

Abstract
Nestled within feeding circuits, the oval (ov) region of the Bed Nucleus of the Stria Terminalis (BNST) may be critical for monitoring energy balance through changes in synaptic strength. Here we report that bidirectional plasticity at ovBNST GABA synapses was tightly linked to the caloric state of male rats, seesawing between long-term potentiation (iLTP, fed) and depression (iLTD, food restricted). L-α-lysophosphatidylinositol (LPI) acting on GPR55 receptors and 2-arachidonoylglycerol (2-AG) through CB1R were respectively responsible for fed (iLTP) and food restricted (iLTD) states. Thus, we have characterized a potential gating mechanism within the ovBNST that may signal metabolic state within the rat brain feeding circuitry.
Subject terms: Feeding behaviour, Synaptic plasticity
Introduction
Feeding behavior is regulated in the brain by bottom-up (e.g., homeostatic) and top-down (e.g., non-homeostatic) processes [1]. The Bed Nucleus of the Stria Terminalis (BNST) is ideally positioned to integrate this information and coordinate the selection of appropriate physiological and behavioral foraging and feeding responses [2–4]. Food consumption activates BNST neurons [5] and optical stimulation of monosynaptic GABA projections from the BNST (including the oval subregion [ovBNST]) to the lateral hypothalamus (LH) and other structures that modulate metabolic processes initiates voracious feeding in mice [6]. This suggests that the ovBNST may in part contribute to the central regulation of energy homeostasis.
Endocannabinoids (eCBs) are widely represented in central and peripheral systems related to energy homeostasis [7, 8]. The most described constituents of the eCB system are probably the type 1 cannabinoid receptor (CB1R) and its main endogenous ligands arachidonoyl-ethanolamide (AEA) and 2-arachidonoylglycerol (2-AG) [9–11]. The eCBs may interact with other lipidic neurochemical systems which includes GPR55 receptors and their potential endogenous ligand L-α-lysophosphatidylinositol (LPI) [12, 13]. GPR55, a rhodopsin-like seven transmembrane G-protein-coupled receptor, responds to cannabinoids despite lacking the classical cannabinoid binding pocket and sharing less than 15% sequence identity with other eCB receptors [13, 14]. Although several ligands acting at classical CB1Rs are also functional at GPR55, LPI is inactive at CB1R and is gradually becoming the most accepted putative endogenous ligand for the GPR55 receptor [12]. Evidence suggests that GPR55 and its ligand(s) contribute peripherally to energy homeostasis but this remains to be investigated centrally [15].
Classical eCBs are important neuromodulators in feeding brain circuits and are known to stimulate feeding behaviors [16–18]. Reported outcomes of GPR55 activation at synapses in the CNS are opposite to that of eCBs-signalling through CB1Rs, mostly favoring neurotransmitter release [19, 20]. We hypothesized that GPR55 and CB1R might act in concert at ovBNST synapses to orchestrate proper integration of energy homeostasis signals and potentially adaptive feeding behaviors.
We report a form of feeding state-dependent bidirectional plasticity at ovBNST GABA synapses mediated by 2-AG acting at CB1R and LPI at GPR55. Depending on the feeding status of rats, GABA synapses toggled between activity-dependent long-term potentiation (iLTP, fed) and depression (iLTD, food restricted). We identified a novel mechanism underlying LTP of GABA synapses mediated by GPR55 and its putative ligand LPI in the ovBNST of free-fed rats. In contrast, 2-AG produced CB1R-dependent iLTD of ovBNST GABA synapses of food restricted rats. Together, our data suggest that ovBNST GABA synapses are capable of encoding energy status through lipid signaling-mediated long-term plasticity.
Methods
Animals
One hundred and eight long evans rats (Charles River Laboratories, St-Constant, QC, Canada) weighing between 75 and 125 g were pair-housed in clear Plexiglas cages. Thirty adult mice were individually housed in clear Plexiglas cages. Cage floors were lined with bedding (Beta Chip, NEPCO, Warrenburg, NY, USA) and located in climate-controlled colony rooms (21 ± 1 °C; humidity 40–70%) on a 12-h reversed light/dark cycle (7:00 AM lights off –7:00 PM lights on). All experimental manipulations occurred at least 2 h into the dark cycle (approximately 9:00 AM). All animals acclimatized for at least 7 days upon arrival from suppliers. Rodent chow (LabDiet rodent feed #5001, PMI Nutrition International, Brentwood, MO, USA) and water were provided in home cages until behavioral portion of testing. All experiments were conducted in accordance with the Canadian Council on Animal Care guidelines for use of animals in experiments and approved by the Queen’s University Animal Care Committee.
Behavioral manipulations
All behavioral manipulations were conducted in colony room home-cages. Free fed (FF): Fifty-seven rats had free access to food at all time in their home-cage while being pair-housed. However, to control for the potential effect of acute social isolation mild stress during food restriction where rats needed to be single housed for 24 h, 9 FF rats were singly housed for 24 h with ad libitum access to food prior to brain extraction and slices preparation. Food restricted (FR): Early in the dark cycle, rats were singly housed and acutely food restricted (no food for 24 h, n = 51). The following morning, brain slices were prepared for electrophysiology 2 h into the dark cycle (9:00 AM). Refed (RE): A subset of FR rats were acutely refed (n = 7) when chow was freely available for 40 min prior to brain extraction and slices preparation. Water restricted (WR): The potential mild stress effect of nutrient deprivation was also tested with acute water restriction (n = 3). Early in the dark cycle, rats were singly housed with their water bottles removed for 24 h. The following morning, brain slices were prepared for electrophysiology 2 h into the dark cycle (9:00 AM).
CB1R transgenic mouse (CB1R−/−)
Eleven male (5 knockouts [KO, homozygous CB1R-deficient mice, CB1R−/−; 6 wild-type littermates [WT, CB1R+/+]) transgenic mice were provided by Dr. Jean-François Bouchard (Université de Montréal, Montréal, QC, Canada). This targeted mutant was created and characterized by the research team of Pr. Beat Lutz (University of Mainz, Mainz, Germany; [21]). Strain development: In brief, floxed-neo allele bearing mice were created and crossed with transgenic mice ubiquitously expressing Cre recombinase. Mice carrying a germ-line transmissible deletion of cnr1 were backcrossed for five generations into C57BL/6N (Charles Rivers Laboratories, St-Constant, QC, Canada). Expression of CB1R: lack of CB1R expression in the central nervous system of KO mice was previously shown by in situ hybridization and by immunohistochemistry [21].
GPR55 transgenic mouse (GPR55−/−)
Fourteen male (7 knockouts [KO, homozygous Gpr55-deficient mice, GPR55−/−; 7 wild-type littermates [WT, GRP55+/+]) transgenic mice were bred by Dr. Ken Mackie from founders obtained from the Texas A&M Institute of Genomic Medicine (TIGM, College Station, TX, USA) and maintained on a C57BL/6J background. Strain development: in brief, a targeted mutation was generated in 129SvEvBrd-derived embryonic stem (ES) cells using a targeting cassette designed to replace a portion of exon 2 of the Gpr55 gene containing the entire coding region of GPR55 protein with a selection cassette. Homologous integration was detected by Southern Analysis using probes internal to the targeting vector on the 5’ side and external to the targeting vector on the 3’ side. Following blastocyst injection, chimeric mice were bred to C57BL/6J mice to generate F1 heterozygous animals. Subsequently, these mice have been backcrossed onto a C57BL/6J background for >10 generations. Validation of GPR55 knockout: To confirm the appropriate deletion from the GPR55 allele, PCR analysis was conducted [22].
Brain slices preparation and electrophysiology
Rats and mice were anesthetized with isoflurane (5% at 5 L/min) for rapid brain removal and brains were kept in iced-cold physiological solution containing (in mM): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 6 CaCl2, 1.2 NaH2PO4, 25 NaHCO3, and 12.5 d-glucose equilibrated with 95%O2/5%CO2. Coronal brain slices (250 µm) were cut with a vibrating blade microtome (Leica VT-1000, Leica Canada, Richmond Hill, ON, Canada) while in the physiological solution (2 °C). ovBNST-containing slices were incubated at 34 °C for 60 min and transferred to a tissue chamber constantly perfused (3 ml/min) with physiological solution held at RT. All recordings were done at RT to facilitate long-lasting high-quality whole-cell voltage clamp recordings required to study long-term synaptic plasticity. The recordings were made using glass microelectrodes (3.5 MOhm) filled with a solution containing (in mM): 70 Cs+MeSO3−, 58 KCl, 0.5 EGTA, 7.5 HEPES, 1.2 MgCl2, 12 NaCl, 1 MgATP, 0.3 GTP, and 1 P-creatine. For detailed methodology for recordings of ovBNST GABAA-IPSC see [23]. Postsynaptic GABAA currents were evoked by local fiber stimulation with tungsten bipolar electrodes (FHC, Bowdoin, ME, USA) using a bipolar stimulus isolator (World Precision Instruments, Sarasota, FL, USA) in the presence of the AMPA antagonist DNQX (50 µM). Electrodes were placed in the ovBNST, 100–500 µm dorsal from the recorded neurons, and paired electrical stimuli (10–100 µA, 0.1 ms duration, 20 Hz) were evoked at 0.1 Hz. Evoked GABAA-IPSCs were titrated to ~50% of the maximum response before baseline recording to allow for bidirectional plasticity. Following 5 min of stable recording, neurons were subjected to low-frequency stimulation (LFS; 1 Hz, 5 min) or bath application of drugs, followed by a minimum of 30 min post manipulation period. Recordings were made using Multiclamp 700B amplifier and Digidata 1440 A (Molecular Devices Scientific, Sunnyvale, CA, USA), sampled at 10 kHz and filtered at 1 kHz. Cell access was tested with 1 mV, 100 ms test pulses before evoking IPSCs [24]. Data were acquired and analyzed with Axograph X (Axographx.com).
Drugs
Stock solutions of AM251 (10 nM), L-α-lyso-phosphatidyl inositol (LPI; 5 µM), YM 26734 (1 µM), AA COCF3 (1 µM), O-2050 (10 nM), O-1602 (100 nM), CID 16020046 (10 µM), RHC 80267 (100 µM), JNJ 16259685 (50 nM), DHPG (5 µM), dioctanoylglycol (DOG, 10 mM), URB 597 (100 nM), and DNQX (100 mM) were prepared in DMSO (100%). Drugs were dissolved in the physiological solutions at the desired concentration. DMSO concentration never exceeded 0.1%. Drugs were obtained from Sigma-Aldrich Canada (Oakville, ON, Canada), R&D Systems (Minneapolis, MN, USA) or EMD Millipore Corp (Billerica, MA, USA).
Statistical analyses
We measured change in total charge (pA × msec) of postsynaptic currents from baseline in percentage (((Chargepost–Chargebaseline)/Chargebaseline) × 100). Data are reported as mean ± s.e.m. Each data point shown in time-courses is the average of 1-min bins (6 evoked GABAA-IPSCs) across recorded neurons. Paired-pulse ratios (PPR) were calculated by dividing the second (S2) by the first (S1) peak amplitude. Peak amplitudes for S1 and S2 were calculated from baseline values measured immediately before stimulus artefacts; when S1 did not fully decay before S2, baselines were re-zeroed before measuring S2 magnitude (see [23, 25] for details). Coefficient of variations (CV) for each cell were calculated [26]. PPRs and 1/CV2 were compared before and after LFS or drug (bath) application using paired t-tests or Wilcoxon signed-rank test when data violated parametric assumptions.
Neuronal responses to LFS or drug application were averaged for each group and graphically displayed. Where appropriate, responses were further categorized as inhibitory long-term potentiation of evoked GABAA-IPSCs (iLTP), inhibitory long-term depression (iLTD) or no change, using previously established criteria [27–29]. In brief, iLTP-induced changes in IPSC charge was defined as a consistent (>20% deviation from baseline for at least 15 min) change in GABAA-IPSC and observable at 25 min post-LFS or drug application. Likewise, iLTD was defined as a >20% deviation below baseline for at least 15 min and observable at 25 min post-LFS or drug application. Neurons failing to reach either iLTP or iLTD criteria were categorized as no change. Repeated measures analyses of variance (ANOVA) one-way (time) and two-way (time x group) compared the effects of experimental manipulations on GABAA-IPSC change in total charge. For all repeated measures ANOVA, trend analyses (contrasts) were done [30]. Post hoc analyses with Bonferroni correction explored significant main effects and interactions. Contingency tables (the Fisher’s Exact Test [2 × 2] with and without the Freeman-Halton extension [2 × 3]) compared response frequencies (iLTP, iLTD, or no change) across groups. Pearson’s correlation coefficient examined the relationship between Δ GABA-IPSC and baseline PPR. Statistical significance was set at p < 0.05. All statistical analyses were done with SPSS 24.0 (IBM, Chicago, IL, USA) or Prism 6.
Results
Feeding state determined the polarity of bidirectional plasticity at ovBNST GABA synapses
First, we identified a form of activity-dependent long-term potentiation of evoked GABAA-IPSCs (iLTP) in ovBNST neurons of free fed (FF) male rats (time [FF, all neurons], F1,9 = 18, p = 0.009; Fig. 1a). iLTP occurred in a majority of ovBNST neurons (90%) following a 5-min low frequency (1 Hz) local fiber stimulation (LFS; Fig. 1a, e). Twenty-four hours of food restriction (FR) eliminated iLTP (time [FR, all neurons], F1,15 = 0.05, p = 0.5) in part via significantly changing the polarity of the response to LFS, unmasking inhibitory long-term depression (Fisher’s Exact Test with Freeman-Halton extension, FF × FR, p = 0.004; Fig. 1b, e). In some neurons LFS did not produce a net change in GABAA-IPSC, indicating a possible intermediate synaptic state that might exist along the continuum of bidirectional GABA plasticity at ovBNST synapses. PPR and CV analyses suggested that both iLTP and iLTD resulted from a presynaptic change in the probability of GABA release (iLTP, Fig. 1a: PPR, t8 = 3.6, p = 0.007; 1/CV2, t8 = −3.5, p = 0.009; iLTP, Fig. 1b: PPR, t3 = 3.3, p = 0.05; 1/CV2, t3 = 5.5, p = 0.01; iLTD, Fig. 1b: PPR, t6 = −5.2, p = 0.002; 1/CV2, Wilcoxon signed-rank test, p = 0.02). iLTP rapidly recovered in FR rats subjected to a 40-min refeed (RE), suggesting that bidirectional plasticity was tightly linked to the rats’ energy state (Fig. 1c; FF vs RE, Fisher’s exact test, p = 0.2; time × group [FF × FR × RE]: time, F1,39 = 30, p < 0.001; group, F2,39 = 6.2, p = 0.004; interaction, F2,39 = 4.5, p = 0.02). This was further confirmed by comparing peak GABAA-IPSCs following LFS for FF, FR, and RE (Fig. 1e; Group, F2,3 = 7.1, p = 0.002; Post hoc: FF × FR, p = 0.004; FF × RE, p = 1; FR × RE, p = 0.02). Metabolic state might also have modulated the probably of GABA release as FR significantly reduced baseline PPRs (group, FF × FR, F1,24 = 4.3, p = 0.05) and a significant correlation arose between LFS-induced change in GABAA-IPSC magnitude and baseline PPRs (Fig. 1f; r = 0.6, p < 0.001). Nutritional status, more than fluid, determined the direction of ovBNST GABA plasticity as 24 h of water restriction (WR) did not significantly affect LFS-induced change in GABAA-IPSC magnitude (time × group [FF × WR]: time, F1,17 = 21, p < 0.001; group, F1,17 = 0.5, p = 0.5; interaction, F1,17 = 0.01, p = 0.9, Fig. 1d) or the percentage of neurons displaying iLTP, iLTD, or no change (WR × FF, Fisher’s exact test, p = 0.1; Fig. 1e).
Fig. 1.

Metabolic state-dependent bidirectional GABA plasticity in the ovBNST. Effect of low-frequency stimulation (LFS) on electrically evoked GABAA-IPSC (change in total charge) in ovBNST neurons of a free-fed rats (FF), b 24-h acute food restricted (FR) and c 24-h FR followed by a 40-min refeed (RE) as a function of time. Insets in a–c show representative evoked GABAA-IPSC before and after LFS followed by paired-pulse ratios (PPR; S1 [first] and S2 [second] peak amplitudes) and 1/CV2 of S1 peak amplitudes at two time points (1) baseline and (2) 25–30 min following LFS. Double arrows represent LFS (5 min, 1 Hz). Scale bar: 100 pA and 25 ms. Number of rats in each figure is denoted by n. Dashed lines indicate ± 20% change in GABAA-IPSC. d Peak change in GABAA-IPSC for each neuron in FF, FR, RE, and WR; color denotes polarity of response. e Histogram summarizing the proportion and number (overlay on bars) of responding neurons to LFS under conditions a–c, and 24 h acute water restriction (WR). f Baseline PPR alone and plotted against peak change in GABAA-IPSC with polarity of response for each neuron in FF, FR, and RE. Black, all neuron responses averaged; Purple, iLTP; Red, iLTD; Gray, no change. *p < 0.05
Acute (24 h) social isolation alone did not change LFS-induced change GABAA-IPSC magnitude (time × group [group–housed FF × social isolation FF]: time, F1,23 = 38, p < 0.001; group, F1,23 = 1.3, p = 0.3; interaction F1,23 = 4.1, p = 0.06, data not shown) or neuronal response distribution (group-housed FF (n = 9) vs social isolation FF (n = 8); Fisher’s exact test, p = 1, data not shown).
iLTP at ovBNST synapses involved LPI acting at GPR55
GPR55 activation facilitates GABA release in the striatum such that we investigated whether GPR55 was responsible for LFS-induced iLTP in the ovBNST [31]. First, we bath applied the phospholipid LPI on brain slices of FF rats (Fig. 2a, f). LPI (5 µM, 10 min) consistently produced long-lasting increases in GABAA-IPSC magnitude (time [all neurons], F1,8 = 14, p = 0.005) due to an increase in the probability of GABA release (PPR, t8 = 3.1, p = 0.02; 1/CV2, t8 = −2.8, p = 0.02; Fisher’s exact test, FF × LPI, p = 1; Fig. 2a, b, e). LPI is an endogenous ligand of GPR55 suggesting that LFS-induced iLTP in the ovBNST might be mediated by this receptor [12]. Consistent with this hypothesis, the GPR55 receptor antagonist CID 16020046 (10 µM; [32]) abolished iLTP and revealed some iLTD in FF rats (time [all neurons], F1,8 = 0.3, p = 0.6; Fisher’s Exact Test with Freeman-Halton extension, LPI × CID 16020046, p < 0.001; Fig. 2c, f). LPI synthesis relies on phospholipase A (PLA) enzymatic activity and accordingly, postsynaptic PLA1/2 inhibition (i.e., drug in the recording pipette; PLA1 inhibitor: AA COCF3 [1 µM], PLA2 inhibitor: YM 26734 [1 µM]) significantly interfered with iLTP (time [all neurons], F1,14 = 3.0, p = 0.1; Fisher’s exact test with Freeman-Halton extension, LPI × AA COCF3/YM 26,734, p = 0.002; Fig. 2d, f). The above significant effects on bidirectional plasticity were further confirmed by comparing LFS-induced change in GABAA-IPSC (FF, with CID 16020046 in the bath or AACOCF3/YM 26734 intracellular) and bath application (LPI); Fig. 2e; group, F3,38 = 9.5, p < 0.001; Post hoc: FF × LPI, p = 1.0; FF × CID 16020046, p = 0.001; FF × AA COCF3/YM 26734, p = 0.008; CID 16020046 × AA COCF3/YM 26734, p = 1). We used GPR55−/− mice to support our pharmacological evidence of GPR55-mediated iLTP at ovBNST GABA synapses. Although LFS did not produce iLTP in C57 or GPR55+/+ mice as reliably as in rats, LPI-induced iLTP was completely abolished in GPR55−/− mice (Fisher’s exact test, GPR55+/+ × GPR55−/−, p = 0.09; time [GPR55−/−], F1,10 = 0.001, p = 1; Fig. 2f).
Fig. 2.

Role of LPI acting at GPR55 LFS-induced iLTP at ovBNST GABA synapses. a Effects of LPI (5 µM, black bar) on electrically evoked GABAA-IPSC in ovBNST neurons of FF rats. b Effect of LPI on paired-pulse ratios (PPR; S1 [first] and S2 [second] peak amplitudes) and 1/CV2 of S1 peak amplitudes at two time points (1) baseline and (2) 25–30 min following the application of LPI. c Effect of LFS on evoked GABAA-IPSC in the presence of the GPR55 antagonist CID 16020046 (10 µM). d Effect of LFS on evoked GABAA-IPSC in the presence of YM 26734 (1 µM) and AA COCF3 (1 µM), respectively, PLA1 and 2 blockers, in the recording pipette (intracellular). Insets show representative ovBNST GABAA-IPSC before and after LPI a application or LFS c. Double arrows represent LFS (5 min, 1 Hz). Scale bar: 250 pA and 25 ms. Number of rats in each figure is denoted by n. Dashed lines indicate ± 20% change in GABAA-IPSC. e Peak change in GABAA-IPSC for each neuron in FF, LPI, CID 16020046, and YM 26734/AACOCF3(intracellular); color denotes polarity of response. f Histogram summarizing the proportion and number (overlay on bars) of responding neurons to LPI or LFS. Black, all neuron responses averaged; Purple, iLTP; Red, iLTD; Gray, no change.*p < 0.05
iLTD at ovBNST synapses resulted from 2-AG acting at CB1R
eCBs reduce inhibitory synaptic transmission through presynaptic CB1R suggesting a mechanism for LFS-induced iLTD in the ovBNST [33, 34]. Similar to rats, LFS resulted in presynaptic iLTD in FR CB1R+/+ mice (time × group [iLTD × no change]: time, F1,10 = 3.8, p = 0.08; group, F1,10 = 28, p < 0.001; interaction, F1,10 = 6.2, p = 0.03; PPR, t4 = 7.2, p = 0.002; 1/CV2, Wilcoxon signed-rank test, p = 0.04; Fig. 3a, b, g). LFS failed to produce any form of change in GABAA-IPSC in FR CB1R−/− mice suggesting that CB1R may be responsible for iLTD in the ovBNST (time, [CB1R−/−], F1,8 = 2.8, p = 0.1). Pharmacological blockade of CB1R with O-2050 (10 nM) in FR rats completely abolished iLTD to reveal iLTP (time, [all neurons], F1,10 = 14, p = 0.004; Fisher’s exact test with Freeman-Halton extension, FR × O-2050, p = 0.006; Fig. 3c, f). We then sought to identify the endogenous eCB(s) responsible for CB1R-dependent iLTD. eCBs (specifically the lipid-molecule 2-arachidonoylglycerol [2-AG]) can be generated via Group 1 mGluRs activating a Gαq-dependent phospholipase C that cleaves phosphatidyl inositol 4,5-bisphosphate into diacylglycerol (DAG). Subsequently, DAG lipase α (DAGLα) hydrolyzes DAG to 2-AG [35]. Accordingly, interfering with the production of 2-AG with a Group 1 mGluR antagonist (JNJ 162 59685, 50 nM) in FR rats completely eliminated iLTD (time, [all neurons], F1,11 = 3.0, p = 0.1; Fisher’s exact test with Freeman-Halton extension, FR × JNJ 162 59685, p = 0.02, Fig. 3d, g). Conversely, promoting 2-AG synthesis by inhibiting DAG kinase with dioctanoylglycerol (DOG, 10 µM) reduced LFS-induced iLTP and slightly promoted iLTD in FF rat (Fisher’s exact test with Freeman-Halton extension, FF × DOG, p = 0.003, Fig. 3g). Furthermore, reducing 2-AG production by inhibiting DAGLα with RHC 80267 (100 µM) significantly interfered with LFS-induced iLTD in FR rats (time, [all neurons], F1,8 = 2.7, p = 0.1; Fisher’s exact test with Freeman-Halton extension, FR × RHC 80267, p = 0.05, Fig. 3e, g). In contrast, inhibiting the catabolism of anandamide (AEA) with the FAAH blocker URB 597 (100 nM) in FF rats failed to reveal iLTD, indicating 2-AG but not AEA may be responsible for LFS-induced iLTD of ovBNST GABA synapses (Fisher’s exact test with Freeman-Halton extension, FF × URB 597, p = 0.3, Fig. 3f). Mechanistic effects on bidirectional plasticity were further confirmed by comparing LFS-induced change in GABAA-IPSCs magnitude in FR, or FR in the presence of O-2050, JNJ 162 59685, and RHC 80267 (Fig. 3f; group, F3,45 = 4.0, p = 0.01; Post hoc: FR × O-2050, p = 0.01; FR × JNJ 162 59685, p = 1.0; FR × RHC 80267, p = 0.8; JNJ 162 59685 × O-2050, p = 0.1). Notably, neither LFS-induced change in GABAA-IPSC magnitude in the presence of JNJ 162 59685 nor RHC 80267 differed from FR; however, both drugs completely blocked iLTD expected in FR rats, supporting a role for 2-AG in iLTD (Fig. 3d–g).
Fig. 3.

Role of 2-AG acting at CB1R in LFS-induced iLTD at ovBNST GABA synapses. a Effect of low-frequency stimulation (LFS) on electrically evoked GABAA-IPSC in ovBNST neurons in brain slices prepared from food restricted CB1R+/+ and CB1R−/− mice. b Effects of LFS on paired-pulse ratios (PPR; S1 [first] and S2 [second] peak amplitudes) and 1/CV2 of S1 peak amplitudes at two time points (1) baseline and (2) 25–30 min following LFS in CB1R+/+ mice. Effect of LFS on electrically evoked GABAA-IPSC in rat brain slices in the presence of c the CB1R antagonist O-2050 (10 nM), d the Group 1 mGluR antagonist JNJ 16529685 (50 nM) and e the DAGLα blocker RHC 80267 (100 µM). Insets in a, c show representative evoked GABAA-IPSCs before and after LFS. Double arrows represent LFS (5-min, 1-Hz). Scale bar: 250 pA and 25 ms. Number of animals in each figure is denoted by n. Dashed lines indicate ± 20% change in GABAA-IPSC. f Peak change in GABAA-IPSC for each neuron following in LFS in FR, and with O-2050, JNJ 16259685, or RHC 80267 present in the bath; color denotes polarity of response. g Histogram summarizing the effects genetic and pharmacological manipulations of endocannabinoids on LFS-induced change in GABAA-IPSC magnitude. Black, all neuron responses averaged; Purple, iLTP; Red, iLTD; Gray, no change. *p < 0.05
Food restriction enhanced CB1R function at ovBNST GABA synapses
Thus far, our data suggest that LPI/GPR55 (iLTP) and 2-AG/CB1R (iLTD) were the underlying mechanisms determining feeding state-dependent bidirectional plasticity at ovBNST GABA synapses. We next sought to determine whether feeding state affected GPR55 and/or CB1R regulation of ovBNST GABA transmission to bias bidirectional plasticity towards iLTP or iLTD. The iLTP resulting from bath application of the synthetic GPR55 agonist O-1602 (100 nM) was unaffected by FR, showing that GPR55 function was not modified by the rats’ feeding state (Fig. 4a; time × group [FF × FR]: time, F1,19 = 24, p < 0.001; group, F1,19 = 0.008, p = 0.9; interaction, F1,19 = 0.002, p = 1.0). In contrast, CB1R-mediated regulation of ovBNST GABA plasticity was significantly affected by FR. Bath application of the CB1R inverse agonists/antagonists AM251 (10 nM, Fig. 4b; time × group [FF × FR]: time, F1,21 = 26, p < 0.001; group, F1,21 = 8.4, p = 0.009; interaction, F1,21 = 5.1, p = 0.03) or O-2050 (10 nM, Fig. 4c; time × group [FF × FR]: time, F1,20 = 43, p < 0.001; group, F1,20 = 7.8, p = 0.01; interaction, F1,20 = 5.9, p = 0.02) resulted in iLTP that was significantly more robust in FR compared to FF rats. Bath application of AM251 produced iLTP at ovBNST GABA synapses in slices prepared from both GPR55+/+ and GPR55−/− mice suggesting a dominant inverse agonist/antagonist effect at CB1R rather than an agonist effect at GPR55 (Fig. 4d; time × group [GPR55+/+ × GPR5−/−]: time, F1,14 = 21, p < 0.001; group, F1,1 = 2.0, p = 0.2; interaction, F1,14 = 1.4, p = 0.2).
Fig. 4.

Effect of metabolic state on GPR55- and CB1R-mediated regulation of GABAA-IPSCs in the ovBNST. Effect of a the GPR55 agonist O-1602 (100 nM) or b, c the CB1R antagonists/inverse agonists AM-251 (10 nM) or O-2050 (10 nM) on GABAA-IPSC magnitude in FF (circles) or FR (squares). d Effect of the CB1R antagonist/inverse agonist AM-251 on GABAA-IPSC in the ovBNST of FR GPR55+/+ and GPR55−/− mice. Number of animals in each figure is denoted by n. Dashed lines indicate ± 20% change in GABAA-IPSC. Black bar indicates drug application. Purple, iLTP; Gray, no change. *p < 0.05
Discussion
Here, we characterized a bidirectional mechanism of GABA plasticity in the ovBNST that was feeding state-dependent and mediated by an extended endogenous eCB system (Fig. 5). First, low-frequency synaptic activity in the ovBNST promoted iLTP through GPR55 receptors and their putative endogenous ligand, LPI. This mechanism worked in concert with classical eCBs, notably 2-AG, wherein the feeding state of the animal determined the polarity of GABA synaptic plasticity. Thus, in an acute food restricted state, CB1R/2-AG-activity dominated so that low frequency synaptic activity resulted in iLTD. This suggests interplay between constituents of the eCB system encoding the rat energy homeostatic state in the ovBNST.
Fig. 5.
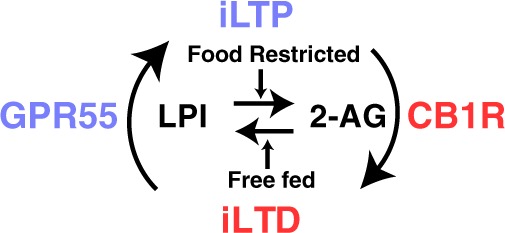
Schematic illustrating a proposed mechanistic interplay between feeding state and bidirectional GABAA-IPSC plasticity in the rat ovBNST
LTP of inhibitory synapses in the ovBNST was LPI- and GPR55 dependent
Our data confirmed that GPR55 and its putative endogenous ligand, LPI, are potent regulators of synaptic transmission in the rat. Short (10 min) bath application of the putative endogenous GPR55 ligand LPI or the synthetic-specific agonist O-1602 produced a long-lasting enhancement of GABAA-IPSC magnitude by increasing the probability of GABA release, identical to that produced by low frequency electrical stimulation. Additionally, pharmacological blockade of GPR55 with CID-160220046 [32] eliminated LFS-induced iLTP, suggesting activity-dependent synthesis of an endogenous ligand acting at GPR55. Accordingly, interfering with LPI synthesis by postsynaptic inhibition of PLA1/2 largely prevented LFS-induced iLTP [36]. Pharmacological blockade of GPR55 or inhibition of LPI synthesis revealed LFS-induced iLTD, strongly suggesting that plasticity at ovBNST GABA synapses was bidirectional. This bidirectional GABA plasticity may occurred at individual neurons and/or synapses, although this needs to be thoroughly confirmed.
We used genetically-engineered mice to support our pharmacological demonstration of activity-dependent synthesis of LPI and GPR55 activation-mediated modulation of ovBNST GABA synapses. However, LFS was notably less effective at inducing iLTP in the ovBNST of C57 or GPR55 WT mice. The reasons for this discrepancy is unknown and potential explanations can only be speculative. One difference is that mice remained singly housed throughout the study such that chronic social isolation may have interfered with LFS-induced iLTP. Another possibility is a specie-specific relationship between bidirectional plasticity of ovBNST GABA synapses and stimulation frequency whereby 1 Hz may not be optimal in mice. Notwithstanding these limitations, we were not able to observe any hint of LFS-induced iLTP in GPR55 null mice (11 neurons in 5 mice), suggesting that GPR55 is indeed critical in activity-dependent iLTP of ovBNST synapses.
Our paired-pulse ratios (PPR) and coefficient of variation (CV) analyses of LFS- or LPI-induced iLTP were consistent with a presynaptic increased in the probability of GABA release [37]. This finding is consistent with the GPR55-dependent enhancement of presynaptic neurotransmitter release found in the hippocampus [19, 20]. Putative postsynaptic blockade of LPI synthesis with intracellular PLA1/2 inhibition suggests a retrograde messenger mode of synaptic modulation for the GPR55 system in the rat ovBNST.
LTD of ovBNST GABA synapses was 2-AG- and CB1R dependent
We next identified that 2-AG acting at CB1R produced iLTD that was uncovered by 24 h of food restriction. In FR male mice (CB1R+/+), LFS produced iLTD that was absent in CB1R null mice (CB1R−/−). Likewise, in FR rats, pharmacologically blocking the CB1R (with O-2050) ablated iLTD. Ubiquitous throughout the brain (including the BNST), CB1Rs have been repeatedly localized on the presynaptic compartment while their lipophilic ligands (e.g., 2-AG) are synthesized on-demand postsynaptically and function as retrograde signals to inhibit glutamate and GABA release [38–40]. Our PPR and CV analyses were consistent with a presynaptic location for CB1Rs and suggested that iLTD resulted from a decrease in GABA release.
Interestingly, our data suggest that 2-AG was the eCB responsible for ovBNST iLTD rather than AEA which mediate LTP/LTD of BNST glutamate synapses [33, 41]. We pharmacologically targeted multiple pathways to pinpoint 2-AG synthesis as the mediator of iLTD at ovBNST synapses. We found that manipulating 2-AG metabolism overrode the effect of food restriction on the polarity of GABA plasticity. For instance, increasing 2-AG synthesis by blocking DAGK with DOG promoted iLTD, whereas decreasing 2-AG production via mGluR blockade or inhibition of DAG lipase completed prevented LFS-induced iLTD. Consistent with a key role for 2-AG in ovBNST iLTD, increasing AEA (via inhibition of FAAH with URB) did not promote the occurrence of iLTD in FF animals.
Together, our data suggest that ovBNST neurons have the ability to synthesize either LPI or 2-AG -depending on the feeding state of the animal- and that both products of intracellular lipid metabolism have the ability to act as retrograde messengers to presynaptically promote or decrease GABA release, respectively, for several minutes.
Feeding state ruled the polarity of bidirectional plasticity at ovBNST GABA synapses
We characterized a mechanism of bidirectional plasticity at ovBNST GABA synapses that was dependent on rats feeding state. We suggest two distinct but complementary molecular mechanisms ruling the polarity of GABA plasticity, but yet, how feeding state orchestrates the biochemical machinery necessary to toggle between on signaling pathway to the other remains to be determined. A positive energy balance (ad libitum feeding) clearly favored the occurrence of LFS-induced iLTP in the ovBNST. However, feeding state did not impact agonist-induced GPR55-mediated change in GABA transmission, suggesting that the regulation of LPI synthesis may be a critical target for energy state variations. Consistent with this hypothesis, 48 h of food restriction affects the levels of circulating plasma LPI [42]. Within the ovBNST, we saw that LPI-dependent iLTP tightly followed the rats’ feeding state being abolished by 24 h of food restriction, but quickly recovering after a short (40 min) access to food. It is possible that feeding (e.g., increased circulating glucose) regulates LPI production via an increase in LPI’s precursor, phosphatidyl inositol [PI] and/or modulating PLA1/2 activity. LPI synthesis may therefore be considered as central satiety signal that might ultimately affect feeding behaviors.
In contrast, food restriction increased CB1R-mediated iLTD in the ovBNST, consistent with the general orexigenic effects of CB1R and 2-AG in the CNS [18]. However, the relationship between eCBs and energy homeostasis is complex as divergent effects on food intake depend on the location and type of synapses (GABA versus glutamate) expressing CB1Rs [16]. In the ovBNST, 24 h of food restriction uncovered iLTD at GABA synapses that was mediated by CB1R activity. Bath application of both CB1R antagonists/inverse agonists (AM251 and O-2050) showed a greater response when the rats were food restricted. We surmised that an increase in GABA was likely due to either inverse agonist actions or GPR55 agonism. We ruled out agonist effects of AM251 at GPR55 by observing identical effects brain slices from FR GPR55−/− and GPR55+/+ mice. Thus, we determined that acute food restriction increased the activity of CB1R in the ovBNST to bias GABA synaptic plasticity toward iLTD. Our data do not allow however to determine whether this effect was mediated by membrane receptor recruitment, enhanced intrinsic CB1R function, or facilitated intracellular signaling as a result of food restriction. Likewise, this functional change of CB1R activity may coincide with increased basal levels of 2-AG, biosynthesis of which is stimulated in the periphery by fasting [43]. Overall, we hypothesize that LPI/GPR55 encodes a satiety signal and conversely, that 2-AG/CB1R signaling is a ‘hunger’ signal in the ovBNST.
The synthetic pathways for both LPI and 2-AG production are intertwined suggesting that both systems, although acting in opposing fashion, work in concert. LPI can be synthesized by PLA1/2 hydrolyzing an acyl group from PI. While 2-AG is classically synthesized by the sequential actions of phospholipase C (PLC) and diacylglycerol lipase on a phospholipid, frequently PI, 2-AG can also be generated by PLA1 hydrolyzing PI to lyso-PI, followed by cleavage of the phosphodiester bond by a lyso-PLC [44]. Corticosteroids inhibit PLA2 activity such that stress states, including acute food restriction, increase circulating corticosteroids level that might inhibit PLA2 ‘unmasking’ postsynaptic production of 2-AG [45]. Therefore, bidirectional plasticity of the ovBNST may in part be mediated by bidirectional regulation of PLA1/2 activity.
It is notable that feeding state may gate plasticity mechanisms in addition to influencing the basal probability of GABA release. We saw a significant increase in the probability of GABA release (i.e. decrease in PPR) in the ovBNST of FR rats. Interestingly, LFS-induced iLTD consistently occurred in neurons with FR-related low PPRs which is consistent with mechanistic hypotheses for homeostatic bidirectional synaptic plasticity [46–48]. Accordingly, it can be speculated that food restriction might reduce threshold for iLTD (or conversely increase threshold for iLTP) through an increase in the probability of ovBNST GABA release.
Importantly, acute stressors such as social isolation and water deprivation did not favor iLTD in the rat ovBNST. One possibility is that these stressors may not increase corticosteroids levels to the same degree as food restriction does. There is evidence that laboratory animals can quickly habituate their corticosteroids response to social isolation [49, 50] and that corticosteroids levels can remain elevated in 48 h fasted but not water-deprived rats [51].
Regardless, bidirectional plasticity at GABA synapses in the ovBNST seems important for energy metabolism-related stress and future experiments should examine the interplay between stressors, corticosteroids levels, and LPI/2-AG synthesis.
Was feeding state-dependent GABA plasticity bidirectional at unitary neuronal populations in the ovBNST?
There is a paucity of studies that explicitly show bidirectional synaptic plasticity within single neurons, and to our knowledge, this has never been shown at GABA synapses [52]. Reasons for this possibly include the challenge of maintaining high quality whole-cell recordings long enough to get both potentiation and depression that can be qualified as long-term within single recordings. It is nonetheless unlikely that a population of synaptic inputs onto a single neuron would not sustain bidirectional plasticity without seriously encroaching the fundamental rules of synaptic homeostasis [46, 48]. Consequently, we cannot claim that GABA inputs onto single ovBNST neurons have the ability to undergo both iLTP and iLTD but this is quite probable. Although impossible to determine whether feeding state can alter the direction of bidirectional plasticity in single neurons in brain slices, future experiments could aim at determining the effect of a second LFS after stable iLTP or iLTD. Nonetheless, we saw iLTP in numerous neurons in this study, but iLTD was restricted to a minority of neurons upon food restriction or pharmacological manipulations. One possibility is that our LFS protocol (1 Hz, 5 min) might not be optimal for iLTD and whether other plasticity protocols (frequency and duration) may affect the direction of bidirectional plasticity at ovBNST GABA synapses remains to be examined. This potential relationship between plasticity protocol and neurophysiological outcome could also have explained the difference between rats and mice that we observed in our study.
One important question is also whether all ovBNST neurons can synaptically detect changes in the animal’s feeding state. It is reasonable to claim that the vast majority of ovBNST neurons can undergo LFS-induced iLTP. However, our data suggest that only a subpopulation of ovBNST neurons switched polarity with caloric status and that iLTP resisted food restriction in some neurons. This is not to say that those neurons resisting food restriction cannot undergo iLTD but perhaps, not in response to an acute caloric challenge. Interestingly, we saw an enhancement in the probability of GABA release (decrease in baseline PPR) in neurons undergoing LFS-induced iLTD in FR rats. This suggests that food restriction strengthened GABA inputs onto a subpopulation of ovBNST neurons which, through an enhancement in CB1R function, became homeostatically primed for iLTD. The identity of these potentially caloric challenge-sensitive ovBNST neurons is currently unknown. There is a prevalence of neurotensin-positive neurons in the ovBNST which largely co-express CRF and the opioid peptide dynorphin [28, 29]. Based on some of our previous work and the work of others, there is a distinct population of enkephalin-positive neurons [28, 53, 54]. Others have suggested subpopulations of neurons with distinct neurophysiological signatures (type I, II, or III) in the rat dorsolateral BNST [55, 56]. Most likely because we are anatomically conservative in our recordings (i.e. recordings strictly restricted to the oval subregion of the dlBNST), we do not typically (rarely) detect neurons with distinct neurophysiological signatures (see methods section). Nonetheless, it will be interesting to determine which subtype(s) of ovBNST neurons might have the ability to detect caloric challenge, i.e. whether they are projection neurons and if so, whether they have specific targets or whether they are interneurons. It would also be important to determine whether neurons with specific neurochemical contents (neurotensin/CRF/dynorphin vs. ENK) have the ability to detect caloric challenges or other type of homeostatic disturbances (e.g. fluids). Neuronal heterogeneity could also explain the striking difference in GABA plasticity we observed between rats and mice.
Conclusions
The ovBNST is ideally organized and connected to regulate diverse motivational states, in part via its synaptic targets [6, 57–60]. Primarily comprising GABAergic neurons, the BNST can regulate feeding circuits through a monosynaptic GABA projection to the lateral hypothalamus (LH) to suppress glutamatergic neurons and promote feeding [6]. Here we show that ovBNST synaptic plasticity was sensitive to and depended on rats feeding state, seesawing between an inhibited sated and an active food restricted state. Thus, GABAergic projection neurons from the ovBNST in food deprived animals could plausibly restrict LH glutamatergic neurons activity to promote feeding. Inevitably, the level of synaptic and neuronal activity that will influence synaptic activity in the ovBNST will be tributary to extrinsic excitatory and inhibitory inputs. In fact, the ovBNST does receive inputs from brain regions that are clearly part of the brain’s energy metabolism circuits such as the PVT (glutamate), insular cortex (glutamate), and the central amygdala (GABA) [61–63]. Exploring the behavioral consequences of altered satiety-state-dependent bidirectional GABA plasticity will be key to uncovering the adaptive and maladaptive function of these circuits.
Funding and disclosure
Dr. Emily R. Hawken was funded by the CIHR Postdoctoral Fellowship (MFE-123712). Dr. Catherine P. Normandeau was funded by the CIHR Vanier Graduate Scholarship (338319). Dr. James Gregory Gardner was funded by the NSERC Vanier Graduate Scholarship (201411DVC-347523-257929). Dr. Jean-François Bouchard was funded by the Natural Science and Engineering Research Council of Canada (NSERC, RGPAS 478115-2015/RGPIN 2015-06582) and the Canadian Institutes of Health Research (PJT-156029). Dr. Ken Mackie was funded by the National Institutes of Health (DA021696). Dr. Éric C. Dumont was funded by the Canadian Institute of Health Research (MOP-25953). The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cameron J, Doucet E. Getting to the bottom of feeding behaviour: who’s on top? Appl Physiol Nutr Metab. 2007;32:177–89. doi: 10.1139/h06-072. [DOI] [PubMed] [Google Scholar]
- 2.Dong HW, Petrovich GD, Watts AG, Swanson LW. Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J Comp Neurol. 2001;436:430–55. doi: 10.1002/cne.1079. [DOI] [PubMed] [Google Scholar]
- 3.Dong HW, Swanson LW. Projections from bed nuclei of the stria terminalis, anteromedial area: cerebral hemisphere integration of neuroendocrine, autonomic, and behavioral aspects of energy balance. J Comp Neurol. 2006;494:142–78. doi: 10.1002/cne.20788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dumont EC. What is the bed nucleus of the stria terminalis? Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1289–90. doi: 10.1016/j.pnpbp.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angeles-Castellanos M, Mendoza J, Escobar C. Restricted feeding schedules phase shift daily rhythms of c-Fos and protein Per1 immunoreactivity in corticolimbic regions in rats. Neuroscience. 2007;144:344–55. doi: 10.1016/j.neuroscience.2006.08.064. [DOI] [PubMed] [Google Scholar]
- 6.Jennings JH, Rizzi G, Stamatakis AM, Ung RL, Stuber GD. The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science. 2013;341:1517–21. doi: 10.1126/science.1241812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Marzo V, Ligresti A, Cristino L. The endocannabinoid system as a link between homoeostatic and hedonic pathways involved in energy balance regulation. Int J Obes. 2009;33(Suppl 2):S18–24. doi: 10.1038/ijo.2009.67. [DOI] [PubMed] [Google Scholar]
- 8.Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev. 2006;27:73–100. doi: 10.1210/er.2005-0009. [DOI] [PubMed] [Google Scholar]
- 9.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–9. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 10.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–4. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 11.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-D. [DOI] [PubMed] [Google Scholar]
- 12.Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun. 2007;362:928–34. doi: 10.1016/j.bbrc.2007.08.078. [DOI] [PubMed] [Google Scholar]
- 13.Sawzdargo M, Nguyen T, Lee DK, Lynch KR, Cheng R, Heng HH, et al. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, PsiGPR53 and GPR55: GPR55 is extensively expressed in human brain. Brain Res Mol Brain Res. 1999;64:193–8. doi: 10.1016/S0169-328X(98)00277-0. [DOI] [PubMed] [Google Scholar]
- 14.Henstridge CM, Balenga NA, Kargl J, Andradas C, Brown AJ, Irving A, et al. Minireview: recent developments in the physiology and pathology of the lysophosphatidylinositol-sensitive receptor GPR55. Mol Endocrinol. 2011;25:1835–48. doi: 10.1210/me.2011-1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tuduri E, Imbernon M, Hernandez-Bautista RJ, Tojo M, Ferno J, Dieguez C, et al. GPR55: a new promising target for metabolism? J Mol Endocrinol. 2017;58:R191–R202. doi: 10.1530/JME-16-0253. [DOI] [PubMed] [Google Scholar]
- 16.Bellocchio L, Lafenetre P, Cannich A, Cota D, Puente N, Grandes P, et al. Bimodal control of stimulated food intake by the endocannabinoid system. Nat Neurosci. 2010;13:281–3. doi: 10.1038/nn.2494. [DOI] [PubMed] [Google Scholar]
- 17.Cristino L, Becker T, Di Marzo V. Endocannabinoids and energy homeostasis: an update. Biofactors. 2014;40:389–97. doi: 10.1002/biof.1168. [DOI] [PubMed] [Google Scholar]
- 18.Crosby KM, Inoue W, Pittman QJ, Bains JS. Endocannabinoids gate state-dependent plasticity of synaptic inhibition in feeding circuits. Neuron. 2011;71:529–41. doi: 10.1016/j.neuron.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hurst K, Badgley C, Ellsworth T, Bell S, Friend L, Prince B, et al. A putative lysophosphatidylinositol receptor GPR55 modulates hippocampal synaptic plasticity. Hippocampus. 2017;27:985–98. doi: 10.1002/hipo.22747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sylantyev S, Jensen TP, Ross RA, Rusakov DA. Cannabinoid- and lysophosphatidylinositol-sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc Natl Acad Sci USA. 2013;110:5193–8. doi: 10.1073/pnas.1211204110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–4. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 22.Wu CS, Zhu J, Wager-Miller J, Wang S, O’Leary D, Monory K, et al. Requirement of cannabinoid CB(1) receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. Eur J Neurosci. 2010;32:693–706. doi: 10.1111/j.1460-9568.2010.07337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krawczyk M, Georges F, Sharma R, Mason X, Berthet A, Bezard E, et al. Double-dissociation of the catecholaminergic modulation of synaptic transmission in the oval bed nucleus of the stria terminalis. J Neurophysiol. 2011;105:145–53. doi: 10.1152/jn.00710.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dumont EC, Williams JT. Noradrenaline triggers GABAA inhibition of bed nucleus of the stria terminalis neurons projecting to the ventral tegmental area. J Neurosci. 2004;24:8198–204. doi: 10.1523/JNEUROSCI.0425-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krawczyk M, Sharma R, Mason X, Debacker J, Jones AA, Dumont EC. A switch in the neuromodulatory effects of dopamine in the oval bed nucleus of the stria terminalis associated with cocaine self-administration in rats. J Neurosci. 2011;31:8928–35. doi: 10.1523/JNEUROSCI.0377-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J. 1991;60:1288–94. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naughton MH. Electrophysiological study of the effects of arginine vasopressin in the rat juxtacapsular nucleus of the bed nucleus of the stria terminalis. MSc thesis, Queen’s University, Queen’s Graduate Theses and Dissertations (2016).
- 28.Normandeau CP, Torruella Suarez ML, Sarret P, McElligott ZA, Dumont EC. Neurotensin and dynorphin Bi-Directionally modulate CeA inhibition of oval BNST neurons in male mice. Neuropharmacology. 2018;143:113–21. doi: 10.1016/j.neuropharm.2018.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Normandeau CP, Ventura-Silva AP, Hawken ER, Angelis S, Sjaarda C, Liu X, et al. A key role for neurotensin in chronic-stress-induced anxiety-like behavior in rats. Neuropsychopharmacol: Off Publ Am Coll Neuropsychopharmacol. 2018;43:285–93. doi: 10.1038/npp.2017.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glass GV, Hopkins KD. Statistical methods in psychology and education, 3rd edn. Allyn and Bacon, MarylandAllyn and Bacon, Maryland (1996).
- 31.Musella A, Fresegna D, Rizzo FR, Gentile A, Bullitta S, De Vito F, et al. A novel crosstalk within the endocannabinoid system controls GABA transmission in the striatum. Sci Rep. 2017;7:7363. doi: 10.1038/s41598-017-07519-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kargl J, Brown AJ, Andersen L, Dorn G, Schicho R, Waldhoer M, et al. A selective antagonist reveals a potential role of G protein-coupled receptor 55 in platelet and endothelial cell function. J Pharmacol Exp Ther. 2013;346:54–66. doi: 10.1124/jpet.113.204180. [DOI] [PubMed] [Google Scholar]
- 33.Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, et al. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci. 2011;14:1542–7. doi: 10.1038/nn.2974. [DOI] [PubMed] [Google Scholar]
- 34.Castillo PE, Younts TJ, Chavez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–8. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 36.Pineiro R, Falasca M. Lysophosphatidylinositol signalling: new wine from an old bottle. Biochim Biophys Acta. 2012;1821:694–705. doi: 10.1016/j.bbalip.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 37.Del Castillo J, Katz B. Quantal components of the end-plate potential. J Physiol. 1954;124:560–73. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Massi L, Elezgarai I, Puente N, Reguero L, Grandes P, Manzoni OJ, et al. Cannabinoid receptors in the bed nucleus of the stria terminalis control cortical excitation of midbrain dopamine cells in vivo. J Neurosci. 2008;28:10496–508. doi: 10.1523/JNEUROSCI.2291-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel NA, Moldow RL, Patel JA, Wu G, Chang SL. Arachidonylethanolamide (AEA) activation of FOS proto-oncogene protein immunoreactivity in the rat brain. Brain Res. 1998;797:225–33. doi: 10.1016/S0006-8993(98)00364-3. [DOI] [PubMed] [Google Scholar]
- 40.Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–82. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- 41.Puente N, Elezgarai I, Lafourcade M, Reguero L, Marsicano G, Georges F, et al. Localization and function of the cannabinoid CB1 receptor in the anterolateral bed nucleus of the stria terminalis. PLoS ONE. 2010;5:e8869. doi: 10.1371/journal.pone.0008869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imbernon M, Whyte L, Diaz-Arteaga A, Russell WR, Moreno NR, Vazquez MJ, et al. Regulation of GPR55 in rat white adipose tissue and serum LPI by nutritional status, gestation, gender and pituitary factors. Mol Cell Endocrinol. 2014;383:159–69. doi: 10.1016/j.mce.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 43.DiPatrizio NV, Igarashi M, Narayanaswami V, Murray C, Gancayco J, Russell A, et al. Fasting stimulates 2-AG biosynthesis in the small intestine: role of cholinergic pathways. Am J Physiol Regul Integr Comp Physiol. 2015;309:R805–813. doi: 10.1152/ajpregu.00239.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murataeva N, Straiker A, Mackie K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br J Pharmacol. 2014;171:1379–91. doi: 10.1111/bph.12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shukla PK, Gautam L, Sinha M, Kaur P, Sharma S, Singh TP. Structures and binding studies of the complexes of phospholipase A2 with five inhibitors. Biochim Biophys Acta. 2015;1854:269–77. doi: 10.1016/j.bbapap.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 46.Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- 47.deBacker J, Hawken ER, Normandeau CP, Jones AA, Di Prospero C, Mechefske E, et al. GluN2B-containing NMDA receptors blockade rescues bidirectional synaptic plasticity in the bed nucleus of the stria terminalis of cocaine self-administering rats. Neuropsychopharmacology. 2015;40:394–405. doi: 10.1038/npp.2014.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abraham WC, Mason-Parker SE, Bear MF, Webb S, Tate WP. Heterosynaptic metaplasticity in the hippocampus in vivo: a BCM-like modifiable threshold for LTP. Proc Natl Acad Sci USA. 2001;98:10924–9. doi: 10.1073/pnas.181342098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hodges TE, McCormick CM. Adolescent and adult male rats habituate to repeated isolation, but only adolescents sensitize to partner unfamiliarity. Horm Behav. 2015;69:16–30. doi: 10.1016/j.yhbeh.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 50.Ravenelle R, Santolucito HB, Byrnes EM, Byrnes JJ, Donaldson ST. Housing environment modulates physiological and behavioral responses to anxiogenic stimuli in trait anxiety male rats. Neuroscience. 2014;270:76–87. doi: 10.1016/j.neuroscience.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tinnikov AA. Responses of serum corticosterone and corticosteroid-binding globulin to acute and prolonged stress in the rat. Endocrine. 1999;11:145–50. doi: 10.1385/ENDO:11:2:145. [DOI] [PubMed] [Google Scholar]
- 52.Royer S, Pare D. Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience. 2002;115:455–62. doi: 10.1016/S0306-4522(02)00455-4. [DOI] [PubMed] [Google Scholar]
- 53.Day HE, Curran EJ, Watson SJ, Jr., Akil H. Distinct neurochemical populations in the rat central nucleus of the amygdala and bed nucleus of the stria terminalis: evidence for their selective activation by interleukin-1beta. J Comp Neurol. 1999;413:113–28. doi: 10.1002/(SICI)1096-9861(19991011)413:1<113::AID-CNE8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 54.Ju G, Han ZS. Coexistence of corticotropin releasing-factor and neurotensin within oval nucleus neurons in the bed nuclei of the stria terminalis in the rat. Neurosci Lett. 1989;99:246–50. doi: 10.1016/0304-3940(89)90454-0. [DOI] [PubMed] [Google Scholar]
- 55.Hammack SE, Mania I, Rainnie DG. Differential expression of intrinsic membrane currents in defined cell types of the anterolateral bed nucleus of the stria terminalis. J Neurophysiol. 2007;98:638–56. doi: 10.1152/jn.00382.2007. [DOI] [PubMed] [Google Scholar]
- 56.Dabrowska J, Hazra R, Guo JD, Li C, Dewitt S, Xu J, et al. Striatal-enriched protein tyrosine phosphatase-STEPs toward understanding chronic stress-induced activation of corticotrophin releasing factor neurons in the rat bed nucleus of the stria terminalis. Biol Psychiatry. 2013;74:817–26. doi: 10.1016/j.biopsych.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jennings JH, Sparta DR, Stamatakis AM, Ung RL, Pleil KE, Kash TL, et al. Distinct extended amygdala circuits for divergent motivational states. Nature. 2013;496:224–8. doi: 10.1038/nature12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim SY, Adhikari A, Lee SY, Marshel JH, Kim CK, Mallory CS, et al. Diverging neural pathways assemble a behavioural state from separable features in anxiety. Nature. 2013;496:219–23. doi: 10.1038/nature12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krawczyk M, Mason X, DeBacker J, Sharma R, Normandeau CP, Hawken ER, et al. D1 dopamine receptor-mediated LTP at GABA synapses encodes motivation to self-administer cocaine in rats. J Neurosci. 2013;33:11960–71. doi: 10.1523/JNEUROSCI.1784-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maracle Amanda C., Normandeau Catherine P., Dumont Éric C., Olmstead Mary C. Dopamine in the oval bed nucleus of the stria terminalis contributes to compulsive responding for sucrose in rats. Neuropsychopharmacology. 2018;44(2):381–389. doi: 10.1038/s41386-018-0149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dong HW, Petrovich GD, Swanson LW. Topography of projections from amygdala to bed nuclei of the stria terminalis. Brain Res Brain Res Rev. 2001;38:192–246. doi: 10.1016/S0165-0173(01)00079-0. [DOI] [PubMed] [Google Scholar]
- 62.Li S, Kirouac GJ. Projections from the paraventricular nucleus of the thalamus to the forebrain, with special emphasis on the extended amygdala. J Comp Neurol. 2008;506:263–87. doi: 10.1002/cne.21502. [DOI] [PubMed] [Google Scholar]
- 63.McDonald AJ, Shammah-Lagnado SJ, Shi C, Davis M. Cortical afferents to the extended amygdala. Ann N Y Acad Sci. 1999;877:309–38. doi: 10.1111/j.1749-6632.1999.tb09275.x. [DOI] [PubMed] [Google Scholar]