

Abstract
Dopamine receptors are important G protein-coupled receptors (GPCRs) with therapeutic opportunities for treating Parkinson’s Disease (PD) motor and cognitive deficits. Biased D1 dopamine ligands that differentially activate G protein over β-arrestin recruitment pathways are valuable chemical tools for dissecting positive versus negative effects in drugs for PD. Here, we reveal an iterative approach toward modification of a D1-selective noncatechol scaffold critical for G protein-biased agonism. This approach provided enhanced understanding of the structural components critical for activity and signaling bias and led to the discovery of several novel compounds with useful pharmacological properties, including three highly GS-biased partial agonists. Administration of a potent, balanced, and brain-penetrant lead compound from this series results in robust antiparkinsonian effects in a rodent model of PD. This study suggests that the noncatechol ligands developed through this approach are valuable tools for probing D1 receptor signaling biology and biased agonism in models of neurologic disease.
Keywords: Parkinson’s disease, D1 dopamine receptor, noncatechol agonists, dyskinesia, β-arrestins, biased agonism, structure–functional selectivity relationship
Graphical Abstract
INTRODUCTION
G protein-coupled receptors (GPCRs) are a superfamily of receptors with seven transmembrane helical domains that carry out a large variety of physiological processes. With over 800 members capable of carrying out a large variety of physiological processes, GPCRs are a critically important class of targets for drug discovery.1 In fact, over 30% of FDA-approved drugs target GPCRs and cover a large number of clinical indications. As such, GPCRs are one of the most successful and promising target classes for drug discovery.2,3
Traditionally, GPCRs were thought to conduct intracellular signaling exclusively through a canonical G protein-dependent pathway. More recently, the field has moved toward appreciating the fact that the GPCR superfamily is actually capable of downstream signaling through multiple G protein-independent pathways.4–7 Perhaps the most notable of these downstream pathways are the β-arrestin-dependent pathways.8 Signaling through β-arrestin-dependent pathways typically proceeds when a family of protein kinases called G protein-coupled receptor kinases (GRKs) phosphorylate the intracellular domains of a GPCR upon activation, thereby allowing the phosphorylated GPCR to recruit β-arrestins.9–12 Previously, β-arrestins were primarily appreciated for their role in desensitizing G protein signaling by interacting with trafficking proteins, such as clathrin, leading to internalization and downregulation.13–15 A new paradigm has been developed, however, whereby β-arrestins are now also appreciated for their ability to act as signal transducers themselves downstream of GPCRs, with the ability to interact with components of the MAPK cascade,16–19 nonreceptor tyrosine kinases,20–22 and other signaling pathways that mediate important cell biological processes.
With the identification of downstream signaling pathways that operate independent of G proteins, such as β-arrestins, it has been shown that these mediators can interact directly with GPCRs to stabilize conformationally distinct active states of the receptor.23 Furthermore, it has been demonstrated that ligands of GPCRs can induce a unique receptor conformation that can subsequently lead to preferential activation of a certain downstream signaling pathway, a phenomenon termed “functional selectivity” or “biased signaling”.13–19 Accumulating evidence in this area has propelled the concept of functional selectivity to the forefront of the GPCR field, with many studies beginning to explore the structural–functional selectivity relationships (SFSRs) that may exist between the structural features of a ligand that may determine its precise binding mode within a GPCR and its observed functional selectivity profile.24–29
Dopamine receptors are important GPCR targets with therapeutic potential in treating motor deficits in diseases, such as Parkinson’s Disease (PD), as well as cognitive deficits resulting from various neuropsychiatric disorders including Alzheimer’s Disease, PD, and schizophrenia.30–33 There are five known subtypes of dopamine receptors that are categorized into two subfamilies, the D1-like (D1R and D5R) and D2-like (D2R, D3R, and D4R) receptors, which signal through GS or Gi/o G proteins, respectively.9,10,34 Despite their effective use in treating several neuropsychiatric disorders, dopaminergic agonists can carry certain debilitating motor side effects including dyskinesias, which can compromise patient function and quality of life, thereby presenting a considerable limitation to their use in diseases, such as PD.35,36 In addition, concerns over clinically significant side effects, such as hypotension and tolerance observed following treatment with D1R agonist candidates have prevented their approval.37,38 Safer and more effective drugs are needed to not only help restore dopaminergic transmission and signaling in these pathophysiological states but also to minimize the motor side effects that may result from chronic dopaminergic therapy.
Recent genetic animal studies support the notion that dyskinesias are associated with increased G protein signaling and also validate β-arrestin2 as a novel target for treating PD without causing dyskinesia. For example, these studies have shown that knocking out β-arrestin2 in rodent and nonhuman primate models of PD worsened dyskinesias following l-DOPA administration, while β-arrestin2 overexpression reduced dyskinesias and increased locomotion via β-arrestin2 signaling.34 These studies, as well as others in the field that have investigated functionally selective signaling at other dopaminergic,24,25,27,39–49 serotonergic,26,50–53 and opioid receptors,54–67 have shown that through the preferential activation of one distinct signaling pathway over another, it is possible to sometimes separate therapeutic from adverse effects at a given receptor. Taken together, evidence in the field suggests that designing biased ligands may be a novel solution for developing safer and more efficacious drugs that retain their therapeutic effects while mitigating the incidence of adverse side effects.
Functionally selective ligands of GPCRs represent valuable chemical tools for elucidating the roles of signaling pathways downstream of the receptor. In 2018, Gray et al. disclosed a novel noncatechol-containing scaffold that was reported to display potent agonism of D1R.68 Several properties of this agonist class make it a particularly interesting candidate for medicinal chemistry exploration. First, this scaffold was characterized as inherently G protein-biased, likely due to its unique putative binding mode within the orthosteric site. Mutagenesis work suggests that this scaffold makes contacts with certain residues in the orthosteric pocket that are distinctly different from those typically made by catechol-containing ligands, including dopamine. Second, a 2018 study by Davoren et al., which described the high throughput screening campaign that ultimately led to the discovery of this noncatechol scaffold, also emphasized the importance of atropisomerism inherent to the structure of these ligands.69 Given these interesting findings, this unique scaffold represents an excellent opportunity to design novel chemical tools for better understanding the structural determinants of D1R agonism and functionally selective signaling.
Previously, we reported a comprehensive SFSR study of this noncatechol D1R agonist scaffold.25 Through the systematic modification of four modifiable regions of the scaffold, we generated over 50 novel derivatives with diverse functional selectivity profiles. By associating certain structural modifications with alterations in functional selectivity, we were able to describe several interesting SFSR trends for this scaffold and gain new insights regarding which regions of the scaffold were critical for maintaining potency and efficacy in both G protein pathway activation and β-arrestin2 recruitment.25
Here, we report our findings from a new exploratory study that takes a significantly different approach to ligand discovery from both our previous systematic SFSR campaign and other studies reporting noncatechol analogues of D1R. Instead, we utilize the lessons and trends learned from previous studies to design novel ligands “from the ground up”. Importantly, ligand design using this approach carries the distinct advantage of enabling a better understanding of precise structural components that are critical for noncatechol D1 potency, efficacy, and signaling bias. As a result of this process, we have discovered several new scaffold derivatives with novel pharmacological properties and robust in vivo activity. We describe the iterative design, synthesis, and pharmacological assessment of these derivatives in both in vitro and in vivo systems. We believe this is a simple, yet powerful approach to GPCR ligand discovery with particular relevance to exploration of the structural components of a given ligand that are critical to enable biased signaling incorporation into D1 drug design.
RESULTS AND DISCUSSION
Previous SFSR studies of the right-hand side (RHS) of the noncatechol scaffold suggest that several of its properties are critical to the activity of the overall ligand at D1R.25,68,69 In particular, our previous SFSR campaign on the RHS of PF-6142 suggested that factors, such as ring size, heteroatom arrangement, and substituent position were of utmost importance in maintaining the efficacy and potency of the scaffold in both G protein and β-arrestin2 pathways. For example, introducing the same imidazopyrazinyl RHS with a different methylation position and attachment point significantly diminished potency in both the GS-cAMP accumulation and β-arrestin2 recruitment assays while maintaining partial agonism in the GS-cAMP accumulation assay. Conversely, omission of the 6-methyl substituent and the 7-position nitrogen of the imidazopyrazinyl ring to give an unsubstituted imidazopyridinyl ring on the RHS resulted in a compound with only mildly compromised potency in the GS-cAMP accumulation and β-arrestin2 recruitment assays but full agonism in the Gs-cAMP accumulation assay.25 Our previous investigation also elucidated that various monocyclic moieties on the RHS region of the noncatechol scaffold also possessed varying degrees of potency and bias relative to the initial bicyclic starting compound.
While these and other findings do implicate the RHS of the noncatechol scaffold as important for various pharmacological properties of the ligand, it is difficult to gain significant insight into which precise factors are responsible for such effects when the ligands comprise part of a diverse set of exploratory modifications, rather than an iterative series of carefully implemented changes. To elucidate the critical structural elements of a potent RHS moiety on the noncatechol scaffold, we took an iterative approach to building the RHS starting from the ground up.
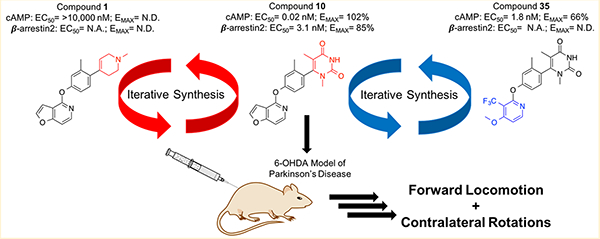
Starting from a very simple monocyclic RHS group, we synthesized a small series of highly related structural analogues and determined their in vitro pharmacological properties in both stimulating D1R-mediated cAMP production in a D1R-Gs GloSensor assay and in activating D1R-mediated β-arrestin2 recruitment in a bioluminescence resonance energy transfer (BRET) platform-based assay. Dopamine was included as a positive full agonist control in both assays. When interpreting results from these assays, it is also important to consider their mechanistic differences as the GloSensor assay involves a signal amplification mechanism, while the BRET assay does not, which may have implications in terms of the receptor occupancy required to achieve an EMAX in each system. With these considerations in mind, compounds from each synthetic iteration were assayed together to infer relative changes to structural, spatial, and electronic properties on ligand bias and potency. The inferences gleaned from this small series of ligands informed the design of subsequent batches of compounds, progressing into compounds with increasingly improved activities and diverse functional selectivity profiles. Upon optimization of the RHS, this same approach was also applied to the left-hand side (LHS), resulting in several novel compounds with interesting pharmacological properties (Figure 1).
Figure 1.

Iterative strategy for generating novel noncatechol analogues of compound 1 with increased agonist activity at D1R and diverse functional selectivity profiles.
Iterative SFSR of the RHS Heterocycle.
To establish a baseline RHS moiety to which we could easily incorporate new structural elements, we began with a relatively simple compound (1) and, subsequently, designed and synthesized the compounds presented in Figure 2. All of the compounds presented in Figure 2 were prepared according to the synthetic strategy shown in the first iteration of Scheme 1. Briefly, these ligands were prepared by reacting the commercially available chlorinated pyridofuran ring with 4-bromo-3-methylphenol to produce the known brominated intermediate. Conversion of the brominated intermediate into the known boronic ester intermediate68 then enabled a subsequent Suzuki coupling of the boronic ester with a series of either custom-made or commercially available brominated heterocycles using catalytic amounts of tetrakis(triphenylphosphine)palladium(0). This strategy afforded the desired compounds in good yields.
Figure 2.

First iteration of analogues (2–5) derived from compound 1 to explore the effects of nitrogen methylation and addition of a carbonyl oxygen and an additional nitrogen into the RHS heterocycle. Full concentration–response curves for the first iteration of RHS analogues measured using the GloSensor assay for GS pathway activation (A) and the BRET assay for β-arrestin2 recruitment (B) as a percentage of the maximal dopamine (DA) response. Curves represent at least three independent experiments performed in triplicate. Data points represent the mean ± SEM.
Scheme 1. Synthesis of the First, Second, and Third Iterations of Compounds for Exploring the RHS Heterocyclic Moietya.

aReagents and conditions: (a) Cs2CO3, DMSO, 125 °C, 48 h; (b) Pd(dppf)Cl2, KOAc, B2(pin)2, dioxane, 80 °C, 18 h; (c) Pd(PPh3)4, K2CO3, dioxane, H2O, 120 °C, 25 min; (d) NaOMe, reflux, 24 h; (e) NaNO2, CuBr2, rt, 66 h; (f) p-TsOH, THF, reflux, 24 h; (g) MeB(OH)2, Pd(dppf)Cl2, Cs2CO3, dioxane, H2O, 110 °C, 2 h.
The ability of each final compound to stimulate D1R-mediated cAMP production and D1R-mediated recruitment of β-arrestin2 was assessed in vitro using a D1R-GS GloSensor assay and a BRET platform-based assay, respectively. Importantly, dopamine was used as a full agonist positive control in both assays.70–72
Compound 1, which featured a simple 1-methyl-1,2,3,6-tetrahydropyridin-4-yl, was selected as the starting point for this investigation. When tested for both GS-cAMP accumulation and β-arrestin2 recruitment, compound 1 was found to be largely inactive in both assays. From here, we made several minor modifications to the RHS of 1 to explore the immediate chemical space around its RHS entity. In particular, we sought to explore the effects of the addition of a carbonyl oxygen on the 6-position of 1’s RHS ring (compound 2), the effects of removing the N-methyl group from 2 (compound 3), and the effects of moving the methyl substituent over one position to the 2-position on the ring (compound 4). Additionally, in the same iteration, we sought to determine how the addition of another nitrogen atom to the ring at the 3-position to convert it into a pyrimidine (compound 5) would affect the pharmacological profile. As shown in Figure 2, the presence of a carbonyl oxygen (compounds 2–4), which may serve as potential point for hydrogen bonding with a polar receptor residue, appeared to substantially increase the activity of those compounds over the baseline 1. Further, this initial mini-series of analogues also suggested that conversion of the tetrahydropyridine ring into its pyrimidine version represented another favorable change that provided an additional boost in compound 5’s activity (GS-cAMP EC50 = 133 nM, EMAX = 96%; β-arrestin2 EC50 > 10,000 nM) compared to compound 3 (GS-cAMP EC50 = 176 nM, EMAX = 81%; β-arrestin2 no activity) (Table S1).
On the basis of these results, we designed our next series of analogue ligands starting from compound 5 (Figure 3). The compounds in this series were created using a similar synthetic strategy (Scheme 1, second iteration). In this series, we sought to explore the effects of removing the 6-position carbonyl group on the RHS ring but retaining the core pyrimidine heterocycle (compound 6), adding a amine substituent between the pyrimidinyl nitrogens at position 2 (compound 7), adding a second nitrogen substituent at the 6-position, which was the original position of the carbonyl group (compound 8), and the effect of changing both of the nitrogen substituents back into carbonyl groups to give an unsubstituted pyrimidine dione moiety (compound 9). On the basis of this series of compounds, we observed a clear trend suggesting that the carbonyl oxygens (compounds 5 and 9) were preferred over nitrogen substituents (compounds 6–8) and that the pyrimidine dione moiety (9) resulted in significantly greater activity in both the GS-cAMP and β-arrestin2 pathways (GS-cAMP EC50 = 9.0 nM, EMAX = 78%; β-arrestin2 EC50 = 2089 nM, EMAX = 37%) compared to the diaminopyrimidine analogue (8) (GS-cAMP EC50 > 10,000 nM; β-arrestin2 no activity) (Table S2).
Figure 3.

Second iteration of analogues (6–9) derived from compound 5 were designed to explore the effects of carbonyl oxygen and amine substituents on the RHS heterocycle. Full concentration–response curves for the second iteration of RHS analogues measured using the GloSensor assay for GS pathway activation (A) and the BRET assay for β-arrestin2 recruitment (B) as a percentage of the maximal dopamine (DA) response. Curves represent at least three independent experiments performed in triplicate. Data points represent the mean ± SEM.
In continuing our optimization of the RHS moiety, we next synthesized a targeted series of compounds based on the structure of 9 (Figure 4). These particular compounds were prepared using similar methods as outlined previously (Scheme 1, third iteration). Starting from 9, we first sought to explore the effect of adding two methyl substituents to the 1- and 5-positions of the pyrimidine dione ring (compound 10), which would be predicted to increase the steric hindrance preventing RHS ring rotation about the bond axis. We also then explored the effect of removing the 4-position carbonyl oxygen from 10 (compound 11). Finally, we constructed analogues featuring variations to the positions of the same structural elements present in 11’s RHS, giving rise to compounds 12 and 13 as structural isomers. Not surprisingly based on the trends outlined by Davoren et al.,69 the dimethyl substituents of 10 resulted in a dramatic increase in its potency and efficacy in both the GS-cAMP and β-arrestin2 pathways relative to the unsubstituted 9 (Figure 4). Compound 10, which was very recently disclosed by Wang et al., was reported to have an EC50 = 0.3 nM and EMAX = 107% in the GloSensor-measured GS-cAMP pathway and an EC50 = 35 nM and EMAX = 86% in recruiting β-arrestin2 as determined in a Tango assay.73 In our assays, however, we observed similar potency and efficacy in the GS-cAMP pathway but slightly increased potency in recruiting β-arrestin2 (GS-cAMP EC50 = 0.02 nM, EMAX = 102%; β-arrestin2 EC50 = 3.1 nM, EMAX = 85%), as well as a slight β-arrestin2 bias (bias factor = 1.8 for β-arrestin2). It is possible that these differences might stem from the divergent assay mechanisms used to quantify β-arrestin2 recruitment in these two studies; the Tango assay involves a signal amplification mechanism, while the BRET assay does not. In comparison to compound 10, the in vitro profiles of compounds 11–13 suggest that neither removing a carbonyl oxygen group (compound 11) nor rearranging the nitrogen atoms within the heterocyclic ring and altering the locations of the carbonyl oxygen entities (compounds 12 and 13) are well-tolerated for maintaining potent, balanced activity at D1R (compound 11 GS-cAMP EC50 = 1.8 nM, EMAX = 88%, β-arrestin2 EC50 = 151 nM, EMAX = 19%; compound 12 GS-cAMP EC50 = 1.1 nM, EMAX = 92%, β-arrestin2 EC50 = 196 nM, EMAX = 63%; compound 13 GS-cAMP EC50 = 205 nM, EMAX = 82%, β-arrestin2 no activity) (Table S3).
Figure 4.

Third iteration of analogues (10–13) derived from compound 9 to explore the effects of heteroatom positioning and methylation of the RHS heterocycle. Full concentration–response curves for the third iteration of RHS analogues measured using the GloSensor assay for GS pathway activation (A) and the BRET assay for β-arrestin2 recruitment (B) as a percentage of the maximal dopamine (DA) response. Curves represent at least three independent experiments performed in triplicate. Data points represent the mean ± SEM.
Modifications of the Pyrimidine Dione RHS.
Given the exquisite potency and efficacy that we observed with compound 10 in both the GS-cAMP and β-arrestin2 assays, we sought to determine whether targeted alterations to readily modifiable portions of the pyrimidine dione RHS could result in an improved compound or a compound with a unique pharmacological profile.
In the first series of modifications, we sought to explore the effects of N-alkylation at the 3-position of the RHS ring (Figure 5). Briefly, these analogues were synthesized by reacting 10 with various iodinated alkyl groups in the presence of sodium hydride (Scheme S1). We created a simple series of analogues consisting of the methyl (compound 14), ethyl (compound 15), and isopropyl (compound 16) derivatives (Figure 5). On the basis of the previous trends we observed in our earlier series in this study (compounds 2 and 3), we hypothesized that N-alkylation at this position would be detrimental to the potency in the β-arrestin2 recruitment pathway. As expected, we did observe diminishing potencies and efficacies in both pathways (Table S4). We also observed a very mild reversal of signaling bias favoring β-arrestin2, relative to dopamine, as we progressed from isopropyl down to methyl (bias factors for β-arrestin2 isopropyl = 4.3, ethyl = 4.6, methyl = 7.3). Unfortunately, given that overall potencies and efficacies also diminished significantly upon alkylation, this would not likely represent a viable strategy for deriving bias from this scaffold.
Figure 5.

Fourth iteration of analogues (14–16) derived from compound 10 to explore the effects of alkylation of the 3-position nitrogen on the RHS heterocycle. Full concentration–response curves for the fourth iteration of RHS analogues measured using the GloSensor assay for GS pathway activation (A) and the BRET assay for β-arrestin2 recruitment (B) as a percentage of the maximal dopamine (DA) response. Curves represent at least three independent experiments performed in triplicate. Data points represent the mean ± SEM.
Next, we explored the possibility of modifying the size of the substituent alkyl groups at the 1-position of the pyrimidine dione RHS (Figure 6). A set of ligands was created starting with a series of commercially available ureas and alkylated 2-cyanoesters (Scheme S2). We explored the effect of changing the 1-position methyl group into ethyl (compound 17), cyclopropyl (compound 18), methylcyclopropyl (compound 19), cyclobutyl (compound 20), n-propyl (compound 21), and n-butyl (compound 22) groups. The trends in this series of compounds suggest a limit in the allowed bulk of the substituent in this location before general ligand activity is compromised. The methyl-containing compound 10 retained the highest potency and efficacy in both signaling pathways (GS-cAMP EC50 = 0.02 nM, EMAX = 102%; β-arrestin2 EC50 = 3.1 nM, EMAX = 85%), while ethyl (compound 17) (GS-cAMP EC50 = 0.08 nM, EMAX = 95%; β-arrestin2 EC50 = 13 nM, EMAX = 65%) and cyclopropyl (compound 18) (GS-cAMP EC50 = 0.06 nM, EMAX = 90%; β-arrestin2 EC50 = 7.4 nM, EMAX = 74%) modifications resulted in modest reductions in potency and efficacy in comparison (Table S5). Further increases in substituent bulk diminished potency even further, as both methylcyclopropyl (compound 19) (GS-cAMP EC50 = 0.5 nM, EMAX = 84%; β-arrestin2 no activity) and cyclobutyl (compound 20) (GS-cAMP EC50 = 0.8 nM, EMAX = 82%; β-arrestin2 no activity) analogues appeared even less active. Interestingly, recruitment in the β-arrestin2 assay appeared to be compromised to a greater extent than G protein pathway activation. As such, compounds in this series trended closer toward strong GS bias as the bulk of the 1-position alkyl substituent increased. Interestingly, when an n-propyl chain was introduced at this position (21), activities in the GS and β-arrestin2 recruitment assays were somewhat improved relative to the bulkier cycloalkyl-containing derivatives (compounds 19 and 20). An n-butyl chain at this position (22), however, was likely too bulky and was not well-tolerated (Figure 6).
Figure 6.

Fifth iteration of analogues (17–22) derived from compound 10 to explore the effects of alkylation of the 1-position nitrogen on the RHS heterocycle. Full concentration–response curves for the fifth iteration of RHS analogues measured using the GloSensor assay for GS pathway activation (A) and the BRET assay for β-arrestin2 recruitment (B) as a percentage of the maximal dopamine (DA) response. Curves represent at least three independent experiments performed in triplicate. Data points represent the mean ± SEM.
Finally, a small set of 5-position ethyl analogues were synthesized based on the top three most active compounds from the previous set of analogues that explored modifications of alkyl size at the 1-position (Figure 7). In particular, we focused on the effect of a slight increase in alkyl bulk at the 5-position in compounds that also contained methyl (compound 23), ethyl (compound 24), and cyclopropyl (compound 25) moieties at the 1-position. These ligands were synthesized starting with a series of commercially available alkyl ureas and reacting them with ethyl 2-cyanobutanoate (Scheme S2). This series of compounds revealed that increasing the steric bulk at the 5-position of the pyrimidine dione RHS did not result in further improvement in ligand activity in either the GS activation and β-arrestin2 recruitment assays. In general, it appeared that additional bulk at the 1- and 5-positions of the RHS together were tolerated but were not favorable for improving ligand activity or in significantly shifting functional selectivity either direction (Table S6).
Figure 7.

Sixth iteration of analogues (23–25) derived from compound 10 to explore the effects of ethylation of the 5-position nitrogen on the RHS heterocycle. Full concentration–response curves for the sixth iteration of RHS analogues measured using the GloSensor assay for GS pathway activation (A) and the BRET assay for β-arrestin2 recruitment (B) as a percentage of the maximal dopamine (DA) response. Curves represent at least three independent experiments performed in triplicate. Data points represent the mean ± SEM.
Modifications of the Middle Phenyl Ring.
After making serial modifications to key areas of the pyrimidine dione RHS and examining the resulting derivatives in assays for both GS activation and β-arrestin2 recruitment, it is clear that increasing substituent bulk at the 1-, 3-, and 5-positions of the ring are not optimal changes for maintaining or improving ligand activity in either downstream pathway. Therefore, we decided to retain the RHS core of compound 10 and instead continue our exploration of other scaffold regions.
Given previous findings that the middle phenyl ring of this noncatechol scaffold is sensitive to dramatic alteration,25,73 we opted to pursue a more targeted modification strategy for this particular region of the scaffold. We sought to create analogue compounds that exploited previous findings in that region to determine if they also held with the pyrimidine dione RHS (Figure 8). All of the compound derivatives that were synthesized to explore this region were synthesized starting with various commercially available brominated phenols using the same general synthetic strategy outlined previously (Scheme S3).
Figure 8.

Compounds 26 and 27 were derived from compound 10 and were designed to explore the importance of the methyl substituent and its position on the middle phenyl ring of the noncatechol scaffold in the presence of the highly active pyrimidine dione RHS moiety. Full concentration–response curves for the middle phenyl ring analogues measured using the GloSensor assay for GS pathway activation (A) and the BRET assay for β-arrestin2 recruitment (B) as a percentage of the maximal dopamine (DA) response. Curves represent at least three independent experiments performed in triplicate. Data points represent the mean ± SEM.
First, on the basis of our previous findings suggesting that moving the methyl substituent over one position to the 3-position on the middle phenyl ring was detrimental to ligand activity,25 we synthesized compound 26 to see what effect this would have on ligand activity and bias compared to compound 10. As expected, we observed a large reduction in potency and efficacy in GS activation and complete abrogation of β-arrestin2 recruitment when 26 was examined. Compound 27, which featured an unsubstituted middle phenyl ring, was synthesized to probe the importance of the biaryl interlocking ring system in this scaffold. The findings outlined in a recent paper by Davoren et al. suggested the importance of a certain degree of steric bulk in the space immediately adjacent to the bond connecting the middle phenyl ring and the RHS because it was believed to help lock the RHS into a fixed position, unable to rotate about its bond axis.69 In comparison to the interlocking ring system in 10, the RHS of compound 27 is expected to be significantly less obstructed and more able to freely rotate. While activity was modestly reduced in both pathways compared to compound 10, compound 27 produced a much milder loss of ligand activity in both pathways (GS-cAMP EC50 = 0.08 nM, EMAX = 96%; β-arrestin2 EC50 = 40 nM, EMAX = 75%) compared to compound 26 (GS-cAMP EC50 = 33 nM, EMAX = 100%; β-arrestin2 no activity) (Table S7). This striking difference is interesting to note in light of the fact that, similar to 27, compound 26 also lacks the steric bulk on the middle phenyl ring immediately proximal to the RHS. Given these findings, it is likely that moving the methyl substituent to the 3-position not only abolishes the interlocking biaryl ring system as in 27 but may also introduce a steric clash with a neighboring residue within D1R, thereby further reducing ligand potency and activity.
Iterative SFSR of the LHS Heterocycle.
Given that the findings in previous studies suggest that the LHS moiety plays a key role in determining the pharmacological properties of the noncatechol ligand at D1R, we opted to explore this region of the scaffold with two different approaches to see if we could obtain some important insights from either method. First, we took an iterative approach to designing an LHS moiety from the ground up, while fixing the RHS and middle phenyl ring as they appear in compound 10 (Figure 9). Derivatives in this initial series were created by following the same general methods previously outlined (Scheme 2).
Figure 9.

First iteration of analogues (28–30) derived from compound 10 to explore the effects of the presence of an LHS heterocycle on the noncatechol scaffold. Full concentration–response curves for the first iteration of LHS analogues measured using the GloSensor assay for GS pathway activation (A) and the BRET assay for β-arrestin2 recruitment (B) as a percentage of the maximal dopamine (DA) response. Curves represent at least three independent experiments performed in triplicate. Data points represent the mean ± SEM.
Scheme 2. Synthesis of the First and Second Iterations of Compounds for Exploring the LHS Heterocyclic Moietya.

aReagents and conditions: (a) Pd(PPh3)4, K2CO3, dioxane, H2O, 120 °C, 25 min; (b) Cs2CO3, DMSO, 125 °C, 48 h; (c) Pd(dppf)Cl2, KOAc, B2(pin)2, dioxane, 80 °C, 18 h.
We began by synthesizing compound 28, which lacked an LHS moiety, to determine if the RHS and middle linker together could form an active ligand on their own. In addition, we synthesized the methoxy analogue 29 to both slightly grow the LHS in size and to cap the polar oxygen in the event that the terminal hydroxyl moiety was problematic for scaffold binding. Finally, we synthesized the pyridine analogue (compound 30) to determine the relative benefit of an aromatic ring at the LHS position. Our findings from this preliminary series of simple LHS moieties suggested the critical importance for an LHS with sufficient size and aromaticity, as both the hydroxy (28) and methoxy (29) analogues were inactive while the unsubstituted pyridine analogue (30) was considerably more active in both the GS activation and β-arrestin2 recruitment assays (GS-cAMP EC50 = 8.1 nM, EMAX = 107%; β-arrestin2 EC50 = 172 nM, EMAX = 40%) (Table S8). Compound 30 was a potent full agonist in stimulating GS activation and served as a partial agonist of β-arrestin2 recruitment.
On the basis of our results from the initial set of LHS derivatives, we synthesized a series of four additional derivatives based on compound 30 (Figure 10). These ligands, which were synthesized with commercially available substituted pyridines (Scheme 2), aimed to explore the effects of various mono- and disubstitution patterns on the LHS pyridine core. In particular, we initially explored the effect of a 4-methoxy substituent (compound 31), a 3-trifluoromethyl substituent (compound 32), 3-cyano-4-methoxy substituents (compound 33), and 3-trifluoromethyl-4-chloro substituents (compound 34) on the LHS pyridine. Interestingly, similar to the unsubstituted pyridine analogue 30, the monosubstituted derivatives 31 and 32 displayed potent full agonism in the GS activation assay as well as partial agonism in the β-arrestin2 recruitment assay (compound 31 GS-cAMP EC50 = 2.0 nM, EMAX = 108%; β-arrestin2: EC50 = 52 nM, EMAX = 27%; compound 32 GS-cAMP EC50 = 0.8 nM, EMAX = 118%; β-arrestin2 EC50 = 68 nM, EMAX = 30%) (Table S9). The disubstituted ligands 33 and 34, however, became completely GS biased, albeit with only relatively mild potency and partial agonism displayed in the GS activation assay (compound 33 GS-cAMP EC50 = 23 nM, EMAX = 59%; β-arrestin2 no activity; compound 34 GS-cAMP EC50 = 105 nM, EMAX = 82%; β-arrestin2 no activity). On the basis of the interesting trends observed in this series of ligands, we sought to determine if we could combine the favorable elements of potency and efficacy obtained from 4-methoxy and 3-trifluoromethyl substituents with the enhanced GS bias observed with pyridine disubstitution on this scaffold to create a highly GS-biased ligand with enhanced potency. The resulting hybrid compound 35, which featured both a 4-methoxy substituent and a 3-trifluoromethyl substituent, was in fact a potent GS-biased ligand. Of note, compound 35 was also characterized as a partial agonist in the GS activation assay (GS-cAMP EC50 = 1.8 nM, EMAX = 66%; β-arrestin2 no activity), while the two monosubstituted analogues 31 and 32 were both full agonists in the GS activation assay (Table S9). This is an interesting finding that warrants further investigation in medicinal chemistry, mutagenesis, and computational modeling experiments. Further, we believe that compounds 31, 32, and 35 together form a powerful set of chemical tools that can be used to further explore the structural determinants of both GS bias and partial agonism in D1R.
Figure 10.

Second iteration of analogues (31–34) derived from compound 30 were designed to explore the effects of substituents on the 3- and 4-positions of the LHS pyridine heterocycle. The interesting trends noted in this mini-series led to the subsequent development of 35. Full concentration–response curves for the second iteration of LHS analogues measured using the GloSensor assay for GS pathway activation (A) and the BRET assay for β-arrestin2 recruitment (B) as a percentage of the maximal dopamine (DA) response. Curves represent at least three independent experiments performed in triplicate. Data points represent the mean ± SEM.
Modifications of the Pyridofuran LHS.
The LHS pyridofuran entity, which has been consistent feature of all of the most active compounds synthesized from this scaffold in both past studies25,68,69 and in the current study (as with compound 10), represents an important element of the noncatechol scaffold and likely contributes to ligand pharmacology through its electronic and spatial properties. Therefore, the second approach we took to explore the critical elements of the LHS region of the scaffold was to make very slight modifications to the pyridofuran entity (Figure 11). Synthesis of these derivatives followed the same general procedures previously discussed (Scheme S4).
Figure 11.

Compounds 36–40 were derived from compound 10 and were designed to explore the effects of methylation, heteroatom type, and heteroatom position within the LHS moiety. Full concentration–response curves for the analogues exploring the LHS pyridofuran moiety measured using the GloSensor assay for GS pathway activation (A) and the BRET assay for β-arrestin2 recruitment (B) as a percentage of the maximal dopamine (DA) response. Curves represent at least three independent experiments performed in triplicate. Data points represent the mean ± SEM.
Briefly, we synthesized two analogues exploring the effects of methylation at various ring positions, including the 2-position of the pyridofuran ring (compound 36) and at the 1-position, which required switching the pyridofuran LHS into a pyrrolopyridine (compound 37). In addition, the effect of removing the 1-position oxygen altogether was explored (compound 38), as was conversion of the 1-position oxygen to a sulfur giving a thienopyridinyl (compound 39). Finally, on the basis of results from our previous study demonstrating that a thieno[2,3-c]pyridine moiety on the LHS could dramatically increase the scaffold’s ability to stimulate β-arrestin2 recruitment,25 we introduced this LHS moiety into this particular set of derivatives (compound 40). Characterization of this series of ligands in GS activation and β-arrestin2 recruitment assays revealed some surprising SFSR trends and yielded several novel compounds with interesting functional selectivity profiles (Figure 11).
First, we observed that simple methylation of the 2-position of the pyridofuran moiety (36) converted the potent, balanced agonist 10 into a less potent partial agonist with complete GS-bias (GS-cAMP EC50 = 4.5 nM, EMAX = 69%; β-arrestin2 no activity). The 1-methyl pyrrolopyridine derivative 37 displayed similar GS-biased partial agonism, albeit with improved potency (GS-cAMP EC50 = 0.8 nM, EMAX = 74%; β-arrestin2 no activity). Compound 38 was also GS-biased (GS-cAMP EC50 = 0.4 nM, EMAX = 115%; β-arrestin2 EC50 > 10 000 nM), though it did retain slightly more activity in β-arrestin2 recruitment compared to 37. Compound 39, which featured a thieno[3,2-c]pyridine moiety on the LHS, displayed diminished activity in both the GS activation and β-arrestin2 recruitment assays (GS-cAMP EC50 = 7.0 nM, EMAX = 104%; β-arrestin2 EC50 = 13 nM, EMAX = 30%) (Table S10). These two compounds underscore the importance of the furan oxygen in the bicyclic LHS for maintaining potent balanced agonism with this scaffold, as neither carbon nor sulfur substitutions were as well-tolerated.
Finally, when the thieno[2,3-c]pyridine moiety was introduced on the scaffold LHS, the activity of the resulting compound 40 in both the GS activation and β-arrestin2 recruitment assays remained roughly similar (GS-cAMP EC50 = 0.02 nM, EMAX = 107%; β-arrestin2 EC50 = 3.5 nM, EMAX = 65%) to that of compound 10 (Table S10). Further work remains to continue understanding the drivers of β-arrestin2 recruitment at D1R and synthesizing compounds with improved β-arrestin2 bias. Nevertheless, the interesting SFSR trends of the LHS suggest that this region of the scaffold is critical for enabling potent, β-arrestin2 recruitment and may potentially be leveraged to design the first β-arrestin2-biased D1R agonist.
Taken together, an iterative design strategy employed at both ends of an interesting noncatechol scaffold yielded several novel compounds with highly diverse functional selectivity profiles and bias factors (Figure 12). In addition, compared to previous SFSR strategies in the field, ligand design from this “ground up” approach enabled the development of better insights and a clearer understanding of precisely which structural components were critical for the scaffold’s potency, efficacy, and signaling bias.
Figure 12.

Summary of the full concentration–response curves of structurally related analogue compounds of the noncatechol scaffold that display dramatically different functional selectivity profiles, as measured using the GloSensor assay for GS pathway activation (GS-cAMP) and the BRET assay for β-arrestin2 (β-arr2) recruitment as a percentage of the maximal dopamine (DA) response. Dopamine (A) is a potent, balanced full agonist of both signaling pathways. Compound 10 (B) is a highly potent full agonist with balanced activity relative to dopamine. Compound 35 (C) is a potent partial agonist that displays complete GS-bias, a similar profile shared with compounds 36 (D) and 37 (E). Compound 40 (F) is a potent agonist of both signaling pathways. Data points represent the mean concentrations ± SEM from at least three independent experiments.
Binding Affinity and Selectivity.
Our ground-up exploration and synthesis of novel noncatechol compounds yielded several interesting new candidates that we subsequently tested in validated radioligand binding affinity assays to assess target selectivity.74 In particular, we selected six ligands from our study that possessed unique structural elements and in vitro functional selectivity profiles and tested them for their affinity across a broad panel of over 30 potential off-target proteins, including Class A aminergic GPCRs, transporters, and ion channels. Compounds were first screened in primary radioligand binding assays to determine if they reached a certain affinity threshold (measured as % inhibition) for any target on the panel. Compounds with a mean inhibition greater than 50% at 10 μM across four independent determinations were considered to have met the affinity threshold and were subsequently examined in secondary radioligand binding assays for Ki determination at each receptor that met the minimum threshold.
We chose to examine the relatively balanced ligands 10 and 40, the highly GS-biased ligands 35, 36, and 37, and the slightly β-arrestin2-biased ligand 18. The results from these experiments clearly indicate that regardless of potency, efficacy, or functional selectivity, derivatives of this noncatechol scaffold retain high selectivity for the D1-like dopamine receptors, D1R and D5R (Table 1).
Table 1.
Radioligand Binding Affinities of Compounds 10, 18, 35, 36, 37, and 40 at Select GPCRs, Ion Channels, and Transportersa
| binding affinity Ki (nM) | ||||||
|---|---|---|---|---|---|---|
| receptor | 10 | 18 | 35 | 36 | 37 | 40 |
| D1 | 5.7 | 10 | 70 | 93 | 14 | 5.1 |
| D2 | NC | NC | NC | NC | NC | NC |
| D3 | NC | NC | NC | NC | NC | NC |
| D4 | NC | NC | NC | NC | NC | NC |
| D5 | 6.7 | 15 | 171 | 185 | 90 | 53 |
| 5-HT1A | NC | NC | NC | NC | NC | NC |
| 5-HT1B | NC | NC | NC | NC | NC | NC |
| 5-HT1D | NC | NC | NC | NC | NC | NC |
| 5-HT1E | NC | NC | NC | NC | NC | NC |
| 5-HT2A | NC | NC | NC | NC | NC | NC |
| 5-HT2B | NC | NC | NC | NC | NC | NC |
| 5-HT3 | NC | NC | NC | NC | NC | NC |
| 5-HT4 | NC | NC | NC | NC | NC | NC |
| 5-HT5A | NC | NC | NC | NC | NC | NC |
| 5-HT6 | NC | NC | NC | NC | NC | NC |
| 5-HT7A | NC | NC | NC | NC | NC | NC |
| Alpha1A | NC | NC | NC | NC | NC | NC |
| Alpha1B | NC | NC | NC | NC | NC | NC |
| Alpha2A | NC | NC | 6900 | NC | NC | NC |
| Beta1 | NC | NC | NC | NC | NC | NC |
| Beta2 | NC | NC | NC | NC | NC | NC |
| MOR | NC | NC | NC | NC | NC | NC |
| KOR | NC | NC | NC | NC | NC | NC |
| M1 | NC | NC | NC | NC | NC | NC |
| M2 | NC | 7700 | 6900 | NC | NC | NC |
| M3 | NC | NC | NC | NC | 4200 | 4600 |
| Sigma1 | NC | NC | NC | NC | NC | NC |
| H1 | NC | NC | NC | NC | NC | NC |
| H2 | NC | NC | NC | NC | NC | NC |
| DAT | NC | NC | NC | NC | NC | NC |
| SERT | NC | NC | NC | NC | NC | NC |
Ki values represent the average of at least three triplicate experiments. SEM < ±20%. NC: Not calculated because minimum affinity threshold was not reached in primary binding assay.
Mouse Pharmacokinetic Studies.
Previous studies of this scaffold suggest that it possesses excellent pharmacokinetic properties and blood–brain barrier (BBB) penetrance, likely owing to its small molecular weight, relative hydrophobicity, and lack of a catechol group.25,68 In this study, we discovered a series of novel compounds with chemical entities that differ significantly from those described in previous studies. In particular, the pyrimidine dione moiety on the RHS is a new addition that distinguishes these series of compounds from those in previous studies that examined pharmacokinetics and BBB penetration. Further, this RHS structure is relatively more polar, and from a chemical perspective, appears to be highly capable of forming hydrogen bonding interactions. Therefore, we were not sure how well these structural modifications would be tolerated when these ligands were tested in vivo.
To adequately assess the in vivo pharmacokinetic properties of a representative set of ligands, we selected compounds 10 and 35 to be administered to mice in triplicate. Each compound was administered to mice through an intraperitoneal (i.p.) injection at a dose of approximately 50 mg/kg. The drug concentrations were monitored in the brain and plasma at three time points, 0.5, 1.5, and 4.0 h after injection. This study revealed that, despite potential concerns that the chemical structures of this series may impede BBB penetrance or lower plasma exposure, the ligands all displayed excellent pharmacokinetic properties and BBB penetrance (Figure 13). For example, compounds 10 and 35 both reached peak concentrations of nearly 100 μM in the brain and plasma after administration at 50 mg/kg. These high concentration levels persisted well up to even 4 hours after administration. Further, despite administration at a relatively high dose, the compounds appeared well-tolerated and none of them appeared to induce any adverse clinical signs in the animals.
Figure 13.

Mouse pharmacokinetic profile of compounds 10 (A) and 35 (B) in the plasma and brain. Both compounds were injected into the peritoneum at 50 mg/kg. Concentrations in the brain and plasma were quantified at 0.5, 1.5, and 4.0 h after administration. Experiments were carried out in biological triplicates, with points representing mean concentrations ± SEM.
Together, these two ligands, which each display unique differences in their potencies, efficacies, and functional selectivity profiles, both possess excellent pharmacokinetic properties and BBB penetrance. These features indicate that these ligands may be valuable tools for the scientific community to further explore the roles of biased and balanced signaling at D1R.
Behavioral Studies in Mice.
The highly selective activity of this class of noncatechol agonists for the D1-like receptors D1R and D5R, as well as the favorable pharmacokinetic properties and high BBB penetrance, encouraged us to examine how a subset of these ligands with diverse functional selectivity profiles could impact various behavioral and physiological outcomes in animal studies. One particularly compelling pathophysiological domain for investigating the role of D1R agonism and functional selectivity is in the treatment of PD and LID.
In a previous study by Gray et al., a GS-biased noncatechol ligand was administered in an acute rodent model of PD.68 Interestingly, compared to dopamine, the ligand was found to produce a more sustained dopaminergic response in the animals. This was believed to be due to the inability of the GS-biased ligands to recruit β-arrestin2, thereby attenuating any β-arrestin2-mediated desensitization and tachyphylaxis which may occur after repeated dosing. Separate studies of D1R signaling in rodent PD models have suggested that the GS-mediated pathway may actually be responsible for the development of LID after long-term treatment with l-DOPA.75 At the same time, genetic models have suggested that β-arrestin2-mediated signaling may attenuate LID while still remedying locomotor deficits.20,21,75 Despite the varying hypotheses of which downstream signaling pathway may be more important to target, the complex pathophysiology underlying these models and disease states make them fascinating systems in which to apply our ligands to study GPCR functional selectivity.
For these reasons, we opted to explore how our ligands might impact animal behaviors in animal models of PD. For our studies, we used 6-hydroxydopamine (6-OHDA)-lesioned mice as an animal model of PD. Unilateral injection of 6-OHDA into medial forebrain bundle in rodents results in supersensitivity of central dopamine receptors, a state conceptualized as similar to PD.76 Similarly, unilaterally 6-OHDA-lesioned rodents treated with L-DOPA develop “dyskinesias” consisting of contralateral turning of the neck and jerking of contralateral forelimbs.77,78 For these reasons, 6-OHDA-lesioned mice represent a relevant model of PD that is widely used in the field of neuroscience for studying PD pathophysiology. In these studies, we chose to focus our experiments on compound 10. We selected compound 10 because it was a balanced agonist with high potency in both signaling assays and displayed highly favorable pharmacokinetic properties and BBB penetrance, with minimal decline in concentrations achieved in the brain or plasma even up to 4 hours after administration.
Compound 10 was administered to 6-OHDA-lesioned mice as a 5 mg/kg intraperitoneal injection (i.p.). SKF81297, a known D1/D5-selective full agonist, was also administered at 5 mg/kg i.p. in a cohort of mice as a positive control, while vehicle was administered as a negative control. The motor activities of the mice were subsequently monitored using an automated tracking system that included overhead recording cameras and sensor beams that detect movement within the arena. The distances traveled by the mice and the number of contralateral rotations they performed were tracked, recorded, and quantified. Both of these motor behaviors are recognized as lines of evidence supportive of central dopaminergic stimulation in 6-OHDA model of PD.79–82
We found that injection of compound 10 into 6-OHDA-lesioned mice produced statistically significant increases in forward locomotion at each measured time interval (Figure 14A) and in the total distance traveled during the observation period (Figure 14B), compared to the vehicle control. Similar effects were observed in mice that were administered SKF81297. We also observed significant increases in the number of contralateral rotations that the mice performed when they were injected with either compound 10 or SKF81297 (Figure 14C and D).
Figure 14.

Compound 10 displays a potent antiparkinsonian-like activity in a motor activity study in 6-OHDA-lesioned mice. 6-OHDA-lesioned mice were given the vehicle, 10 (5 mg/kg), or SKF81297 (5 mg/kg) intraperitoneally and monitored for 90 min. (A) Locomotor activities are shown as binned 10 min intervals. The repeated measures analysis of variance (RMANOVA) are provided: [time F(8, 224) = 3.17, p = 0.0020; treatment F(2, 28) = 12.42, p = 0.0001; time by treatment F(16, 224) = 7.825, p < 0.0001]. (B) Cumulative locomotor activities from the 0–60 and 61–90 min intervals. A RMANOVA found: [prepost F(1, 28) = 89.06, p < 0.0001; treatment F(2, 28) = 12.42, p = 0.0001; prepost by treatment F(2, 28) = 17.42, p < 0.0001]. (C) Contralateral rotation activities are shown as binned 10 min intervals. The repeated measures analysis of variance (RMANOVA) are provided: [time F(8, 224) = 5.23, p < 0.0001; treatment F(2, 28) = 5.482, p = 0.0098; time by treatment F(16, 224) = 4.212, p < 0.0001]. (D) Cumulative contralateral rotation activities from the 0–60 min and 61–90 min intervals. A RMANOVA found: [prepost F(1, 28) = 18.52, p = 0.0002; treatment F(2, 28) = 5.482, p = 0.0098; prepost by treatment F(2, 28) = 6.013, p = 0.0067]. N = 10–11 mice/group; Bonferroni-corrected pairwise comparisons—*p < 0.05, **p < 0.0001.
Interestingly, in both of these experiments, we noted a gradual decline in the behavioral efficacy of the SKF81297 control compound over the 90 min study interval, particularly starting around the 50 min mark, while this effect was not observed with compound 10. As such, SKF81297-treated mice no longer displayed significantly increased forward locomotion compared to vehicle after 1 h, while compound 10-treated mice did (Figure 14B). While it is possible that this may be related to the pharmacokinetic differences between the ligands, such as a longer half-life or better BBB penetration, future investigation should be performed to better determine the factors that may be contributing to this phenomenon. Regardless, these studies suggest that compound 10 is a potent dopaminergic agonist that demonstrates robust in vivo activity, comparable to other known D1 agonists, and is suitable for in vivo studies of dopamine signaling and transmission.
CONCLUSION
We conducted a novel exploratory study that takes a significantly different approach to ligand discovery from both our previous systematic SFSR campaign and other studies reporting non-catechol analogues of D1R. Here, we utilize the lessons and trends learned from previous studies to design novel ligands “from the ground up”. From this approach, we obtained a better understanding of precisely which structural components are critical for the scaffold’s potency, efficacy, and signaling bias. In addition, we report the discovery of several new scaffold derivatives with bias signaling properties and robust in vivo activity. In particular, compound 10 is a balanced full agonist at D1R with picomolar potency, while compounds 35, 36, and 37 are completely GS-biased partial agonists with low nanopolar potency. We also report the discovery of compound 40, an analogue of 10 with low nanomolar potency and balanced activity in stimulating the β-arrestin2 signaling pathway.
Despite the exquisite potency of these compounds at D1R, these ligands also remained highly selective for the D1-like receptors, D1R and D5R, when tested across a broad panel of potential off-targets, including Class A aminergic GPCRs, ion channels, and transporters. Importantly, compounds 10 and 35 were also demonstrated to have good BBB penetrance and maintain stable levels in the brain and plasma for at least 4 hours after administration. Upon intraperitoneal administration in a 6-OHDA mouse model of PD, compound 10 demonstrated antiparkinsonian and pro-dyskinetic potential through restoration of locomotion and potentiation of behaviors indicative of dyskinesia. This is the first in vivo characterization of a potent, unbiased noncatechol D1R agonist. Taken together, the results from this study suggest that the ligands developed herein have strong potential to serve as chemical tools for future work investigating the effects of biased signaling at D1R. Finally, we believe that this study may also serve as a valuable template for an iterative approach to functionally selective GPCR ligand discovery, carrying particular relevance to exploring the structural determinants of a ligand that are critical for biased signaling.
METHODS
Chemistry General Procedures.
Each of the reagents in this study were of commercial grade and were used without additional purification. Dry solvents were purchased from Sigma-Aldrich and were anhydrous. Procedures necessitating microwave heating involved the use of a Discover SP microwave system with an Explorer 12 Hybrid Autosampler by CEM (Buckingham, UK). Reactions performed via microwave heating were conducted at 125 °C for 25 min using 250 W and a pressure less than 300 psi. Analytical thin-layer chromatography (TLC) was carried out using EMD Millipore 210–270 μm 60-F254 silica gel plates. TLC plate visualization was done using 254 μm UV light. Flash column chromatography was implemented with a Teledyne (Thousand Oaks, CA) ISCO CombiFlash Rf+ system. This system was equipped with normal phase RediSep Rf silica columns, a UV detector, and a fraction collector apparatus. A preparative high-performance liquid chromatography (HPLC) system was used to purify all final compounds. Preparative HPLC was performed using an Agilent Prep 1200 series with a Phenomenex Luna 750 mm × 30 mm, 5 μm, C18 column, with column injection proceeding at room temperature. HPLC solvent flow rate was set to 40 mL/min while the UV detector was set to 254 nm. A linear gradient was set for the purification program and consisted of 10% (or 50%) MeOH (A) in H20) (with 0.1% TFA) (B) to 100% MeOH (A). HPLC was used to establish the purity of each target compound. All biologically evaluated compounds had >95% purity after purification via the HPLC methods described. A liquid chromatography–mass spectrometer (LC-MS) was used to characterize all compounds. Liquid chromatography–high-resolution mass spectrometry (LC-HRMS) was used to further assess key compounds, as follows. An Agilent 1200 series system, fully equipped with a DAD detector, was used to acquire HPLC spectra for all compounds. This system contained a 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column and the flow rate was set to 0.4 mL/min. The solvents used in the separation gradient program were as follows: solvent A included water containing 0.1% formic acid and solvent B included acetonitrile containing 0.1% formic acid. The separation gradient program included 1% B from 0 to 1 min, followed by 1–99% B from 1 to 4 min, and finally 99% B from 4 to 8 min. The resulting spectra were used in reporting the HPLC retention times for the compounds below. HRMS data was also obtained for key compounds below. To obtain HRMS data, samples were ionized by electrospray ionization (ESI) in positive mode. HRMS analysis was conducted using a G1969A high-resolution API-TOF mass spectrometer by Agilent Technologies. To this spectrometer was attached the 1200 HPLC system described previously. Nuclear magnetic resonance (NMR) spectroscopy was also implemented for the characterization of all compounds. NMR spectra were acquired on a Bruker (Billerica, MA) DRX-600 spectrometer. The chemical shifts reported below are given in parts per million (ppm, δ) and are relative to residual solvent peaks (CD3OD, 1H 3.31 ppm; CDCl3, 1H 7.26 ppm). Data below reported from the 1H NMR spectra are given as chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, app = apparent), coupling constant, and integration.
4-(3-Methyl-4-(1-methyl-1,2,3,6-tetrahydropyridin-4-yl)-phenoxy)furo[3,2-c]pyridine (1).
To an oven-dried microwavable tube was added a stir bar, tetrakis(triphenylphosphine)palladium(0) (26 mg, 0.023 mmol), potassium carbonate (44 mg, 0.32 mmol), 4-bromo-1-methyl-1,2,3,6-tetrahydropyridine (27 mg, 0.15 mmol), and the known intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine25,68,69 (60 mg, 0.17 mmol) in dioxane (1 mL) and water (0.2 mL). Microwave irradiation was then applied to the mixture at 125 °C for 25 min, as described in the General Chemistry Procedures section. The resulting mixture was subsequently filtered through Celite; the filter was washed with several times with ethyl acetate, and the organic filtrate diluted with water and extracted three times with ethyl acetate. Brine and saturated aqueous NaHCO3 were then used to wash the combined organic layers. The organic layers were then dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was taken up in 2 mL of methanol and purified by HPLC using the conditions described previously. This procedure yielded compound 1 as a clear oil (5.6 mg, 11%). 1H NMR (500 MHz, methanol-d4): δ 7.98 (d, J = 5.9 Hz, 1H), 7.90 (d, J = 2.2 Hz, 1H), 7.41 (dd, J = 5.9, 1.0 Hz, 1H), 7.23 (d, J = 8.2 Hz, 1H), 7.10 (d, J = 2.4 Hz, 1H), 7.05 (dd, J = 8.3, 2.5 Hz, 1H), 6.84 (p, J = 1.1 Hz, 1H), 5.73–5.68 (m, 1H), 4.08 (d, J = 16.5 Hz, 1H), 3.84 (dd, J = 16.7, 3.1 Hz, 1H), 3.72 (dd, J = 12.3, 5.9 Hz, 1H), 3.45 (td, J = 11.6, 4.9 Hz, 1H), 3.06 (s, 3H), 2.97 (d, J = 9.1 Hz, 1H), 2.64 (d, J = 18.5 Hz, 1H), 2.37 (s, 3H). MS (ESI): m/z 321.2 [M + 1]+.
4-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-1-methylpyridin-2(1H)-one (2).
Compound 2 was prepared as a clear oil using the same procedure outlined above for the preparation of compound 1 starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine and 4-bromo-1-methylpyridin-2(1H)-one, yield = 25%. 1H NMR (600 MHz, methanol-d4): δ 8.02 (s, 1H), 7.93 (s, 1H), 7.75 (s, 1H), 7.44 (s, 1H), 7.34 (s, 1H), 7.20 (s, 1H), 7.14 (s, 1H), 6.83 (s, 1H), 6.55 (s, 1H), 6.49 (s, 1H), 3.36–3.29 (m, 3H), 2.36 (s, 3H). MS (ESI): m/z 333.1 [M + 1]+.
4-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)pyridin-2(1H)-one (3).
Compound 2 was prepared as a clear oil using the same procedure as was used for the preparation of compound 1 starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaboro-lan-2-yl)phenoxy)furo[3,2-c]pyridine and 4-bromopyridin-2(1H)-one, yield = 64%. 1H NMR (600 MHz, methanol-d4): δ 7.99 (s, 1H), 7.92 (s, 1H), 7.71 (s, 1H), 7.44 (s, 1H), 7.35 (s, 1H), 7.19 (s, 1H), 7.16 (s, 1H), 6.86 (s, 1H), 6.52 (s, 1H), 6.51 (s, 1H), 2.37 (s, 3H). MS (ESI): m/z 319.1 [M + 1]+.
4-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-6-methylpyridin-2(1H)-one (4).
Compound 2 was prepared as a clear oil using the same procedure as was used for the preparation of compound 1 starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine and 4-bromo-6-methylpyridin-2(1H)-one, yield = 51%. 1H NMR (600 MHz, methanol-d4): δ 8.03 (d, J = 5.9 Hz, 1H), 7.94 (d, J = 2.3 Hz, 1H), 7.47 (d, J = 6.0 Hz, 1H), 7.37 (d, J = 8.3 Hz, 1H), 7.23 (s, 1H), 7.17 (d, J = 8.2 Hz, 1H), 6.86 (s, 1H), 6.61 (s, 1H), 6.56 (s, 1H), 2.48 (s, 3H), 2.38 (s, 3H). MS (ESI): m/z 333.1 [M + 1]+.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)pyrimidin-4(3H)-one (5).
Compound 2 was prepared as a clear oil using the same procedure as was used for the preparation of compound 1 starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine and 6-bromopyrimidin-4(3H)-one, yield = 5%. 1H NMR (600 MHz, methanol-d4): δ 8.32 (s, 1H), 8.00 (d, J = 5.9 Hz, 1H), 7.91 (d, J = 2.3 Hz, 1H), 7.52 (d, J = 8.3 Hz, 1H), 7.41 (d, J = 5.9 Hz, 1H), 7.18 (d, J = 2.4 Hz, 1H), 7.14 (dd, J = 8.4, 2.5 Hz, 1H), 6.91 (d, J = 2.2 Hz, 1H), 6.56 (s, 1H), 2.46 (s, 3H). MS (ESI): m/z 320.1 [M + 1]+.
4-(3-Methyl-4-(5-methylpyrimidin-4-yl)phenoxy)furo[3,2-c]-pyridine (6).
Compound 2 was prepared as a clear oil using the same procedure as was used for the preparation of compound 1 starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaboroan-2-yl)phenoxy)furo[3,2-c]pyridine and 4-bromo-5-methylpyrimidine, yield = 78%. 1H NMR (500 MHz, methanol-d4): δ 9.16 (s, 1H), 8.07 (d, J = 6.0 Hz, 1H), 7.96–7.92 (m, 1H), 7.50–7.47 (m, 2H), 7.34 (d, J = 8.3 Hz, 1H), 7.28 (d, J = 2.3 Hz, 1H), 7.23–7.20 (m, 1H), 6.82 (dt, J = 2.3, 1.1 Hz, 1H), 2.27 (s, 3H), 2.17 (s, 3H). MS (ESI): m/z 318.1 [M + 1]+.
4-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-5-methylpyrimidin-2-amine (7).
Compound 2 was prepared as a clear oil using the same procedure as was used for the preparation of compound 1 starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine and 4-bromo-5-methylpyrimidin-2-amine, yield = 99%. 1H NMR (500 MHz, methanol-d4): δ 8.51 (s, 1H), 8.03 (d, J = 5.8 Hz, 1H), 7.92 (d, J = 2.2 Hz, 1H), 7.47–7.42 (m, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.28 (d, J = 2.3 Hz, 1H), 7.22 (dd, J = 8.4, 2.4 Hz, 1H), 6.93 (dd, J = 2.2, 1.0 Hz, 1H), 2.28 (s, 3H), 2.12 (s, 3H). MS (ESI): m/z 333.1 [M + 1]+.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)pyrimidine-2,4-diamine (8).
Compound 2 was prepared as a clear oil using the same procedure as was used for the preparation of compound 1 starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine and 6-bromopyrimidine-2,4-diamine, yield = 19%. 1H NMR (500 MHz, methanol-d4): δ 7.99 (dd, J = 5.9, 1.5 Hz, 1H), 7.93 (d, J = 2.5 Hz, 1H), 7.49 (d, J = 7.9 Hz, 1H), 7.43 (dd, J = 5.9, 1.7 Hz, 1H), 7.27 (d, J = 2.4 Hz, 1H), 7.22 (d, J = 8.3 Hz, 1H), 6.97 (d, J = 2.3 Hz, 1H), 6.18–6.14 (m, 1H), 2.43 (s, 3H). MS (ESI): m/z 334.1 [M + 1]+.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)pyrimidine-2,4(1H,3H)-dione (9).
Compound 2 was prepared as a clear oil using the same procedure as was used for the preparation of compound 1 starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine and 6-bromopyrimidine-2,4(1H,3H)-dione, yield = 26%. 1H NMR (600 MHz, methanol-d4): δ 7.97 (d, J = 11.0 Hz, 1H), 7.91 (d, J = 14.9 Hz, 1H), 7.43 (d, J = 17.0 Hz, 2H), 7.19 (d, J = 13.3 Hz, 1H), 7.15 (s, 1H), 6.95 (d, J = 14.2 Hz, 1H), 5.61 (d, J = 14.6 Hz, 1H), 2.42 (s, 3H). MS (ESI): m/z 336.1 [M + 1]+.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (10).
A 4.4 M solution of NaOMe (11.9 mmol) in methanol was added to a solution of 1-methylurea (0.83 g, 10.9 mmol) and ethyl 2-cyanopropanoate (9.96 mmol) in methanol (7.5 mL). The reaction was then heated at reflux for 18 h, and then cooled to room temperature. The solvent was then removed under vacuum, and the residue was taken up in acetonitrile and repeatedly evaporated. The residue was then partitioned in 1:1 acetonitrile: H2O and 6 M HCl was added until the pH was measured to be 2. At this point, the mixture was stirred for 1 h to allow precipitate formation. The precipitate was then collected, filtered, and washed with diethyl ether to generate the intermediate 6-amino-1,5-dimethylpyrimidine-2,4-(1H,3H)-dione in good yields and was used in subsequent reactions without further purification. 6-Amino-1,5-dimethylpyrimidine-2,4-(1H,3H)-dione (6.7 mmol) was then reacted with NaNO2 (10.2 mmol) and CuBr2 (13.4 mmol) in solution of acetonitrile (7 mL) and H2O (7 mL) at room temperature for 66 h. After it was stirred for 66 h, approximately 20 mL of 1 N H2SO4 and 10 mL of ethyl acetate were added to enable precipitation to occur. The resulting precipitate was then collected by filtration and washed with ethyl acetate and H2O before being dried under vacuum. This yielded the intermediate 6-bromo-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (10i) in excellent yield and the material was used in subsequent reactions without further purification. Compound 10 was prepared as a clear oil using the same procedure as was used for the preparation of compound 1 starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine and 6-bromo-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (10i), yield = 31%. 1H NMR (600 MHz, methanol-d4): δ 8.01 (d, J = 5.8 Hz, 1H), 7.93 (d, J = 2.4 Hz, 1H), 7.43 (d, J = 5.9 Hz, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 8.5 Hz, 1H), 6.96 (s, 1H), 3.08 (h, J = 2.5, 1.7 Hz, 3H), 2.24 (s, 3H), 1.67 (h, J = 2.7, 2.0 Hz, 3H). HPLC 99% pure, tR = 4.745 min. HRMS: m/z [M + H]+ calculated for C20H18N3O4+ 364.1292, found 364.1299.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-1,5-dimethylpyrimidin-2(1H)-one (11).
Compound 2 was prepared as a clear oil using the same procedure as was used for the preparation of compound 1 starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine and 6-bromo-1,5-dimethylpyrimidin-2(1H)-one, yield = 56%. 1H NMR (600 MHz, methanol-d4): δ 8.72 (s, 1H), 8.02 (d, J = 5.9 Hz, 1H), 7.96 (d, J = 2.2 Hz, 1H), 7.47–7.44 (m, 1H), 7.39 (s, 1H), 7.37–7.31 (m, 2H), 7.03 (d, J = 2.1 Hz, 1H), 3.45 (s, 3H), 2.24 (s, 3H), 1.96 (s, 3H). MS (ESI): m/z 348.1 [M + 1]+.
5-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-4-methylpyridazin-3(2H)-one (12).
To an oven-dried flask was added 4,5-dimethylpyridazin-3-ol (4.2 g, 25 mmol), 3,4-dihydro-2H-pyran (16.8 g, 200 mmol), p-toluenesulfonic acid monohydrate (0.95 g, 5 mmol), and tetrahydrofuran (200 mL). The solution was stirred overnight to give 4,5-dichloro-2-(tetrahydro-2H-pyran-2-yl)pyridazin-3(2H)-one (5.7 g, 22.88 mmol), which was subsequently reacted with methylboronic acid (1.37 g, 22.88) in the presence of cesium carbonate (22.36 g, 68.64 mmol), [1,1′-bis(diphenylphosphino)ferrocene]-dichloropalladium(II) (0.83 g, 1.14 mmol), dioxane (40 mL), and water (4 mL) at 110 °C for 2 h. Chromatography and mass spectrometry revealed that this reaction resulted in production of both of the structural isomers, 5-chloro-4-methyl-2-(tetrahydro-2H-pyran-2-yl)pyridazin-3(2H)-one (12i) and 4-chloro-5-methyl-2-(tetrahydro-2H-pyran-2-yl)pyridazin-3(2H)-one (13i). Compound 12 was prepared as a light yellow residue using a modified Suzuki coupling procedure starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine and 5-chloro-4-methyl-2-(tetrahydro-2H-pyran-2-yl)pyridazin-3(2H)-one (12i). In this modified procedure, after the Suzuki coupling reaction was performed as described for the preparation of compound 1, the resulting reaction mixture was filtered over Celite, slowly acidified by the dropwise addition of 4.0 M HCl in dioxanes, and stirred overnight to allow full deprotection prior to HPLC purification. This method afforded the desired compound 12, yield = 34%. 1H NMR (600 MHz, methanol-d4): δ 7.98 (d, J = 23.5 Hz, 1H), 7.91 (d, J = 13.6 Hz, 1H), 7.79–7.73 (m, 1H), 7.65 (s, 1H), 7.42 (s, 1H), 7.24 (s, 1H), 7.17 (s, 1H), 6.89 (s, 1H), 2.19 (s, 3H), 2.02 (s, 3H). MS (ESI): m/z 334.1 [M + 1]+.
4-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-5-methylpyridazin-3(2H)-one (13).
Compound 13 was prepared as a light yellow residue using the same modified procedure as was used for the preparation of compound 12 above starting with the intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo-[3,2-c]pyridine and 4-chloro-5-methyl-2-(tetrahydro-2H-pyran-2-yl)-pyridazin-3(2H)-one (13i), yield = 24%. 1H NMR (600 MHz, methanol-d4): δ 7.99 (d, J = 5.9 Hz, 1H), 7.96 (s, 1H), 7.88 (d, J = 2.3 Hz, 1H), 7.39 (d, J = 5.9 Hz, 1H), 7.19 (d, J = 2.4 Hz, 1H), 7.17 (d, J = 8.2 Hz, 1H), 7.14–7.10 (m, 1H), 6.87 (s, 1H), 2.16 (s, 3H), 2.10 (s, 3H). MS (ESI): m/z 334.1 [M + 1]+.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-1,3,5-trimethylpyrimidine-2,4(1H,3H)-dione (14).
To an oven-dried flask was added a stir bar, compound 10 (40 mg, 0.11 mmol), lithium bromide (11.5 mg, 0.13 mmol), sodium bis(trimethylsilyl)amide (24.3 mg, 0.13 mmol), and anhydrous N,N-dimethylformamide (1.0 mL). The reaction was allowed to stir at room temperature under N2 for 30 min before the dropwise addition of methyl iodide (0.143 mmol). The reaction was then stirred overnight, then quenched with H2O, and neutralized to pH 7 with 1 M HCl. The solution was then extracted three times with ethyl acetate, dried over anhydrous sodium sulfate, concentrated in vacuo, and purified by HPLC to give compound 14 as a tan oil, yield = 46%. 1H NMR (600 MHz, methanol-d4): δ 8.02 (s, 1H), 7.94 (s, 1H), 7.44 (s, 1H), 7.30 (s, 2H), 7.24 (s, 1H), 6.96 (s, 1H), 3.43 (s, 3H), 3.14 (s, 3H), 2.24 (s, 3H), 1.71 (s, 3H). MS (ESI): m/z 378.1 [M + 1]+.
3-Ethyl-6-(4-(furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (15).
Compound 15 was prepared as a tan oil using the same procedure as was used for the preparation of compound 14 starting with compound 10 and instead using ethyl iodide, yield = 56%. 1H NMR (600 MHz, methanol-d4): δ 8.02 (s, 1H), 7.94 (s, 1H), 7.43 (s, 1H), 7.34–7.26 (m, 2H), 7.24 (s, 1H), 6.96 (s, 1H), 4.11 (s, 2H), 3.13 (s, 3H), 2.24 (s, 3H), 1.70 (s, 3H), 1.29 (s, 3H). MS (ESI): m/z 392.2 [M + 1]+.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-3-isopropyl-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (16).
Compound 16 was prepared as a tan oil using the same procedure as was used for the preparation of compound 14 starting with compound 10 and instead using isopropyl iodide, yield = 23%. 1H NMR (600 MHz, methanol-d4): δ 8.01 (d, J = 7.7 Hz, 1H), 7.92 (s, 1H), 7.42 (s, 1H), 7.27 (d, J = 9.0 Hz, 2H), 7.21 (s, 1H), 6.95 (s, 1H), 3.09 (s, 3H), 2.23 (s, 3H), 1.67 (s, 3H), 1.57 (d, J = 6.9 Hz, 1H), 1.53 (t, 6H). MS (ESI): m/z 406.2 [M + 1]+.
1-Ethyl-6-(4-(furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-5-methylpyrimidine-2,4(1H,3H)-dione (17).
Compound 17 was prepared in an analogous fashion to the preparation of compound 10, using the same procedure but starting with 1-ethylurea and ethyl 2-cyanopropanoate to obtain the 1-ethyl analogue of compound 10. The desired product was obtained as an off-white residue, yield = 11%. 1H NMR (600 MHz, methanol-d4): δ 8.03 (d, J = 10.9 Hz, 1H), 7.94 (s, 1H), 7.45 (s, 1H), 7.36 (d, J = 9.9 Hz, 1H), 7.30 (s, 1H), 7.25 (s, 1H), 6.92 (d, J = 13.5 Hz, 1H), 3.87 (s, 2H), 2.25 (s, 3H), 1.64 (s, 3H), 1.10 (s, 3H). MS (ESI): m/z 378.1 [M + 1]+.
1-Cyclopropyl-6-(4-(furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-5-methylpyrimidine-2,4(1H,3H)-dione (18).
Compound 18 was prepared in an analogous fashion to the preparation of compound 10, using the same procedure but starting with 1-cyclopropylurea and ethyl 2-cyanopropanoate to obtain the 1-cyclopropyl analogue of compound 10. The desired product was obtained as an off-white residue, yield = 6%. 1H NMR (600 MHz, methanol-d4): δ 8.03 (s, 1H), 7.92 (s, 1H), 7.43 (d, J = 12.3 Hz, 1H), 7.36 (s, 1H), 7.25 (s, 1H), 7.19 (s, 1H), 6.91 (s, 1H), 2.57 (s, 1H), 2.27 (s, 3H), 1.69 (s, 3H), 0.78–0.62 (m, 4H). HPLC 99% pure, tR = 4.577 min. HRMS: m/z [M + H]+ calculated for C22H20N3O4+ 390.1448, found 390.1456.
1-(Cyclopropylmethyl)-6-(4-(furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-5-methylpyrimidine-2,4(1H,3H)-dione (19).
Compound 19 was prepared in an analogous fashion to the preparation of compound 10, using the same procedure but starting with 1-methylcyclopropyllurea and ethyl 2-cyanopropanoate to obtain the 1-methylcyclopropyl analogue of compound 10. The desired product was obtained as an off-white residue, yield = 9%. 1H NMR (500 MHz, methanol-d4): δ 8.14 (s, 1H), 7.92 (d, J = 2.2 Hz, 1H), 7.46 (d, J = 4.8 Hz, 1H), 7.37 (d, J = 8.3 Hz, 1H), 7.28 (d, J = 2.3 Hz, 1H), 7.23 (dd, J = 8.3, 2.3 Hz, 1H), 6.92 (dd, J = 2.2, 0.9 Hz, 1H), 3.66 (dd, J = 14.4, 7.2 Hz, 1H), 3.41–3.35 (m, 2H), 2.25 (s, 3H), 1.66 (s, 3H), 0.23 (dd, J = 4.7, 3.2 Hz, 1H), 0.16 (dd, J = 4.8, 3.5 Hz, 1H). MS (ESI): m/z 404.2 [M + 1]+.
1-Cyclobutyl-6-(4-(furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-5-methylpyrimidine-2,4(1H,3H)-dione (20).
Compound 20 was prepared in an analogous fashion to the preparation of compound 10, using the same procedure but starting with 1-cyclobutylurea and ethyl 2-cyanopropanoate to obtain the 1-cyclobutyl analogue of compound 10. The desired product was obtained as an off-white residue, yield = 5%. 1H NMR (500 MHz, methanol-d4): δ 8.03 (d, J = 5.8 Hz, 1H), 7.94 (d, J = 2.2 Hz, 1H), 7.43 (dd, J = 5.9, 0.9 Hz, 1H), 7.30 (s, 1H), 7.23 (d, J = 1.4 Hz, 2H), 7.00 (dd, J = 2.2, 1.0 Hz, 1H), 4.28–4.19 (m, 1H), 3.21–3.11 (m, 2H), 3.11–3.02 (m, 2H), 2.22 (s, 3H), 2.05–1.96 (m, 2H), 1.61 (s, 3H). MS (ESI): m/z 404.2 [M + 1]+.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-5-methyl-1-propylpyrimidine-2,4(1H,3H)-dione (21).
Compound 21 was prepared in an analogous fashion to the preparation of compound 10, using the same procedure but starting with 1-propylurea and ethyl 2-cyanopropanoate to obtain the 1-propyl analogue of compound 10. The desired product was obtained as an off-white residue, yield = 19%. 1H NMR (500 MHz, methanol-d4): δ 8.04 (d, J = 5.8 Hz, 1H), 7.92 (d, J = 2.2 Hz, 1H), 7.47–7.43 (m, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.30 (d, J = 2.4 Hz, 1H), 7.23 (dd, J = 8.3, 2.4 Hz, 1H), 6.93 (dd, J = 2.3, 1.0 Hz, 1H), 3.81–3.72 (m, 1H), 3.22 (ddd, J = 13.7, 10.4, 5.5 Hz, 1H), 2.24 (s, 3H), 1.65 (s, 3H), 1.63–1.55 (m, 1H), 1.50 (d, J = 10.4 Hz, 1H), 0.77 (t, J = 7.4 Hz, 3H). MS (ESI): m/z 392.2 [M + 1]+.
1-Butyl-6-(4-(furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-5-methylpyrimidine-2,4(1H,3H)-dione (22).
Compound 22 was prepared in an analogous fashion to the preparation of compound 10, using the same procedure but starting with 1-butylurea and ethyl 2-cyanopropanoate to obtain the 1-butyl analogue of compound 10. The desired product was obtained as an off-white residue, yield = 15%. 1H NMR (500 MHz, methanol-d4): δ 8.04 (d, J = 5.9 Hz, 1H), 7.92 (d, J = 2.2 Hz, 1H), 7.44 (dd, J = 5.9, 0.9 Hz, 1H), 7.35 (d, J = 8.3 Hz, 1H), 7.32–7.28 (m, 1H), 7.24 (dd, J = 8.3, 2.4 Hz, 1H), 6.92 (dd, J = 2.2, 1.0 Hz, 1H), 3.83–3.75 (m, 1H), 3.27 (ddd, J = 13.6, 10.6, 5.3 Hz, 1H), 2.25 (s, 3H), 1.66 (s, 3H), 1.56 (dd, J = 8.0, 4.9 Hz, 1H), 1.46 (q, J = 6.0 Hz, 1H), 1.19 (dq, J = 17.9, 8.0, 7.5 Hz, 2H), 0.81 (t, J = 7.4 Hz, 3H). MS (ESI): m/z 406.2 [M + 1]+.
5-Ethyl-6-(4-(furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-1-methylpyrimidine-2,4(1H,3H)-dione (23).
Compound 23 was prepared in an analogous fashion to the preparation of compound 10, using the same procedure but starting with 1-methylurea and ethyl 2-cyanobutanoate to obtain the 5-ethyl analogue of compound 10. The desired product was obtained as an off-white residue, yield = 36%. 1H NMR (600 MHz, methanol-d4): δ 8.01 (s, 1H), 7.92 (s, 1H), 7.43 (s, 1H), 7.32 (s, 1H), 7.29 (s, 1H), 7.22 (s, 1H), 6.93 (s, 1H), 3.30 (d, J = 5.3 Hz, 2H), 3.05 (d, J = 3.0 Hz, 3H), 2.23 (d, J = 15.0 Hz, 3H), 0.94 (s, 3H). MS (ESI): m/z 378.1 [M + 1]+.
1,5-Diethyl-6-(4-(furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-pyrimidine-2,4(1H,3H)-dione (24).
Compound 24 was prepared in an analogous fashion to the preparation of compound 10, using the same procedure but starting with 1-ethylurea and ethyl 2-cyanobutanoate to obtain the 1,5-diethyl analogue of compound 10. The desired product was obtained as an off-white residue, yield = 10%. 1H NMR (600 MHz, methanol-d4): δ 8.03 (s, 1H), 7.92 (d, J = 2.3 Hz, 1H), 7.43 (d, J = 5.9 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H), 7.28 (s, 1H), 7.22 (d, J = 8.2 Hz, 1H), 6.95 (s, 1H), 3.98 (d, J = 7.2 Hz, 1H), 3.88 (dd, J = 14.0, 7.0 Hz, 1H), 2.24 (s, 3H), 1.91 (dd, J = 13.7, 7.2 Hz, 1H), 1.25 (t, J = 7.1 Hz, 1H), 1.09 (d, J = 7.7, 7.0 Hz, 3H), 0.93 (d, J = 7.4 Hz, 3H). MS (ESI): m/z 392.2 [M + 1]+.
1-Cyclopropyl-5-ethyl-6-(4-(furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)pyrimidine-2,4(1H,3H)-dione (25).
Compound 25 was prepared in an analogous fashion to the preparation of compound 10, using the same procedure but starting with 1-cyclopropylurea and ethyl 2-cyanobutanoate to obtain the 5-ethyl, 1-cyclopropyl analogue of compound 10. The desired product was obtained as an off-white residue, yield = 5%. 1H NMR (600 MHz, methanol-d4): δ 8.04 (s, 1H), 7.92 (s, 1H), 7.67 (s, 1H), 7.58 (s, 1H), 7.43 (s, 1H), 7.38 (s, 1H), 6.90 (s, 1H), 2.53 (s, 1H), 2.29 (s, 3H), 2.02 (s, 1H), 1.32 (s, 1H), 0.94 (d, J = 10.4 Hz, 3H), 0.75 (s, 2H), 0.68 (s, 2H). MS (ESI): m/z 404.2 [M + 1]+.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)-3-methylphenyl)-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (26).
To an oven-dried microwavable tube was added a stir bar, tetrakis(triphenylphosphine)palladium(0) (21 mg, 0.018 mmol), potassium carbonate (25 mg, 0.18 mmol), the previously described intermediate 6-bromo-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (10i) (20 mg, 0.09 mmol), and the known intermediate 4-(2-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine25 (35 mg, 0.10 mmol) in dioxane (1 mL) and water (0.2 mL). Microwave heating was then applied to the mixture for 25 min, as described in the General Chemistry Procedures. The resulting mixture was subsequently filtered through Celite; the filter was washed with several times with ethyl acetate, and the organic filtrate diluted with water and extracted three times with ethyl acetate. Brine and saturated NaHCO3 were then used to wash the combined organic layers. The organic layers were then dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was taken up in 2 mL of methanol and purified by HPLC using the conditions described previously. This procedure yielded compound 26 as a clear oil (9.8 mg, 30%). 1H NMR (500 MHz, methanol-d4): δ 7.93 (d, J = 2.2 Hz, 1H), 7.55 (t, J = 7.3 Hz, 1H), 7.41–7.38 (m, 1H), 7.34 (s, 1H), 7.31 (d, J = 8.2 Hz, 1H), 7.25 (dd, J = 8.2, 2.1 Hz, 1H), 7.00 (dd, J = 2.2, 1.0 Hz, 1H), 3.14 (s, 3H), 2.27 (s, 3H), 1.73 (s, 3H). MS (ESI): m/z 364.1 [M + 1]+.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)phenyl)-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (27).
Compound 27 was prepared in an analogous fashion to the preparation of compound 10, using the same procedure but starting with 4-bromophenol to obtain the desmethyl intermediate 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine. From here, synthesis of the desired product was carried out using the same Suzuki coupling methods previously described. The desired product was obtained as an off-white residue, yield = 16%. 1H NMR (600 MHz, methanol-d4): δ 8.02 (d, J = 5.9 Hz, 1H), 7.94 (d, J = 2.3 Hz, 1H), 7.43 (d, J = 5.2 Hz, 5H), 7.02–6.99 (m, 1H), 3.13 (s, 3H), 1.73 (s, 3H). MS (ESI): m/z 350.1 [M + 1]+.
6-(4-Hydroxy-2-methylphenyl)-1,5-dimethylpyrimidine-2,4-(1H,3H)-dione (28).
To an oven-dried microwavable tube was added a stir bar, tetrakis(triphenylphosphine)palladium(0) (25 mg, 0.021 mmol), potassium carbonate (29 mg, 0.21 mmol), the previously described intermediate 6-bromo-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (10i) (23 mg, 0.11 mmol), and commercially available (4-hydroxyphenyl)boronic acid (18 mg, 0.12 mmol) in dioxane (1 mL) and water (0.2 mL). Microwave heating was then applied to the mixture for 25 min, as described in the General Chemistry Procedures. The resulting mixture was subsequently filtered through Celite; the filter washed with several times with ethyl acetate, and the organic filtrate diluted with water and extracted three times with ethyl acetate. Brine and saturated NaHCO3 were then used to wash the combined organic layers. The organic layers were then dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was taken up in 2 mL of methanol and purified by HPLC using the conditions described previously. This procedure yielded compound 28 as a beige residue, yield = 46%. 1H NMR (500 MHz, methanol-d4): δ 7.00 (d, J = 8.3 Hz, 1H), 6.84 (d, J = 2.4 Hz, 1H), 6.80 (dd, J = 8.3, 2.5 Hz, 1H), 3.00 (s, 3H), 2.13 (s, 3H), 1.61 (s, 3H). MS (ESI): m/z 247.1 [M + 1]+.
6-(4-Methoxy-2-methylphenyl)-1,5-dimethylpyrimidine-2,4-(1H,3H)-dione (29).
To an oven-dried microwavable tube was added a stir bar, tetrakis(triphenylphosphine)palladium(0) (25 mg, 0.021 mmol), potassium carbonate (29 mg, 0.21 mmol), the previously described intermediate 6-bromo-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (10i) (23 mg, 0.11 mmol), and commercially available (4-methoxyphenyl)boronic acid (20 mg, 0.12 mmol) in dioxane (1 mL) and water (0.2 mL). Microwave heating was then applied to the mixture for 25 min, as described in the General Chemistry Procedures. The resulting mixture was subsequently filtered through Celite; the filter was washed with several times with ethyl acetate, and the organic filtrate diluted with water and extracted three times with ethyl acetate. Brine and saturated NaHCO3 were then used to wash the combined organic layers. The organic layers were then dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was taken up in 2 mL of methanol and purified by HPLC using the conditions described previously. This procedure yielded compound 29 as a beige residue, yield = 50%. 1H NMR (500 MHz, methanol-d4): δ 7.12 (d, J = 8.3 Hz, 1H), 6.99 (d, J = 2.5 Hz, 1H), 6.96 (dd, J = 8.4, 2.6 Hz, 1H), 3.87 (s, 3H), 3.00 (s, 3H), 2.19 (s, 3H), 1.60 (s, 3H). MS (ESI): m/z 261.1 [M + 1]+.
1,5-Dimethyl-6-(2-methyl-4-(pyridin-2-yloxy)phenyl)pyrimidine-2,4(1H,3H)-dione (30).
The intermediate 2-(4-bromo-3-methylphenoxy)pyridine (174.1 mg, 0.66 mmol) was prepared using 4-bromo-3-methylphenol (120 mg, 0.64 mmol) and 2-bromopyridine (92 mg, 0.58 mmol) using the same conditions previously described.25,68 2-(4-Bromo-3-methylphenoxy)pyridine (174.1 mg, 0.66 mmol) was then converted to the boronic ester intermediate 2-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)-pyridine (227 mg, 0.73 mmol, 100%) with the same conditions described previously. Compound 30 was prepared using the same procedure as preparing compound 10 starting with 2-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)pyridine (23 mg, 0.07 mmol). The title compound was obtained as a beige residue, yield = 33%. 1H NMR (500 MHz, methanol-d4): δ 7.91 (t, J = 7.7 Hz, 1H), 7.27 (d, J = 8.3 Hz, 2H), 7.20 (s, 2H), 7.14 (d, J = 8.6 Hz, 2H), 3.07 (s, 3H), 2.23 (d, J = 1.8 Hz, 3H), 1.68–1.64 (m, 3H). MS (ESI): m/z 324.1 [M + 1]+.
6-(4-((4-Methoxypyridin-2-yl)oxy)-2-methylphenyl)-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (31).
Starting from 2-chloro-4-methoxypyridine and 4-bromo-3-methylphenol, compound 31 was prepared according to the same procedures and synthetic route as 30. The title compound was obtained as a beige residue, yield = 49%. 1H NMR (500 MHz, methanol-d4): δ 7.32–7.28 (m, 1H), 7.24 (s, 1H), 7.18 (d, J = 8.4 Hz, 1H), 6.93 (s, 1H), 6.69 (s, 1H), 3.98–3.93 (m, 3H), 3.08–3.03 (m, 3H), 2.24 (s, 3H), 1.68–1.62 (m, 3H). MS (ESI): m/z 354.1 [M + 1]+.
1,5-Dimethyl-6-(2-methyl-4-((3-(trifluoromethyl)pyridin-2-yl)-oxy)phenyl)pyrimidine-2,4(1H,3H)-dione (32).
Starting from 2-bromo-3-(trifluoromethyl)pyridine and 4-bromo-3-methylphenol, compound 32 was prepared according to the same procedures and synthetic route as 30. The title compound was obtained as a beige residue, yield = 42%. 1H NMR (500 MHz, methanol-d4): δ 8.36 (dd, J = 4.8, 1.9 Hz, 1H), 8.22 (d, J = 7.4 Hz, 1H), 7.34–7.30 (m, 1H), 7.25 (d, J = 2.3 Hz, 1H), 7.19 (dd, J = 8.3, 2.5 Hz, 1H), 3.07 (s, 3H), 2.25 (s, 3H), 1.67 (s, 3H). MS (ESI): m/z 392.1 [M + 1]+.
2-(4-(3,5-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)-3-methylphenoxy)-4-methoxynicotinonitrile (33).
Starting from 2-chloro-4-methoxynicotinonitrile and 4-bromo-3-methylphenol, compound 33 was prepared according to the same procedures and synthetic route as 30. The title compound was obtained as a beige residue, yield 48%. 1H NMR (500 MHz, Methanol-d4) δ 8.23 (d, J = 6.1 Hz, 1H), 7.32 (s, 1H), 7.30 (s, 1H), 7.24 (d, J = 8.3 Hz, 1H), 7.05 (d, J = 6.1 Hz, 1H), 4.13 (s, 3H), 3.07 (s, 3H), 2.26 (s, 3H), 1.66 (s, 3H). MS (ESI) m/z 379.1 [M + 1]+.
6-(4-((4-Chloro-3-(trifluoromethyl)pyridin-2-yl)oxy)-2-methyl-phenyl)-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (34).
Starting from 2,4-dichloro-3-(trifluoromethyl)pyridine and 4-bromo-3-methylphenol, compound 34 was prepared according to the same procedures and synthetic route as 30. The title compound was obtained as a beige residue, yield = 16%. 1H NMR (500 MHz, methanol-d4): δ 8.42 (d, J = 5.8 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.30 (d, J = 2.4 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 5.8 Hz, 1H), 3.08–3.04 (m, 3H), 2.27 (s, 3H), 1.68–1.64 (m, 3H). MS (ESI) m/z 426.1 [M + 1]+.
6-(4-((4-Methoxy-3-(trifluoromethyl)pyridin-2-yl)oxy)-2-methylphenyl)-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (35).
Starting from 2-chloro-4-methoxy-3-(trifluoromethyl)pyridine and 4-bromo-3-methylphenol, compound 35 was prepared according to the same procedures and synthetic route as 30. The title compound was obtained as a beige residue, yield = 52%. 1H NMR (600 MHz, chloroform-d): δ 8.40 (s, 1H), 8.21 (s, 1H), 7.14 (s, 1H), 7.13 (s, 1H), 6.77 (d, J = 5.7 Hz, 1H), 4.01 (s, 3H), 3.06 (s, 3H), 2.20 (s, 3H), 1.69 (s, 3H). HPLC 99% pure, tR = 4.753 min. HRMS m/z [M + H]+ calculated for C20H19F3N3O4+ 4 422.1322, found 422.1321.
1,5-Dimethyl-6-(2-methyl-4-((2-methylfuro[3,2-c]pyridin-4-yl)-oxy)phenyl)pyrimidine-2,4(1H,3H)-dione (36).
Starting from 4-chloro-2-methylfuro[3,2-c]pyridine and 4-bromo-3-methylphenol, compound 36 was prepared according to the same procedures and synthetic route as 30. The title compound was obtained as a beige residue, yield = 32%. 1H NMR (500 MHz, methanol-d4): δ 8.05 (s, 1H), 7.42 (d, J = 5.6 Hz, 1H), 7.33–7.29 (m, 2H), 7.23 (dd, J = 8.3, 2.4 Hz, 1H), 6.45–6.43 (m, 1H), 3.08 (s, 3H), 2.53 (s, 3H), 2.25 (s, 3H), 1.67 (s, 3H). HPLC 99% pure, tR = 4.672 min. HRMS m/z [M + H]+ calculated for C21H20N3O4+ 378.1448, found 378.1442.
1,5-Dimethyl-6-(2-methyl-4-((1-methyl-1H-pyrrolo[3,2-c]pyridin-4-yl)oxy)phenyl)pyrimidine-2,4(1H,3H)-dione (37).
Starting from 4-chloro-1-methyl-1H-pyrrolo[3,2-c]pyridine and 4-bromo-3-methylphenol, compound 37 was prepared according to the same procedures and synthetic route as 30. The title compound was obtained as a beige residue, yield = 50%. 1H NMR (500 MHz, methanol-d4): δ 8.04 (s, 1H), 7.72 (d, J = 6.4 Hz, 1H), 7.52 (d, J = 3.3 Hz, 1H), 7.47 (d, J = 2.2 Hz, 1H), 7.44–7.38 (m, 2H), 6.12 (d, J = 3.4 Hz, 1H), 3.99 (s, 3H), 3.09 (s, 3H), 2.28 (s, 3H), 1.68 (s, 3H). HPLC 99% pure, tR = 3.834 min. HRMS: m/z [M + H]+ calculated for C21H21N4O3+ 377.1608, found 377.1610.
6-(4-(Isoquinolin-1-yloxy)-2-methylphenyl)-1,5-dimethylpyrimidine-2,4(1H,3H)-dione (38).
Starting from 1-chloroisoquinoline and 4-bromo-3-methylphenol, compound 38 was prepared according to the same procedures and synthetic route as 30. The title compound was obtained as a beige residue, yield = 40%. 1H NMR (500 MHz, methanol-d4): δ 8.46 (d, J = 8.3 Hz, 1H), 7.95 (dd, J = 14.2, 6.9 Hz, 2H), 7.89–7.83 (m, 1H), 7.78–7.72 (m, 1H), 7.54 (d, J = 5.8 Hz, 1H), 7.36–7.26 (m, 3H), 3.11 (d, J = 2.7 Hz, 3H), 2.29–2.25 (m, 3H), 1.69 (d, J = 2.5 Hz, 3H). MS (ESI): m/z 374.1 [M + 1]+.
1,5-Dimethyl-6-(2-methyl-4-(thieno[3,2-c]pyridin-4-yloxy)-phenyl)pyrimidine-2,4(1H,3H)-dione (39).
Starting from 4-chlorothieno[3,2-c]pyridine and 4-bromo-3-methylphenol, compound 39 was prepared as a white solid according to the same procedures and synthetic route as 30. The title compound was obtained as a beige residue, yield = 29%. 1H NMR (500 MHz, methanol-d4): δ 7.96 (d, J = 5.7 Hz, 1H), 7.78 (d, J = 5.5 Hz, 1H), 7.75 (dd, J = 5.7, 0.9 Hz, 1H), 7.48 (s, 1H), 7.31 (dd, J = 5.4, 3.0 Hz, 2H), 7.25 (dd, J = 8.3, 2.4 Hz, 1H), 3.09 (s, 3H), 2.26 (s, 3H), 1.68 (s, 3H). MS (ESI): m/z 380.1 [M + 1]+.
1,5-Dimethyl-6-(2-methyl-4-(thieno[2,3-c]pyridin-7-yloxy)-phenyl)pyrimidine-2,4(1H,3H)-dione (40).
Starting from 7-chlorothieno[2,3-c]pyridine and 4-bromo-3-methylphenol, compound 40 was prepared according to the same procedures and synthetic route as 30. The title compound was obtained as a beige residue, yield = 49%. 1H NMR (600 MHz, chloroform-d): δ 8.54 (s, 1H), 8.07 (s, 1H), 7.76 (d, J = 5.3 Hz, 1H), 7.50 (s, 1H), 7.43 (d, J = 5.3 Hz, 1H), 7.28 (d, J = 5.4 Hz, 1H), 7.15 (d, J = 8.2 Hz, 1H), 3.06 (s, 3H), 2.20 (s, 3H), 1.69 (s, 3H). HPLC 99% pure, tR = 4.775 min. HRMS: m/z [M + H]+ calculated for C20H18N3O3S+ 380.1063, found 380.1064.
Experimental Procedures for In Vitro Pharmacology Assays.
General Procedures.
The protocols that were used to carry out the cAMP biosensor and bioluminescent resonance energy transfer (BRET) assays are provided below.
D1R GS-Mediated GS-cAMP Accumulation Assay.
D1R-mediated accumulation of cAMP was measured in HEK293T cells cotransfected with human D1 and the cAMP biosensor GloSensor-22F (Promega) in a ratio of 1:15. The next day 50 000 cells per well in cMEM + 2% dialyzed FBS were plated in PDK-treated 96 well plates and incubated overnight. The next day media was removed and cells were washed with 100 μL of HBSS + HEPES and 25 μL of luciferin (Gold Biotechnology) was added per well. Plates were incubated for 2 h at room temperature. Compounds were prepared in HBSS/HEPES/0.01% BSA/0.1% ascorbate at 10× and 10 μL per well was added. After 15 min, cAMP accumulation was measured using a BMG Clariostar plate reader. Net response was determined by subtracting the reading without compound from the reading with compound added. Values were normalized to percent of maximum dopamine response and analyzed using GraphPad Prism 7 (Graphpad Software, Inc., San Diego, CA).
Bioluminescence Resonance Energy Transfer (BRET) β-Arrestin2 Assay.
To measure D1R mediated recruitment of β-arrestin2, HEK293T cells were cotransfected with human D1 fused with C-terminal Renilla luciferase (RLuc8) and N-terminal Venus β-arrestin 2 in a ratio of 1:20. The next day transfected cells were plated at 50 000 cells per well into 96-well PDK-coated plates in 100 μL of cMEM + 2% dialyzed FBS and incubated overnight. The next day media was replaced with 80 μL of HBSS, 20 mM HEPES, and 10 μL of coelentrazine h (Nanolight Technology, 5 μM final concentration) was added. Compounds were prepared in HBSS/HEPES/.01%BSA/.01% ascorbate at 10× and 10 μL per well was added. Plates were incubated 15 min and then the luminescence at 485 nm and fluorescence at 530 nm were both measured for 1 s per well using a BMG Clariostar plate reader. The ratio of eYFP/RLUC was calculated per well and the net BRET ratio was determined by subtracting the net BRET without drug from the net BRET in wells containing drug. The data were normalized to the percent of the maximal response with dopamine stimulation. The net BRET was plotted as a function of compound concentration using Graphpad Prism 7 (Graphpad Software Inc., San Diego, CA).
Bias Calculation.
The Black and Leff operational model, in which τ is agonist efficacy and KA is the equilibrium dissociation constant, was implemented to determine the transduction coefficients (log (τ/KA)). Transduction coefficients for GS activity and β-arrestin2 recruitment were calculated and averaged across experiments, using dopamine as the full agonist reference. The method outlined by Kenakin et al. was used in the calculation of bias factors, where the Δlog(τ/KA) was calculated relative to the reference and the ΔΔlog(τ/KA) was calculated by subtracting the GS from β-arrestin2 transduction coefficient.83 This analysis was performed in Graphpad Prism 7.0.
Radioligand Binding Affinity Assays.
Compounds were screened in primary radioligand binding assays to determine if they reached a certain affinity threshold (measured as% inhibition) across a wide panel of over 30 other potential targets. Compounds with a mean inhibition greater than 50% across four determinations are considered to have surpassed the affinity threshold and were subsequently examined in secondary radioligand binding assays for Ki determination at each receptor over the threshold. Primary and secondary assays were conducted using the same protocols and procedures that have been previously described.74 Additional information and details concerning assay methods can be found online at https://pdspdb.unc.edu/pdspWeb/?site=assays.
Experimental Procedures for In Vivo Pharmacokinetic Studies.
Compounds 10 and 35 were taken up in 5% NMP, 5% Solutol HS-15, 30% PEG-400, and 60% normal saline for formulation. A formulated solution of compounds 10 and 35 were each administered intraperitoneally at a 50 mg/kg dose to a designated group of nine male Swiss Albino mice. Approximately, 60 μL of blood were acquired from a set of three mice within the group at 0.5, 1.5, and 4 h under light isoflurane anesthesia. Blood samples were centrifuged to harvest plasma. The plasma was then stored at −70 ± 10 °C until analysis. Three mice were sacrificed immediately after blood collection to obtain brain samples for each time point at 0.5, 1.5, and 4 h. Brain samples were homogenized using ice-cooled phosphate buffer saline (pH 7.4) in a ratio of 2 (buffer): 1(brain). Homogenates were stored below −70 ± 10 °C until analysis. Total homogenate volume was three times the brain weight. Pharmacokinetic analysis was conducted using the plasma and brain concentration–time data of compounds 10 and 35. The NCA module of Phoenix WinNonlin (version 7.0) was used to perform the pharmacokinetic analysis. A fit-for-purpose LCMS/MS method was used to quantify plasma and brain samples (LLOQ = 1.01 ng/mL for plasma and 3.03 ng/g for brain).
Experimental Procedures for In Vivo Behavioral and Locomotion Studies in 6-Hydroxydopamine (6-OHDA)-Lesioned Animals.
Animals.
Male and female C57Bl/6 mice were used in these experiments. They were housed 3–5 animals per cage on a 14: 10 h light: dark cycle (lights on 07:00 h), with food and water ad libitum, and they were tested at 4–6 months of age. Experiments took place between 09:00 and 17:00 h.
Drugs.
SKF81297 (Tocris) was dissolved in sterile saline and compound 10 in 5% NMP, 5% Kolliphor, and 90% sterile saline. Drugs were injected i.p., in a volume of 5 mL/kg. 6-Hydroxydopamine hydrobromide (6-OHDA, Sigma) was dissolved in 0.02% ascorbic acid to a concentration of 3.6 mg/mL.
Surgeries.
Mice were anesthetized with a mixture of ketamine/zylazine 100/10 mg/kg and placed in the stereotaxic frame. A hole was made in the right side of the skull at coordinates −0.8, L 1.1 relative to Bregma. A 2 μL syringe was lowered at −4.75 mm below skull and 1 μL of 6-OHDA was delivered over 5 min into the right medial forebrain bundle (Paxinos, 2007). The needle remained in place for 5 additional minutes and was then slowly withdrawn. Animals were allowed to recover for 15–19 days after surgery until their body weights stabilized. During this time, they received high fat diets and condensed milk supplements. Sterile 5% dextrose solution was administered subcutaneously daily to prevent dehydration.
Apparatus.
Clear plastic arenas 20 cm in diameter were placed in the open field apparatus (Omnitech Electronics Inc., Columbus, OH) measuring 42 × 42 × 40. Overhead cameras recorded videos of the sessions. These videos were analyzed off-line by Ethovision software (Noldus) as distance moved by the center point of the mouse’s body or contralateral rotations around center point (away from lesioned side).
Procedure.
After recovery, mice were given vehicle, SKF 81297 (5 mg/kg), or compound 10 (5 mg/kg) and immediately placed in the plastic arenas for 90 min. These experiments were performed in accordance with all national and local guidelines and regulations.
Statistics.
Data are means and SEMs. Two-way ANOVA was used for analysis of time as a within-subject factor and treatment as a between-subject factor. In instances of significant interactions (P < 0.05), Bonferroni posthoc tests were subsequently applied.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the U.S. National Institutes of Health through F30AG062054, awarded to M.L.M. and R01NS100930, awarded to M.G.C. and J.J. M.L.M. also acknowledges the support by the National Institute of General Medical Sciences (NIGMS)-funded Integrated Pharmacological Sciences Training Program T32GM062754 and the Medical Scientist Training Program (MSTP) training grant T32GM007280 at the Icahn School of Mount Sinai, both from the U.S. National Institutes of Health. Ki determinations and receptor binding profiles were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract HHSN-271-2018-00023-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth, MD, PhD, at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA.
ABBREVIATIONS
- GPCR
G protein-coupled receptor
- SFSR
structure–functional selectivity relationship
- D1R
dopamine D1 receptor
- GS
stimulatory G protein
- cAMP
cyclic adenosine monophosphate
- RHS
right-hand side
- LHS
left-hand side
- BRET
bioluminescence resonance energy transfer
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- D4R
dopamine D4 receptor
- D5R
dopamine D5 receptor
- 5-HT
5-hydroxytryptamine
- H1
histamine receptor 1
- H2
histamine receptor 2
- M1
muscarinic receptor 1
- M2
muscarinic receptor 2
- M3
muscarinic receptor 3
- MOR
mu opioid receptor
- KOR
kappa opioid receptor
- SERT
serotonin transporter
- DAT
dopamine transporter
- TFA
trifluoroacetic acid
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.9b00410.
Synthetic schemes for the fourth, fifth, and sixth iterations of compounds exploring the RHS, compounds exploring the middle phenyl moiety, and compounds exploring the LHS pyridofuran moiety; compound potencies, efficacies, and bias factors for all compounds in the D1R GloSensor and D1R BRET cell-based assays; 1H NMR spectra of compounds 10, 35, and 40; and molecular formula strings for all compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Lagerstrom MC, and Schioth HB (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discovery 7, 339–357. [DOI] [PubMed] [Google Scholar]
- (2).Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, and Overington JP (2017) A comprehensive map of molecular drug targets. Nat. Rev. Drug Discovery 16, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, and Gloriam DE (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discovery 16, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, and Caron MG (2005) An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273. [DOI] [PubMed] [Google Scholar]
- (5).Urs NM, Daigle TL, and Caron MG (2011) A dopamine D1 receptor-dependent beta-arrestin signaling complex potentially regulates morphine-induced psychomotor activation but not reward in mice. Neuropsychopharmacology 36, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lefkowitz RJ, and Shenoy SK (2005) Transduction of receptor signals by beta-arrestins. Science 308, 512–517. [DOI] [PubMed] [Google Scholar]
- (7).Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, Snyder SH, Caron MG, and Lefkowitz RJ (1992) Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J. Biol. Chem 267, 17882–17890. [PubMed] [Google Scholar]
- (8).Reiter E, Ahn S, Shukla AK, and Lefkowitz RJ (2012) Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol 52, 179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Beaulieu JM, and Gainetdinov RR (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 63, 182–217. [DOI] [PubMed] [Google Scholar]
- (10).Spano PF, Govoni S, and Trabucchi M (1978) Studies on the pharmacological properties of dopamine receptors in various areas of the central nervous system. Adv. Biochem Psychopharmacol 19, 155–165. [PubMed] [Google Scholar]
- (11).Oakley RH, Laporte SA, Holt JA, Barak LS, and Caron MG (1999) Association of beta-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J. Biol. Chem 274, 32248–32257. [DOI] [PubMed] [Google Scholar]
- (12).Goodman OB Jr., Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, and Benovic JL (1996) Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 383, 447–450. [DOI] [PubMed] [Google Scholar]
- (13).Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, and Lefkowitz RJ (2001) Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc. Natl. Acad. Sci. U. S. A 98, 2449–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, and Bunnett NW (2000) beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol 148, 1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, and Lefkowitz RJ (2000) Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290, 1574–1577. [DOI] [PubMed] [Google Scholar]
- (16).Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, and Luttrell LM (2003) The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J. Biol. Chem 278, 6258–6267. [DOI] [PubMed] [Google Scholar]
- (17).Imamura T, Huang J, Dalle S, Ugi S, Usui I, Luttrell LM, Miller WE, Lefkowitz RJ, and Olefsky JM (2001) beta -Arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. J. Biol. Chem 276, 43663–43667. [DOI] [PubMed] [Google Scholar]
- (18).Barlic J, Andrews JD, Kelvin AA, Bosinger SE, DeVries ME, Xu L, Dobransky T, Feldman RD, Ferguson SS, and Kelvin DJ (2000) Regulation of tyrosine kinase activation and granule release through beta-arrestin by CXCRI. Nat. Immunol 1, 227–233. [DOI] [PubMed] [Google Scholar]
- (19).Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, and Lefkowitz RJ (1999) Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283, 655–661. [DOI] [PubMed] [Google Scholar]
- (20).Fisone G, and Bezard E (2011) Molecular mechanisms of l-DOPA-induced dyskinesia. Int. Rev. Neurobiol 98, 95–122. [DOI] [PubMed] [Google Scholar]
- (21).Jenner P (2008) Molecular mechanisms of l-DOPA-induced dyskinesia. Nat. Rev. Neurosci 9, 665–677. [DOI] [PubMed] [Google Scholar]
- (22).Macpherson T, Morita M, and Hikida T (2014) Striatal direct and indirect pathways control decision-making behavior. Front. Psychol 5, 1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Park PS-H (2012) Ensemble of G protein-coupled receptor active states. Curr. Med. Chem 19, 1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chen X, McCorvy JD, Fischer MG, Butler KV, Shen Y, Roth BL, and Jin J (2016) Discovery of G Protein-Biased D2 Dopamine Receptor Partial Agonists. J. Med. Chem 59, 10601–10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Martini ML, Liu J, Ray C, Yu X, Huang XP, Urs A, Urs N, McCorvy JD, Caron MG, Roth BL, and Jin J (2019) Defining Structure-Functional Selectivity Relationships (SFSR) for a Class of Non-Catechol Dopamine D1 Receptor Agonists. J. Med. Chem 62, 3753–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).McCorvy JD, Wacker D, Wang S, Agegnehu B, Liu J, Lansu K, Tribo AR, Olsen RHJ, Che T, Jin J, and Roth BL (2018) Structural determinants of 5-HT2B receptor activation and biased agonism. Nat. Struct. Mol. Biol 25, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).McCorvy JD, Butler KV, Kelly B, Rechsteiner K, Karpiak J, Betz RM, Kormos BL, Shoichet BK, Dror RO, Jin J, and Roth BL (2018) Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat. Chem. Biol 14, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shen Y, McCorvy JD, Martini ML, Rodriguiz RM, Pogorelov VM, Ward KM, Wetsel WC, Liu J, Roth BL, and Jin J (2019) D2 Dopamine Receptor G Protein-Biased Partial Agonists Based on Cariprazine. J. Med. Chem 62, 4755–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chen X, Sassano MF, Zheng L, Setola V, Chen M, Bai X, Frye SV, Wetsel WC, Roth BL, and Jin J (2012) Structure-functional selectivity relationship studies of beta-arrestin-biased dopamine D(2) receptor agonists. J. Med. Chem 55, 7141–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kim YC, Alberico SL, Emmons E, and Narayanan NS (2015) New therapeutic strategies targeting D1-type dopamine receptors for neuropsychiatric disease. Front. Biol. (Beijing, China) 10, 230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kehne JH, Andree TH, and Heinrich JN (2008) D2 receptor partial agonists: treatment of CNS disorders of dopamine function. Curr. Top. Med. Chem 8, 1068–1088. [DOI] [PubMed] [Google Scholar]
- (32).Davis KL, Kahn RS, Ko G, and Davidson M (1991) Dopamine in schizophrenia: a review and reconceptualization. Am. J. Psychiatry 148, 1474–1486. [DOI] [PubMed] [Google Scholar]
- (33).Shapiro DA, Renock S, Arrington E, Chiodo LA, Liu LX, Sibley DR, Roth BL, and Mailman R (2003) Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 28, 1400–1411. [DOI] [PubMed] [Google Scholar]
- (34).Greengard P (2001) The neurobiology of dopamine signaling. Biosci. Rep 21, 247–269. [DOI] [PubMed] [Google Scholar]
- (35).Porras G, Berthet A, Dehay B, Li Q, Ladepeche L, Normand E, Dovero S, Martinez A, Doudnikoff E, Martin-Negrier ML, Chuan Q, Bloch B, Choquet D, Boue-Grabot E, Groc L, and Bezard E (2012) PSD-95 expression controls l-DOPA dyskinesia through dopamine D1 receptor trafficking. J. Clin. Invest 122, 3977–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ahmed MR, Berthet A, Bychkov E, Porras G, Li Q, Bioulac BH, Carl YT, Bloch B, Kook S, Aubert I, Dovero S, Doudnikoff E, Gurevich VV, Gurevich EV, and Bezard E (2010) Lentiviral overexpression of GRK6 alleviates L-dopa-induced dyskinesia in experimental Parkinson’s disease. Sci. Transl. Med 2, 28ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Conroy JL, Free RB, and Sibley DR (2015) Identification of G protein-biased agonists that fail to recruit beta-arrestin or promote internalization of the D1 dopamine receptor. ACS Chem. Neurosci 6, 681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Rashid AJ, So CH, Kong MM, Furtak T, El-Ghundi M, Cheng R, O’Dowd BF, and George SR (2007) D1–D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc. Natl. Acad. Sci. U. S. A 104, 654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, Jensen NH, Che X, Bai X, Frye SV, Wetsel WC, Caron MG, Javitch JA, Roth BL, and Jin J (2011) Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U. S. A 108, 18488–18493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Park SM, Chen M, Schmerberg CM, Dulman RS, Rodriguiz RM, Caron MG, Jin J, and Wetsel WC (2016) Effects of beta-Arrestin-Biased Dopamine D2 Receptor Ligands on Schizo-phrenia-Like Behavior in Hypoglutamatergic Mice. Neuropsychopharmacology 41, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wang S, Che T, Levit A, Shoichet BK, Wacker D, and Roth BL (2018) Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 555, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Urs NM, Gee SM, Pack TF, McCorvy JD, Evron T, Snyder JC, Yang X, Rodriguiz RM, Borrelli E, Wetsel WC, Jin J, Roth BL, O’Donnell P, and Caron MG (2016) Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc. Natl. Acad. Sci. U. S. A 113, E8178–E8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Urban JD, Vargas GA, von Zastrow M, and Mailman RB (2007) Aripiprazole has functionally selective actions at dopamine D2 receptor-mediated signaling pathways. Neuropsychopharmacology 32, 67–77. [DOI] [PubMed] [Google Scholar]
- (44).Hiller C, Kling RC, Heinemann FW, Meyer K, Hubner H, and Gmeiner P (2013) Functionally selective dopamine D2/D3 receptor agonists comprising an enyne moiety. J. Med. Chem 56, 5130–5141. [DOI] [PubMed] [Google Scholar]
- (45).Möller D, Kling RC, Skultety M, Leuner K, Hubner H, and Gmeiner P (2014) Functionally selective dopamine D(2), D(3) receptor partial agonists. J. Med. Chem 57, 4861–4875. [DOI] [PubMed] [Google Scholar]
- (46).Möller D, Banerjee A, Uzuneser TC, Skultety M, Huth T, Plouffe B, Hubner H, Alzheimer C, Friedland K, Muller CP, Bouvier M, and Gmeiner P (2017) Discovery of G Protein-Biased Dopaminergics with a Pyrazolo[1,5-a]pyridine Substructure. J. Med. Chem 60, 2908–2929. [DOI] [PubMed] [Google Scholar]
- (47).Shonberg J, Herenbrink CK, Lopez L, Christopoulos A, Scammells PJ, Capuano B, and Lane JR (2013) A structure-activity analysis of biased agonism at the dopamine D2 receptor. J. Med. Chem 56, 9199–9221. [DOI] [PubMed] [Google Scholar]
- (48).Bonifazi A, Yano H, Ellenberger MP, Muller L, Kumar V, Zou MF, Cai NS, Guerrero AM, Woods AS, Shi L, and Newman AH (2017) Novel Bivalent Ligands Based on the Sumanirole Pharmacophore Reveal Dopamine D2 Receptor (D2R) Biased Agonism. J. Med. Chem 60, 2890–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Weïwer M, Xu Q, Gale JP, Lewis M, Campbell AJ, Schroeder FA, Van de Bittner GC, Walk M, Amaya A, Su P, DĐordevic L, Sacher JR, Skepner A, Fei D, Dennehy K, Nguyen S, Faloon PW, Perez J, Cottrell JR, Liu F, Palmer M, Pan JQ, Hooker JM, Zhang YL, Scolnick E, Wagner FF, and Holson EB (2018) Functionally Biased D2R Antagonists: Targeting the beta-Arrestin Pathway to Improve Antipsychotic Treatment. ACS Chem. Biol 13, 1038–1047. [DOI] [PubMed] [Google Scholar]
- (50).Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, and Clarke WP (1998) Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol. Pharmacol 54, 94–104. [PubMed] [Google Scholar]
- (51).Cheng J, McCorvy JD, Giguere PM, Zhu H, Kenakin T, Roth BL, and Kozikowski AP (2016) Design and Discovery of Functionally Selective Serotonin 2C (5-HT2C) Receptor Agonists. J. Med. Chem 59, 9866–9880. [DOI] [PubMed] [Google Scholar]
- (52).Zhang G, Cheng J, McCorvy JD, Lorello PJ, Caldarone BJ, Roth BL, and Kozikowski AP (2017) Discovery of N-Substituted (2-Phenylcyclopropyl)methylamines as Functionally Selective Serotonin 2C Receptor Agonists for Potential Use as Antipsychotic Medications. J. Med. Chem 60, 6273–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Stroth N, Niso M, Colabufo NA, Perrone R, Svenningsson P, Lacivita E, and Leopoldo M (2015) Arylpiperazine agonists of the serotonin 5-HT1A receptor preferentially activate cAMP signaling versus recruitment of beta-arrestin-2. Bioorg. Med. Chem 23, 4824–4830. [DOI] [PubMed] [Google Scholar]
- (54).DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, and Violin JD (2013) A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther 344, 708–717. [DOI] [PubMed] [Google Scholar]
- (55).Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, and Lin FT (1999) Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286, 2495–8. [DOI] [PubMed] [Google Scholar]
- (56).Raehal KM, Walker JK, and Bohn LM (2005) Morphine side effects in beta-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther 314, 1195–1201. [DOI] [PubMed] [Google Scholar]
- (57).Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang XP, Sassano MF, Giguère PM, Löber S, Duan D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, and Shoichet BK (2016) Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Hill R, Disney A, Conibear A, Sutcliffe K, Dewey W, Husbands S, Bailey C, Kelly E, and Henderson G (2018) The novel mu-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol 175, 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Bohn LM, and Aube J (2017) Seeking (and Finding) Biased Ligands of the Kappa Opioid Receptor. ACS Med. Chem. Lett 8, 694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Dunn AD, Reed B, Guariglia C, Dunn AM, Hillman JM, and Kreek MJ (2018) Structurally Related Kappa Opioid Receptor Agonists with Substantial Differential Signaling Bias: Neuroendocrine and Behavioral Effects in C57BL6Mice. Int. J. Neuropsychopharmacol 21, 847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Brust TF, Morgenweck J, Kim SA, Rose JH, Locke JL, Schmid CL, Zhou L, Stahl EL, Cameron MD, Scarry SM, Aube J, Jones SR, Martin TJ, and Bohn LM (2016) Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci. Signaling 9, No. ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Dogra S, and Yadav PN (2015) Biased agonism at kappa opioid receptors: Implication in pain and mood disorders. Eur. J. Pharmacol 763, 184–190. [DOI] [PubMed] [Google Scholar]
- (63).Frankowski KJ, Ghosh P, Setola V, Tran TB, Roth BL, and Aube J (2010) N-Alkyl-octahydroisoquinolin-1-one-8-carboxamides: a Novel Class of Selective, Nonbasic, Nitrogen-Containing kappa-Opioid Receptor Ligands. ACS Med. Chem. Lett 1, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Frankowski KJ, Hedrick MP, Gosalia P, Li K, Shi S, Whipple D, Ghosh P, Prisinzano TE, Schoenen FJ, Su Y, Vasile S, Sergienko E, Gray W, Hariharan S, Milan L, Heynen-Genel S, Mangravita-Novo A, Vicchiarelli M, Smith LH, Streicher JM, Caron MG, Barak LS, Bohn LM, Chung TD, and Aube J (2012) Discovery of Small Molecule Kappa Opioid Receptor Agonist and Antagonist Chemotypes through a HTS and Hit Refinement Strategy. ACS Chem. Neurosci 3, 221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodder P, Madoux F, Cameron MD, Prisinzano TE, Aube J, and Bohn LM (2013) Development of functionally selective, small molecule agonists at kappa opioid receptors. J. Biol. Chem 288, 36703–36716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).White KL, Scopton AP, Rives ML, Bikbulatov RV, Polepally PR, Brown PJ, Kenakin T, Javitch JA, Zjawiony JK, and Roth BL (2014) Identification of novel functionally selective kappa-opioid receptor scaffolds. Mol. Pharmacol 85, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, and Roth BL (2015) The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J. Pharmacol. Exp. Ther 352, 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Gray DL, Allen JA, Mente S, O’Connor RE, DeMarco GJ, Efremov I, Tierney P, Volfson D, Davoren J, Guilmette E, Salafia M, Kozak R, and Ehlers MD (2018) Impaired beta-arrestin recruitment and reduced desensitization by non-catechol agonists of the D1 dopamine receptor. Nat. Commun 9, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Davoren JE, Nason DM, Coe J, Dlugolenski K, Helal CJ, Harris AR, LaChapelle E, Liang S, Liu Y, O’Connor RE, Orozco CC, Rai BK, Salafia M, Samas BM, Xu W, Kozak R, and Gray D (2018) Discovery and Lead Optimization of Atropisomer D1 Agonists with Reduced Desensitization. J. Med. Chem 61, 11384–11397. [DOI] [PubMed] [Google Scholar]
- (70).Hanson BJ, Wetter J, Bercher MR, Kopp L, Fuerstenau-Sharp M, Vedvik KL, Zielinski T, Doucette C, Whitney PJ, and Revankar C (2009) A homogeneous fluorescent live-cell assay for measuring 7-transmembrane receptor activity and agonist functional selectivity through beta-arrestin recruitment. J. Biomol. Screening 14, 798–810. [DOI] [PubMed] [Google Scholar]
- (71).Wang T, Li Z, Cvijic ME, Krause C, Zhang L, and Sum CS (2004) Measurement of beta-Arrestin Recruitment for GPCR Targets In Assay Guidance Manual (Sittampalam GS, Coussens NP, Brimacombe K, Grossman A, Arkin M, Auld D, Austin C, Baell J, Bejcek B, Caaveiro JMM, Chung TDY, Dahlin JL, Devanaryan V, Foley TL, Glicksman M, Hall MD, Haas JV, Inglese J, Iversen PW, Kahl SD, Kales SC, Lal-Nag M, Li Z, McGee J, McManus O, Riss T, Trask OJ Jr., Weidner JR, Wildey MJ, Xia M, and Xu X, Eds.) Bethesda, MD. [Google Scholar]
- (72).Zhang R, and Xie X (2012) Tools for GPCR drug discovery. Acta Pharmacol. Sin 33, 372–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Wang P, Felsing DE, Chen H, Raval SR, Allen JA, and Zhou J (2019) Synthesis and Pharmacological Evaluation of Noncatechol G Protein Biased and Unbiased Dopamine D1 Receptor Agonists. ACS Med. Chem. Lett 10, 792–799. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (74).Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FR, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, and Hopkins AL (2012) Automated design of ligands to polypharmacological profiles. Nature 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Urs NM, Bido S, Peterson SM, Daigle TL, Bass CE, Gainetdinov RR, Bezard E, and Caron MG (2015) Targeting beta-arrestin2 in the treatment of l-DOPA-induced dyskinesia in Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A 112, E2517–E2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Costall B, Naylor RJ, and Pycock C (1976) Non-specific supersensitivity of striatal dopamine receptors after 6-hydroxydopamine lesion of the nigrostriatal pathway. Eur. J. Pharmacol 35 (2), 275–283. [DOI] [PubMed] [Google Scholar]
- (77).Henry B, Crossman AR, and Brotchie JM (1998) Characterization of enhanced behavioral responses to l-DOPA following repeated administration in the 6-hydroxydopamine-lesioned rat model of Parkinson’s disease. Exp. Neurol 151, 334–342. [DOI] [PubMed] [Google Scholar]
- (78).Francardo V, Recchia A, Popovic N, Andersson D, Nissbrandt H, and Cenci MA (2011) Impact of the lesion procedure on the profiles of motor impairment and molecular responsiveness to l-DOPA in the 6-hydroxydopamine mouse model of Parkinson’s disease. Neurobiol. Dis 42, 327–340. [DOI] [PubMed] [Google Scholar]
- (79).Carey RJ (1992) Factors in amphetamine-induced contralateral rotation in the unilateral 6-OHDA lesion rat model during the first-week postoperative: implications for neuropathology and neural grafting. Brain Res 570, 11–20. [DOI] [PubMed] [Google Scholar]
- (80).Schwarting RK, Bonatz AE, Carey RJ, and Huston JP (1991) Relationships between indices of behavioral asymmetries and neurochemical changes following mesencephalic 6-hydroxydopamine injections. Brain Res 554, 46–55. [DOI] [PubMed] [Google Scholar]
- (81).Oberlander C, Euvrard C, Dumont C, and Boissier JR (1979) Circling behaviour induced by dopamine releasers and/or uptake inhibitors during degeneration of the nigrostriatal pathway. Eur. J. Pharmacol 60, 163–170. [DOI] [PubMed] [Google Scholar]
- (82).Anden NE, Bedard P, Fuxe K, and Ungerstedt U (1972) Early and selective increase in brain dopamine levels after axotomy. Experientia 28, 300–302. [DOI] [PubMed] [Google Scholar]
- (83).Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, and Novick S (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci 3, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.