

Abstract
Cytomegalovirus (CMV) infects a majority of adult humans. During early development and in the immunocompromised adult, CMV causes neurological deficits. We used recombinant murine cytomegalovirus (mCMV) expressing either green fluorescent protein (GFP) or β-galactosidase under control of human elongation factor 1 promoter or CMV immediate early-1 promoter as reporter genes for infected brain cells. In vivo and in vitro studies revealed that neurons and glial cells supported strong reporter gene expression after CMV exposure. Brain cultures selectively enriched in either glia or neurons supported viral replication, leading to process degeneration and cell death within 2 d of viral exposure. In addition, endothelial cells, tanycytes, radial glia, ependymal cells, microglia, and cells from the meninges and choroid were infected. Although mCMV showed no absolute brain cell preference, relative cell preferences were detected. Radial glia cells play an important role in guiding migrating neurons; these were viral targets in the developing brain, suggesting that cortical problems including microgyria that are a consequence of CMV may be caused by compromised radial glia. Although CMV is a species-specific virus, recombinant mCMV entered and expressed reporter genes in both rat and human brain cells, suggesting that mCMV might serve as a vector for gene transfer into brain cells of non-murine species. GFP expression was sufficiently strong that long axons, dendrites, and their associated spines were readily detected in both living and fixed tissue, indicating that mCMV reporter gene constructs may be useful for labeling neurons and their pathways.
Keywords: virus, neuron, GFP, development, mouse CMV, gene therapy, neuropathology
Cytomegaloviruses (CMVs) are a widespread group of double-stranded DNA viruses that infect many different mammals in a species-specific manner. Human cytomegalovirus (hCMV) is commonly found in humans where virus distribution among adults ranges from 50 to 90% of the population (Mocarski, 1996;Johnson, 1998). Similarly, murine CMV (mCMV) is ubiquitous in wild mice. Both CMVs have large genomes consisting of ∼230 kilobase pairs (kbp) exhibiting low level nucleotide sequence homology outside theie1 region transcriptional enhancer and genome packaging signals but retaining colinear genome organization. Although similar in virion morphology, effect on cells, pathogenesis, and biology, all CMVs exhibit striking species specificity (Osborn, 1982;Ho, 1991; Mocarski, 1996).
CMV is the leading viral cause of congenital birth defects (White and Fenner, 1994; Alford and Britt, 1996). CMV can cause substantial brain damage when infection occurs during early human development, resulting in epilepsy, microencephaly, microgyria, hydrocephalus, deafness, and decreased IQ (Bray et al., 1981; Bale et al., 1985; Hicks et al., 1993;Perez-Jiminez et al., 1998). CMV infections are found in ∼1% of human births, and clear neurological damage is found in 10% of those infected (Hicks et al., 1993) with a higher percentage of more subtle neurological damage probable (Johnson, 1998). The range of cell types that are involved remains poorly defined (Ho, 1991).
In contrast to early development, children and adults control infection with little apparent effect on the nervous system, in part because of an effective immune response that maintains the virus in a lifelong latency. In immunocompromised individuals, CMV can contribute to neurological deficits and mental disorders (Navia et al., 1986;Chimelli et al., 1992; d'Arminio Monforte et al., 1992; Fiala et al., 1993; Arribas et al., 1995). Increased levels of virus in the brain are often associated with later stages of HIV infection (Kalayjian et al., 1993), sometimes with cellular colocalization of CMV and HIV (Nelson et al., 1988).
Microgyri found in cases of congenital disease (Diezel, 1954) have been suggested to be caused by CMV-mediated deficits in blood supply possibly related to endothelial cell infection (Tsutsui, 1995) rather than a direct damage to neurons (Marques-Dias et al., 1984). Others have suggested that the primary cell involved is a glial cell, either microglia as studied in guinea pig (Booss et al., 1988) or possibly a monocyte-derived macrophage/microglia as described in humans (Pulliam, 1991). Neuron and astrocyte involvement has also been suggested in developing rodents (Tsutsui, 1995) and HIV-infected humans (Wiley et al., 1986; Wiley and Nelson, 1988). In many cases, a single cell type has been suggested as the primary substrate for infection, with little detectable involvement of other cell types.
The species specificity of this group of viruses (Mocarski, 1996) suggests that rodent CMVs, replication incompetent in human cells (Ho, 1991), might serve as vectors to introduce foreign genes into brain or brain cells. To this end we tested the hypothesis that mCMV would serve to introduce foreign genes into mouse, rat, and human brain cells. Although a previous analysis had suggested that neither human embryonic brain cells nor human cell lines were susceptible to mCMV entry or infection (Kim and Carp, 1971), more recent advances in molecular biology of recombinant viruses and reporter gene induction provide a more sensitive assay, as described here. We generated mCMVs expressing an enhanced green fluorescent protein (GFP) or β-galactosidase (β-gal) to study CMV infection and reporter gene expression in vitro and in vivo in live and fixed brain cells.
MATERIALS AND METHODS
Recombinant CMV. Recombinant viruses derived from the K181 strain of mCMV capable of expressing β-gal (RM427+) or enhanced GFP (EGFP) (MC.55 and RM4503) were used in these studies (Fig.1). RM4503, like RM427 (Manning et al., 1992), was constructed to express a marker gene insert under control of a chimeric promoter-enhancer composed of an hCMV promoter-enhancer fragment (−242 to +7 relative to the transcription start site) inserted adjacent to the mCMV enhancer in the mCMV genome. This construct included an SV40-derived polyadenylation signal downstream of the marker gene and directed constitutive high-level expression of the marker throughout infection (Manning et al., 1992; . The EGFP-puro construct, a gift of Mark Prichard and Greg Pari (Hybridon Corporation, Cambridge, MA), fuses the entire EGFP protein coding sequence to the puromycin resistance protein coding sequence with an intervening five amino acid spacer. The expression cassette was inserted into the mCMV ie2 gene, which has been shown to be completely dispensable for viral growth in cell culture as well as for growth, latency, and pathogenesis in BALB/c mice (Saederup et al., Cardin et al., 1995). pON4503 was constructed by digesting pON4457, which carries a 6.6 kbp DraI/EcoRI fragment of mCMV DNA (nucleotide 183086–189674), with HpaI, cleaving at two closely spaced sites in the ie2 promoter and cloning a 1.7 kbp SnaBI/HpaI fragment from EGFP-puro in the same transcriptional orientation as ie2 (Fig. 1). Recombinant virus was created by co-transfection of NIH 3T3 cells with RM427+ DNA and PacI/AflII digested pON4503 with Superfect (Life Technologies, Gaithersburg, MD) in OptiMEM (Life Technologies) followed by addition of standard DMEM (Life Technologies) supplemented with 10% NuSerum (Fisher Scientific, Pittsburgh, PA) and antibiotics as described (Vieira et al., 1994). Recombinant viruses were isolated by plaque assay from supernatants collected at 7 d after transfection. Recombinant virus was enriched by replacing standard medium with medium containing puromycin (5 μg/ml) (Sigma, St. Louis, MO) for the final 2 d and during one additional round of growth after which ∼80% of plaques exhibited green fluorescence. Enriched pools were used to inoculate BALB/c mice by the intraperitoneal route; progeny virus was recovered from salivary glands 14 d after inoculation and was followed by two rounds of limiting dilution purification at which time RM4503 was judged pure and authentic by the absence of any β-gal-positive virus and by DNA blot hybridization of viral DNA restriction fragments. Green fluorescence was detectable as early as 6 hr after infection with plaque-pure RM4503 in NIH 3T3 cells. RM4503 grew as well as wild type in cell culture and, after intraperitoneal inoculation, reached titers similar to wild type in salivary glands.
Fig. 1.

Map of the HindIII K, L, and J fragments of murine CMV (K181+ strain). The murine CMVie1/ie2/ie3 transcriptional enhancer and the arrangement of ie1,ie2, ie3, sgg1, andMCK transcripts are also shown, with splicing patterns indicated on the arrows depicting individual transcripts as they appear in wild-type murine CMV. The lacZ insertion mutation in RM427+, the EGFP-puro insertion in RM4503–1, and the GFP–GPT insertion in MC.55 are depicted below the map. Expression of the lacZ gene in RM427+ was regulated by a 199 bp human CMV ie1/ie2 promoter (αHCMV) fragment (−219 to −19 relative to the transcription start site) (Manning et al., 1992). Expression of theEGFP-puro gene in RM4503 was regulated by a 249 bp human CMV ie1/ie2 promoter (−242 to + 7 relative to the transcription start site). These promoters display immediate early expression kinetics when placed adjacent to the murine CMV enhancer (Enh) in the HpaI sites of the ie2 gene (Manning et al., 1992; our unpublished observations). Expression of EGFP in MC.55 was regulated by the human elongation factor 1a promoter (EF-1α) (Uetsuki et al., 1989), whereas the E. coli GPT was under the control of the mouse phosphoglycerate kinase (PGK) promoter.
A second recombinant mCMV (Fig. 1, MC.55) containing the GFP gene was constructed using pON488, which is composed of theDraI (position 183086) to ClaI (position 188569) fragment of mouse CMV (Rawlinson et al., 1996) containing the immediate early-2 gene cloned into pUC21, which had been cut with PstI (made blunt with Klenow fragment and all four deoxynucleotides) andClaI. A cassette consisting of the EGFP gene (Clontech, Palo Alto CA) under the control of human elongation factor 1a promoter (Uetsuki et al., 1989), and the Escherichia coliguanosine-hypoxanthine phosphoribosyltransferase gene (GPT) under the control of the mouse phosphoglycerate kinase (PGK) promoter, was inserted between the HpaI (position 184236) andNheI (position 187119) sites of pON488 to create pQ55. For the transfection of NIH 3T3 cells, pQ55 was digested withSalI, extracted with phenol/chloroform (24:1) and chloroform, ethanol-precipitated, dried, and suspended in STE (5 mm NaCl, 5 mm Tris-HCl, pH 7.5, 1 mm EDTA) at a concentration of 1–2 mg/ml. For electroporation, 2 × 106 to 5 × 106 NIH 3T3 cells plus 20 mg DNA in 0.4 ml of electroporation buffer [a 1:3 mixture of OptiMEM I (Life Technologies) and cytomix (van der Hoff et al., 1992) (120 mm KCl, 0.15 mm CaCl2, 10 mmK2HPO4/KH2PO4, p H7.6, 5 mm MgCl2)] were electroporated with a BTX ECM 600 instrument set at 275 V and 1075 mF in a 4 mm cuvette at room temperature. Cells were plated after electroporation and infected with mCMV at an multiplicity of infection (MOI) of 2–5 at 18 hr after electroporation. Progeny virus from the transfection/infection cultures were grown for two rounds under selection for the GPT gene as described (Vieira et al., 1994) to enrich for recombinant virus. Recombinant virus, identified by green fluorescent plaques under 488 nm illumination, were ultimately purified by twofold limiting dilution. In this MC.55 recombinant virus theie2 gene, which is dispensable for growth (Cardin et al., 1995), is disrupted. Viability of mCMV was expressed as plaque-forming units (pfu) determined before the virus was frozen in stock solutions at −80° for future use.
Viral replication. If virus replicates in the brain cultures, then as time progresses, the culture supernatant should show an increased concentration of infectious virus. To test this, rat (Sprague Dawley) neurons from hippocampus, hypothalamus, and cortex were mixed and plated in 35-mm-diameter wells at a density of 106 cells/well. Parallel experiments were performed with mouse (BALB/c) neurons. Each condition was performed in triplicate. Brain cultures were infected with either of two different GFP-expressing viruses. The MC.55 virus was used at 106 pfu, and RM4503 was used at 5 × 105 pfu. At 6 hr after inoculation, brain cultures were washed four times to remove the unadsorbed inoculum and placed in 2 ml growth medium. At 18 hr after the initial infection, all tissue culture medium supernatant (2 ml) was collected and replaced with 2 ml sterile culture medium. Culture medium contained DMEM supplemented with 10% fetal bovine serum (Hyclone, Logan, UT). Additional collections were made at successive 12 hr intervals. Tissue culture supernatant containing CMV was frozen within minutes after collection and kept at −80°C.
To determine relative viral concentration, NIH 3T3 cells were grown on 12-mm-round coverslips until the cells were 60% confluent. Then a 500 μl aliquot from each of the test conditions was added to the 3T3 cells. After 18 hr, all 3T3 cells were killed with 3% paraformaldehyde overnight. After washing in phosphate buffer, 3T3 cells were examined in an inverted Nikon epifluorescent microscope using a 20× Olympus objective. Green fluorescent 3T3 cells were counted in eight visual fields on each 3T3-containing coverslip.
Because CNS cultures contain a mixture of glia and neurons, in a second set of experiments we used more selective cultures enriched in glia or neurons to infect with MC.55, as described above. Glia were obtained from postnatal day (P) 5 mouse cortex. Glia were plated at low density and then allowed to replicate until they were 75% confluent in a 35 mm culture dish. Neurons were obtained from a P6 cerebellum. These cultures consist primarily of cerebellar granule cells, neurons that undergo their final mitosis at approximately this stage of development, substantially later than most neurons. A more detailed description of granule cell (Liljelund et al., 1994) and astrocyte (van den Pol et al., 1992) enriched cultures is found elsewhere. Granule cells were also plated at 75% confluency. Both cell types in these experiments were maintained with Neurobasal media (Life Technologies) for 4 d before viral contact. To avoid glial proliferation after viral infection, fetal bovine serum was not used during those 4 d or thereafter. Medium was harvested and replenished every 12 hr, as described above, and experiments were performed in duplicate.
Virus in CNS. To determine whether mCMV would infect brain cells in vivo, 500 nl-1.5 μl (103 pfu/nl) of virus was injected with a Hamilton microsyringe directly into the brain. Mice from P1 to adults were used. Adult rats also received intracerebral injections at the same virus concentration. Injections were aimed so that the tip of the syringe needle would pass through the cerebral cortex and end in the striatum, hippocampus, or hypothalamus. To reduce a sudden increase in local pressure, the virus was injected slowly over 2–3 min.
Microscopy. Photomicrographs were taken on an Olympus IX70 inverted fluorescent microscope with a Spot 2 digital camera (Diagnostic Instruments) interfaced with a Macintosh computer. Contrast was adjusted with Adobe Photoshop, and images were printed on a Kodak 8650 dye sublimation digital printer.
RESULTS
Viral infection of neurons and glia in vitro
To examine the course of CMV infection of brain cells, developing brains were cultured on glass coverslips and studied after incubation with GFP-expressing mCMV (either RM4503 or MC.55) or β-gal expressing mCMV. Within 6 hr of viral addition to the culture (MOI = 15), some neurons and glia began to show GFP-mediated green fluorescence or β-gal staining with each of the three viruses used. In cultures containing both glial cells and neurons, a greater number of astrocytes showed GFP labeling than did neurons at earlier time points. Within the next 24 hr, a greater number of cells showed expression of the reporter genes. In parallel, the expression of both GFP and β-gal became much stronger. In cultures containing roughly equal numbers of astrocytes and neurons, 96 astrocytes were labeled and only four neurons were found after 18 hr. The ratio changed over time, with greater numbers of neurons showing evidence of infection. Mouse brain cultures contained a mixture of neurons, astrocytes, and oligodendrocytes, as identified with immunolabeling for L1 (neurons), glial fibrillary acidic protein (astrocytes), and myelin basic protein (oligodendrocytes) antisera, as described previously (van den Pol et al., 1992; van den Pol and Kim, 1993; Liljelund et al., 1994).
Some cultures of hypothalamus, cortex, and hippocampus (n = 6 each) were infected with mCMV at an MOI of 15. Interestingly, all cells, including neurons, astrocytes, and oligodendrocytes, died within 48 hr. This finding was based both on fluorescent microscopy and the use of video-enhanced differential interference contrast (DIC) microscopy used to detect nonfluorescent cells. Ethidium homodimer (Molecular Probes, Eugene, OR), which labels the nuclei of dead cells, was also used to assess cell viability. In cultures made from postnatal day 2 glial cells from hippocampus, by 30 hr after infection, most astrocytes that had a typical sheet-like flat appearance were green, indicating infection with mCMV (Fig.2A). After 48 hr, cells either were detached and floating or broken down with no cellular continuity; the cells still attached to the substrate were bright green and round, exhibiting viral cytopathic effects (Fig.2B). In contrast, noninfected control cultures generated at the same time showed little evidence of cell death, suggesting that all types of brain cells can be killed by a high concentration of virus. When a lower dose of virus (MOI = 1) was used, cells survived much longer, with some neurons and glia still viable after 7 d. With time, however, all mouse brain cells died, whereas same-age noninfected controls remained viable. Although all infected cells expressed the reporter genes, consistent with infection, the level of expression varied with time and dose of virus. The highest level of reporter gene expression came with longer survival times achieved with lower MOIs. This may have been because cells infected with high MOIs (>14) died before the higher levels of gene expression could be achieved.
Fig. 2.
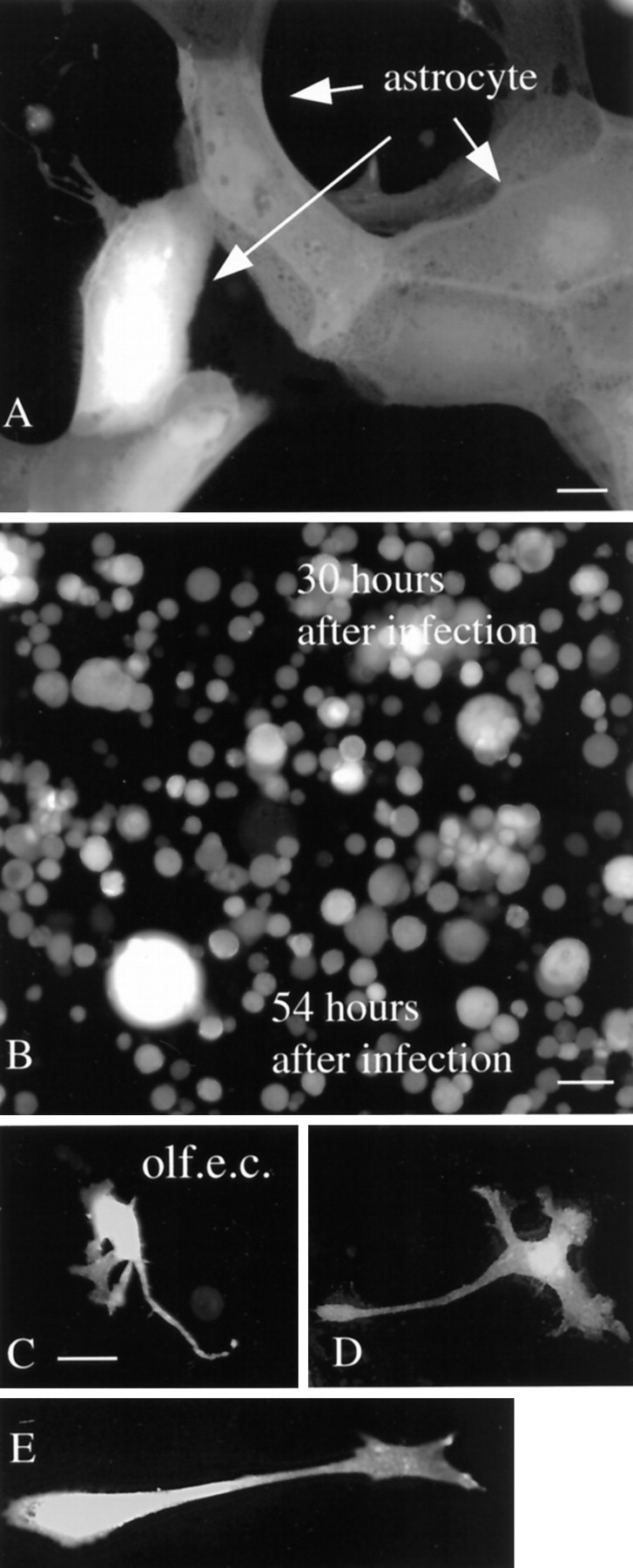
Glia infection in vitro.A, After 30 hr, most astrocytes in vitrowere green, indicating infection with mCMV. Scale bar, 8 μm.B, By 54 hr, most glia were dead or dying and had a round featureless shape. Scale bar, 25 μm.C–E, Olfactory ensheathing cells (olf.e.c.) became green 30 hr after infection with CMV. Scale bar, 12 μm.
Earlier work on virus penetration into the brain suggested that the olfactory mucosa might be a potential portal of entry for virus into the brain by spread along the olfactory nerve as it entered the olfactory bulb. A primary cell type that encircles the olfactory axons is the olfactory ensheathing cell, a type of glial cell. Cultures of mouse olfactory ensheathing cells showed strong GFP expression after infection with mCMV (Fig. 2C–E). That the cultured cells were primarily ensheathing cells was confirmed with immunostaining for P75, the low-affinity NGF receptor that these cells selectively express (Vickland et al., 1991; Ramon-Cueto and Valverde, 1995).
In control cultures, and in early stages of viral infection, neurons showed long processes that included both dendrites several hundred micrometers in length and axons exceeding 1000 μm in length. After mCMV infection, neurons initially began to turn green with no obvious sign of pathology (Fig. 3). However, soon after the neurites showed a green coloration, they began showing unusual dilations and constrictions in processes, some reaching several micrometers in diameter. Green processes were only found after the parent cell body showed reporter gene expression indicative of viral infection and gene expression. At a later stage of infection, processes began to break down and become strongly segmented (Fig.4). At late stages of infection, parts of green processes could be found without connection to the cell body of origin (Fig. 4), and round cells devoid of processes were common (Fig.4). These process dilations were not found in noninfected control cultures. With mCMV MOI of 15, the time course for neuronal degeneration was complete within 2 d, as determined in >20 cultures; with lower MOI (= 1), the time course could be extended.
Fig. 3.

Mouse neurons in vitro: brief infection. A, A montage of a neuron in vitro for 5 d, 2 d after mCMV infection. Processes are labeled. Scale bar, 22 μm. B, A neuron with wide dendrites and a thin axon-like process in box.C, Higher magnification of box inB, showing a bright GFP-expressing axonal process growing on top of the dendrite. D, Axonal arbor 2 d after mCMV introduction.
Fig. 4.
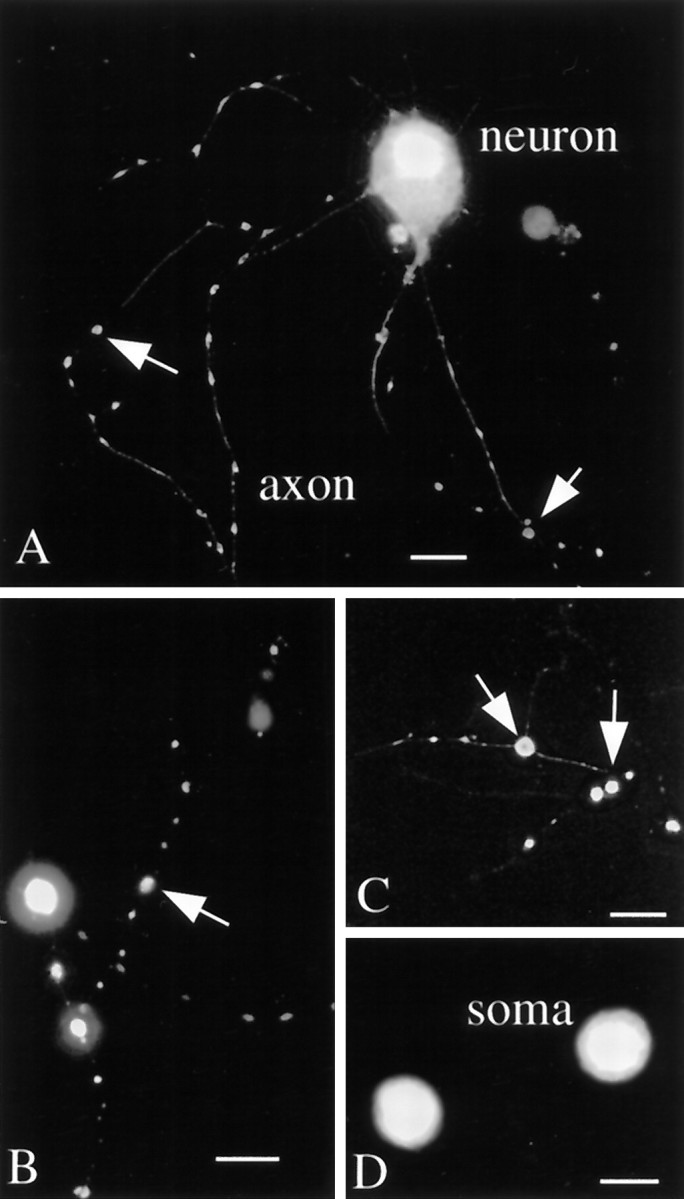
Mouse neurons in vitro: extended infection. A, Four days after infection axons show a greater degree of beading than seen in control cultures.B, C, Some axons in late stages of degeneration show only a few poorly connected swellings that are fluorescent. Scale bar, 10 μm. D, Two somata are seen with unusual round shapes and are devoid of processes 5 d after infection. Scale bar, 12 μm.
During later stages of infection, some groups of cells appeared to fuse together and had the appearance of giant multinucleated cells. These giant “cells” had diameters from 40 to 60 μm, with large ones approaching 200 μm. The largest of these appeared to be composed of >30 cells or nuclei (Fig. 5C,D). These cells showed continuous strong green fluorescence, suggesting that the outer membrane was intact. No giant cells were found that were not green, underlining the importance of viral infection and subsequent GFP expression. At later stages of infection, and after cell lysis, GFP was liberated into the culture medium, and the remaining cellular debris showed only faint green fluorescence. Giant multinucleated cells were not found in any control cultures. In culture dishes that contained giant cells, other groups of cells were found where the individual cells were not contained with a single membrane and therefore were not considered giant cells (Fig.5A,B). Parallel detection of giant cells after CMV infection in humans has been reported (Belec et al., 1990; Horn et al., 1992). The presence of giant cells suggests that CMV mediated fusion of the outer plasma membranes of different cells.
Fig. 5.

Giant cells after mCMV infection. DIC images are shown on the left, and on the right are shown the same field with fluorescent microscopy for detecting GFP.A, B, Three days after infection of a mixed neuronal–glial culture, a small group of cells are tightly packed together but do not show characteristics of a giant cell.C, D, A giant cell appears to be composed of many other cells fused together and is seen with a few cells attached on the outside to the left. InC, the DIC image shows a relatively smooth outer membrane (arrows). Within the giant cell, a heterogeneous GFP-mediated fluorescence is seen, suggesting that there is not total cytoplasmic continuity at this point. Scale bar, 16 μm.
Viral replication
To assess viral replication in neurons and glia, tissue culture supernatant was taken at intervals after initial infection of brain cell cultures and complete washout of mCMV. The relative density of viral production was assessed by incubation of the extracted media with a second set of cultures containing NIH 3T3 fibroblasts, which are a fully permissive cell line for plaque assay and virus propagation. GFP-labeled 3T3 cells were then counted after 18 hr. 3T3 cells were used here because they are a common permissive host cell for raising CMV. The number of green 3T3 cells incubated with virus for a fixed period (18 hr) increased with time (Fig.6). In the initial test 18 hr after virus introduction into brain cell cultures, little evidence of virus was found in the brain culture supernatant. As the infection proceeded, the relative number of viral progeny that were produced increased dramatically, as deduced from the proportion of 3T3 cells that became green. Eight circular fields of 500 μm diameter were counted per 3T3-containing coverslips, which had a total area of 113 × 106μm2.
Fig. 6.

3T3 test cultures used for viral replication test.A, DIC image of NIH 3T3 culture incubated with tissue culture supernatant from brain cultures that had been infected for 18 hr. Arrow identifies a single cell. B, Same field as in A, showing a single fluorescent 3T3 cell (arrow). C, In parallel cultures, DIC imaging shows a group of 3T3 cells, one identified with anarrow to facilitate recognition in DIC and fluorescence.D, With fluorescent microscopy of the same field, most of the cells express GFP-mediated fluorescence. Tissue culture supernatant was taken from brain cultures 42 hr after mCMV infection. In both sets of experiments, 3T3 cells were examined 18 hr after their initial incubation with brain culture supernatant. Scale bar, 25 μm.
Viral replication of two GFP-expressing mCMVs (RM4503 and MC.55) was compared in rat and mouse brain cultures (Fig.7). When a three-way ANOVA was used to test for statistical differences between conditions (virus × brain species × survival after virus infection), a significant difference was found (p < 0.001). To make more discrete comparisons, a Tukey test was used. This revealed that each of the viruses used (RM4503 and MC.55) showed an increase in number over time (p < 0.05), with a slow initial rise time, followed by substantial increases in virus. Although the rate of replication was substantially lower in rat cells than in mouse cells (p < 0.05), viral replication was observed in rat brain cultures infected with the MC.55. However, little GFP was expressed in 3T3 cells incubated with tissue culture supernatant from rat cultures infected with a second virus, RM4503. This may be attributable to strain differences between RM4503 and MC.55. It could also be attributable in part to a lower general level of fluorescence found with RM4503, reducing detection in infected cells, or to a lower initial concentration of virus.
Fig. 7.

mCMV replication enhanced in mouse brain cultures.A, Using MC.55 that was engineered to express GFP under control of elongation factor 1 promoter, viral replication in brain cell cultures was studied by harvesting culture supernatant and determining the number of 3T3 cells that showed GFP expression. Mouse brain culture supernatant generated strong GFP label in an increasing number of 3T3 cells, to the point that all 3T3 cells in culture were labeled with medium from cultures 3 d after mCMV infection. Rat cultures showed a low level of viral replication. Forty-eight thousand 3T3 cells were plated per coverslip; the plateau between the last two mouse points was not attributable to a decrease in viral replication but rather to the fact that the maximum number of test 3T3 cells (48,000) had turned green. B, Parallel experiments were performed with the mCMV under control of the human CMV promoter (RM4503). Replication was found in mouse brain cultures but at a lower rate than found with MC.55. With this viral strain, replication in rat brain cultures as deduced by GFP expression in 3T3 cells was minimal.
The cultures described above contained a mixture of glia and neurons. To examine viral proliferation in more selective cultures, glial-enriched cultures were compared with cultures enriched in cerebellar granule cell neurons (Fig. 8), each growing at roughly 75% confluency, and tested with neuron- and astrocyte-specific antisera, L1 and GFAP, respectively. After inoculation with MC.55 (106 pfu per dish), the glia cultures showed a more rapid cell death than occurred in the neuronal cultures (Fig. 9). Analysis of virus replication showed that culture supernatant had low levels of virus during the first 18 hr, but then began to rapidly increase. By 42 hr, sufficient virus was produced to infect almost all of the 3T3 test cells. By 54 hr, approximately half of the glial cells showed signs of lethal infection, determined by cell swelling, detachment, and loss of cell number. At that time, most of the cells began to detach from the substrate, some even retaining high levels of GFP expression, suggesting that the plasma membrane was still intact. Because less than half of the astrocytes remained after 54 hr, samples were not tested further for release of virus progeny.
Fig. 8.
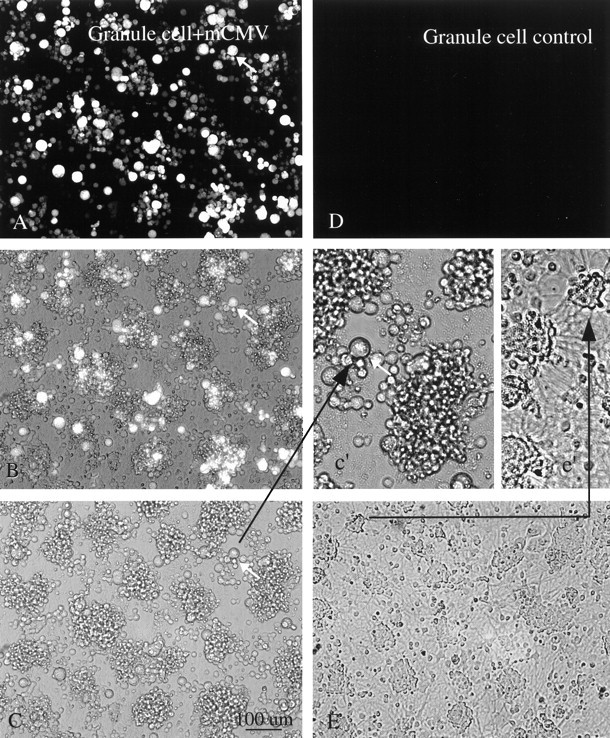
Cerebellar granule neurons. A, Four days after mCMV (MC.55) infection of a culture enriched with granule cells, many of the cells show bright GFP fluorescence. The same cell is indicated by an arrow in A–c'to facilitate recognition. B, Same field as inA but with partial fluorescence, partial phase contrast. Some cells seen in phase contrast are not fluorescent.C, Same field as in A but only with phase contrast. Scale bar, 100 μm. A higher magnification ofC is shown in c' (arrow). Neurites are not found in these infected neurons. D, Control granule cell culture not infected with mCMV shows no fluorescence. E, Phase-contrast photomicrograph of culture 2 d after infection. Neurites are commonly found spreading out from groups of neurons, shown in higher magnification ine'.
Fig. 9.

mCMV replication in cultures enriched in astrocytes or neurons. Cultures enriched in cerebellar granule cells (neurons, ●) or astrocytes (○) were infected with MC.55. Viral replication was more rapid in the cultures enriched in astrocytes. Neuronal cultures also showed viral replication, but at a slower rate. Forty-eight thousand 3T3 cells were plated per coverslip.
In contrast, the neuronal-enriched cultures showed a substantial delay of reporter gene expression with ∼25% of the neurons showing detectable GFP after 54 hr, a time when all glial cells were infected and more than half the glia were dead. However, with time the number of neurons showing GFP expression increased. After 3 d of CMV infection, both infected and noninfected control cultures of granule cells showed strong neuritic extension. These were primarily composed of long bundles of granule cell axons and characteristically short granule cell dendrites. Interestingly, 5 d after mCMV infection, little evidence of neurites was found in the infected culture, whereas control cultures showed vigorous neuritic outgrowth, suggesting that mCMV caused a profound reduction in neurites (Fig. 8). Cells in the neuronal enriched cultures survived for a longer time than cells in the glial enriched cultures. Analysis of viral replication in 3T3 cells showed that 102 hr (4 d) after initial infection, sufficient virus was produced to turn all 3T3 test cells green. The rate of viral replication was significantly slower in neuronal cultures than found in glial cultures prepared at the same time (ANOVA, p < 0.01).
Viral infection of mouse brain in vivo
mCMV was microinjected at a single site in neonatal or adult mouse brains. After fixation and sectioning, the presence of mCMV was detected by the presence of either fluorescent GFP or x-gal staining in the presence of β-galactosidase. Peroxidase immunostaining with antisera against GFP was also used as a sensitive means of detection. Of nine adult mice injected (5 × 105 pfu, 500 nl), none died in the 1–6 d after the injection. In contrast, of 17 mice injected at P1 or P2, nine (53%) died in the following 4 d. mCMV injections into P3 mice were lethal to 4 (40%) of 10 mice by 6 d after infection.
Many different cell types in the brain showed evidence of viral infection. Robust expression of β-gal and GFP was seen in the meninges (Fig.10A,B) and choroid plexus (Fig. 10F). Some of the ependymal cells lining the third and lateral ventricles also were β-gal or GFP positive (Fig. 10D,E). A recent report has suggested that ventricular ependymal cells may serve a role as neuronal stem cells (Johansson et al., 1999). If CMV causes loss of these cells, this may reduce potential neurogenesis, exacerbating problems in the developing brain. Endothelial cells lining the blood vessels showed indication of viral presence, particularly in the region of the injection (Fig. 10C). In addition, virus-infected endothelial cells were found at a considerable distance from the injection site, suggesting spread of the virus through the vasculature.
Fig. 10.
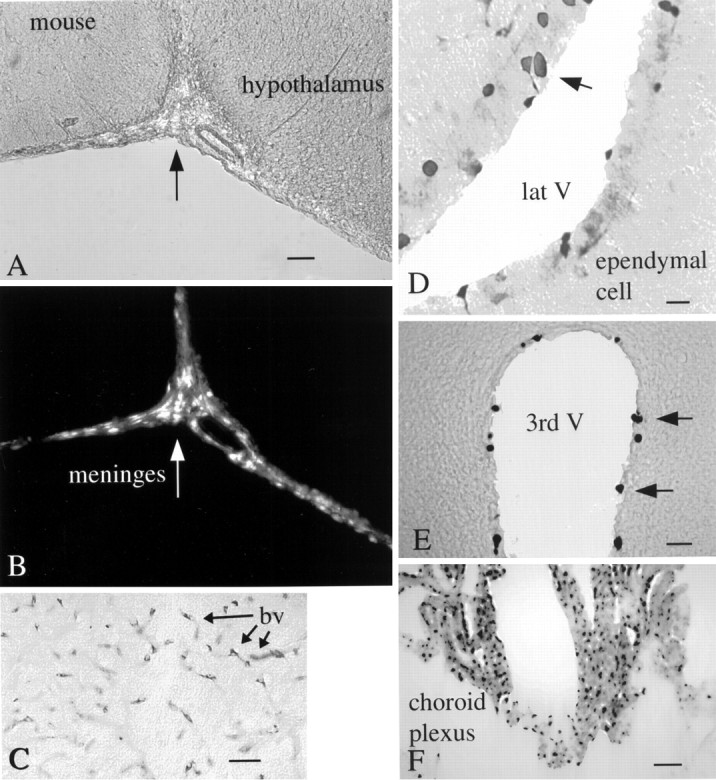
Intracerebral injections into mouse brain. mCMV injections were made into the lateral hypothalamus, with the syringe needle passing through and releasing virus into the ventricular system.A, The ventral region of the brain that includes the lateral hypothalamus (right of arrow). Scale bar, 65 μm. B, Same field as inA; shows GFP labeling of the meninges (arrow). C, Cells surrounding the blood vessels (arrows, bv) of the mammillary bodies after nearby injection of mCMV; the GFP label was stained with immunoperoxidase. D, Ependymal cells (arrows) of the lateral ventricle (lat V) are labeled (arrow) after immunoperoxidase staining of GFP. E, Some of the ependymal cells of the third ventricle (3rd V) show viral infection after x-gal staining for β-gal.F, Many cells of the choroid plexus are labeled after injection of the β-gal expressing mCMV.
In older mice, in the third ventricle, infected cells included tanycytes, long radial glia that are present in both developing and mature brain with the cell body in the wall of the ventricle. In developing mice, dramatic expression of GFP was found in radial glia in many brain areas, particularly near a site of injection. Radial glia serve as important cellular guides for neuronal migration during early development (Rakic, 1978). In the cerebral cortex, label was found in cell bodies of the radial glia in the deeper regions of the cortex and in the long processes that stretched outward, dividing into small endfeet at the pial surface of the cortex surface (Fig.11A,B). Some infected radial glia showed signs of degeneration, characterized by abnormal swelling along the long glial process in both the inner (Fig.11C) and outer (Fig. 11D) cortex. These process dilations appear similar to process degeneration found in infected cultures. Although neurons and astrocytes greatly outnumber radial glial in general numbers in normal mice, in some developing mice (<5 d old) these radial glia accounted for most of the cells infected by mCMV in affected areas of the cortex.
Fig. 11.
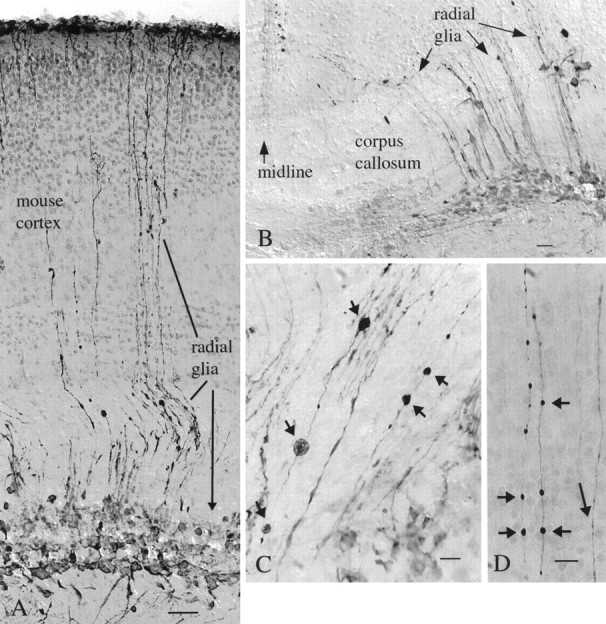
Preferential infection of radial glia in developing mouse cortex. A, In mouse cortex, radial glia appear to be the primary cells infected after mCMV injections into the P2 mouse. Long processes travel from the cell body layer (arrow) and ascend to the outer cortical surface. The syringe needle passed through the cortex and into the hippocampus, and cells were labeled in both places. Scale bar, 50 μm.B, In the same brain, near the midline, a number of processes cross the corpus callosum and continue through the developing gray matter. Scale bar, 15 μm. C, D, Processes of mCMV-infected radial glia in the deep cortex (C) and outer cortex (D) show dilations (small arrows) suggestive of pathology. Some dilations reach almost 10 μm in diameter. Other processes (long arrow) show no dilations. Scale bars: 20 μm. All micrographs here are after peroxidase immunostaining of GFP.
Some intracerebral injections of virus were made into different brain regions, including cortex, hippocampus, striatum, and hypothalamus. Particularly striking was the strong presence of virus in glial cells with the appearance of astroglia around the injection site. In addition, neurons were infected with virus; these cells had dendritic and axonal processes typical of neurons (Fig.12). A large number of cases were found in many parts of the brain where neurons appeared to be the primary cell infected. Isolated neurons were found in the cortex (Fig.12A), hypothalamus (Fig. 12B), and hippocampus (Fig. 12C). Of potential importance, some GFP-expressing neurons were found at considerable distances from the injection site. This could have been caused in part by diffusion of the virus at the time of injection. However, in a number of cases, a random distribution pattern of the virus in distal regions of the brain was not found. Instead, a GFP-filled process could be followed from the labeled cell into the zone of brain damage elicited by penetration of the injecting microsyringe needle. This suggests that the virus may be transported within the neuron process back to the cell body where the virus entered the nucleus to express GFP. Many labeled axons were found far from the injection site (Fig.12D–F), including in the white matter of the corpus callosum (Fig. 12E).
Fig. 12.
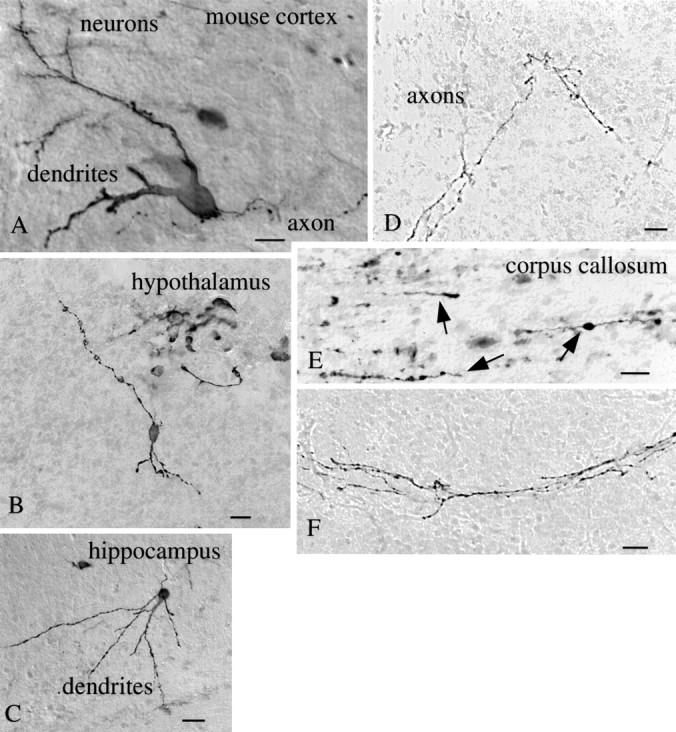
Mouse neuronal infection by mCMV. Neurons in the mouse brain were infected with mCMV as shown after immunoperoxidase staining of GFP in the cerebral cortex (A, scale bar, 12 μm), hypothalamus (B, scale bar, 20 μm), and hippocampus (C, scale bar, 25 μm). Axons show GFP label with no nearby neuronal cell body labeling, as shown inD (scale bar, 8 μm), E (scale bar, 5 μm), and F (scale bar, 8 μm). In Ethe GFP-expressing axons run parallel to the axis of many other axons running in the corpus callosum.
Similar to neurons in culture, neurons in vivo that showed GFP expression several days after viral infection showed severe symmetrical dilations and constrictions along long axons. Some of the dilations reached 5 or 6 μm in diameter; such large dilations were not seen in noninfected control axons. This type of dilation was also not found in control brain tissue labeled with DiI or horseradish peroxidase injections, suggesting that it was not an artifact of fixation or histological processing.
In developing mouse brains, intracerebral injections of mCMV can cause strong labeling of hippocampus. As shown in Figure13A, cells surrounding the neonatal hippocampus show strong viral infection. Within the hippocampus, neurons and their processes are infected with the virus (Fig. 13B).
Fig. 13.
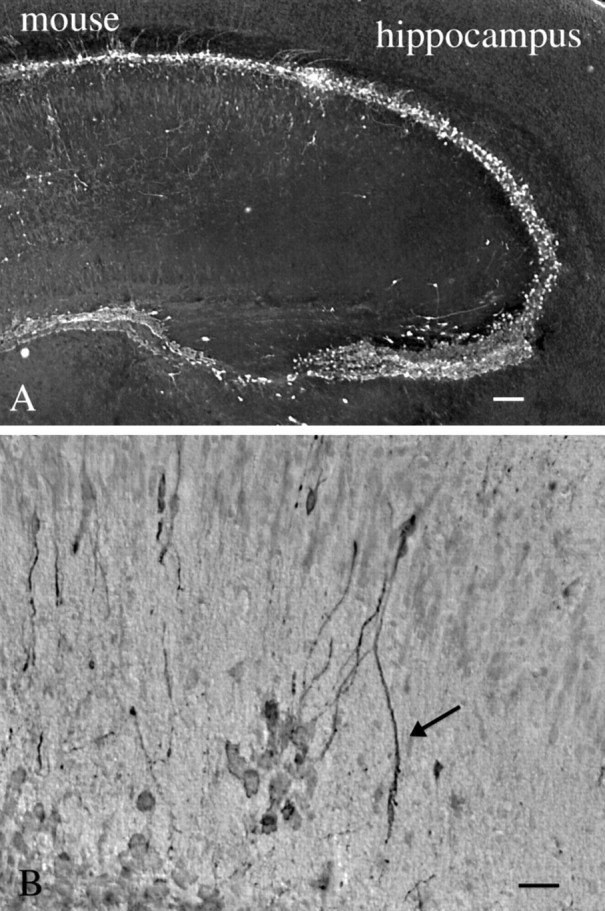
mCMV in neonatal mouse hippocampus.A, After intracerebral injection into the P2 mouse, cells surrounding the hippocampus show strong evidence of infection 3 d later. Scale bar, 45 μm. B, Higher magnification of the same brain shows labeling of some dendritic arbors (arrow) ventral to the pyramidal cell layer. Scale bar, 20 μm.
We found a number of cells that were clearly infected with mCMV, detected with expression of the viral GFP, but the infected cell showed little evidence of cytomegaly, the characteristic pathological determinant of CMV infection. This was true both in cultured cells and in histological sections from the brain. Thus some of the earlier reports suggesting a selective lack of infection of one type or another in the brain may simply be due to the absence of cytomegaly and the subsequent inability to detect infected cells. Histopathological detection of cytomegaly in tissues is insensitive but in the past has been used widely to search for evidence of CMV infection. The use of cytopathology as a marker for CMV infection at the cellular level is further complicated by the finding that induction of apoptosis in noninfected neurons can be generated by a secondary signal from CMV-infected cells in mice (Kosugi et al., 1998). The use of reporter genes as described here appears to be a more sensitive mode of assessment of mCMV presence than the cytomegaly commonly associated with CMV. Unlike approaches based on histological processing that may also be sensitive, for instance in situ hybridization (Myerson et al., 1984; Slobedman and Mocarski, 1999), our approach, particularly with GFP, does not require fixation or histochemical treatment.
Gene transfer to rat and human
The experiments with mouse brain cells suggested that mCMV might be a potentially useful vector for gene transfer into mouse brain cells. Given the potential toxicity of the virus to mouse cells, the virus would probably not be ideal for any beneficial action. However, because replication of CMVs is to a large degree species specific, we sought to determine whether CMV could be used for gene transfer to other species. To this end, we used rat and human cells in vitro and rat brain in vivo.
Rat brain
After mCMV infection, cultures of rat brain showed GFP expression in both neurons and astrocytes. Microglia, identified by immunolabeling with an antibody against rat CD11b/c (Cedarlane Labs, Hornby, Ontario, Canada) that binds to brain microglia and macrophages (Robinson et al., 1986), also showed GFP expression. There was also a low level of viral replication in rat brain cultures, as described earlier.
In an in vivo set of experiments, mCMV was directly injected into rat brains (n = 12). After periods ranging from 1 to 7 d, animals (eight adult rats, four P5 rats) appeared healthy; no rat death was attributed to the mouse CMV. At or near the site of mCMV injection, strongly labeled neurons were found in hypothalamus, hippocampus, striatum, and cerebral cortex (Fig.14A,B,D,E). In the example in Figure14A,B,D, adult striatal neurons showed very strong GFP labeling with little detectable GFP in the surrounding glial cells. Single axons and bundles of long axons could be followed from the striatal area several thousand micrometers toward the midbrain (Fig. 14C), and local axons were abundant around labeled cells. Spiny neurons in the striatum could be identified by the strong labeling in the dendritic spines (Fig.14B). Despite the strong neuronal labeling in the striatum, relatively few cells with the morphology of glial cells could be found. Although the injection site included both cortex and striatum, the level of neuronal labeling in the striatum was dramatically stronger than in the cortex, suggesting some preferential infection of the virus.
Fig. 14.
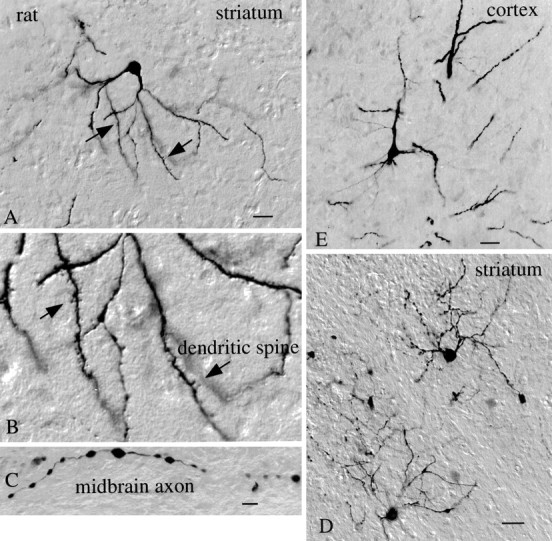
mCMV infects neurons of the rat brain.A, A medium-size spiny cell of the rat striatum is labeled 3 d after nearby mCMV administration and immunoperoxidase staining for GFP. Scale bar, 25 μm. B, Higher magnification of A shows high density of dendritic spines (arrows) on labeled cell. C, Axon in the midbrain, from the same brain asA–D, shows labeling. Scale bar, 8 μm.D, On an adjacent section to A, two more striatal neurons with their dendrites and axons show evidence of mCMV infection. Scale bar, 30 μm. E, Neuron from cerebral cortex was infected with mCMV. Scale bar, 20 μm.
In its role as a vehicle for gene transfer, recombinant mCMV that expresses GFP may prove useful for some aspects of exploring neuronal circuitry and cell structure. This would not be limited to mouse, because we found strong labeling in rat brain and cultured human brain cells, suggesting that mCMV might also work in other species. Previous work with pseudorabies virus (Card et al., 1990, 1991, 1999), herpes simplex virus (Ho et al., 1995; Meier et al., 1998), and lentiviruses based on HIV vectors with CMV promoters (Blomer et al., 1997; Miyoshi et al., 1997) has demonstrated the effectiveness of viral vectors in the brain.
Human brain
Although earlier studies suggested that mCMV did not infect human cells (Kim and Carp, 1971), in our hands human cells cultured from a brain astrocytoma showed intense green labeling after introduction of MC.55 (MOI = 2), indicating robust expression of the transgene under control of the human elongation factor 1a promoter (Fig.15A–C). Most of the human cells that showed infection on the basis of GFP expression showed little evidence of cytomegaly. The longest experiments in this series lasted 1 week, but because human cells continued to show strong fluorescence at this time, it is likely that expression would have continued for a longer period. In contrast, the number of mouse brain cells expressing GFP increased over time, so that after 1 week all mouse cells were dead or green, suggesting viral replication andde novo infection of additional generations of brain cells. In contrast to mouse brain cell cultures, there was no striking increase over time in the number of human brain cells that showed GFP expression, consistent with a lack of replication of the mCMV. Previous work with human CMV has demonstrated its presence in human astrocytes and astrocyte-related tumors (Duclos et al., 1989; Ho et al., 1991;McCarthy et al., 1995). MC.55 (MOI = 2) also infected cultures of a cell line of human neuroblastoma cells (gift of Dr. K. Vives, Yale University). These cells also showed strong levels of GFP expression (Fig. 15D,E).
Fig. 15.
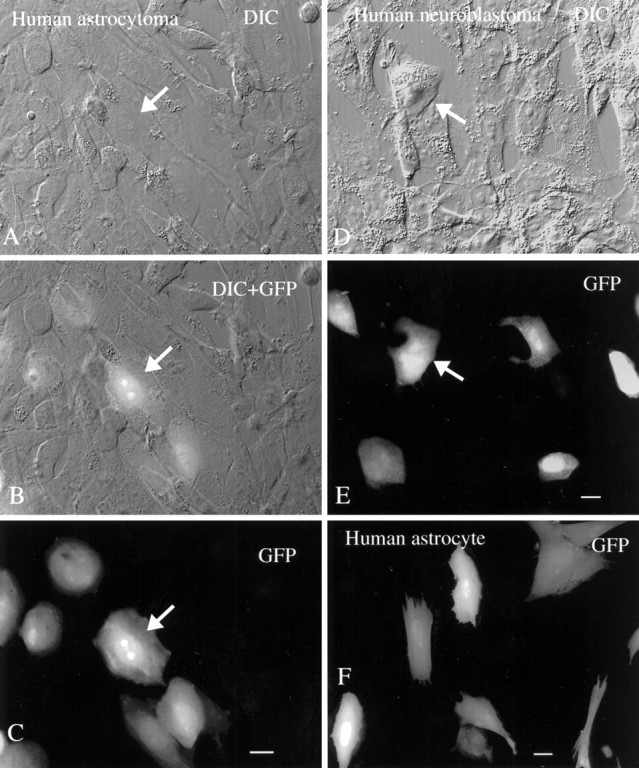
Human brain cells are infected with mouse CMV. A–C, DIC, DIC + GFP, and GFP images of the same microscope field containing glial cells from a human astrocytoma are shown. A subset of cells shows strong GFP-mediated fluorescence. Scale bar, 10 μm. D, E, The same field shows the DIC image (D) and the GFP image (E). A subset of human neuroblastoma cells, 2 d after plating with mCMV, is infected with virus. Scale bar, 10 μm. F, “Normal” glial cells with an astrocyte morphology were cultured from human brain and show GFP expression 30 hr after infection. Scale bar, 10 μm.
Both of the experiments above were performed with human cells derived from a tumor and show abnormally high rates of cell division in vitro. Cells were also cultured from “normal” human cortex, removed surgically to provide access to a more ventral lesion. These cells replicated very slowly and appeared normal in culture. When these cultures were infected with mCMV (MOI = 2), cells with a flat sheet-like morphology typical of astrocytes showed GFP expression indicating viral infection and gene expression. In a typical culture well 2 weeks after low-density culturing of the human cells, and within 30 hr of CMV addition, all 47 cells with the flat sheet-like morphology typical of astrocytes showed GFP expression, indicating viral gene expression (Fig. 15F). At 2 weeks after infection, the longest interval studied, green cells were still found in these cultures.
DISCUSSION
CMV cell preference is relative, not absolute
In the present study we found that all different types of cells in the brain can be infected with mCMV. This includes neurons, astrocytes, ependymal cells lining the ventricles, ventricular tanycytes, radial glia, endothelial cells of the capillaries, ensheathing cells of the olfactory bulb, meninges, microglia, and cells of the choroid plexus. These data support the view that mCMV in the brain is an opportunistic virus, with no single cellular target. However, there was strong evidence that related subsets of cells showed a higher incidence of GFP expression, suggesting some viral preference. An example of this was the high probability of striatal neuron infection coupled with the low incidence of infection in glial cells in the same brain region, and the strong infection of cortical radial glia, discussed below. This could be attributable to differential cell-virus adhesion and cellular uptake, viral replication, or differences in cellular activators of the promoters for the gene reporters used. Some neurotrophic viruses, for instance pseudorabies virus (Card et al., 1990, 1991, 1999) or herpes simplex-1 (Dash et al., 1996), may be selective for subsets of neurons. In the present study we found evidence for infection of various neuronal types from cortex, hippocampus, striatum, and hypothalamus.
In our studies examining viral replication, mouse cultures enriched in either astrocytes or neurons showed evidence of strong mCMV replication. Virus number peaked more rapidly in astrocyte cultures than in neuronal cultures. This could be attributable in part to a greater level of replication in glial cells, although this was minimized by the use of defined medium not including serum and not favorable for astrocyte mitosis. Alternatively, although the cultures showed similar levels of confluency at the time of infection and were infected with equal amounts of virus, the glial cultures received a higher MOI, given the larger size of astrocytes compared with the small granule cell neurons. The larger membrane area of the cell body of astrocytes compared with neurons would also contribute to the increased probability of viral infection. The more rapid expression of GFP in astrocytes than in neurons, coupled with the higher rate of replication in glia, is consistent with the concept that relative differences in viral infection exist.
Strong viral expression was found in radial glia in the developing mouse brain. One result of early CMV infection in human brains is the occurrence of microgyri (Diezel, 1954; Wolf and Cowen, 1972; Ho, 1991), a reduction in the size of cortical gyri. This has been previously attributed to vascular problems arising from CMV infection (Marques-Dias et al., 1984). However, we suggest another mechanism. Radial glia play an important role in serving as a pathway for the migration of neurons that are born in the ventricular zone and migrate outward along the radial glia in the developing rodent and primate brain (Rakic, 1978). Thus our data demonstrating CMV infection in radial glia in the developing brain suggest that after CMV-mediated radial glial degeneration, later outward neuronal migration may be blocked or misdirected. Thus microgyri or other anomalies of organization in CMV-infected human brains may hypothetically result from a selective loss of radial glia and subsequent abnormal neuronal migration. Further corroborating evidence is provided by studies in which mCMV was found to alter cortical neuronal migration in developing mice (Shinmura et al., 1997). Our finding of a high level of infection by mCMV in cells surrounding the hippocampus, with involvement of neurons within the hippocampus, may in some cases underlie hippocampal deficits, including epilepsy (Perez-Jiminez et al., 1998).
CMV may enter the brain in several ways. Our data showing CMV infection of the endothelial cells of the vascular system is consistent with a previous report suggesting that blockade of the vascular system may cause brain deficits (Marques-Dias et al., 1984; Wiley and Nelson, 1988). Another potential site of entry is the olfactory mucosa. Olfactory ensheathing cells that surround the olfactory nerve as it penetrates the brain can be infected with CMV. After intraperitoneal infection with CMV, the virus was detected in cells of the olfactory mucosa (Trgovcich et al., 1998), and intranasal administration leads to viral infection (Mannini and Medearis, 1961). The olfactory mucosa to the olfactory bulb has been suggested as one route of entry into the CNS for a number of viruses, including polio virus, semliki forest virus, and vesicular stomatitis virus (Johnson, 1998). Experimental injection of CMV in the guinea pig (Booss et al., 1989) or mouse (Tsutsui et al., 1995) peritoneal cavity results in brain infection a few days later. The finding of infection of vascular endothelial cells suggests that mCMV could use these cells as a potential means of entering or exiting the neuropil of the brain. CMV-infected lymphocytes may provide a means of facilitating entry into the brain. Another possibility for viral penetration into the brain may be through neuronal axons. Axonal transport is a critical mode of viral translocation for some other virus such as polio, rabies, and herpes simplex-1 (Johnson, 1998). Transport of the virus through processes would serve as a partial explanation for the selective GFP expression in striatal neurons. It is unlikely that the promoters used expressed reporter genes in only one cell type, because in culture all brain cells showed GFP expression after virus addition to the culture medium.
Both in vivo and in vitro GFP-labeled axons showed severe dilations, and in culture, segments of labeled axons that had separated from the parent cell body. The lack of processes in later stages of infection of mouse neurons probably results from a combination of retraction of short processes and virally induced deterioration of the longer processes. Whether the virus may exert a direct and local effect on process deterioration or whether this process degeneration is secondary to infection in the cell body remains to be determined.
mCMV-mediated gene transfer
Although previous reports have suggested that mCMV does not infect human cells (Kim and Carp, 1971), by using the more sensitive recombinant mCMV described here we found robust GFP expression in human glial cells after infection. No obvious signs of cell death were found in mCMV-infected human cultures, suggesting that the mouse CMV may be a good candidate for further studies relating to use as a vector for gene introduction in human brain cells. A number of viruses, including herpes simplex-1, adenovirus, and adeno-associated virus, have been used for gene transfer in the CNS (Ho et al., 1995; Bartlett et al., 1998; Meier et al., 1998). Some viruses, for instance the Moloney murine leukemia retrovirus, may work well for gene transfer to dividing cells but work poorly in postmitotic neurons (Sena-Esteves et al., 1996). Although CMV has often provided its major immediate early gene promoter as a strong promoter of choice for expression of foreign genes in the brain, it has generally been adapted to other viruses such as adeno-associated virus (Lowenstein et al., 1996; Bartlett et al., 1998), and its expression from recombinant CMV has not been previously evaluated. The large size of the CMV genome can be an advantage or disadvantage. On the positive side, large inserts can be engineered into mCMV, unlike some other viruses such as adeno-associated virus, which has a size restriction of a few kilobases. On the other hand, the CMV genome codes for >200 proteins the expression of which might have consequences for the infected cell. Molecular deletions can remove viral genes that may prove deleterious to the use of these viruses as vectors (Saederup et al., 1999). Future experimentation using replication competent or defective mCMV or hCMV should examine these possibilities.
The fact that mCMV generated expression of GFP in neuronal cell bodies, dendrites, and axons suggests that these genetically engineered viruses may be useful as a tool for labeling neurons and their pathways. Labeled axons were found that extended thousands of micrometers away from perikarya near a zone of mCMV administration, indicating that GFP diffusion or transport is considerable. This is consistent with our previous work with transfection with plasmids coding for GFP under the regulation of the human major immediate early CMV promoter (van den Pol et al., 1998) or in transgenic mice expressing GFP under the control of a human CMV promoter (van den Pol and Ghosh, 1998). Different sequences of the CMV promoter may result in reporter gene expression in different neurons of the CNS (Koedood et al., 1995; Baskar et al., 1996; Fritschy et al., 1996; van den Pol and Ghosh, 1998); differential activation of CMV promoters may serve as a partial explanation for why the virus may infect different regions of the brain and cause various neurological problems. With transgenic mice expressing reporter genes controlled by different regions of the immediate early promoter, neurons were commonly labeled, and labeled glia were rare; in contrast, in the present experiments we found that the mCMV retained the ability to express foreign genes in mouse, rat, and human glia and neurons.
Footnotes
This work was supported by National Institutes of Health Grants NS10174, NS31573, AI30363, NS37788, and NS34887, and the National Science Foundation. We thank Y. Yang, and P. Gip for excellent technical assistance, and Drs. H. C. Heller, R. G. Phillips, J. Peipmeier, C. Duncan, and Ken Vives for suggestions, help, or cells used in these experiments.
Correspondence should be addressed to Anthony N. van den Pol, Department of Neurosurgery, 333 Cedar Street, Yale University Medical School, New Haven, CT 06520. E-mail:anthony.vandenpol@yale.edu.
REFERENCES
- 1.Alford CA, Britt WJ. Cytomegalovirus. In: Fields BN, Knipe DM, Howley PM, editors. Fields virology. Lippincott-Raven; New York: 1996. pp. 2493–2534. [Google Scholar]
- 2.Arribas JR, Clifford DB, Fichtenbaum CJ, Commins DL, Powderly WG, Storch GA. Level of cytomegalovirus (CMV) DNA in cerebrospinal fluid of subjects with AIDS and CMV infection of the central nervous system. J Infect Dis. 1995;172:527–531. doi: 10.1093/infdis/172.2.527. [DOI] [PubMed] [Google Scholar]
- 3.Bale JF, Jr, Bray PF, Bell WE. Neuroradiographic abnormalities in congenital cytomegalovirus infection. Pediat Neurol. 1985;1:42–47. doi: 10.1016/0887-8994(85)90008-6. [DOI] [PubMed] [Google Scholar]
- 4.Bartlett JS, Samulski RJ, McCown TJ. Selective and rapid uptake of adeno-associated virus type 2 in brain. Hum Gene Ther. 1998;9:1181–1186. doi: 10.1089/hum.1998.9.8-1181. [DOI] [PubMed] [Google Scholar]
- 5.Baskar JF, Smith PP, Nilaver G, Jupp RA, Hoffmann S, Peffer NJ, Tenney DJ, Colberg-Poley AM, Ghazal P, Nelson JA. The enhancer domain of the human cytomegalovirus major immediate-early promoter determines cell type-specific expression in transgenic mice. J Virol. 1996;70:3207–3214. doi: 10.1128/jvi.70.5.3207-3214.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belec L, Gray F, Mikol J, Scaravelli F, Mhiri C, Sobel A, Poirier J. Cytomegalovirus (CMV) encephalomyeloradiculitis and human immunodeficiency virus (HIV) encephalitis: presence of HIV and CMV co-infected multinucleated giant cells. Acta Neuropathol. 1990;81:99–104. doi: 10.1007/BF00662645. [DOI] [PubMed] [Google Scholar]
- 7.Blomer U, Naldini L, Kafri T, Trono D, Verma IM, Gage FH. Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J Virol. 1997;71:6641–6649. doi: 10.1128/jvi.71.9.6641-6649.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Booss J, Dann PR, Griffith BP, Kim JH. Glial nodule encephalitis in the guinea pig: serial observations following cytomegalovirus infection. Acta Neuropathol. 1988;75:465–473. doi: 10.1007/BF00687133. [DOI] [PubMed] [Google Scholar]
- 9.Booss J, Winkler SR, Griffith BP, Kim JH. Viremia and glial nodule encephalitis after experimental systemic cytomegalovirus infection. Lab Invest. 1989;61:644–649. [PubMed] [Google Scholar]
- 10.Bray PF, Bale JF, Anderson RE, Kern ER. Progressive neurological disease associated with chronic cytomegalovirus infection. Ann Neurol. 1981;9:499–502. doi: 10.1002/ana.410090513. [DOI] [PubMed] [Google Scholar]
- 11.Card JP, Rinaman L, Schwaber JS, Miselis RR, Whealy ME, Robbins AK, Enquist LW. Neurotropic properties of pseudorabies virus: uptake and transneuronal passage in the rat central nervous system. J Neurosci. 1990;10:1974–1994. doi: 10.1523/JNEUROSCI.10-06-01974.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Card JP, Whealy ME, Robbins AK, Moore RY, Enquist LW. Two alpha-herpesvirus strains are transported differentially in the rodent visual system. Neuron. 1991;6:957–969. doi: 10.1016/0896-6273(91)90236-s. [DOI] [PubMed] [Google Scholar]
- 13.Card JP, Enquist LW, Moore RY. Neuroinvasiveness of pseudorabies virus injected intracerebrally is dependent on viral concentration and terminal field density. J Comp Neurol. 1999;407:438–452. doi: 10.1002/(sici)1096-9861(19990510)407:3<438::aid-cne11>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 14.Cardin RD, Abenes GB, Stoddart CA, Mocarski ES. Murine cytomegalovirus IE2, an activator of gene expression, is dispensable for growth and latency in mice. Virology. 1995;209:236–241. doi: 10.1006/viro.1995.1249. [DOI] [PubMed] [Google Scholar]
- 15.Chimelli L, Rosemberg S, Hahn MD, Lopes MB, Netto MB. Pathology of the central nervous system in patients infected with the human immunodeficiency virus (HIV): a report of 252 autopsy cases from Brazil. Neuropathol Appl Neurobiol. 1992;18:477–488. doi: 10.1111/j.1365-2990.1992.tb00815.x. [DOI] [PubMed] [Google Scholar]
- 16.Dash R, Lawrence M, Ho D, Sapolsky R. A herpes simplex virus vector overexpressing the glucose transporter gene protects the rat dentate gyrus from an antimetabolite toxin. Exp Neurol. 1996;137:43–48. doi: 10.1006/exnr.1996.0005. [DOI] [PubMed] [Google Scholar]
- 17.Diezel PB. Mikorgyrie infolge cerebraler Speicheldrusenvirus Infektion in Rahmen einer geneialisierten Cytomegalie bei einen Saugling. Zugleich ein Beitrag zur Theorie der Windungsbildung. Virchows Arch. 1954;325:109–130. doi: 10.1007/BF00954506. [DOI] [PubMed] [Google Scholar]
- 18.d'Arminio Monforte A, Vago L, Lazzarin A, Boldorini R, Bini T, Guzzetti S, Antinori S, Moroni M, Costanzi J. AIDS-defining diseases in 250 HIV-infected patients: a comparative study of clinical and autopsy diagnoses. AIDS. 1992;6:1159–1164. doi: 10.1097/00002030-199210000-00015. [DOI] [PubMed] [Google Scholar]
- 19.Duclos H, Elfassi E, Michelson S, Arenzana Seisdedos F, Hazan U, Munier A, Virelizier JL. Cytomegalovirus infection and trans-activation of HIV-1 and HIV-2 LTRs in human astrocytoma cells. AIDS Res Hum Retroviruses. 1989;5:217–224. doi: 10.1089/aid.1989.5.217. [DOI] [PubMed] [Google Scholar]
- 20.Fiala M, Singer EJ, Graves MC, Tourtellotte WW, Stewart JA, Schable CA, Rhodes RH, Vinters HV. AIDS dementia complex complicated by cytomegalovirus encephalopathy. J Neurol. 1993;240:223–231. doi: 10.1007/BF00818709. [DOI] [PubMed] [Google Scholar]
- 21.Fritschy JM, Brandner S, Aguzzi A, Koedood M, Luscher B, Mitchell PJ. Brain cell type specificity and gliosis-induced activation of the human cytomegalovirus immediate-early promoter in transgenic mice. J Neurosci. 1996;16:2275–2282. doi: 10.1523/JNEUROSCI.16-07-02275.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hicks T, Fowler K, Richardson M, Dahle A, Adams L, Pass R. Congenital cytomegalovirus infection and neonatal auditory screening. J Pediatr. 1993;123:779–782. doi: 10.1016/s0022-3476(05)80859-5. [DOI] [PubMed] [Google Scholar]
- 23.Ho DY, Fink SL, Lawrence MS, Meier TJ, Saydam TC, Dash R, Sapolsky RM. Herpes simplex virus vector system: analysis of its in vivo and in vitro cytopathic effects. J Neurosci Methods. 1995;57:205–215. doi: 10.1016/0165-0270(94)00150-f. [DOI] [PubMed] [Google Scholar]
- 24.Ho KL, Gottlieb C, Zarbo RJ. Cytomegalovirus infection of cerebral astrocytoma in an AIDS patient. Clin Neuropathol. 1991;10:127–133. [PubMed] [Google Scholar]
- 25.Ho M. Cytomegalovirus: biology and infection, Ed 2. Plenum; New York: 1991. [Google Scholar]
- 26.Horn M, Schlote W, Herrman G, Gungor T, Jacobi G. Immunocytochemical characterization of cytomegalovirus (CMV) infected giant cells in perinatal acquired human immunodeficiency virus (HIV) infection. Acta Histochem [Suppl] 1992;42:115–122. [PubMed] [Google Scholar]
- 27.Johansson CB, Momma S, Clarke DL, Risling M, Lendahl U, Frisen J. Identification of a neural stem cell in the adult mammalian central nervous system. Cell. 1999;96:25–34. doi: 10.1016/s0092-8674(00)80956-3. [DOI] [PubMed] [Google Scholar]
- 28.Johnson RT. Viral infections of the nervous system. Lippincott-Raven; New York: 1998. [Google Scholar]
- 29.Kalayjian RC, Cohen ML, Bonomo RA, Flanigan TP. Cytomegalovirus ventriculoencephalitis in AIDS. A syndrome with distinct clinical and pathologic features. Medicine (Baltimore) 1993;72:67–77. [PubMed] [Google Scholar]
- 30.Kim KS, Carp RI. Growth of cytomegalovirus in various cell lines. J Virol. 1971;7:720–725. doi: 10.1128/jvi.7.6.720-725.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koedood M, Fichtel A, Meier P, Mitchell PJ. Human cytomegalovirus (CMV) immediate-early enhancer/promoter specificity during embyrogenesis defines target tissue of congenital HCMV infection. J Virol. 1995;69:2194–2207. doi: 10.1128/jvi.69.4.2194-2207.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kosugi I, Shirmura Y, Li RY, Aiba-Masago S, Baba S, Miura K, Tsutsui Y. Murine cytomegalovirus induces apoptosis in non-infected cells of the developing mouse brain and blocks apoptosis in primary neuronal culture. Acta Neuropathol. 1998;96:239–247. doi: 10.1007/s004010050890. [DOI] [PubMed] [Google Scholar]
- 33.Liljelund P, Ghosh P, van den Pol AN. Expression of the neural axon adhesion molecule L1 in the developing and adult rat brain. J Biol Chem. 1994;269:32886–32895. [PubMed] [Google Scholar]
- 34.Lowenstein PR, Wilkinson GWG, Castro MG, Shering AF, Fooks AR, Bain D. Non-neurotropic adenovirus: a vector for gene transfer to the brain and possible gene therapy of neurological disorders. In: Latchman D, editor. Genetic manipulation of the nervous system. Academic Press; New York: 1996. pp. 11–39. [Google Scholar]
- 35.Manning WC, Stoddart CA, Lagenaur LA, Abenes GB, Mocarski ES. Cytomegalovirus determinant of replication in salivary glands. J Virol. 1992;66:3794–3802. doi: 10.1128/jvi.66.6.3794-3802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mannini A, Medearis DN. Mouse salivary gland virus infections. Am J Hygiene. 1961;73:329–343. doi: 10.1093/oxfordjournals.aje.a120192. [DOI] [PubMed] [Google Scholar]
- 37.Marques-Dias MJ, Harmant-van Rijckevorsel G, Landrieu P, Lyon G. Prenatal cytomegalovirus disease and cerebral microgyria: evidence for perfusion failure, not disturbance of histogenesis, as the major cause of fetal cytomegalovirus encephalopathy. Neuropediatrics. 1984;15:18–24. doi: 10.1055/s-2008-1052334. [DOI] [PubMed] [Google Scholar]
- 38.McCarthy M, Wood C, Fedoseyeva L, Whittemore SR. Media components influence viral gene expression assays in human fetal astrocyte cultures. J Neurovirol. 1995;1:275–285. doi: 10.3109/13550289509114024. [DOI] [PubMed] [Google Scholar]
- 39.Meier TJ, Ho DY, Park TS, Sapolsky RM. Gene transfer of calbindin D28k cDNA via herpes simplex virus amplicon vector decreases cytoplasmic calcium ion response and enhances neuronal survival following glutamatergic challenge but not following cyanide. J Neurochem. 1998;71:1013–1023. doi: 10.1046/j.1471-4159.1998.71031013.x. [DOI] [PubMed] [Google Scholar]
- 40.Miyoshi H, Takahashi M, Gage FH, Verma IM. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc Natl Acad Sci USA. 1997;94:10319–10323. doi: 10.1073/pnas.94.19.10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mocarski E. Cytomegaloviruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fields virology. Lippincott-Raven; New York: 1996. pp. 2447–2492. [Google Scholar]
- 42.Myerson D, Hackman RC, Nelson JA, Ward DC, McDougall JK. Widespread presence of histologically occult cytomegalovirus. Hum Pathol. 1984;15:430–439. doi: 10.1016/s0046-8177(84)80076-3. [DOI] [PubMed] [Google Scholar]
- 43.Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex: II. Neuropathology. Ann Neurol. 1986;19:525–535. doi: 10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- 44.Nelson JA, Reynolds-Kohler C, Oldstone MBA, Wiley CA. HIV and HCMV coinfect brain cells in patients with AIDS. Virology. 1988;165:286–290. doi: 10.1016/0042-6822(88)90685-x. [DOI] [PubMed] [Google Scholar]
- 45.Osborn JE. Cytomegalovirus and other herpesviruses. In: Foster HL, Small JD, Fox JG, editors. The mouse in biomedical research. II. Diseases. Academic; New York: 1982. pp. 267–292. [Google Scholar]
- 46.Perez-Jiminez A, Colamaria V, Franco A, Grimau-Merino R, Darra R, Fontana E, Zullini E, Beltramello A, Dalla-Bernardina B. Epilepsy and disorders of cortical development in children with congenital cytomegalovirus infection. Rev Neurol. 1998;26:42–49. [PubMed] [Google Scholar]
- 47.Pulliam L. Cytomegalovirus preferentially infects a monocyte derived macrophage/microglial cell in human brain cultures: neuropathology differs between strains. J Neuropathol Exp Neurol. 1991;50:432–440. doi: 10.1097/00005072-199107000-00004. [DOI] [PubMed] [Google Scholar]
- 48.Rakic P. Neuronal migration and contact guidance in the primate telencephalon. Postgrad Med J [Suppl 54] 1978;1:25–40. [PubMed] [Google Scholar]
- 49.Rawlinson WD, Farrell HE, Barrell BG. Analysis of the complete DNA sequence of murine cytomegalovirus. J Virol. 1996;12:8833–8849. doi: 10.1128/jvi.70.12.8833-8849.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramon-Cueto A, Valverde F. Olfactory bulb ensheathing glia: a unique cell type with axonal growth-promoting properties. Glia. 1995;14:163–173. doi: 10.1002/glia.440140302. [DOI] [PubMed] [Google Scholar]
- 51.Robinson AP, White TM, Mason DW. Macrophage heterogeneity in the rat as delineated by two monoclonal antibodies MRC OX-41 and MRC OX-42, the latter recognizing complement receptor type 3. Immunology. 1986;57:239–247. [PMC free article] [PubMed] [Google Scholar]
- 52.Saederup N, Lin YC, Dairaghi DJ, Schall TJ, Mocarski ES. Cytomegalovirus-encoded beta chemokine promotes monocyte-associated viremia in the host. Proc Natl Acad Sci USA. 1999;96:10881–10886. doi: 10.1073/pnas.96.19.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sena-Esteves M, Aghi M, Pechan PA, Kaye EM, Breakefield XO. Gene delivery to the nervous system using retroviral vectors. In: Latchman D, editor. Genetic manipulation of the nervous system. Academic Press; New York: 1996. pp. 149–180. [Google Scholar]
- 54.Shinmura Y, Kosugi I, Aiba-Masago S, Baba S, Yong LR, Tsutsui Y. Disordered migration and loss of virus-infected neuronal cells in developing mouse brains infected with murine cytomegalovirus. Acta Neuropathol (Berl) 1997;93:551–557. doi: 10.1007/s004010050651. [DOI] [PubMed] [Google Scholar]
- 55.Slobedman B, Mocarski ES. Quantitative analysis of latent human cytomegalovirus. J Virol. 1999;73:4806–4812. doi: 10.1128/jvi.73.6.4806-4812.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trgovcich J, Pernjak-Pugel E, Tomac J, Koszinowski UH, Jonjic S. Pathogenesis of murine cytomegalovirus infection in neonatal mice. In: Scholz M, Rabenau HF, Doerr HW, Cinatl J, editors. CMV related immunopathology. Karger; Basel: 1998. pp. 42–53. [Google Scholar]
- 57.Tsutsui Y. Developmental disorders of the mouse brain induced by murine cytomegalovirus: animal models for congenital cytomegalovirus infection. Pathol Int. 1995;45:91–102. doi: 10.1111/j.1440-1827.1995.tb03428.x. [DOI] [PubMed] [Google Scholar]
- 58.Tsutsui Y, Kashiwai A, Kawamura N, Aiba-Masago SA, Kosugi I. Prolonged infection of mouse brain neurons with murine cytomegalovirus after pre- and perinatal infection. Acta Virol. 1995;140:1725–1736. doi: 10.1007/BF01384337. [DOI] [PubMed] [Google Scholar]
- 59.Uetsuki T, Naito A, Nagata S, Kaziro Y. Isolation and characterization of the human chromosomal gene for polypeptide chain elongation factor-1 alpha. J Biol Chem. 1989;264:5791–5798. [PubMed] [Google Scholar]
- 60.van den Pol AN, Ghosh PK. Selective neuronal expression of green fluorescent protein with cytomegalovirus promoter reveals entire neuronal arbor in transgenic mice. J Neurosci. 1998;18:10640–10651. doi: 10.1523/JNEUROSCI.18-24-10640.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van den Pol AN, Kim WT. NILE/L1 and NCAM-polysialic acid expression on growing axons of isolated neurons. J Comp Neurol. 1993;332:237–257. doi: 10.1002/cne.903320208. [DOI] [PubMed] [Google Scholar]
- 62.van den Pol AN, Finkbeiner SM, Cornell-Bell AH. Calcium excitability and oscillations in suprachiasmatic nucleus neurons and glia in vitro. J Neurosci. 1992;12:2648–2664. doi: 10.1523/JNEUROSCI.12-07-02648.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van den Pol AN, Obrietan K, Belousov AB, Yang Y, Heller HC. Early synaptogenesis in vitro: role of axon target distance. J Comp Neurol. 1998;399:541–560. doi: 10.1002/(sici)1096-9861(19981005)399:4<541::aid-cne7>3.3.co;2-u. [DOI] [PubMed] [Google Scholar]
- 64.van der Hoff MJ, Moorman AF, Lamers WF. Electroporation in “intracellular” buffer increases cell survival. Nucleic Acids Res. 1992;20:2902. doi: 10.1093/nar/20.11.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vickland H, Westrum LE, Kott JN, Patterson SL, Bothwell MA. Nerve growth factor receptor expression in the young and adult rat olfactory system. Brain Res. 1991;565:269–279. doi: 10.1016/0006-8993(91)91659-o. [DOI] [PubMed] [Google Scholar]
- 66.Vieira J, Farrell HE, Rawlinson WD, Mocarski ES. Genes in the HindIII J fragment of the murine cytomegalovirus genome are dispensable for growth in cultured cells: insertion mutagenesis with a lacZ/gpt cassette. J Virol. 1994;68:4837–4846. doi: 10.1128/jvi.68.8.4837-4846.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.White DO, Fenner FJ. Medical virology. Academic; New York: 1994. [Google Scholar]
- 68.Wiley CA, Nelson JA. Role of human immunodeficiency virus and cytomegalovirus in AIDS encephalitis. Am J Pathol. 1988;133:73–81. [PMC free article] [PubMed] [Google Scholar]
- 69.Wiley CA, Schrier RD, Denaro FJ, Nelson JA, Lampert PW, Oldstone MB. Localization of cytomegalovirus proteins and genome during fulminant central nervous system infection in an AIDS patient. J Neuropathol Exp Neurol. 1986;45:127–139. doi: 10.1097/00005072-198603000-00003. [DOI] [PubMed] [Google Scholar]
- 70.Wolf A, Cowen D. Perinatal infections of the central nervous system. In: Minckler J, editor. Pathology of the nervous system. McGraw-Hill; New York: 1972. pp. 2565–2611. [Google Scholar]