine and epinephrine. Cell. 1997c;91:583–592. doi: 10.1016/s0092-8674(00)80446-8. [DOI] [PubMed] [Google Scholar]
- 69.Thomas SA, Matsumoto AM, Palmiter RD. Noradrenaline is essential for mouse fetal development. Nature. 1995;374:643–646. doi: 10.1038/374643a0. [DOI] [PubMed] [Google Scholar]
- 70.Thomas SA, Marck BT, Palmiter RD, Matsumoto AM. Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine β-hydroxylase. J Neurochem. 1998;70:2468–2476. doi: 10.1046/j.1471-4159.1998.70062468.x. [DOI] [PubMed] [Google Scholar]
- 71.Trottier S, Lindvall O, Chauvel P, Bjorklund A. Facilitation of focal cobalt-induced epilepsy after lesions of the noradrenergic locus coeruleus system. Brain Res. 1988;454:308–314. doi: 10.1016/0006-8993(88)90831-1. [DOI] [PubMed] [Google Scholar]
- 72.Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro ES. Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse. 1989;3:154–171. doi: 10.1002/syn.890030207. [DOI] [PubMed] [Google Scholar]
- 73.Waterhouse BD. Electrophysiological assessment of monoamine synaptic function in neuronal circuits of seizure susceptible brains. Life Sci. 1986;39:807–818. doi: 10.1016/0024-3205(86)90459-5. [DOI] [PubMed] [Google Scholar]
- 74.White LE, Price JL. The functional anatomy of limbic status epilepticus in the rat. 1. Patterns of [14C]2-deoxyglucose uptake and Fos immunocytochemistry. J Neurosci. 1993;13:4787–4809. doi: 10.1523/JNEUROSCI.13-11-04787.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Woodbury DM. Convulsant drugs: mechanism of action. Adv Neurol. 1980;27:249–303. [PubMed] [Google Scholar]
- 76.Yacobi R, Burnham WM. The effect of tricyclic antidepressants on cortex- and amygdala-kindled seizures in the rat. Can J Neurol Sci. 1991;18:132–136. doi: 10.1017/s0317167100031589. [DOI] [PubMed] [Google Scholar]