

Abstract
CD4 T cells from HIV-1 infected patients die at excessive rates compared to those from uninfected patients, causing immunodeficiency. We previously identified a dominant negative ligand which antagonizes the TNF-Related Apoptosis-Inducing Ligand (TRAIL) dependent pathway of cell death, that we called TRAILshort. Because the TRAIL pathway has been implicated in CD4 T cell death occurring during HIV-1 infection, we used shRNA knockdown, CRISPR deletion, or antibodies specific for TRAILshort to determine the effect of inhibiting TRAILshort on the outcome of experimental acute HIV infection in vitro. Strikingly, all three approaches to TRAILshort deletion/inhibition enhanced HIV induced death of both infected and uninfected human CD4 T cells. Thus TRAILshort impacts T cell dynamics during HIV infection, and inhibiting TRAILshort causes more HIV infected and uninfected bystander cells to die. TRAILshort is therefore a host-derived, host-adaptive mechanism to limit the effects of TRAIL induced cell death. Further studies on the effects of TRAILshort in other disease states are warranted.
Keywords: TNF-Related Apoptosis-Inducing Ligand, TNF-Related Apoptosis-Inducing Ligand Receptors, Human Immunodeficiency Virus, CD4-Positive T-Lymphocytes, viral replication
INTRODUCTION
The Human Immunodeficiency Virus (HIV) causes disease by killing CD4 T cells and other immune cells, ultimately leading to immunodeficiency. Through evolution, humans have developed innate immune sensing mechanisms to respond to viruses, many of which are mediated by Toll-like receptor (TLR) pathways. TLR activation induces transcriptional upregulation of interferon-stimulated genes (ISG), which act to enhance innate host defense mechanisms. One such immune effector pathway is the TRAIL-TRAIL receptor axis. Importantly, uninfected CD4 T cells undergo HIV-induced death in addition to infected cells. While the death of infected cells is advantageous by controlling viral replication, the death of uninfected CD4 T cells is disadvantageous to the host and contributes to immunodeficiency.
Diverse mechanisms contribute to the death of uninfected cells, including inappropriate immune activation, cytotoxic effects of viral proteins, and enhanced expression of pro-apoptotic ligands such as tumor necrosis factor (TNF), Fas Ligand and TNF-Related Apoptosis-Inducing Ligand (TRAIL) (1).
The normal function of TRAIL is as an immune effector mechanism, to promote the eradication of malignant or infected cells (2), and therefore has been proposed as an immunotherapeutic for malignant and infectious diseases. TRAIL can bind to four membrane-bound receptors, yet ligation to only two of these receptors, TRAIL-R1 (DR4) and TRAIL-R2 (DR5), trigger activation of cell death (3–6). TRAIL binding TRAIL-R3 and TRAIL-R4 fails to induce apoptosis, due to the absence of a cytoplasmic Death Domain (DD) in TRAIL-R3 and truncation of the cytoplasmic DD in TRAIL-R4 (7, 8). TRAIL and the death inducing TRAIL-R1 and -R2 belong to the ISG family, and consequently are expressed following stimulation of TLRs, interferons (9, 10), and a variety of pro-inflammatory cytokines (e.g., IFN-α, IFN-β, IFN-γ, IL-2, TNF-α) (11–14).
There is an increasing body of literature which suggests that TRAIL is involved in the immunopathogenesis of HIV induced immunodeficiency. The HIV envelope protein gp120 promotes acquired sensitivity of T cells to TRAIL-mediated apoptosis through upregulation of TRAIL-R1 and R2 (15). In humanized mouse models, HIV infection in the presence of an anti-TRAIL antibody causes reduced uninfected CD4 T cell apoptosis (16), suggesting that TRAIL mediates uninfected CD4 T cell death. Internally consistent with that finding and the known association of high viremia with accelerated CD4 losses (17), plasma levels of soluble TRAIL are increased in HIV-infected patients, proportional to viral load (18). In patients who experience immune recovery in response to antiretroviral therapy (ART), increases in CD4 T cell counts correlate with decreased expression of TRAIL and TRAIL-R2 on CD4 T cells (18, 19), while poor CD4 T cell recovery following ART is associated with high TRAIL R1 expression (20). Furthermore, TRAIL has been implicated in the apoptotic death of uninfected CD8 T cells and B cells in HIV-infected patients (20–24). Thus, TRAIL-mediated signaling likely contributes to the cytopenias which occur in untreated HIV infection.
HIV-infected cells may also acquire resistance to TRAIL-induced killing. CD4 T cells from HIV-infected patients and been shown to acquire a TRAIL-resistant phenotype despite expression of death-inducing TRAIL receptors R1 and R2 (25–27). Despite this, treatment of cells from HIV positive patients with high doses of TRAIL can overcome this resistance and eradicate HIV infected cells (23, 28, 29). These observations led us to discover TRAILshort, a splice variant of TRAIL that is produced following HIV infection and acts as a decoy ligand and TRAIL antagonist (30). TRAILshort is abundant in the supernatant of HIV-infected cell cultures and in the plasma of untreated HIV-infected patients (30). TRAILshort is expressed by both HIV-infected cells and uninfected cells, in response to both type 1 interferon stimulation and Toll like receptor stimulation (30, 31). In plasma, TRAILshort is found within circulating extracellular vesicles, affording it the ability to not only protect cells producing TRAILshort from TRAIL-mediated killing (31), but to confer TRAIL resistance upon neighboring, non-TRAILshort-producing, cells (31). Because TRAILshort is both necessary and sufficient to cause TRAIL resistance, we have speculated that its presence might limit death of HIV infected cells, thereby favoring HIV persistence, and also limit death of uninfected cells (30, 31).
In the current report we evaluate, using three independent means, the effect of TRAILshort on immune cell homeostasis during HIV infection, and discover that knockdown, knockout or inhibition of TRAILshort increases HIV-induced CD4 T cell death, of both infected and uninfected cells.
METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to, and will be fulfilled by the corresponding author Andrew D Badley (Badley.andrew@mayo.edu).
Cell Culture
All cells were cultured at 37°C in an atmosphere containing 5% CO2. Jurkat human T cells were obtained from American Type Culture Collection (Manassas, VA) and maintained in RPMI 1640 (Gibco) with 10% fetal bovine serum and 2 mM glutamine at a concentration of <106 cells/mL. Uninfected primary peripheral blood mononuclear cells (PMBC) were isolated from donor apheresis cones using density gradient centrifugation (32). Primary uninfected CD4 T cells were isolated by negative selection using RosetteSep™ Human CD4 T cell Enrichment Kit (StemCell Technologies, Seattle, WA) according to the manufacturer’s protocol.
Ethics Statement
All human samples (PBMCs) were obtained according to institutional review board (IRB)-approved protocols in compliance with institutional and federal regulations.
TRAILshort Knockdown
Jurkat T cells were transfected with the pcDNA 6.2-GW/EmGFP-miR plasmid carrying the Blasticidin resistance gene (Invitrogen, Carlsbad, CA) expressing shRNA targeting exon 1 of TRAIL (GATCACGATCAGCACGCAGGTCGTTTTGGCCACTGACTGACGACCTGCGCTGATCGTGAT), or control (GAAATGTACTGCGCGTGGAGACGTTTTGGCCACTGACTGACGTCTCCAC GCAGTACATTT), supplied by Invitrogen or transduced with lentivirus (Mission pLK0.1-puro, Sigma, St. Louis, MO) carrying puromycin resistance and containing an siRNA that is specific for TRAILshort by targeting the exon 2/5 splice junction of TRAIL (GAAAGACTCCAAGAATGAA) or a non-targeting control (Sigma Mission pLK0.1-puro Non-Target shRNA Control, SHC016V). This shRNA control is predicted not to target any known mammalian protein. After transfection, the TRAIL KD shRNA cells were cultured in RPMI 1640 with 10% FBS and 2 mM glutamine plus 10 μg/mL blasticidine (ThermoFisher) for 14 days before culturing in a 96 well plate at an average cell density of 0.5 cells/well. After 20 days, one clone (E3) was selected and assessed for GFP expression (>90% by fluorescent microscopy). The cells were cultured and maintained in the presence of 10 μg/mL blasticidine, which was removed from the medium 48 hours before any experimental procedure. For lentivirus transduction, Jurkat cells were incubated with the lentivirus at a multiplicity of infection (MOI) ~10 for 4 hours at 37°C, then washed 2× with medium and cultured for 14 days in RPMI with 10% FBS and 2 mM glutamine plus 2 μg/mL puromycin (Millpore Sigma). The cultures were found to be greater than 95% GFP+. The cultures were maintained in a medium containing 2 μg/mL of puromycin, which was removed from the medium at least 48 hours before any experimental procedures.
TRAILshort Knockout by CRISPR-Cas9
Genome editing of TRAILshort (TNFSF10 gene, NM_003810) knockout mutation of Jurkat cell lines (ATCC # TIB-152) was contracted through Synthego (Synthego, Redwood City, CA). Briefly cells were test negative for mycoplasma. Guide RNAs (gRNAs) were selected based on high specificity, high activity and propensity to create premature stop codons via frame-shifts in the coding region though insertions or deletions on exon 2 of TRAIL. Based on off-target analysis, modified gRNAs were chosen: TNFSF10+172514928 [TCATACTCTCTTCGTCATTG (GGG)-PAM] or TNFSF10+172514997 [TGGAGTACTTGTCCTGCATC (TGG)-PAM), and transfected using nucleofection protocol. The editing efficiency of Jurkat transfected cells confirmed via PCR and Sanger sequencing followed by then single-cell colonies were grown for three to four weeks. After single cells colonies expansion, cells genomic DNA extracted PCR amplify the edited site, sequence using Sanger sequencing and analysis the data using Synthego Inference of CRISPR Edits (ICE) tool. Based ICE analysis two knockout jurkat clone [Clone I14 contains a homozygous (−13) knockout and Clone B20 contains a homozygous (+1) knockout] is selected and confirmed the knockout within the TRAIL exon 2. CRISPR knockout and parental wild type cells was validated by flow cytometry using TRAILs-CF555 (TRAILs clone 2.2)(30) and also using western blot with specific antibodies.
In vitro HIV-1 Infections
Jurkat T cells were infected with either the laboratory-adapted strain HIV-IIIB (obtained from the NIH AIDS Reagent Program) or VSVG-pseudotyped pNL4.3-nef-Vpr-Δ env-GFP virus (33) for 18 hours at 37°C at a concentration of 24 ng HIV p24 for 10×106 cells, then washed with medium 2× and cultured in RPMI 1640 with 10% FBS and 2mM glutamine. “Low MOI” experiments were performed with 5 to 10 fold less virus. Cultures were monitored for viability using trypan blue exclusion, and supernatants and/or cells were harvested at regular intervals for p24 analysis and proviral DNA content. Culture medium was replaced as needed. Primary CD4 T cells or primary PBMCs were infected in a similar manner after activation with phytohemagglutinin (PHI, 2 μg/mL) and interleukin-2 (IL2, 50 IU/mL). Cell culture supernatants were assayed for HIV-1 p24 levels by ELISA (Zeptometrix, Buffalo, NY) according to the manufacturer’s protocols. HIV-1 cell-associated proviral DNA was measured using a digital droplet (dd) PCR assay, which measures total cell-associated HIV DNA, and has a detection limit of 45 copies/million cells (34), and results were expressed as HIV polymerase copies per 106 viable cells.
Cell Mixing Experiment
106 parental Jurkat cells were added separately to 106 of Jurkat cells expressing the control shRNA (GFP+) or TRAILshort-KD shRNA (GFP+). The cells were infected with HIV IIIB and cultured at 37°C. Cells were harvested at regular intervals, washed with PBS, and stained with Live/Dead aqua stain (Invitrogen) before fixation with 2% paraformaldehyde. The percent of viable GFP+ cells in the total viable population was assessed by flow cytometry for each culture.
Flow Cytometry
PBMCs were stained for cell subsets using the following antibodies: Alexa Fluor 700 mouse anti-human CD3 (BD Pharmingen™/Clone: SP34–2), FITC mouse anti-human CD4 (BD Pharmingen™/Clone: OKT4), Pacific Blue mouse anti-human CD8 (BD Pharmingen™/Clone: RPA-T8), BUV805 mouse anti-human CD14 (BD Horizon™/Clone: M5E2), BUV 395 mouse anti-human CD19 (BD Horizon™/Clone: SJ25C1), APC anti-Hu CD56 (NCAM) (Invitrogen/Clone: TULY56). The mouse monoclonal anti-TRAILshort-specific antibody was developed in the Mayo Antibody Core facility as previously described (30). Intracellular staining for active caspase 3 was performed as described (33). Additional antibodies include mouse anti-TRAIL-R1 (B-N36 PE Cell Sciences) and mouse anti-TRAIL-R2 (B-K29 PE Cell Sciences). Flow cytometry was performed on BD LSR Fortessa flow cytometer. The results were analyzed with FlowJo v10.2 software (Tree Star, Inc, Ashland OR).
Viability Assays
Cell viability, assayed by FACS analysis, was performed using TUNEL staining (Roche In Situ Cell Death Detection TMR Red) or Live/Dead stain (Invitrogen) per the manufacturer’s instructions. Cell viability by ATP content was measured using the Cell Titer-GloViability kit (Promega, Madison, WI) per the manufacturer’s instructions. Live cell imaging was performed using the IncuCyte System (Essen Bioscience) by plating 10,000 cells/well for Jurkat T cells or 20,000 cells/well for primary CD4 T cells; cell death was assessed using either the Cytotox Red reagent or Active Caspase 3/7-Green reagent (Essen Bioscience, Ann Arbor, MI). Live cell imaging data was analyzed using IncuCyte Zoom software (v 2016B).
Western Blot and Immunoprecipitation Assays
Jurkat cells were harvested from culture, centrifuged at 200×g for 5 minutes, washed with cold PBS, centrifuged again at 200×g for 5 minutes, then placed on ice and resuspended in cold lysis buffer (20 mM Tris/HCl pH 7.2, 150 mM NaCl, 0.1% NP40, 0.1% CHAPS, plus protease inhibitors aprotinin, leupeptin, pepstatin, and PMSF) for 5 to10 minutes. The lysate was then centrifuged at 400×g for 5 minutes to pellet nuclei. The supernatant was transferred to a new tube. For Western blotting, the following antibodies were used: mouse anti-TRAILs clone 2 (Mayo Hybridoma Core); mouse anti-human CD253 (TRAIL) (BD Pharmingen, San Jose, CA); rabbit anti-DR5 (D4E9, Cell Signaling Technology, Danvers, MA); goat anti-Actin (I-19, HRP, Santa Cruz Biotechnology, San Jose, CA). 400 μg of cytosol was used for each immunoprecipitation reaction (2 μg anti-caspase 8, Millipore, Burlington, MA) or pull down (2 μg DR5-Fc, R&D systems, Minneapolis, MN) with 10 μL Gammabind Plus protein A/G agarose in 200 μL of lysis buffer. Reactions proceeded overnight at 4°C. The agarose beads were centrifuged and washed 2× with cold lysis buffer. Wash solution was aspirated, 10 μL of 2× Laemmli sample loading buffer was added, and beads were heated at 90°C for 3 minutes to release bound proteins. The proteins were harvested and run on PAGE before transferring to PVDF membrane for Western blotting.
QUANTIFICATION AND STATISTICAL ANALYSIS
Descriptive statistics are generally presented as means +/− standard deviation (SD) unless otherwise noted. Parametric or non-parametric statistical tests were used as appropriate and are listed in the respective figure legends. Statistical significance was accepted when P<0.05. Statistical analysis was performed using GraphPad Prism 6 (GraphPad, Inc).
RESULTS
TRAILshort Knockdown Enhances TRAIL Sensitivity and Alters T Cell Viability Following Acute HIV Infection in vitro.
Having previously shown that TRAILshort is expressed by HIV-infected and uninfected cells, and prevents TRAIL from killing TRAIL receptor expressing cells (30, 31), we hypothesized that TRAILshort production limits HIV-induced cell death. To assess the impact of TRAILshort on T cell dynamics during acute in vitro HIV infection, we used Jurkat T cells transfected with lentiviral shRNA constructs to inhibit the expression of TRAILshort. The mRNA encoding TRAILshort consists of exon 1 and exon 2 of the TRAIL gene, along with a 30 nucleotide sequence from exon 5 (30). Because of the sequence overlap between full-length TRAIL and TRAILshort, we specifically targeted shRNA against the splice junction to achieve specific knockdown of TRAILshort expression as against full-length TRAIL. We identified a construct that reduced TRAILshort protein expression by about 60%, as estimated by densitometry, yet minimally affected expression of full-length TRAIL protein (Figure 1A). In addition the TRAILshort knockdown (TRAILshort KD) did not affect expression of TRAIL, nor TRAIL receptors 1 and 2 (Figure 1B). The resulting TRAILshort KD cells displayed enhanced susceptibility to TRAIL-mediated killing compared to cells expressing the control shRNA (Control LV), as evidenced by TUNEL staining and reduced ATP content upon treatment with 2ng/ml SuperKiller® TRAIL (skTRAIL, a recombinant, soluble human TRAIL oligomer, Enzo Life Sciences) (Figure 1C and D). However, we observed no altered susceptibility to TRAIL receptor-independent death when Jurkat T cells were exposed to hydrogen peroxide, confirming the specificity of TRAILshort in TRAIL-receptor mediated cell death (Figure 1E).
Fig 1. TRAILshort Knockdown Enhances TRAIL Sensitivity and Alters T Cell Viability Following Acute HIV Infection in vitro.
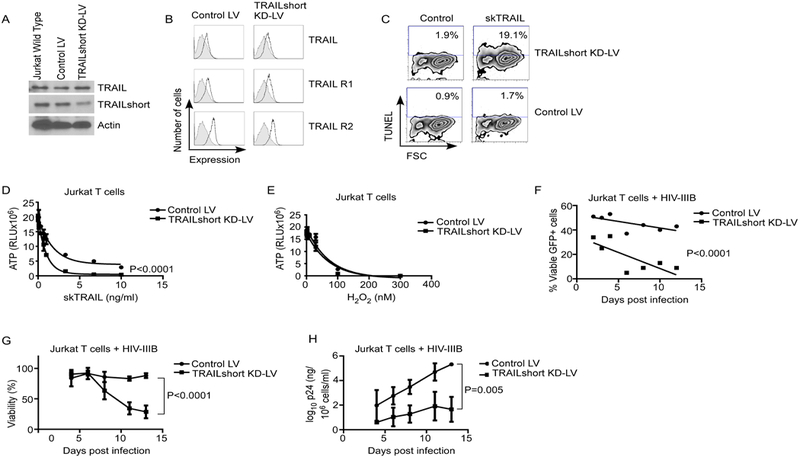
(A) Jurkat T cells were transduced with lentiviral vectors containing control or TRAILshort-targeting shRNA, and assessed for expression of TRAIL and TRAILshort by western blot. (B) Surface expression of full-length TRAIL, TRAIL-R1 and TRAIL-R2 was assessed by flow cytometry. Shaded area indicates isotype control staining. (C) Control LV or TRAILshort KD Jurkat T cells were treated with control or skTRAIL and assessed for apoptosis by TUNEL staining. (D) Transduced cells treated as in (C) assessed for ATP content in the presence of increasing concentrations of skTRAIL. (E) Transduced cells were assessed for ATP content in the presence of increasing concentrations of hydrogen peroxide. (F) Control LV cells or TRAILshort KD cells were mixed 50:50 with parental Jurkat cells, infected with HIV-IIIB, and assessed for survival over time by flow cytometry. (G) Control LV cells or TRAILshort KD cells were infected with a low MOI of HIV-IIIB and viability assessed over time by LIVE/Dead staining flow cytometry. (H) Culture supernatant HIV-1 p24 was measured from cells treated as in (G). Data represent mean values for 2 (panels C, D, and E) or 3 (F and G) independent experiments. P values were determined by linear regression.
Next, we assessed whether expression of TRAILshort affects susceptibility to HIV-induced death. We used a bicistronic vector to co-express GFP with either TRAILshort KD or Control LV. The resultant transfected cells showed normal viability (>95%) and similar doubling times as parental Jurkat T cells, (parental Jurkat T cells = 26.6 hours; TRAILshort KD = 22.1 hours; Control LV = 29.4 hours). The cells co-expressing GFP and either TRAILshort KD or Control LV were mixed in equal proportions with non-transfected GFP-parental cells, and the pooled cells were infected with HIV (Figure 1F). We reasoned that if TRAILshort did not alter cell survival following HIV infection, then, given the similar doubling times, the proportion of transfected GFP+ cells relative to non-transfected cells should remain constant at ~50% over time. Conversely, a change in the proportion of GFP+ cells over time would indicate an increased susceptibility of either cell types to HIV-induced death. Over time, the proportion of GFP+ cells in the mixture remained nearly constant at 50% ±10%. In contrast, the proportion of GFP+ viable cells steadily decreased to <10% on day 7 post infection (P<0.0001), indicating that the cells containing TRAILshort KD were more susceptible to death than either parental or control transfected cells upon acute HIV infection. Thus, expression of TRAILshort confers a survival advantage on cells infected with HIV.
We next assessed cell viability over time in GFP+ TRAILshort KD or Control LV cells infected with a low MOI of HIV-IIIB. Mock infected TRAILshort KD and Control LV cells maintained viabilities of >95% over the entire period of observation. However, TRAILshort KD cells infected with HIV-IIIB died at significantly greater rates than infected Control LV cells (P<0.0001, Figure 1G). Furthermore, culture supernatant from HIV-infected TRAILshort KD cells produced significantly less HIV-1 p24 compared to HIV-infected Control LV cells (P=0.005, Figure 1H).
Antibody Neutralization of TRAILshort Increases T Cell Death Following Acute in vitro Infection
Since genetic approaches to inhibit protein production can incur off-target effects, we extended the results obtained with shRNA-based approach by neutralizing TRAILshort using a monoclonal antibody specific for the C-terminus of TRAILshort (30, 31). We then assessed cell death using Incucyte real time live cell imaging and a GFP labeled HIV pseudovirus (see Methods). In this assay, cells are monitored every 2 hours over the 4 day period of observation, and assessed for Green (productive HIV infection) and death using a cyanine red nucleic acid which stains cells that have lost plasma membrane integrity. These experiments were performed in the presence or absence of an anti-TRAILshort antibody or an isotype control (Figure 2A). As expected, acute HIV infection increased the number of dead cells compared to mock infection (Figure 2B). The addition of anti-TRAILshort antibodies to HIV-infected cultures increased the number of dead cells compared to untreated or isotype control treatment (Figure 2C, P<0.0001). We then took advantage of the GFP-labeled virus to discern whether TRAILshort influences the death of cells infected with HIV as opposed to bystander cells. Addition of anti-TRAILshort antibody significantly enhanced death of both infected (GFP+) cells (Figure 2D, P<0.0001) and uninfected (GFP-) HIV-exposed cells (Figure 2E, P<0.0001), implicating the involvement of TRAIL:TRAIL receptor axis in the death of both HIV-infected and uninfected T cells.
Fig 2. Antibody Neutralization of TRAILshort Increases T Cell Death in Productive in vitro HIV Infection.
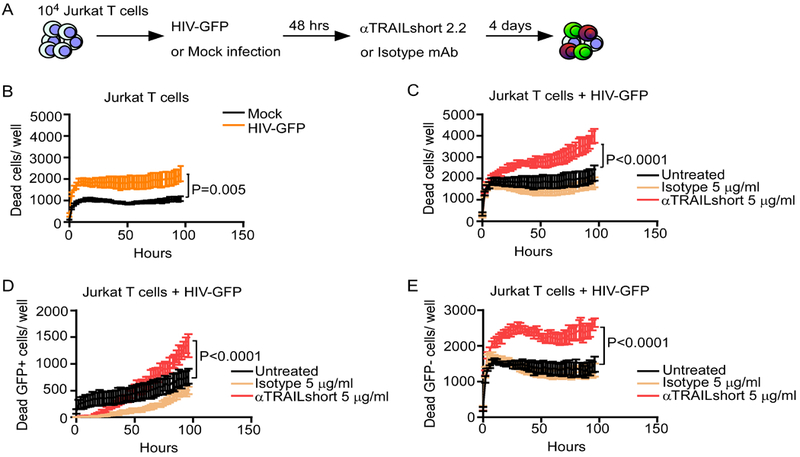
(A) Jurkat T cells were mock infected or infected with a VSVG-pseudotyped HIV expressing GFP, and treated with anti-TRAILshort antibody (clone 2.2) or isotype control, and incubated for 4 days. (B) Cell death was measured in mock- and HIV-infected cells over time by live cell imaging. (C) Cell death was measured in HIV-infected cells in the presence and absence of anti-TRAILshort antibody or isotype control. (D) Cell death in GFP+ (infected) cells was measured as in (C). (E) Cell death in uninfected cells was measured as in (C). Data represents mean + SD for 5–6 replicates from one experiment, representative of at least two independent experiments. P values were determined by linear regression.
TRAILshort knockout Jurkat T cells have heightened sensitivity to TRAIL mediated killing, absent TRAILshort production, and die at enhanced rates following acute HIV infection in vitro.
Using CRISPR-Cas9, we generated TRAILshort knockout Jurkat T cells using sgRNA guides targeting Exon 2 of TNFSF10 (Figure 3A). Expression of TRAILshort in response to treatment with PMA was assessed in parental Jurkat cells (Jurkat-wt), and two knockout clones (Jurkat-B20 and Jurkat-I14) by western blot (Figure 3B) and flow cytometry (Figure 3C). For instance, 24 hour treatment of WT cells with PMA resulted in an increase in TRAILshort expression from 6% to 15% (MFI increase from 644 to 784), but not in TRAILshort KO cells (Figure 3C). This confirmed effective knockout of TRAILshort expression in these two clones. We then compared sensitivity of cells to TRAIL mediated killing between TRAILshort knockout cells and parental controls (Figure 3D). TRAILshort-KO clone Jurkat-I14 demonstrated enhanced death in response to exogenous TRAIL (P<0.0001, Figure 3D) compared to parental WT cells.
Fig 3. TRAILshort Knockout Increases TRAIL Sensitivity and T Cell Apoptosis Following Acute HIV Infection in vitro.
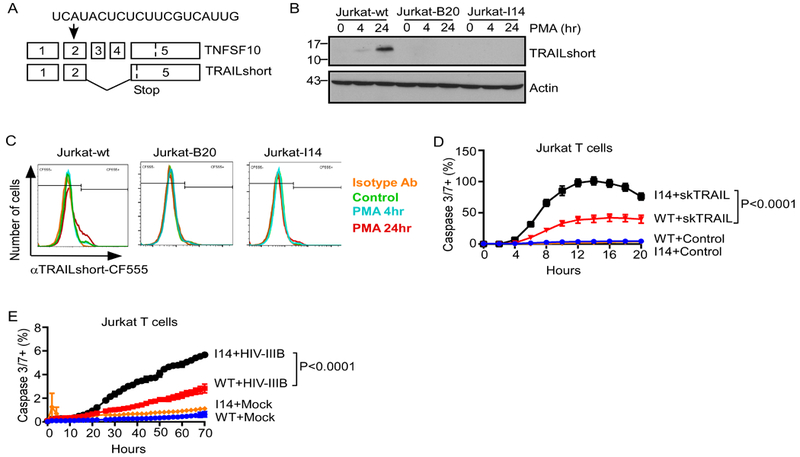
(A) Strategy for CRISPR interruption of Exon 2 of TRAIL (TNFSF10). (B) TRAILshort expression in response to stimulation with phorbol 12-myristate 13-acetate (PMA) was assessed by western blot in parent Jurkat cells (Jurkat-wt) or two TRAILshort-KO clones (B20 and I14). (C) Surface TRAILshort expression in similarly treated cells was assessed by flow cytometry. (D) Jurkat-wt and Jurkat-I14 cells were treated with vehicle control or skTRAIL (1 ng/ml) and assessed for active Caspase 3/7 expression over time by live cell imaging. (E) Jurkat-wt and Jurkat-I14 cells were mock-infected or infected with HIV-IIIB, and and assessed for active Caspase 3/7 expression over time by live cell imaging. Data represents mean + SD for 5–6 replicates from one experiment, representative of at least three independent experiments. P values were determined by linear regression.
We next questioned whether genetic knockout of TRAILshort would impact cell death in acute HIV infection. Consistent with the knockdown approach noted above (see Figure 1), and the antibody neutralization (see Figure 2), Jurkat-I14 clone cells, which cannot make TRAILshort, expressed more active Caspase 3/7 over time in response to HIV infection compared to infected, parental control Jurkat-WT cells (P<0.0001, Figure 3E).
Anti-TRAILshort treatment of primary CD4 T cells infected with HIV-IIIB augments cell death
Using three independent methods, we have now shown that inhibition of TRAILshort in the setting of acute HIV infection of a T cell line increases HIV-induced, and TRAIL-induced, apoptosis. We next evaluated whether the same effect occurs in primary CD4 T cells (Figure 4A). Importantly, spontaneous apoptosis of uninfected primary CD4 T cells treated with anti-TRAILshort antibody at doses up to 20 μg/ml did not differ compared to isotype control treated cells (Figure 4B). On the other hand, treatment of HIV-IIIB infected primary CD4 T cells with increasing doses of anti-TRAILshort antibody significantly increased apoptosis (Figure 4C, P<0.0001). We next questioned whether this increased apoptosis was associated with altered viral replication. Conceivably, increased apoptotic death of infected cells might lead to fewer progeny virions being produced; alternatively, cell death associated with caspase 8 activation (such as the TRAIL pathway) is known to activate NFkB through the BCL10, MALT1, CARMA pathway (35–37) which would increase HIV replication. Interestingly, HIV-1 p24 antigen concentrations in cell culture supernatant over time did not differ from cultures treated with anti-TRAILshort antibody or isotype control (Figure 4D).
Fig 4. Antibody Neutralization of TRAILshort Increases primary CD4 T Cell Death in Productive and Non-Productive in vitro HIV Infection.
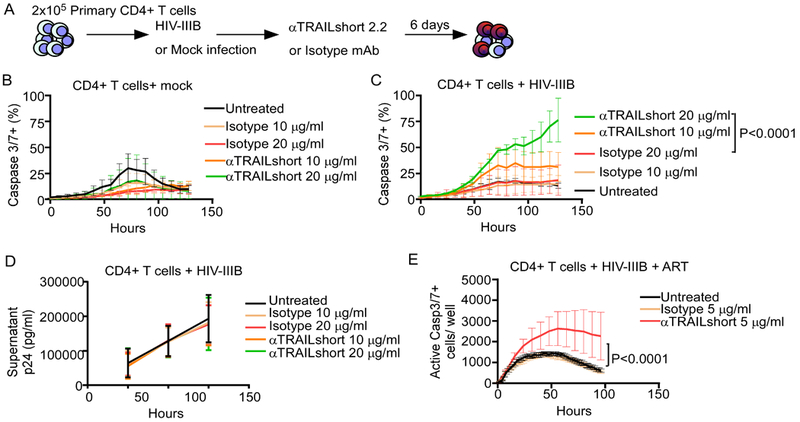
(A) Primary CD4 T cells were mock infected or infected with HIV-IIIB, and treated with anti-TRAILshort antibody (clone 2.2) or isotype control for 6 days. (B) Cell death was measured in mock infected cells with or without TRAILshort antibody or isotype control over time by live cell imaging. (C) Cell death was measured in similarly treated, HIV-IIIB infected cells. (D) HIV-1 p24 antigen was measured in cell culture supernatants over time. (E) Cell death in HIV-infected CD4 T cells, in the presence of ART (T20, EFV, RAL), treated with anti-TRAILshort antibody, isotype control, or vehicle, was measured as in (B). Data represents mean + SD for 5–6 replicates from one experiment, representative of at least three independent experiments. P values were determined by linear regression.
We next infected primary CD4 T cells with HIV-IIIB, and 24 hours later added a combination of ART including enfuvirtide (T20), efavirenz (EFV), and raltegravir (RAL) to inhibit viral replication and reduce exposure to soluble HIV proteins. Cell death was monitored over time after the addition of the anti-TRAILshort antibody or isotype control. The addition of anti-TRAILshort to the ART-treated, HIV-infected CD4 T cells caused more cell death compared to untreated or isotype antibody treated cultures (Figure 4E, P<0.0001). Altogether, these data suggest TRAILshort functions to limit autocrine CD4 T cell apoptosis that is mediated by the TRAIL:TRAIL receptor pathway.
DISCUSSION
Multiple pathways enable the human host to sense and respond to invading pathogens. Long-term HIV infection is associated with sustained upregulation of type I IFN, which drives transcription of interferon-stimulated genes (ISG) that regulate immune cell phenotype and function. The innate and adaptive cellular immune systems function in large part through NK cells and T cells; these cell types kill infected cells through three non-exclusive effector pathways: Fas Ligand, TRAIL and Perforin/granzyme B. TRAIL expression can be induced on immune effector cells such as T and NK cells, while death-inducing TRAIL-R1 and -R2 can be induced on virtually all cell types in response to infection and other activating stimuli (2). Together, the TRAIL:TRAIL receptor axis results in the destruction of infected (and cancerous) cells (38). The relative contribution of these pathways in various disease states remains unclear, yet several observations suggest that the TRAIL pathway is critical in both infectious diseases and in cancer: TRAIL deficient mice have enhanced rates of lymphoid and stromal tumor development, increased rates of metastasis (39–41) and worsened outcomes from infections with influenza (42), pneumococcus (43) and West Nile Virus (44). Furthermore, TRAIL has been implicated in HIV immunopathogenesis through killing of both infected and uninfected CD4 T cells (reviewed in (2)).
A plethora of cellular survival mechanisms have been proposed to counter the effects of TRAIL, including the expression of decoy versus death-inducing receptors (31, 45, 46), elevated expression of antiapoptotic molecules such as FLIP and/or BCL2 family members (47, 48), among others. It is recognized that some cells from HIV infected-donors that express TRAIL R1 and R2 are paradoxically resistant to TRAIL (25–27), an effect that may be mediated by TRAILshort (30, 31). TRAILshort is a transmembrane protein with an extracellular C terminus, and is present within plasma and tissue culture supernatants within extracellular vesicles(31). This latter property confers the ability of a cell producing TRAILshort to protect a neighboring cell (that does not produce TRAILshort) from TRAIL-induced killing (31).
In the current report, we have determined using three independent means that inhibiting TRAILshort production or function during acute HIV infection reduces survival of HIV-infected and bystander (uninfected) cells, extending our previous observations and suggesting a physiologic function of increased TRAILshort production. On the one hand, expression of TRAILshort may reduce CD4 T cell depletion, delaying HIV disease progression, and therefore be a protective host response. On the other hand, TRAILshort-mediated increased survival of HIV-infected cells could contribute to viral persistence, preserving viral production, and at the same time ensuring survival of target CD4 T cells for continued viral replication. The relevant contribution of these two potential functions of TRAILshort to the natural history of HIV infection in vivo warrants further investigation.
Also, understanding the expression of TRAILshort fundamentally impacts the interpretation of TRAIL and TRAIL receptor expression, and argues that in different experimental systems and disease states, functional assessment of TRAILshort involvement is required. Specifically in the case of HIV infection, it is now apparent that multiple stimuli induce TRAIL expression on CD4 T cells, monocytes and plasmacytoid dendritic cells (9–14). Similarly, immune activation causes upregulation of death inducing TRAIL receptors 1 and 2 on both HIV infected and uninfected cells (25–27). Alone these data would suggest that TRAIL dependent pathways might kill and eliminate HIV infected cells, thereby eradicating them. Only with the understanding that TRAILshort is also produced does it make sense that such infected cells are not killed, and thus HIV infected cells persist.
KEY POINTS.
TRAIL binding to TRAIL receptor (R)1 and R2 contributes to CD4 death during HIV infection.
Blocking TRAILshort, a TRAIL antagonist increases death of HIV infected and uninfected cells.
Homeostatic production of TRAILshort may protect cells from HIV-induced cell death.
GRANT FUNDING
A.C.P. and N.W.C. were supported through the Mayo Clinic Foundation. S.R.L. was supported by the National Institutes of Health (NIH) Delaney AIDS Research Enterprise (DARE U19 AI096109 and UM1 AI126611–01) and the National Health and Medical Research Council (NHMRC) of Australia (NHMRC program grant and practitioner fellowship to SRL). A.D.B was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health grant numbers (AI110173 and AI120698). H.-P.K. is a Markey Molecular Medicine Investigator and received support as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Fred Hutch Endowed Chair for Cell and Gene Therapy. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Cummins NW, and Badley AD. 2010. Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis 1: e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cummins N, and Badley A. 2009. The TRAIL to viral pathogenesis: the good, the bad and the ugly. Curr Mol Med 9: 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pan G, Ni J, Wei YF, Yu G, Gentz R, and Dixit VM. 1997. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277: 815–818. [DOI] [PubMed] [Google Scholar]
- 4.Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, and Dixit VM. 1997. The receptor for the cytotoxic ligand TRAIL. Science 276: 111–113. [DOI] [PubMed] [Google Scholar]
- 5.Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, Timour MS, Gerhart MJ, Schooley KA, Smith CA, Goodwin RG, and Rauch CT. 1997. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J 16: 5386–5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu GS, Burns TF, McDonald ER 3rd, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, and el-Deiry WS. 1997. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 17: 141–143. [DOI] [PubMed] [Google Scholar]
- 7.MacFarlane M, Ahmad M, Srinivasula SM, Fernandes-Alnemri T, Cohen GM, and Alnemri ES. 1997. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J Biol Chem 272: 25417–25420. [DOI] [PubMed] [Google Scholar]
- 8.LeBlanc HN, and Ashkenazi A. 2003. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ 10: 66–75. [DOI] [PubMed] [Google Scholar]
- 9.Gong B, and Almasan A. 2000. Genomic organization and transcriptional regulation of human Apo2/TRAIL gene. Biochem Biophys Res Commun 278: 747–752. [DOI] [PubMed] [Google Scholar]
- 10.Sato K, Hida S, Takayanagi H, Yokochi T, Kayagaki N, Takeda K, Yagita H, Okumura K, Tanaka N, Taniguchi T, and Ogasawara K. 2001. Antiviral response by natural killer cells through TRAIL gene induction by IFN-alpha/beta. Eur J Immunol 31: 3138–3146. [DOI] [PubMed] [Google Scholar]
- 11.Kayagaki N, Yamaguchi N, Nakayama M, Eto H, Okumura K, and Yagita H. 1999. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J Exp Med 189: 1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sedger LM, Shows DM, Blanton RA, Peschon JJ, Goodwin RG, Cosman D, and Wiley SR. 1999. IFN-gamma mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J Immunol 163: 920–926. [PubMed] [Google Scholar]
- 13.Liu S, Yu Y, Zhang M, Wang W, and Cao X. 2001. The involvement of TNF-alpha-related apoptosis-inducing ligand in the enhanced cytotoxicity of IFN-beta-stimulated human dendritic cells to tumor cells. J Immunol 166: 5407–5415. [DOI] [PubMed] [Google Scholar]
- 14.Kemp TJ, Elzey BD, and Griffith TS. 2003. Plasmacytoid dendritic cell-derived IFN-alpha induces TNF-related apoptosis-inducing ligand/Apo-2L-mediated antitumor activity by human monocytes following CpG oligodeoxynucleotide stimulation. J Immunol 171: 212–218. [DOI] [PubMed] [Google Scholar]
- 15.Lum JJ, Schnepple DJ, and Badley AD. 2005. Acquired T-cell sensitivity to TRAIL mediated killing during HIV infection is regulated by CXCR4-gp120 interactions. AIDS 19: 1125–1133. [DOI] [PubMed] [Google Scholar]
- 16.Miura Y, Misawa N, Maeda N, Inagaki Y, Tanaka Y, Ito M, Kayagaki N, Yamamoto N, Yagita H, Mizusawa H, and Koyanagi Y. 2001. Critical contribution of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to apoptosis of human CD4+ T cells in HIV-1-infected hu-PBL-NOD-SCID mice. J Exp Med 193: 651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reichelderfer PS, and Coombs RW. 1996. Cartesian coordinate analysis of viral burden and CD4+ cell count in HIV disease: implications for clinical trial design and analysis. Antiviral Res 29: 83–86. [DOI] [PubMed] [Google Scholar]
- 18.Herbeuval JP, Boasso A, Grivel JC, Hardy AW, Anderson SA, Dolan MJ, Chougnet C, Lifson JD, and Shearer GM. 2005. TNF-related apoptosis-inducing ligand (TRAIL) in HIV-1-infected patients and its in vitro production by antigen-presenting cells. Blood 105: 2458–2464. [DOI] [PubMed] [Google Scholar]
- 19.Herbeuval JP, Grivel JC, Boasso A, Hardy AW, Chougnet C, Dolan MJ, Yagita H, Lifson JD, and Shearer GM. 2005. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood 106: 3524–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansjee N, Kaufmann GR, Strub C, Weber R, Battegay M, Erb P, and Swiss HIVCS. 2004. Persistent apoptosis in HIV-1-infected individuals receiving potent antiretroviral therapy is associated with poor recovery of CD4 T lymphocytes. J Acquir Immune Defic Syndr 36: 671–677. [DOI] [PubMed] [Google Scholar]
- 21.Lichtner M, Maranon C, Vidalain PO, Azocar O, Hanau D, Lebon P, Burgard M, Rouzioux C, Vullo V, Yagita H, Rabourdin-Combe C, Servet C, and Hosmalin A. 2004. HIV type 1-infected dendritic cells induce apoptotic death in infected and uninfected primary CD4 T lymphocytes. AIDS Res Hum Retroviruses 20: 175–182. [DOI] [PubMed] [Google Scholar]
- 22.Katsikis PD, Garcia-Ojeda ME, Torres-Roca JF, Tijoe IM, Smith CA, Herzenberg LA, and Herzenberg LA. 1997. Interleukin-1 beta converting enzyme-like protease involvement in Fas-induced and activation-induced peripheral blood T cell apoptosis in HIV infection. TNF-related apoptosis-inducing ligand can mediate activation-induced T cell death in HIV infection. J Exp Med 186: 1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lum JJ, Pilon AA, Sanchez-Dardon J, Phenix BN, Kim JE, Mihowich J, Jamison K, Hawley-Foss N, Lynch DH, and Badley AD. 2001. Induction of cell death in human immunodeficiency virus-infected macrophages and resting memory CD4 T cells by TRAIL/Apo2l. J Virol 75: 11128–11136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Grevenynghe J, Cubas RA, Noto A, DaFonseca S, He Z, Peretz Y, Filali-Mouhim A, Dupuy FP, Procopio FA, Chomont N, Balderas RS, Said EA, Boulassel MR, Tremblay CL, Routy JP, Sekaly RP, and Haddad EK. 2011. Loss of memory B cells during chronic HIV infection is driven by Foxo3a- and TRAIL-mediated apoptosis. J Clin Invest 121: 3877–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibellini D, Re MC, Ponti C, Maldini C, Celeghini C, Cappellini A, La Placa M, and Zauli G. 2001. HIV-1 Tat protects CD4+ Jurkat T lymphoblastoid cells from apoptosis mediated by TNF-related apoptosis-inducing ligand. Cell Immunol 207: 89–99. [DOI] [PubMed] [Google Scholar]
- 26.Chehimi J, Papasavvas E, Tomescu C, Gekonge B, Abdulhaqq S, Raymond A, Hancock A, Vinekar K, Carty C, Reynolds G, Pistilli M, Mounzer K, Kostman J, and Montaner LJ. 2010. Inability of plasmacytoid dendritic cells to directly lyse HIV-infected autologous CD4+ T cells despite induction of tumor necrosis factor-related apoptosis-inducing ligand. J Virol 84: 2762–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melki MT, Saidi H, Dufour A, Olivo-Marin JC, and Gougeon ML. 2010. Escape of HIV-1-infected dendritic cells from TRAIL-mediated NK cell cytotoxicity during NK-DC cross-talk--a pivotal role of HMGB1. PLoS Pathog 6: e1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shepard BD, De Forni D, McNamara DR, Foli A, Rizza SA, Abraham RS, Knutson K, Wettstein PJ, Lori F, and Badley AD. 2008. Beneficial effect of TRAIL on HIV burden, without detectable immune consequences. PLoS One 3: e3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lum JJ, Schnepple DJ, Nie Z, Sanchez-Dardon J, Mbisa GL, Mihowich J, Hawley N, Narayan S, Kim JE, Lynch DH, and Badley AD. 2004. Differential effects of interleukin-7 and interleukin-15 on NK cell anti-human immunodeficiency virus activity. J Virol 78: 6033–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schnepple DJ, Shepard B, Bren GD, Cummins NW, Natesampillai S, Trushin S, Algeciras-Schimnich A, Meng XW, Sainski AM, Rizza SA, Kaufmann SH, and Badley AD. 2011. Isolation of a TRAIL antagonist from the serum of HIV-infected patients. J Biol Chem 286: 35742–35754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nie Z, Aboulnasr F, Natesampillai S, Burke SP, Krogman A, Bren GD, Chung TDY, Anderson JR, Smart MK, Katzmann DJ, Rajagopalan G, Cummins NW, and Badley AD. 2018. Both HIV-Infected and Uninfected Cells Express TRAILshort, Which Confers TRAIL Resistance upon Bystander Cells within the Microenvironment. J Immunol 200: 1110–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dietz AB, Bulur PA, Emery RL, Winters JL, Epps DE, Zubair AC, and Vuk-Pavlovic S. 2006. A novel source of viable peripheral blood mononuclear cells from leukoreduction system chambers. Transfusion 46: 2083–2089. [DOI] [PubMed] [Google Scholar]
- 33.Sainski AM, Dai H, Natesampillai S, Pang YP, Bren GD, Cummins NW, Correia C, Meng XW, Tarara JE, Ramirez-Alvarado M, Katzmann DJ, Ochsenbauer C, Kappes JC, Kaufmann SH, and Badley AD. 2014. Casp8p41 generated by HIV protease kills CD4 T cells through direct Bak activation. J Cell Biol 206: 867–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cummins NW, Rizza S, Litzow MR, Hua S, Lee GQ, Einkauf K, Chun TW, Rhame F, Baker JV, Busch MP, Chomont N, Dean PG, Fromentin R, Haase AT, Hampton D, Keating SM, Lada SM, Lee TH, Natesampillai S, Richman DD, Schacker TW, Wietgrefe S, Yu XG, Yao JD, Zeuli J, Lichterfeld M, and Badley AD. 2017. Extensive virologic and immunologic characterization in an HIV-infected individual following allogeneic stem cell transplant and analytic cessation of antiretroviral therapy: A case study. PLoS Med 14: e1002461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bren GD, Whitman J, Cummins N, Shepard B, Rizza SA, Trushin SA, and Badley AD. 2008. Infected cell killing by HIV-1 protease promotes NF-kappaB dependent HIV-1 replication. PLoS One 3: e2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lemmers B, Salmena L, Bidere N, Su H, Matysiak-Zablocki E, Murakami K, Ohashi PS, Jurisicova A, Lenardo M, Hakem R, and Hakem A. 2007. Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. J Biol Chem 282: 7416–7423. [DOI] [PubMed] [Google Scholar]
- 37.Su H, Bidere N, Zheng L, Cubre A, Sakai K, Dale J, Salmena L, Hakem R, Straus S, and Lenardo M. 2005. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science 307: 1465–1468. [DOI] [PubMed] [Google Scholar]
- 38.Ishikawa E, Nakazawa M, Yoshinari M, and Minami M. 2005. Role of tumor necrosis factor-related apoptosis-inducing ligand in immune response to influenza virus infection in mice. J Virol 79: 7658–7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takeda K, Smyth MJ, Cretney E, Hayakawa Y, Kayagaki N, Yagita H, and Okumura K. 2002. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J Exp Med 195: 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takeda K, Smyth MJ, Cretney E, Hayakawa Y, Yamaguchi N, Yagita H, and Okumura K. 2001. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in NK cell-mediated and IFN-gamma-dependent suppression of subcutaneous tumor growth. Cell Immunol 214: 194–200. [DOI] [PubMed] [Google Scholar]
- 41.Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, and Smyth MJ. 2002. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol 168: 1356–1361. [DOI] [PubMed] [Google Scholar]
- 42.Brincks EL, Katewa A, Kucaba TA, Griffith TS, and Legge KL. 2008. CD8 T cells utilize TRAIL to control influenza virus infection. J Immunol 181: 4918–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steinwede K, Henken S, Bohling J, Maus R, Ueberberg B, Brumshagen C, Brincks EL, Griffith TS, Welte T, and Maus UA. 2012. TNF-related apoptosis-inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. J Exp Med 209: 1937–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shrestha B, Pinto AK, Green S, Bosch I, and Diamond MS. 2012. CD8+ T cells use TRAIL to restrict West Nile virus pathogenesis by controlling infection in neurons. J Virol 86: 8937–8948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang X, Liu Y, Zhang Q, Gao L, Han L, Ma C, Zhang L, Chen YH, and Sun W. 2007. Hepatitis B virus sensitizes hepatocytes to TRAIL-induced apoptosis through Bax. J Immunol 178: 503–510. [DOI] [PubMed] [Google Scholar]
- 46.Liu YG, Liu SX, Liang XH, Zhang Q, Gao LF, Han LH, Cao YL, Hou N, Du J, and Sun WS. 2007. Blockade of TRAIL pathway ameliorates HBV-induced hepatocyte apoptosis in an acute hepatitis model. Biochem Biophys Res Commun 352: 329–334. [DOI] [PubMed] [Google Scholar]
- 47.Mirandola P, Ponti C, Gobbi G, Sponzilli I, Vaccarezza M, Cocco L, Zauli G, Secchiero P, Manzoli FA, and Vitale M. 2004. Activated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicity. Blood 104: 2418–2424. [DOI] [PubMed] [Google Scholar]
- 48.Swingler S, Mann AM, Zhou J, Swingler C, and Stevenson M. 2007. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog 3: 1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]