

Abstract
The glycopeptide analysis field is tightly constrained by a lack of effective tools that translate mass spectrometry data into meaningful chemical information, and perhaps the most challenging aspect of building effective glycopeptide analysis software is designing an accurate scoring algorithm for MS/MS data. Herein, we provide the glycoproteomics community with two tools to address this challenge. The first tool, a curated set of 100 expert-assigned CID spectra of glycopeptides, contains a diverse set of spectra from a variety of glycan types; the second tool, Glycopeptide Decoy Generator, is a new software application that generates glycopeptide decoys de novo. We developed these tools so that emerging methods of assigning glycopeptides’ CID spectra could be rigorously tested. Software developers or those interested in developing skills in expert (manual) analysis can use these tools to facilitate their work. We demonstrate the tools’ utility in assessing the quality of one particular glycopeptide software package, GlycoPep Grader, which assigns glycopeptides to CID spectra. We first acquired the set of 100 expert assigned CID spectra; then we used the Decoy Generator (described herein) to generate 20 decoys per target glycopeptide. The assigned spectra and decoys were used to test the accuracy of GlycoPep Grader’s scoring algorithm; new strengths and weaknesses were identified in the algorithm using this approach. Both newly-developed tools are freely available to interested parties. The software can be downloaded at http://glycopro.chem.ku.edu/GPJ.jar
Keywords: Glycopeptide, Decoys, False Discovery Rate, Collision Induced Dissociation, Liquid Chromatography-Mass Spectrometry
Graphical Abstract

INTRODUCTION
Glycosylation is a common but complex post-translational modification that occurs on proteins during their biosynthesis, and it is known to regulate several biological processes such as cell signaling,1,2 protein folding,3,4 transportation,3,5 and degradation.5 Changes in the glycosylation profiles of endogenous glycoproteins can serve as biomarkers for diseases diagnosis and progression.6,7 In addition, glycosylation can impact the biological activity,8,9 immunogenicity,9,10 and stability11 of glycoprotein-based drugs. Hence, extensive characterization of glycosylation on glycoproteins is vital in understanding important biological events and diseases, as well as the pharmacodynamics and pharmacokinetics of glycoprotein-based drugs.
Mass spectrometry (MS) has become an invaluable analytical tool for glycosylation characterization due to its high sensitivity, high resolution, and complementary fragmentation techniques.12 Two main methods for mass spectrometric glycosylation analysis of glycoproteins are the glycan-based approach13,14 and the glycopeptide-based approach.15,16 While the former approach gives information about the total glycan pool on a glycoprotein, the latter approach provides glycosylation site-specific information. Because glycopeptide analysis is the method of choice for glycosylation profiling of proteins containing more than one glycosylation site, we focus on it herein. Although advances in mass spectrometry instrumentation, sample preparation,17,18 and data acquisition methods19,20 have contributed to advances in glycopeptide analyses, interpretation of the resulting mass spectrometry data from tandem MS experiments remains an additional ongoing field of development.
An area of increasing interest in glycopeptide analysis is, therefore, the development of bioinformatics tools for rapid and automated assignment of glycopeptides to MS/MS data. Glycopeptides are typically analyzed by tandem mass spectrometry using fragmentation methods such as collision induced dissociation (CID), electron transfer dissociation (ETD), and higher energy collision dissociation (HCD). Manual analysis of glycopeptide data generated by these fragmentation methods provides the most confident glycopeptide assignments, but it is extremely time-consuming and requires extensive experience in data analysis. Hence, several bioinformatics tools have been developed for the interpretation of glycopeptide MS/MS data. Examples include GlycoPep Grader,21 GlycoPeptideSearch,22 and MAGIC23 that assign glycopeptides to CID spectra; GPQuest24 and pGlyco25 for assignment of glycopeptides to HCD spectra; and GlycoPep Detector26 and GlycoPep Evaluator27 for assignment of glycopeptides to ETD spectra. Other tools such as GlycoFragwork28 and GlycoMaster DB29 assign glycopeptide spectra based on a combination of two or more fragmentation techniques. A number of reviews that describe these tools, and others, in detail have been reported.30–32 In general, bioinformatics tools match glycopeptides to MS/MS data by scoring potential glycopeptide candidates against a particular MS/MS spectrum, and the candidate with the highest score is assigned to the spectrum.21,26,27 However, automated glycopeptide assignments can be problematic, as the best match for a spectrum can sometimes be an incorrect match. Therefore, it is vital for researchers to assess the accuracy of algorithms that assign glycopeptides to MS/MS data in order to ensure confidence of the results; this rigorous testing also affords developers valuable information that can be used to improve the algorithms.
Herein, we release two new bioinformatics tools to the community; they support glycopeptide analysis software developers and those assigning CID spectra of glycopeptides, either manually, or by an automated tool. The first product, Glycopeptide Decoy Generator (GDG), rapidly generates abundant decoy glycopeptides de novo, and enables determination of the accuracy of tools that assign glycopeptides to CID data. Large numbers of decoys can be easily generated for glycopeptides using our tool. GDG generates abundant decoys for any target glycopeptide, and all the decoys have biologically relevant glycan components. The second product we provide herein is a dataset of 100 expert-assigned CID spectra of a diverse set of glycosylated peptides. The dataset contains all major N-glycosylation types, including sialylated and fucosylated glycoforms. We demonstrate the tools’ utility in assessing Glycopep Grader’s scoring algorithm; this tool assigns glycopeptides to CID spectra. Both newly-developed tools described herein are freely available to interested parties.
EXPERIMENTAL SECTION
Materials and Reagents
Avidin, IgG1, bovine ribonuclease B (RNAse B), bovine fetuin, human apo-transferrin, Tris(hydroxylmethyl)aminomethane (Trizma) base, urea, dithiotreitol (DTT), iodoacetamide (IAM), and formic acid were purchased from Sigma-Aldrich (St. Louis, MO). IgG2 and IgG3 were from Fitzgerald (Acton, MA), and sequencing grade trypsin was from Promega (Madison, WI). HIV-1 envelope glycoprotein samples, C.97ZA012 gp140 and A244-V1V2, were from the Duke Human Vaccine Institute (Durham, NC). Ultrapure water was obtained via a Direct-Q water purification system (MilliporeSigma, Darmstadt, Germany).
Sample Preparation
For the HIV-1 Env proteins, the samples were prepared as reported in Reference 36. Briefly, about 100 μg of each of the proteins were dissolved in 100 mM Tris buffer (pH 8.0), and urea was added to a final concentration of 6 M to denature the proteins. Subsequently, DTT was added to reduce the disulfide bonds, and the reaction was allowed to proceed for 1 h at room temperature. After disulfide bond reduction, IAM was added to a final concentration of 10 mM and incubated for 1 h in the dark to cap free cysteine residues; followed by addition of excess DTT to react with excess IAM. The excess salt (urea and DTT) were either diluted to less than 1M (for gp140) or removed by centrifugal filtration of the samples using 10 kDa molecular weight cut-off filters (for A244-V1V2), and the samples were reconstituted in Tris buffer (pH 8.0) to a final concentration of 2 μg/μL. Finally, trypsin was added at an enzyme-to-protein ratio of 1:30 (w/w) and incubated for 18 h at 37 ◦C, followed by a second trypsin addition at an enzyme-to-protein ratio of 1:30 (w/w) under the same conditions.
For all other glycoproteins, the sample preparation was the same as described above, but with a slight modification during the digestion step. Trypsin digestion was done at 1:30 (w/w) enzyme-to-protein ratio for 18 h at 37 ◦C, followed by a second trypsin addition at an enzyme-to-protein ratio of 1:100 (w/w) for additional 3 h. After digestion, all samples were quenched by addition of 1 μL formic acid for every 100 μL of sample. The samples were analyzed immediately after digestion and/or aliquoted and stored at −20 C until analysis.
LC Separation and MS Data Acquisition
LC-MS analysis was conducted on a Waters Acquity Ultra Performance Liquid Chromatography instrument (Waters Acquity, Milford, MA) coupled to either a LTQ Velos Linear Ion Trap or a LTQ Orbitrap Velos Pro hybrid Mass Spectrometer (Thermo Scientific, San Jose, CA). For avidin and the HIV-1 Env protein, C.97ZA012 gp140, data was acquired on the LTQ Velos Linear Ion Trap, and the column dimensions, gradient, and CID data acquisition settings are the same as those reported in Reference 27. For the remaining proteins, data was acquired on the LTQ Orbitrap Velos Pro. The Mobile Phase A was 99.9% LC/MS-grade water containing 0.1% formic acid and Mobile Phase B was 99.9% acetonitrile with 0.1% formic acid. A C18 Aquasil Gold column (100 × 1 mm i.d, 175 Å, Thermos Scientific, San Jose, CA) was used for reversed phase separation. Sample solutions of 5 μL were injected onto the column and separated at a flow rate of 50 μL/min as follows: The mobile phase B was initially maintained at 2% for 5 min followed by an increase to 35% in 60 min, and then ramped to 60% in 15 min. Mobile phase B was held at 95% for 10 min prior to re-equilibration of the column at 2% B for 10 min.
For both the LTQ Velos and the Orbitrap Velos Pro mass spectrometers, data-dependent acquisition was performed in the positive ion mode, and the acquisition parameters were optimized for each protein. The ESI source spray voltage was maintained at 3.0 kV and the capillary temperature was 200 ◦C for HIV-1 C.97ZA012 gp140, 260 ◦C for RNAse B, 275 ◦C for IgG2, and 250 ◦C for all other proteins. In all experiments, a survey MS scan was obtained from m/z 400 or 500 to 2000 prior to CID fragmentation in the linear ion trap. For MS scans obtained in the Orbitrap mass analyzer, the resolution was set at 30,000 (at m/z 400). CID spectra were obtained by selecting the top 5 ions (top 8 for RNAse B) for CID fragmentation in the linear ion trap. The CID normalization collision energy was 35% (30% for HIV-1 C.97ZA012 gp140) with an activation time of 10 ms and a 3 Da isolation window.
Glycopeptide Spectral Library
A library of glycopeptides’ CID spectra was generated using the following proteins: IgG1, IgG2, IgG3, bovine fetuin, RNAse B, avidin, transferrin, and two HIV-1 Env proteins (C.97ZA012 gp140 and A244-V1V2). These glycoproteins, with the exception of A244-V1V2, have been well-characterized and reported in literature.33–37 For the glycopeptides that have been reported in literature, the dataset of manually characterized CID spectra was generated as follows: For each glycoprotein, a list of previously assigned glycopeptide compositions was compiled, and the theoretical monoisotopic m/z values at different charge states were computed and searched for in the MS data file of the glycopeptide digest of the protein. When a match was found, we determined if the peak was selected for CID fragmentation. If a corresponding CID spectrum was found within 1 minute of the retention time of the peak in the full-MS scan, and if characteristic glycan oxonium ions (e.g ions at m/z 366, 528, and 690) were identified, the spectrum was manually assigned based on knowledge of glycopeptide fragmentation under CID conditions. For A244-V1V2, which has not been reported in the literature, the glycopeptides were assigned using a previously described workflow for characterizing glycosylation on complex glycoproteins.36,38 Briefly, compositional analysis of glycopeptides was carried out by first doing an in silico digestion of the protein to find peptides with the N-X-S/T glycosylation site motif; then CID spectra were identified that contained an abundant ion consistent with the Y1 ions39,40 (ions which are typically used to identify the peptide portions of glycopeptide compositions) that would be generated from these glycopeptides. Once candidate CID spectra were identified in this way, plausible glycopeptide compositions were obtained using high-resolution MS data and GlycoPep DB.41 Potential glycopeptide candidates with experimental monoisotopic m/z values within 10 ppm from the theoretical m/z of the glycopeptide reported by GlycoPep DB were confirmed manually by annotating the glycosidic cleavages observed in the CID data. Overall, for all the proteins, each confirmed glycopeptide assignment in the dataset met the following criteria: (1) the experimental monoisotopic mass of the precursor ion closely matched the theoretical monoisotopic mass of the assigned glycopeptide (within 10 ppm for Orbitrap data and 30 ppm for LTQ Velos data); (2) the CID spectrum contained an intense Y1 ion of the glycopeptide; (3) the CID spectrum contained all or some of the following characteristic oxonium ion peaks: m/z 366, 528, 690, and 657 (for sialylated glycopeptides); and (4) glycosidic cleavages consistent with neutral losses of the monosaccharides present in the glycopeptide were observed. The Supplemental Data includes 13 example annotated spectra. Furthermore, the peaklists for all one hundred CID spectra are supplied in the Supplemental Data, along with their assigned glycopeptide compositions. These spectral data can be used by other software developers who wish to test CID algorithms against expert-verified, pre-assigned data.
RESULTS AND DISCUSSION
Tool 1: The Glycopeptide Decoy Generator
Glycopeptide Decoy Generator (GDG) is a free tool designed to generate abundant glycopeptide decoys for accurate assessment of glycopeptide scoring algorithms that match CID spectra to glycopeptide compositions. Figure 1A shows the graphical user interface of the decoy generator. GDG contains two main menus, the “Input Data” menu and the “Result” menu. To generate decoys, the user enters the monoisotopic m/z value and charge state of the target glycopeptide composition (target), the number of decoys to generate, as well as the desired mass tolerance (in ppm) of the decoys from the target. In addition, the peptide and glycan portions of the target glycopeptide are entered in two adjacent windows with each peptide portion aligned with its glycan portion. The decoys generated by GDG can be divided into three categories based on their glycan compositions, and the user may enter the number of decoys required for each category (“Num from Each Category”). The three decoy categories are: (I) decoys containing [HexNAc]2[Hex]1-n[Fuc]0–2, where n is any integer greater than 1; (II) decoys containing [NeuNAc], and (III) decoy glycopeptides not belonging to categories (I) or (II). If the sum of the numbers entered for each decoy category is lower than the total number of decoys entered by the user, the software randomly adds decoys from all three categories to make up the total number of decoys required. Finally, if the peptide portion contains a cysteine (Cys) residue, the user must specify whether or not the cysteine residue is modified. The current version of GDG has options for cysteine modification using iodoacetamide, iodoacetic acid, and vinyl pyridine, which are the commonly used Cys alkylating agents. For any other modification, including cysteine modification with other alkylating agents like N-ethylmaleimide, the mass of the modification can be entered in brackets after the amino acid residue that is modified. For example, if the peptide DETMFNASQR has Met oxidation, it would be entered as DETM(+15.99)FNASQR. The “Input tips” field at the bottom of the software provides guides with regards to the aforementioned parameters that have to be entered in the “input data” menu. In this study, decoys were generated at a target-to-decoy ratio of 1:20, the “mass/charge tolerance” was set at 20 ppm, the number of decoys from each category was 3, and iodoacetamide was selected for Cys modification of all Cys-containing target glycopeptides.
Figure 1.
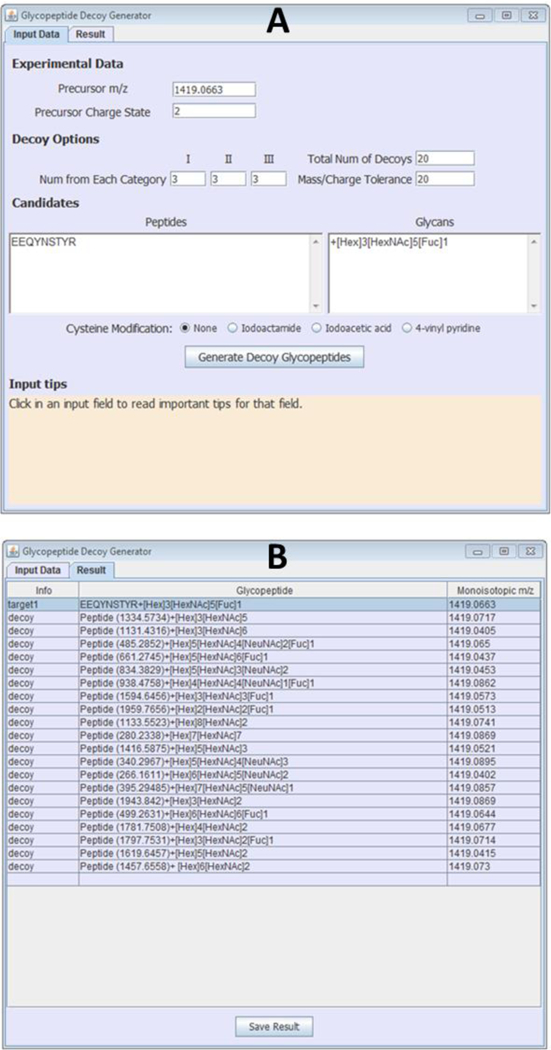
Graphical user interface of Glycopeptide Decoy Generator showing (A) the “Input Data” menu and parameters to generate 20 decoys for a target glycopeptide; and (B) the “Result” menu showing 20 decoys generated for the target glycopeptide in A.
Once all the “Input Data” parameters have been entered, and the user clicks the “Generate Decoy Glycopeptides” button, the software generates the decoys. The decoy list is displayed in the “Result” page of the software, and an example output file is shown in Figure 1B. The figure shows 20 glycopeptide decoys generated for the target glycopeptide and input parameters displayed in Figure 1A. The decoys have varying glycan compositions, and their monoisotopic m/z’s are close (within 20 ppm) to that of the target glycopeptide.
How GDG Generates Glycopeptide Decoys
Figure 2 shows a schematic representation of the approach used by GDG to generate decoys. To create a decoy for any target glycopeptide, GDG uses two main steps. First, a glycan is randomly selected from a library of over 300 biologically relevant N-linked glycans, and secondly, a peptide mass is generated. To choose a glycan for the decoy, the software queries a library of glycans that has been parsed into three categories (described in the preceding section), so that decoys of diverse glycan compositions could be easily generated. After a random glycan is selected, the algorithm determines if the selected glycan had been previously picked to generate a decoy for the same target. If so, the software discards the glycan and selects a new glycan. After selecting a non-redundant glycan from the library, the second step of decoy generation is to identify an appropriate peptide mass. The mass that represents the peptide portion of the decoy is computed such that the m/z of the entire decoy (peptide + glycan portions) is within the user-specified mass tolerance from the m/z of the precursor ion. The randomly selected glycan plus its arbitrary “peptide” mass represents the decoy. One final restriction is placed on the decoy: The mass appended to the glycan must not be smaller than the sum of the monoisotopic masses of asparagine and lysine amino acid residues. Once a decoy is generated, it is added to a restriction list to avoid duplication, and more decoys are subsequently generated until the user-specified number of decoys has been created. GDG can generate up to 45 decoys per target. The decoys are generated irrespective of the glycan type on the target or the enzyme used for digestion of the glycoprotein.
Figure 2.

Schematic representation of the decoy generation approach used by GDG. Decoys are generated via two main steps: First, a glycan is randomly selected from a pool of about 300 biologically relevant N-linked glycans separated in three categories (see text); and second, an arbitrary mass, representing the decoy’s peptide portion, is added to the glycan so that the total mass of the decoy is within a user-specified mass tolerance from the target glycopeptide mass.
Tool 2: The Glycopeptide spectral library.
We collected a significant dataset of 100 CID spectra of known glycopeptide compositions; these spectra can be used for testing any glycopeptide scoring algorithm that accepts CID spectra. Figure 3 shows one example CID spectrum in the dataset. This CID spectrum is from IgG1 monoclonal antibody, and the data was assigned to the glycopeptide composition EEQYNSTYR+[Hex]3[HexNAc]5[Fuc]1. This glycopeptide had been previously assigned as being present in this particular protein.33 Along with all spectra in the dataset, the monoisotopic m/z of this glycopeptide matches the theoretical mass quite closely. The doubly charged precursor ion, m/z 1419.0697, is within 2 ppm from the theoretical value, m/z 1419.0663. In addition to matching the high-resolution mass, inspection of the CID data indicates that oxonium ions at m/z 528.2 and 690.1 are present, further confirming that the precursor ion is indeed a glycopeptide. By also identifying the Y1 ion, and confirming neutral loses of monosaccharide residues from the potential glycopeptide, the spectrum was assigned as EEQYNSTYR+[Hex]3[HexNAc]5[Fuc]1. In a similar manner, the 100 CID spectra in the MS/MS dataset were unambiguously assigned to their known glycopeptide compositions. The dataset consists of spectra from 33 high-mannose glycopeptides, 34 fucosylated glycopeptides and 33 non-fucosylated complex/hybrid glycopeptides. The glycopeptide compositions assigned to these spectra are henceforth referred to as target glycopeptides (or targets). The peaklists for all CID spectra, and their assigned glycopeptide compositions, are available in the Supplemental Data. In addition, 13 of the spectra are annotated. See Supplemental Figures S1 to S13.
Figure 3.
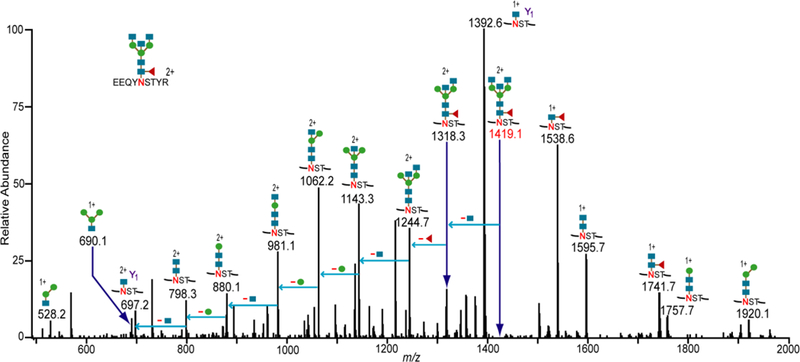
Representative CID spectrum showing the assignment of an IgG1glycopeptide (shown in figure). Glycosidic cleavages of the glycan portion and glycan oxonium ions are observed. The Y1 product ion and neutral monosaccharide losses from the potential glycopeptide candidate (at m/z 1419.1) were used to confirm the assignment. The 100 spectra in the “Glycopeptide Spectral Library” were assigned to their correct glycopeptide compositions in the same way. The blue squares, green circles and red triangles denote N-acetylhexoseamine, hexose, and fucose monosaccharide residues, respectively.
Application of these tools for evaluating the accuracy of CID scoring algorithms for glycopeptides
The dataset of 100 CID spectra and known targets was used in conjunction with the Decoy Generator to demonstrate how these tools are helpful in testing and refining glycopeptide analysis software. Specifically, the scoring algorithm of GlycoPep Grader21 was evaluated herein. GlycoPep Grader uses monosaccharide neutral loses from glycopeptide compositions to score target and decoy glycopeptides against a CID spectrum, and the candidate with the highest score is assigned to the spectrum in question.21 For each of the 100 target glycopeptide compositions, and the 100 assigned spectra, 20 decoys were generated using the Decoy Generator, and GlycoPep Grader was used to score each target and its 20 decoys against the known CID spectrum of the target. The scores were interrogated to determine whether GlycoPep Grader consistently matched the CID spectra to their correct glycopeptide compositions or to decoy glycopeptides. A summary of the results is shown in Figure 4A. As shown, all the 33 spectra originating from high-mannose glycopeptides were matched to their correct targets; one out of the 33 spectra (3%) from non-fucosylated glycopeptides was matched to a decoy glycopeptide (32 spectra matched the correct targets), while 12 out of the 34 spectra (35%) from fucosylated glycopeptides were matched to decoys (22 spectra matched to the correct targets). The results clearly indicate that GlycoPep Grader accurately scores spectra of high-mannose and non-fucosylated complex/hybrid glycopeptide compositions, but it has some weaknesses in scoring spectra of fucosylated glycopeptides. Hence, GlycoPep Grader’s scoring algorithm can be modified to improve its accuracy in scoring spectra of fucosylated glycopeptide compositions. The annotated CID spectra of the 13 glycopeptides that were incorrectly assigned to decoys are shown in Supplementary Figures S1 to S13.
Figure 4.
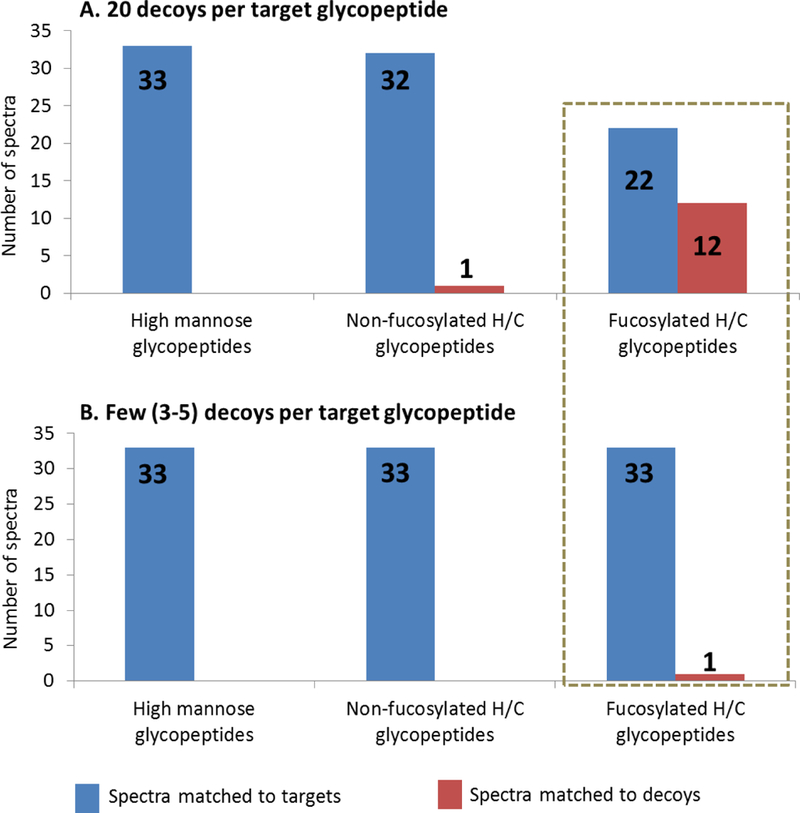
Bar graphs showing results of scoring 33 high mannose, 33 non-fucosylated hybrid/complex (H/C), and 34 fucosylated hybrid/complex glycopeptide spectra scored against the known target glycopeptide compositions along with (A.) abundant decoys (20 decoys) per target glycopeptide and (B.) few decoys (three to five) decoys per target glycopeptide. More spectra of fucosylated glycopeptides were matched to decoys when abundant decoys were used (box).
Are abundant decoys needed for accurate evaluation of glycopeptide scoring algorithms?
After using abundant decoys, generated by the Decoy Generator, to identify the weakness in GlycoPep Grader’s scoring algorithm, we wanted to determine why the limitation was not identified during the development of the software. In the original publication describing GlycoPep Grader,21 a total of 79 glycopeptides were scored using the software, and all 79 glycopeptides, including 17 fucosylated glycopeptides, were correctly assigned to the known glycopeptide spectra, even when scoring each spectrum against decoys. In that work, typically 3–5 decoys were used. Hence, for this case study, we replicated the procedure used to generate decoys during the initial development of GlycoPep Grader, and we scored those decoys and targets against our new dataset of 100 CID spectra. We used the same number of decoys per target glycopeptide (three to five decoys), and we generated the decoys in the same manner as described previously: For each of the 100 target glycopeptide compositions, either three, four, or five decoys were generated from Titin, a glycoprotein containing about 50,000 amino acid residues, and a database of about 200 glycans that were multiplexed to the protein in silico, as described by Woodin et al.21 Each target and its decoys were scored against the known CID spectrum of the target using GlycoPep Grader. The results are shown in Figure 4B. The 33 spectra of high-mannose glycopeptides and the 33 spectra of non-fucosylated glycopeptides were all matched to their correct glycopeptide compositions, and only one out of the 34 spectra of fucosylated glycopeptides was matched to a decoy. Hence, of the 100 target glycopeptides scored with limited numbers of decoys, 99 were matched to the correct target glycopeptides and only one spectrum was matched to a decoy. This result is contrary to the aforementioned case when the target-to-decoy ratio was 1:20, and up to 13 spectra (12 of which were of fucosylated glycopeptides) were assigned to decoys. A comparison of the scores of the 12 spectra from fucosylated glycopeptides that were assigned to decoys when scored at a target-to-decoy ratio of 1:20 and the scores of the same spectra when scored against fewer decoys are provided in Supplemental Tables 1 to 12. Overall, the results indicate that GlycoPep Grader’s limitation in scoring spectra from fucosylated glycopeptides could not be determined by scoring target glycopeptide spectra against a limited number of decoys, which explains why GlycoPep Grader’s weakness was not identified during the development of the tool, when between three to five decoys per target were used. Hence, abundant decoys are indeed needed for accurate assessment of tools that assign glycopeptides to MS/MS spectra.
Given the above results, it is imperative for software developers to use large numbers of decoys to test their scoring algorithms during the development of software designed to match glycopeptides to MS/MS data. By so doing, the probability of decoy matches increases, and when decoys outscore glycopeptide candidates that are known to be correct, software developers can easily make changes to improve the scoring algorithm. Similarly, end-users of glycopeptide software can assess the quality of the output from various tools by testing them against large spectral libraries of known glycopeptide compositions and large numbers of decoys per target glycopeptide. However, generating a large spectral library is time consuming. Hence, the peak lists of all 100 spectra used in this study have been provided in the supplementary information.
CONCLUSION
To simplify the task of building effective glycopeptide software, we developed two new tools, Glycopeptide Decoy Generator (GDG) and an expert-assigned dataset of 100 CID spectra. GDG rapidly generates glycopeptide decoys de novo, and these decoys can be used to assess the quality of tools that assign glycopeptides to CID data. As a secondary contribution, we provide herein peak lists for 100 validated CID spectra that can be used to test any existing software tool or any new tool under development. Using large numbers of decoys generated by our newly developed tool, and our set of 100 validated CID spectra, we evaluated the accuracy of existing software that assigns glycopeptides to CID data. We demonstrate that limitations in the scoring algorithm of the software can be identified when testing it against large sets of decoys, and these limitations could not be identified when only a few decoys were scored.
Our tool is the first software that automatically generates abundant decoys on demand for the assessment of algorithms that assign glycopeptides to CID spectra. The approach for decoy generation is simple; it can be used as-is, or the software can be easily incorporated into other bioinformatics tools designed to match glycopeptides to CID data. The software can be downloaded at http://glycopro.chem.ku.edu/GPJ.jar
Supplementary Material
AKNOWLEDGMENT
The authors acknowledge funding from the National Institutes of Health, grant R01GM103547 to HD.
Footnotes
SUPPORTING INFORMATION
The following files are available free of charge at ACS website http://pubs.acs.org: 1) Supplementary Information File 1. This file contains 13 assigned CID spectra of glycopeptides and 12 tables showing scoring results for fucosylated glycopeptides and their decoys. 2) Supplementary Information File 2. This Excel file contains peaklists for 100 CID spectra of the 100 assigned glycopeptides used for this study.
The authors declare no conflict of interest.
REFERENCES
- 1.Boscher C;James WD; Nabi IR Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol. 2011, 23, 383–392. [DOI] [PubMed] [Google Scholar]
- 2.Haines N;Irvine KD Glycosylation regulates Notch signaling. Nat. Rev. Mol. Cell Biol. 2003, 4, 786–797. [DOI] [PubMed] [Google Scholar]
- 3.Gallagher P;Henneberry J;Wilson I;Sambrook J;Gething MJ Addition of carbohydrate side chains at novel sites on influenza virus hemagglutinin can modulate the folding, transport, and activity of the molecule. J. Cell. Biol. 1988, 107, 2059–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu C;Ng DT Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell. Biol. 2015, 16, 742–752 [DOI] [PubMed] [Google Scholar]
- 5.Helenius A;Aebi M. Intracellular functions of N-linked glycans. Science. 2001, 291, 2364–2369. [DOI] [PubMed] [Google Scholar]
- 6.Yan L.;Yuan T.;Taha R;Prakash A;Lopez MF; Chan DW Zhang H. Simultaneous analysis of glycosylated and sialylated PSA reveals differential distribution of glycosylated PSA Isoforms in prostate cancer tissues. Anal. Chem. 2011, 83, 240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peracaula R;Tabarés G;Royle L;Harvey DJ; Dwek RA; Rudd PM; De Llorens R. Altered glycosylation pattern allows the distinction between prostate-specific antigen (PSA) from normal and tumor origins. Glycobiology. 2003, 13, 457–470. [DOI] [PubMed] [Google Scholar]
- 8.Jefferis R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol. Sci. 2009, 30, 356–362. [DOI] [PubMed] [Google Scholar]
- 9.Liu L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J. Pharm. Sci. 2015, 104, 1866–1884. [DOI] [PubMed] [Google Scholar]
- 10.Chung CH; Mirakhur B;Chan E;Le QT; Berlin J;Morse M;Murphy BA; Satinover SM; Hosen J;Mauro D;Slebos RJ; Zhou Q;Gold D;Hatley T;Hicklin DJ; Platts-Mills T. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N. Engl. J. Med. 2008, 358, 1109–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solá RJ; Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. J. Pharm. Sci. 2009, 98, 1223–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leymarie N;Zaia J. Effective use of mass spectrometry for glycan and glycopeptide structural analysis. Anal Chem. 2012, 84, 3040–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morelle W;Michalski JC Analysis of protein glycosylation by mass spectrometry. Nat. Protoc. 2007, 2, 1585–1602. [DOI] [PubMed] [Google Scholar]
- 14.Aich U;Lakbub J;Liu A. State-of-the-art technologies for rapid and high-throughput sample Preparation and analysis of N-glycans from antibodies. Electrophoresis. 2016, 37, 1468–1488. [DOI] [PubMed] [Google Scholar]
- 15.Dalpathado DS; Desaire H. Glycopeptide analysis by mass spectrometry. Analyst. 2008. 133, 731–738. [DOI] [PubMed] [Google Scholar]
- 16.Zhu Z;Desaire H. Carbohydrates on proteins: site-specific glycosylation analysis by mass spectrometry. Annu. Rev. Anal. Chem. 2015, 8, 463–483. [DOI] [PubMed] [Google Scholar]
- 17.Chen-Chun C;Su Wan-Chih.; Huang Bao-Yu.; Chen Yu-Ju.; Tai Hwan-Ching.; Obena R. Interaction modes and approaches to glycopeptide and glycoprotein enrichment. Analyst. 2014, 139, 688–704. [DOI] [PubMed] [Google Scholar]
- 18.Bodnar E;Perreault H. Qualitative and quantitative assessment on the use of magnetic nanoparticles for glycopeptide enrichment. Anal. Chem. 2013, 85, 10895–10903. [DOI] [PubMed] [Google Scholar]
- 19.Froehlich JW; Dodds ED; Wilhelm M;Serang O;Steen JA; Lee RS A classifier based on accurate mass measurements to aid large scale, unbiased glycoproteomics. Mol. Cell Proteomics. 2013, 12, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu W;Su X;Zhu Z;Go EP; Desaire H. GlycoPep MassList: software to generate massive inclusion lists for glycopeptide analyses. Anal. Bioanal. Chem. 2017, 409, 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woodin CL; Hua D;Maxon M;Rebecchi KR; Go EP; Desaire H. GlycoPep grader: a web-based utility for assigning the composition of N-linked glycopeptides. Anal. Chem. 2012, 84, 4821–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandler KB; Pompach P;Goldman R;Edwards N. Exploring site-specific N-glycosylation microheterogeneity of haptoglobin using glycopeptide CID tandem mass spectra and glycan database search. J. Proteome Res. 2013, 12, 3652–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lynn KS; Chen CC; Lih TM; Cheng CW; Su WC; Chang CH; Cheng CY; Hsu WL; Chen YJ; Sung TY MAGIC: an automated N-linked glycoprotein identification tool using a Y1-ion pattern matching algorithm and in silico MS² approach. Anal Chem. 2015, 87, 2466–2473. [DOI] [PubMed] [Google Scholar]
- 24.Toghi ES; Shah P;Yang W;Li X;Zhang H. GPQuest: A spectral library matching algorithm for site-specific assignment of tandem mass spectra to intact N-glycopeptides. Anal Chem. 2015, 87, 5181–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng WF; Liu MQ; Zhang Y;Wu JQ; Fang P;Peng C;Nie A;Yan G;Cao W;Liu C;Chi H;Sun RX; Wong CC; He SM; Yang P. pGlyco: A pipeline for the identification of intact N-glycopeptides by using HCD-and CID-MS/MS and MS3. Sci. Rep. 2016, 6, 25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu Z;Hua D;Clark DF; Go EP; Desaire H. GlycoPep Detector: A tool for assigning mass spectrometry data of N-linked glycopeptides on the basis of their electron transfer dissociation spectra. Anal. Chem. 2013, 85, 5023–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu Z;Su X;Go EP; Desaire H. New glycoproteomics software, GlycoPep Evaluator, generates decoy glycopeptides de novo and enables accurate false discovery rate analysis for small data sets. Anal. Chem. 2014, 86, 9212–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayampurath A;Yu CY; Song E;Balan J;Mechref Y;Tang H. Computational framework for identification of intact glycopeptides in complex samples. Anal. Chem. 2014, 86, 453–463. [DOI] [PubMed] [Google Scholar]
- 29.He L;Xin L;Shan B;Lajoie GA; Ma B. GlycoMaster DB: Software to assist the automated identification of N-linked glycopeptides by tandem mass spectrometry. J. Proteome Res. 2014, 13, 3881–3895. [DOI] [PubMed] [Google Scholar]
- 30.Hu H;Khatri K;Zaia J;Algorithms and design strategies towards automated glycoproteomics analysis. Mass Spectrom. Rev. 2016, January 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woodin CL; Maxon M;Desaire H. Software for automated interpretation of mass spectrometry data from glycans and glycopeptides. Analyst. 2013, 138, 2793–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu H;Khatri K;Klein J;Leymarie N;Zaia J. A review of methods for interpretation of glycopeptide tandem mass spectral data. Glycoconj. J. 2016, 33, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wuhrer M;Stam JC; Van de Geijn FE; Koeleman CA; Verrips CT; Dolhain RJ; Hokke CH; Deelder AM Glycosylation profiling of immunoglobulin G (IgG) subclasses from human serum. Proteomics. 2007, 7, 4070–4081. [DOI] [PubMed] [Google Scholar]
- 34.Alley WR; Mechref Y;Novotny MV Characterization of glycopeptides by combining collision-induced dissociation and electron-transfer dissociation mass spectrometry data. Rapid Commun. Mass Spectrom. 2009, 2, 161–170. [DOI] [PubMed] [Google Scholar]
- 35.Brown KJ; Vanderver A;Hoffman EP; Schiffmann R;Hathout Y. Characterization of transferrin glycopeptide structures in human cerebrospinal fluid. Int. J. Mass Spectrom. 2012, 312, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Go EP; Chang Q;Liao HX; Sutherland LL; Alam SM; Haynes BF; Desaire H. Glycosylation site-specific analysis of clade C HIV-1 envelope proteins. J. Proteome Res. 2009, 8, 4231–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X;McNally DJ; Nothaft H;Szymanski CM; Brisson JR; Li J. Mass spectrometry-based glycomics strategy for exploring N-linked glycosylation in eukaryotes and bacteria. Anal. Chem. 2006, 78, 6081–6087. [DOI] [PubMed] [Google Scholar]
- 38.Go EP; Herschhorn A.;, Gu C;Castillo-Menendez L;Zhang S;Mao Y;Chen H;Ding H;Wakefield JK; Hua D;Liao HX; Kappes JC; Sodroski J;Desaire H. Comparative analysis of the glycosylation profiles of membrane-anchored HIV-1 envelope glycoprotein trimers and soluble gp140. J. Virol. 2015, 89, 8245–8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Domon B;Costello CE A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glyucoconjugate J. 1988, 5, 397–409. [Google Scholar]
- 40.Jiang H;Desaire H;Butnev VY; Bousfield GR Glycoprotein profiling by electrospray mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 750–758. [DOI] [PubMed] [Google Scholar]
- 41.Go EP; Rebecchi KR; Dalpathado DS; Bandu ML; Zhang Y;Desaire H. GlycoPep DB: a tool for glycopeptide analysis using a “smart search”. Anal. Chem. 2007, 79, 1708–1713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.