

Abstract
INTRODUCTION:
Depression is an episodic form of mental illness, yet the circuit-level mechanisms driving the induction, remission, and recurrence of depressive episodes over time are not well understood. Keta-mine relieves depressive symptoms rapidly, providing an opportunity to study the neuro-biological substrates of transitions from depression to remission and to test whether mechanisms that induce antidepressant effects acutely are distinct from those that sustain them.
RATIONALE:
Contrasting changes in dendritic spine density in prefrontal cortical pyramidal cells have been associated with the emergence of depression-related behaviors in chronic stress models and with ketamine’s antidepressant effects. But whether and how dendritic spine remodeling is causally involved, or whether it is merely correlated with these effects, is unclear. To answer these questions, we used two-photon imaging to study how chronic stress and keta-mine affect dendritic spine remodeling and neuronal activity dynamics in the living prefrontal cortex (PFC), as well as a recently developed optogenetic tool to manipulate the survival of newly formed spines after ketamine treatment.
RESULTS:
The induction of depression-related behavior in multiple chronic stress models was associated with targeted, branch-specific elimination of postsynaptic dendritic spines and a loss of correlated multicellular ensemble activity in PFC projection neurons. Antidepressant-dose keta-mine reversed these effects by selectively rescuing eliminated spines and restoring coordinated activity in multicellular ensembles that predicted motivated escape behavior. Unexpectedly, ketamine’s effects on behavior and ensemble activity preceded its effects on spine formation, indicating that spine formation was not required for inducing these effects acutely. However, individual differences in the restoration of lost spines were correlated with behavior 2 to 7 days after treatment, suggesting that spinogenesis may be important for the long-term maintenance of these effects. To test this, we used a photoactivatable probe to selectively reverse the effects of ketamine on spine formation in the PFC and found that the newly formed spines play a necessary and specific role in sustaining ketamine’s antidepressant effects on motivated escape behavior. By contrast, optically deleting a random subset of spines unrelated to ketamine treatment had no effect on behavior.
CONCLUSION:
Prefrontal cortical spine formation sustains the remission of specific depression-related behaviors after ketamine treatment by restoring lost spines and rescuing coordinated ensemble activity in PFC microcircuits. Pharmacological and neurostimulatory interventions for enhancing and preserving the rescue of lost synapses may therefore be useful for promoting sustained remission. ■
The neurobiological mechanisms underlying the induction and remission of depressive episodes over time are not well understood. Through repeated longitudinal imaging of medial prefrontal microcircuits in the living brain, we found that prefrontal spinogenesis plays a critical role in sustaining specific antidepressant behavioral effects and maintaining long-term behavioral remission. Depression-related behavior was associated with targeted, branch-specific elimination of postsynaptic dendritic spines on prefrontal projection neurons. Antidepressant-dose ketamine reversed these effects by selectively rescuing eliminated spines and restoring coordinated activity in multicellular ensembles that predict motivated escape behavior. Prefrontal spinogenesis was required for the long-term maintenance of antidepressant effects on motivated escape behavior but not for their initial induction.
Graphical Abstract
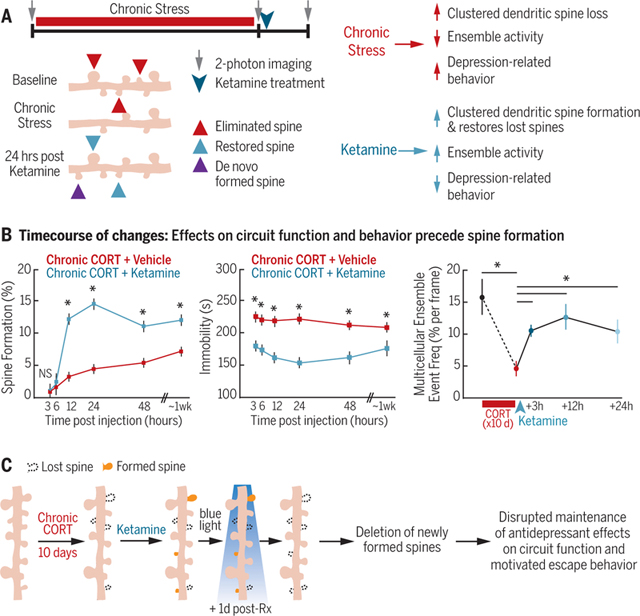
Prefrontal spinogenesis is required for sustaining—but not inducing—ketamine’s effects on behavior and circuit function. (A) Complementary effects of stress and ketamine on spine remodeling, circuit function, and behavior. (B) The time course of changes indicates that spine formation is not required for inducing these effects initially. * indicates significantly different post hoc contrast, Holm-Bonferroni corrected (P < 0.002, P < 0.0006, and P < 0.05 for spine formation, immobility, and ensemble event frequency, respectively). NS, not significant. Error bars indicate SEM. (C) Optogenetic deletion of newly formed spines interferes with the long-termmaintenance of these effects. Rx, treatment.
Depression is an episodic form of mental illness featuring discrete symptomatic periods interposed between periods of apparent wellness. The circuit-level mechanisms driving the induction, remission, and recurrence of depressive episodes over time are not well understood. Ketamine, a rapidly acting antidepressant, relieves depressive symptoms within hours (1–3), providing an experimentally tractable opportunity to study the mechanisms that underlie transitions from depression to remission and recurrence.
Sustaining remission after ketamine treatment is a particular challenge. Antidepressant effects of a single dose of ketamine eventually dissipate, and the long-term safety and efficacy of repeated dosing are not well understood. Whether the antidepressant effects that emerge hours after ketamine treatment are driven by mechanisms distinct from those that sustain the effects in the days after treatment is also unclear. Increasing evidence suggests that signaling pathways (4–6) may converge to increase spine density in prefrontal cortical pyramidal cells (4, 7, 8), potentially accelerating remission by directly and selectively reversing prefrontal cortical synapse loss, a putative neurobiological substrate of depression that occurs in multiple chronic stress models of depression-related behavior (9–12). Alternatively, ketamine may indirectly compensate for synapse loss by inducing synapse formation nonspecifically.
Until recently, it has been difficult to investigate how spine remodeling contributes to transitions between depression-like behavioral states, because of technical obstacles in quantifying synaptic remodeling longitudinally in the living prefrontal cortex (PFC). Using recently developed technologies for obtaining high-resolution optical access to deep brain structures in vivo (13–16), quantifying neocortical activity dynamics in awake, behaving animals (17–19), and optically manipulating the survival of newly formed synapses (20), we tested the hypothesis that keta-mine accelerates the remission of a depression-like behavioral state by restoring lost spines in the PFC.
Results
Targeted, branch-specific spine remodeling underlies behavioral state transitions
We began by testing whether the induction of depression-related behavior in chronic stress models is associated with targeted effects on specific dendritic spine populations. To this end, we imaged the PFC through a prism (13,16) chronically implanted in the contralateral hemisphere (Fig. 1A). Using two-photon (2P) laser-scanning microscopy, we obtained high-resolution images of the apical dendrites of yellow fluorescent protein (YFP)-expressing projection neurons in the medial PFC (mPFC) [in Thy1/YFP-H mice (Jackson Labs)] before and after 21 days of exposure to corticosterone (CORT) (0.10 mg/ml in the drinking water) (21), the principal murine stress hormone (Fig. 1A). CORT is a critical mediator of chronic stress effects on behavior, and chronic CORT treatment recapitulates important aspects of the neuroendocrine response to chronic stress (21–26). The prism implantation procedure elicits a reactive gliosis limited to a volume of up to 50 μm from the face of the prism, whereas at distances of >50 μm, there is no evidence of gliosis or effects on spine remodeling (13,16). Therefore, we limited our analysis to distances of >50 μm from the prism face.
Fig. 1. Targeted, branch-specific spine remodeling underlies behavioral state transitions.

(A) 2P spine imaging of YFP-expressing neurons through a microprism implanted across the midline in the mPFC before and after 21 days of chronic CORT exposure and after antidepressant-dose ketamine (post-ket) (10 mg/kg ip). Red and blue arrows indicate eliminated and formed spines, respectively. FrA, frontal association cortex; ACg, anterior cingulate cortex; PL, prelimbic cortex; IL, infralimbic cortex. (B) Spine elimination increased (*P = 0.0043, Wilcoxon W = 15.0) after 21 days of chronic CORT (n = 6 mice) compared with that in controls (n = 5 mice), whereas ketamine had no effect on spine elimination (n = 7 mice) compared with that in vehicle-treated controls (n = 6 mice). Throughout, box plots depict the median, interquartile range, and minimum and maximum excluding outliers, and outliers are plotted individually. NS, not significant. (C) Spine formation decreased (*P = 0.0087, Wilcoxon W = 44.5) after 21 days of chronic CORT, whereas ketamine increased spine formation. (D)Spine elimination per dendritic branch segment was significantly elevated (t = 8.05, df = 297, P = 1.9 × 10−14) and bimodally distributed in the chronic CORT-exposed group (n = 149 branches from 5 mice) compared with that in controls (n = 150 branches from 6 mice), such that 39.6% of dendritic branch segments in the chronic CORT-exposed group exhibited spine elimination rates that were >2 standard deviations above the mean for controls. (E) Spatially clustered spine elimination after chronic CORT exposure. The average distance between an eliminated spine and its nearest eliminated neighbor (2.5 μm) (red circle) was significantly reduced (P < 0.001), relative to what would be expected by chance given the observed rate of spine elimination, in simulations denoted by the box plot (see supplementary materials). (F) Spine formation per dendritic branch segment was significantly elevated (t = 9.48, df = 205, P = 6.7 × 10−18) and bimodally distributed in the ketamine-treated group (n = 103 branches from 7 mice) compared with that in vehicle-treated controls (n = 104 branches from 7 mice), such that 51.5% of dendritic branch segments in the ketamine-treated group exhibited spine formation rates that were highly elevated compared with those in vehicle-treated controls. (G) The average distance between a newly formed spine and its nearest eliminated neighbor (2.8 μm) (blue circle) was significantly reduced (P < 0.001) relative to what would be expected by chance given the observed rate of spine formation, as estimated in simulated data.
Chronic CORT recapitulated established depression- and anxiety-related behaviors (27–31), including increased immobility on the tail suspension test (TST), decreased sucrose preference, and decreased open-arm exploration in an elevated plus maze (fig. S1). Chronic CORT also increased spine elimination rates (Fig. 1B) and decreased spine formation rates (Fig. 1C). To assess the time course and stability of these changes, we quantified spine remodeling in mice imaged after both 10 and 21 days of CORT exposure (fig. S2). Similar effects were observed at the 10-day time point, and the vast majority (88.8%) of spines that were eliminated after 10 days did not recur spontaneously at the 21-day time point. To test whether these effects generalized to other forms of chronic stress, we repeated these experiments in mice exposed to 21 days of repeated restraint stress, a commonly used psychosocial stress paradigm, and found comparable effects on both behavior (fig. S3, A to D) and spine remodeling (fig. S3, E and F).
Stress-induced dendritic atrophy is limited to specific prefrontal projections (32), and functionally homogeneous synaptic inputs cluster on individual branch segments of the apical dendritic arbor in cortical pyramidal cells (33–40). We therefore examined the spatial distribution of stress-related spine elimination in order to assess whether spine loss occurred randomly throughout the apical dendritic tree or targeted specific dendritic branch segments. Eliminated spines were not randomly distributed. Whereas most dendritic branches exhibited spine elimination rates comparable to those observed in controls, spine elimination rates were two to four times as high (~35 to 40%) in a subset of branches, yielding a bimodal distribution across dendritic branch segments (Fig. 1D). Furthermore, within individual branches, spine loss was strongly spatially clustered to a significantly greater degree than would be expected by chance, given the observed rates of spine loss (Fig. 1E). Similar, spatially clustered effects were also observed after repeated restraint stress (fig. S3G), indicating that the emergence of depression-related behavior in multiple models is associated with spatially clustered, branch-specific spine loss.
Next, we tested whether the remission of depression-related behavior after ketamine treatment was associated with a targeted reversal of stress-induced spine loss. We used 2P imaging to visualize dendritic spine remodeling before and after chronic CORT exposure and again 24 hours after a single subanesthetic, anti-depressant dose of ketamine [10 mg per kilogram of body weight, intraperitoneally (ip)] (Fig. 1A). Ketamine had rapid effects on depression-related behavior, reversing CORT effects on immobility in the TST, open-arm exploration in an elevated plus maze, and sucrose preference, relative to those in vehicle-treated controls (fig. S4) (4–6). Ketamine also increased spine formation rates to 16.8% over 24 hours but did not significantly alter spine elimination rates (Fig. 1, B and C). Newly formed spines were also clustered and branch specific. Ketamine effects on spine formation were bi-modally distributed across individual dendritic branches and driven by a subset of branches (51.5%) for which spine formation rates were 5 to 10 times as high as the mean formation rate in vehicle-treated controls (Fig. 1F). Likewise, ketamine’s effects on spine formation were spatially clustered relative to controls and to a significantly greater degree than would be expected by chance given the observed rates of spine formation (Fig. 1G and fig. S3, H to J).
Antidepressant-dose ketamine selectively restores lost spines
Rather than compensating for stress-related synapse loss by increasing spine formation indiscriminately, ketamine may instead restore precisely the same synaptic connections lost during chronic CORT exposure. To test this, we began by assessing whether spines formed after ketamine treatment appeared in the same dendritic location as spines lost during chronic CORT exposure (Fig. 2A). Of the newly formed spines, 47.7% were located <2 μm from a spine lost during chronic CORT exposure, a proportion significantly greater than the 14.5% observed in vehicle-treated controls (Fig. 2B), as well as the 17.0% expected by chance if new spines were randomly distributed (Fig. 2C), given the observed rates of CORT-induced spine elimination and ketamine-induced spine formation. These selective reversal effects were not observed in vehicle-treated controls (Fig. 2B) or in CORT-treated mice imaged repeatedly after 10 and 21 days of exposure (fig. S2), indicating that the observed rescue of eliminated spines was not attributable to technical obstacles in resolving spines at a single time point. At the same time, whereas spinogenesis after ketamine treatment fully reversed the effects of CORT on spine density (Fig. 2D), many of these newly formed spines appeared in new positions where a spine did not previously exist (Fig. 2A, de novo formed spines). This meant that a majority (mean = 51.7%) of spines lost during chronic CORT exposure were not restored by ketamine treatment (Fig. 2E). Thus, these longitudinal imaging experiments reveal that ketamine only partially restores the baseline configuration of spines on PFC projection neurons and that substantial CORT-induced spine loss persists after treatment, despite the normalization of depression-related behaviors tested at the same time point (fig. S4).
Fig. 2. Ketamine selectively restores spines lost during chronic CORT exposure.

(A) Experimental timeline (identical to that in Fig. 1A). Restored spines (blue) were defined as spines that formed in the 24-hour period after ketamine treatment in a position <2 μm from a previously eliminated spine. (B) Of newly formed spines, 47.7% were located <2 μm from a spine lost during chronic CORT exposure (n = 7 mice), a proportion significantly greater than the 14.5% observed in vehicle-treated controls (n = 6 mice; *P = 0.0012, Wilcoxon W = 21.0). (C) The observed fraction of restored spines (blue circle) was significantly (P < 0.001) greater than expected by chance given the observed rate of spine formation in simulated data in which formed spines were randomly distributed (box plot) (see supplementary materials). (D) The change in spine density from day 1 (baseline) to day 21 (post-chronic CORT) or day 22 (postketamine) was quantified by subtracting the spine elimination rate from the spine formation rate during the specified imaging interval. Ketamine reversed chronic CORT effects on spine density [Kruskal-Wallis analysis of variance (ANOVA), **P = 5.6 × 10−4, χ2 = 14.96]. (E) Of spines lost during chronic CORT, 48.3% were restored after ketamine treatment (n = 7 mice) versus 3.3% in vehicle-treated controls (n = 6 mice; **P = 0.0012, Wilcoxon W = 21.0). (F) Representative confocal image of PSD-95 immuno-fluorescence in the mPFC. Scale bar, 30 μm. (G) PSD-95 density (z-scored with respect to the mean density in controls; N = 18 samples from n = 6 subjects per group) varied by experimental condition (mixed-effects ANOVA, F2,45 = 10.39, P = 0.0019). In the dorsal mPFC, PSD-95 density was significantly lower in the chronic CORT-treated group than in untreated controls (t10 = 2.43, *P = 0.035). Ketamine reversed this effect (t10= 4.15, **P = 0.002). (For other PFC subregions, see fig. S6.)
Two additional experiments revealed that ketamine not only restored lost spines but also generated functional synapses. First, motivated by prior work showing that essentially all new spines that persist for at least 4 days contain functional synapses (41, 42), we imaged the same dendritic segments 4 days later (fig. S5A). Ketamine increased the formation of these long-lasting spines (fig. S5B), which are highly likely to contain functional synapses. Furthermore, restored spines (those that formed <2 μm from a previously lost spine) were significantly more likely to persist than de novo formed spines (fig. S5C). On the basis of these data, we estimate that ketamine restored 20.4 to 31.0% of lost synapses in their original positions after 4 days, versus the restoration of 0 to 2.4% in vehicle-treated controls (fig. S5D). Second, we tested for corresponding changes in spine density in fixed tissue and synapse density as quantified by immunofluorescence labeling of the postsynaptic density protein PSD-95. Chronic CORT treatment decreased both PSD-95 density and dendritic spine density throughout the PFC, including the anterior cingulate, prelimbic, and infralimbic subregions, whereas ketamine had the opposite effect (Fig. 2, F and G and fig. S6).
Ketamine rescues PFC microcircuit dysfunction
How CORT- and ketamine-induced spine re-modeling relates to prefrontal microcircuit function is not well understood. We hypothesized that the CORT-induced elimination of excitatory synapses within dendritic spines would be associated with a loss of functional coupling between PFC neurons. We therefore used 2P calcium imaging to characterize functional connectivity in PFC microcircuits before and after 10 days of chronic CORT exposure and again 24 hours after ketamine treatment (Fig. 3, A to C). Using vascular landmarks, the contours of the prism, and measurements of depth from the cortical surface, we were able to record activity repeatedly from the same PFC cell populations, which were stably expressing a genetically encoded calcium sensor (GCaMP6s) (17) over this 11-day period. We selected this interval to avoid the potentially toxic accumulation of GCaMP that can occur after prolonged, virally driven expression and on the basis of spine imaging experiments showing comparable effects at 10 and 21 days (fig. S2). We tested for CORT- and ketamine-induced changes in functional connectivity between PFC neurons, as indexed by correlation matrices quantifying correlated activity across all cells in a field of view (Fig. 3D). Chronic CORT significantly reduced functional connectivity within PFC microcircuits (Fig. 3, D and E). At baseline, most activity in a given field of view occurred at discrete time points involving many simultaneously active cells (Fig. 3, B and C), reminiscent of the multicellular ensemble activity that has been implicated in pattern formation and stimulus encoding and is disrupted in animal models of neuropsychiatric disease (43–45). After chronic CORT exposure, the frequency of these multicellular ensemble events (see supplementary materials) was significantly reduced (Fig. 3F), and they tended to involve significantly fewer cells (Fig. 3G). Ketamine rescued both effects 24 hours after treatment (Fig. 3, E to G). By contrast, the correlated activity and multicellular ensemble events were stable over time in untreated controls (fig. S7).
Fig. 3. Ketamine rescues CORT-induced microcircuit dysfunction.
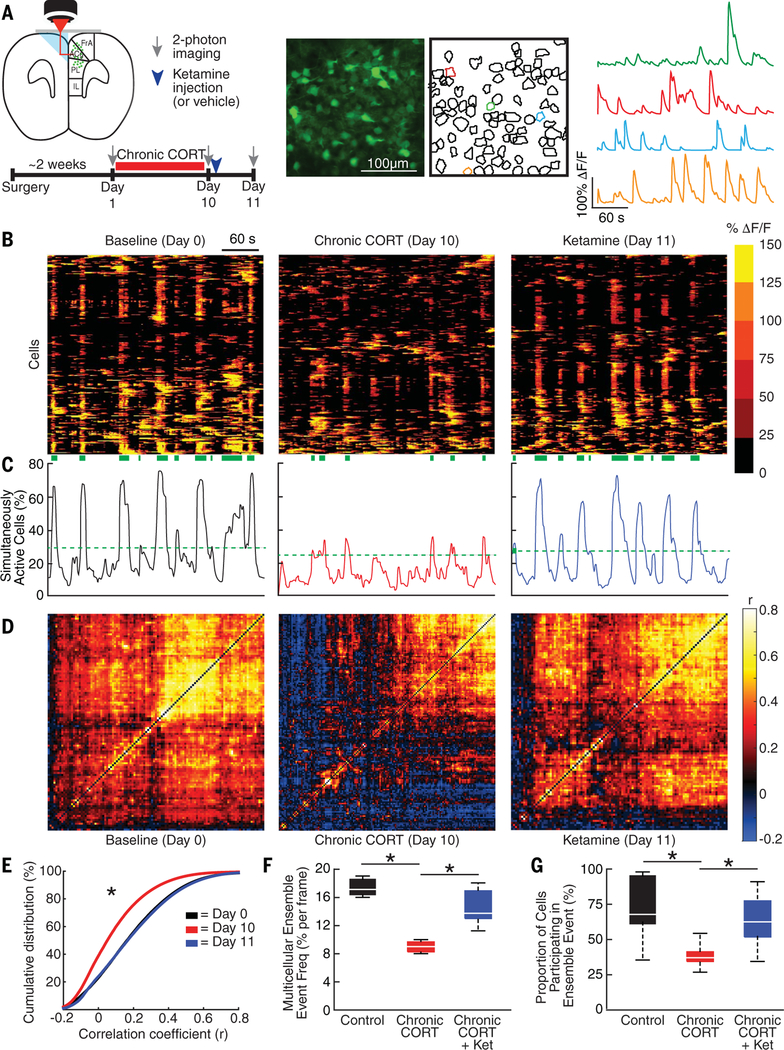
(A) 2P calcium imaging of GCaMP6s-expressing neurons through a microprism implanted across the midline in the mPFCs of head-fixed, awake mice. Calcium imaging occurred before and after 10 days of chronic CORT treatment and 24 hours after ketamine treatment (10 mg/kg ip). (B) Raster plots depicting changes in activity (ΔF/F) over time for a representative neuronal population before and after chronic CORT and 1 day after ketamine treatment. Each row represents a single cell. Green bands below denote statistically significant multicellular ensemble events. (C) Representative traces depicting the proportion of simultaneously active cells over time for the neuronal population depicted in (B).The green dashed line delineates the upper end of a 99.9% confidence interval for the proportion of simultaneously active cells in shuffled data (see supplementary materials). (D) Representative correlation matrices quantifying functional connectivity between each cell and every other cell. (E) Cumulative distribution plot of cell-cell correlations for all cells across all animals in each condition (N = 98,878 correlations at baseline, N = 145,467 after chronic CORT, N = 114,593 after ketamine from n = 3 mice). Mixed-effects ANOVA (subject = random effect) showed that the distribution shifted left (decreased) after chronic CORT (mean correlation coefficient r = 0.084 versus 0.188 at baseline; t = 3.02; P = 0.0025 for the difference between chronic CORT and baseline), and ketamine rescued this effect (mean r = 0.187; t = 0.39; P = 0.698 for the difference between ketamine and baseline). (F) The frequency of multicellular ensemble events (expressed as the probability of an ensemble event occurring in any given time frame) decreased after chronic CORT (median, 8.4% versus 17.4% at baseline) and was rescued after ketamine treatment (chronic CORT + Ket) (median, 14.7%; mixed-effects ANOVA, F2,15 = 17.1, P = 0.00013 for main effect of group; N = 8 observations per experimental condition). (G) The proportion of cells participating in a multicellular ensemble event also decreased after chronic CORT (mean = 37.7% versus 74.1% at baseline for N = 39 and N = 28 multicellular ensemble events in n = 3 mice) and was rescued after ketamine treatment (mean = 63.8% for N = 35 events; mixed-effects ANOVA, F2,93 = 10.8, P = 0.024). By contrast, in untreated controls, no significant changes occurred over time in any of the measures in (E) to (G) (fig. S5). *P < 0.05 (significantly different post hoc contrast, Holm-Bonferroni corrected).
To better understand how ketamine’s effects on spine formation and microcircuit activity relate to its antidepressant behavioral effects, we used fiber photometry (18,19) to record the activity of GCaMP6s-expressing neurons in the ventromedial PFC (vmPFC) during tail suspension (Fig. 4A and fig. S8). The vmPFC has been implicated in driving motivated escape behavior during similar behavioral challenge assays (46) in which mice alternate between periods of passive immobility and active struggling to escape. Prolonged immobility is a consistent finding in chronic stress models (47), and anti-depressants tend to reverse this behavior (48,49). PFC activity was significantly elevated during periods of struggling (fig. S9), an effect that was likely driven in part by differences in locomotor activity (46). However, we also found that increased PFC activity preceded the onset of struggling, predicting the shift from immobility to struggling behavior within ~1 s on average (Fig. 4B). This effect was not due to systematic delays in recording the onset of struggling (fig. S10). PFC activity during bouts of immobility tended to occur at the end of a bout (<1 s before struggling) and predicted a significantly shorter latency to the onset of struggling than randomly shuffled data (Fig. 4C). Larger calcium transients predicted progressively shorter latencies to struggling, such that the largest PFC activity events predicted a shift from immobility to struggling within <1 s >80% of the time (Fig. 4D).
Fig. 4. Ketamine rescues CORT effects on coordinated PFC activity and active avoidance behavior.

(A) (Top) Experimental timeline and schematic showing optical fiber placement in the vmPFC and representative fiber photometry recording of GCaMP6s-expressing neurons during tail suspension. (Bottom) A representative photometry trace in which pink bands indicate periods of struggling to escape. Insets show representative examples of GCaMP6s activity increasing before the onset of struggling. (B) Mean vmPFC activity (ΔF/F) ± SEM (gray band) time locked to the onset of struggling (time = 0), averaged over all bouts of struggling across all mice. Increased vmPFC activity preceded the onset of struggling by ~1 s. (C) Distribution of calcium-transient events during bouts of immobility by latency to the onset of the next bout of struggling. Monte Carlo simulation showed that during bouts of immobility, calcium transients predicted the onset of struggling: The median latency to the onset of struggling (4.0 s) was significantly shorter than would be expected by chance (7.0 s) in randomly shuffled data (P < 0.0001 across 10,000 iterations), and 37.1% of all transients occurred <1 s before the onset of struggling. (D) Cumulative distribution of calcium transients by latency to the onset of struggling, grouped by transient amplitude. Linear mixed-effects modeling showed that latency to the onset of struggling varied with transient amplitude (t303 = 3.84, P = 0.0015), such that larger transients predicted shorter latencies. The gray band denotes the 99% confidence interval for the median latency in shuffled data. The median latency for transients in all four groups was less than that expected by chance in shuffled data, with the largest effects occurring for transients in the upper quartile (median latency = 0.69 s). (E) Chronic CORT increased the total duration of immobility during tail suspension, whereas ketamine rescued this effect [n = 8 control mice, n = 10 chronic CORT-treated mice, n = 14 chronic CORT-plus-ketamine (CORT+ket)-treated mice; Kruskal-Wallis ANOVA, *P = 0.0003, χ2 = 15.93]. (F) Chronic CORT significantly reduced the frequency of vmPFC calcium transients during tail suspension, and ketamine rescued this effect (n = 8 control mice, n = 10 chronic CORT-treated mice, n = 11 ketamine-treated mice; Kruskal-Wallis ANOVA, *P = 0.0083, χ2 = 9.59). (G) Cumulative distribution of calcium transients by latency since the onset of immobility showing the accumulation of transients overtime, sorted by experimental condition. Linear mixed-effects modeling showed that vmPFC activity accumulated more slowly over time after chronic CORT exposure (median latency, 10.2 s for n = 378 transients versus 7.2 s for n = 305 transients in controls), and ketamine rescued this effect (median latency, 6.8 s for n = 378 transients; t983 = 2.21).
Chronic CORT exposure significantly reduced the frequency of calcium transients during the TST (Fig. 4F). Calcium transients also accumulated more slowly over time during bouts of immobility and were associated with longer immobility periods and increased immobility overall (Fig. 4, E and G). Ketamine reversed these deficits.
Prefrontal spinogenesis sustains the remission of depression-related behavior over time
Lastly, we investigated whether PFC spino-genesis is required for—or merely correlated with—changes in behavior and circuit function after antidepressant treatment. We began by characterizing the time course of these changes in the hours and days after ketamine treatment. As in (5), we observed rapid effects on motivated avoidance behavior in the TST, beginning just 3 hours after ketamine treatment (10 mg/kg ip), peaking 24 hours postinjection, and persisting for up to a week (Fig. 5, A and B). However, ketamine effects on spine formation were slower: Formation rates were not significantly altered at 3 to 6 hours posttreatment but were markedly elevated at 12 to 24 hours (Fig. 5C). Increased spine formation in Thy1/YFP-expressing projection neurons is thus not required for the induction of ketamine’s acute effects on immobility behavior 3 to 6 hours after treatment. In accordance with this interpretation, we found no association between spine formation and immobility in the first day after treatment (fig. S11). By contrast, individual differences in the restoration of spines lost during chronic CORT exposure were correlated with immobility behavior 2 to 7 days after treatment (Fig. 5D).
Fig. 5. Prefrontal spinogenesis is not required for inducing ketamine’s effects on behavior or circuit function.

(A) Experimental timeline for data in (B) to (D). To avoid habituation in the TST due to repeated testing on the same day, each subject in (B) was tested only once in this assay (between-subjects design; N = 12 subjects per time point per group). For 2P imaging in (C), each subject was assessed repeatedly at each specified time point (within-subjects design; N = 12 subjects per group). (B) Rapid effect of ketamine on immobility in the TST (main effect of treatment, F1,132 = 149.1, P < 0.0001; main effect of time, F5,132 = 1.20, P = 0.314; interaction, F5,132 = 1.24, P = 0.295). *P < 0.002 (significant post hoc linear contrast). (C) Delayed effect of ketamine on spine formation (main effect of treatment, F1,108 = 145.7, P < 0.0001; main effect of time, F5,108 = 81.6, P < 0.0001; interaction, F5,108 = 17.4, P < 0.000l). *P < 0.0006 (significant post hoc linear contrast). NS, not significant. (D) The survival of restored spines (the percentage of spines lost after chronic CORT and restored after ketamine) was significantly correlated with immobility behavior 2 to 7 days after treatment (Spearman’s rho = 0.770, P = 0.0034). (E) Experimental timeline for data in (F) to (K). To test whether ketamine effects on microcircuit activity preceded or followed effects on spines, 2P calcium imaging was performed before and after chronic CORT and 3, 12, and 24 hours after ketamine treatment (within-subjects design). (F) Raster plots depicting changes in activity (ΔF/F) over time for a representative neuronal population before and after chronic CORT and 1 day after ketamine treatment, as in Fig. 3B.(G) Representative traces depicting the proportion of simultaneously active cells over time for the neuronal population depicted in (F). The green dashed line delineates the upper end of a 99.9% confidence interval for the proportion of simultaneously active cells in shuffled data (see supplementary materials). (H) Representative correlation matrices quantifying functional connectivity between each cell and every other cell. (I) There was a reduction in functional connectivity (mean correlation) after chronic CORT, and ketamine rescued this effect within 3 hours of treatment (N = 5 to 8 samples per condition from n = 3 subjects; mixed-effects ANOVA, subject = random effect; main effect of time, F4,32 = 3.05, P = 0.032). For cumulative distribution functions for all correlation coefficients, see fig. S12. (J) The frequency of multicellular ensemble events (expressed as the probability of an ensemble event occurring in any given time frame) decreased after chronic CORT and was rescued within 3 hours of ketamine treatment (mixed-effects ANOVA; main effect of time, F4,32 = 9.28, P = 4.27 × 10−5). (K) The proportion of cells participating in a multicellular ensemble event also decreased after chronic CORT and was rescued within 3 hours of ketamine treatment (mixed-effects ANOVA; main effect of time, F4,341 = 7.65, P = 6.55 × 10−6). For (I) to (K), *P < 0.05 (significantly different post hoc contrast, Holm-Bonferroni corrected).
To investigate the temporal relationship between spinogenesis and ketamine’s effects on PFC microcircuit dynamics, we repeated the 2P calcium imaging experiment in Fig. 3, imaging before and after chronic CORT exposure and 3,12, and 24 hours after ketamine treatment (Fig. 5E). As in Fig. 3, chronic CORT interfered with correlated activity, reduced the frequency of multicellular ensemble events, and decreased the proportion of cells participating in these events (Fig. 5, F to K). Ketamine reversed all three effects within 3 hours of treatment (Fig. 5, I to K, and fig. S12), ~9 hours before effects on spine formation were detectable in Fig. 5C. This suggests that ketamine-induced spine remodeling may be an activity-dependent consequence, rather than a cause of changes in circuit function. In conjunction with the data in Fig. 5D, these results indicate that prefrontal spino-genesis is not required for inducing ketamine’s effects on circuit function and behavior, but it may be important for sustaining these effects over time.
To test this hypothesis, we used a photo-activatable probe—activated synapse-targeting photoactivatable Rac1 (AS-PaRac1)—to optically manipulate the survival of newly formed spines (20). This construct is selectively expressed in newly formed and recently potentiated synapses, and upon prolonged photoactivation by blue light, it causes spine shrinkage and collapse (20). To validate this tool for manipulating the survival of spines formed after ketamine treatment, we injected a viral vector driving AS-PaRac1 expression (AAV1-SARE-Arc-PSDdelta1.2-mVenus-PARac1) into the mPFC and implanted a microprism for visualizing changes in AS-PaRac1 expression over time. As in (20), AS-PaRac1 expression was sparse and punctate in the mPFC (fig. S13, A to C) and increased significantly 1 day after ketamine treatment [mean increase in AS-PaRac1-positive (AS-PaRac1+) puncta = 92.3%] (fig. S13, D and E). Furthermore, keta-mine induced a spatially clustered pattern of AS-PaRac1 expression (fig. S13, F to H) that resembled its spatially clustered effects on spine formation, with 41.4% of AS-PaRac1+ puncta occurring in spatially clustered pairs or triplets, versus <5% in simulated data in which an equal number of puncta were randomly distributed throughout an equivalent imaging volume (fig. S13, I and J) (see Materials and methods for details).
To test whether prefrontal spinogenesis is required for maintaining ketamine’s effects on PFC circuit function and behavior, we injected viral vectors driving AS-PaRac1 and GCaMP6s expression bilaterally in the mPFC, implanted dual-core optical fibers, and treated the mice with CORT for 10 days, followed by ketamine on day 11, photoactivation (20-mW, 150-ms pulses at 1 Hz for 1 hour) 1 day after ketamine treatment, and behavioral testing 2 days after ketamine treatment (Fig. 6A). To validate this strategy for deleting newly formed spines, a separate cohort of Thy1/YFP-H transgenic mice was injected with AS-PaRac1 but not GCaMP6s.
Fig. 6. Prefrontal spinogenesis sustains antidepressant-induced behavioral recovery over time.

(A) Experimental time course schematic. To control for individual differences in TST behavior as in a recently published report (72), we tested subjects before chronic CORT exposure and again 2 days after ketamine treatment (Rx) to determine whether ketamine reversed CORT effects on immobility and whether maintaining this effect required spine formation. (B) Photoactivating AS-PaRac1 (N = 7 mice) blocked ketamine effects on spine formation (Wilcoxon W = 50.0, *P = 0.0025) (right) and increased spine elimination (Wilcoxon W = 18.0, *P = 0.0177) (left) compared with that in controls not expressing AS-PaRac1 (N = 5 mice). (C) Photoactivating AS-PaRac1 interfered with the maintenance of ketamine’s effects on immobility behavior (F1,17 = 9.96, *P = 0.0058) and (D) calcium-transient frequency (F1,17 = 16.6, *P = 0.019) during tail suspension 2 days after treatment. (E) No effect of photoactivating AS-PaRac1 on sucrose preference behavior 2 days after ketamine treatment (N = 8 mice per group; Wilcoxon W = 63.0, P = 0.645). (F) Photoactivating AS-PaRac1 3 hours after ketamine treatment (before the onset of ketamine’s effects on spine formation) had no effect on TST behavior 2 days after treatment (F1,17 = 0.201, P = 0.660, N = 10 AsPaRac1 mice and N = 9 controls). NS, not significant.
Photoactivating AS-PaRac1 increased spine elimination and blocked the effects of ketamine on spine formation (Fig. 6B) relative to spine elimination and spine formation in controls exposed to the same ketamine and photoactivation paradigm but not expressing AS-PaRac1. Behavioral testing 2 days after ketamine treatment showed that deleting newly formed spines by photoactivating AS-PaRac1 blocked the long-term effects of ketamine on PFC activity events (calcium transients) and on motivated avoidance behavior in the TST (Fig. 6, C and D), a behavior that is linked specifically to PFC function (Fig. 4) (46). This effect on behavior was selective: Ketamine’s long-term effect on sucrose preference was preserved (Fig. 6E), indicating that spine remodeling in other brain regions or spine-independent processes may be involved in sustaining ketamine’s effects on other depression-related behaviors. To evaluate the effect of eliminating a random subset of spines unrelated to ketamine treatment, we also tested mice after activating AS-PaRac1 3 hours after ketamine treatment—before the onset of ketamine-induced spine formation. This manipulation had no effect on immobility behavior at 2 days after treatment (Fig. 6F).
Discussion
A growing body of work has begun to elucidate the molecular mechanisms underlying stress and antidepressant effects on depression-related behavior, including neurotrophic factors, inflammatory signals, and regulators of neuronal excitability (4,5,50–59). By contrast, the circuit-level mechanisms mediating transitions between depression-related behavioral states over time are not well understood. Our findings delineate one such mechanism. The emergence of depression-related behavior in multiple chronic stress models was associated with spatially clustered spine loss and a loss of functional connectivity within PFC microcircuits, driven in part by the disruption of coordinated activity in multicellular ensembles, which play an important role in multiple computational processes (43–45). Ketamine facilitated the transition to recovery by rescuing these effects in both stress models. Varying stress mediators (60–66) and circuit-level mechanisms (51, 67–76) may contribute to model-specific stress effects on behavior and circuit function. Synaptic remodeling in PFC projection neurons may also influence depression-related behavior indirectly, through effects on social hierarchical interactions, which are modulated by synaptic efficacy and thala-mocortical inputs to the PFC (77–79). Distinct processes may also be involved in sustaining the long-term effects of conventional antidepressants. These caveats notwithstanding, our convergent findings implicate complementary effects on spine remodeling and multicellular ensemble activity in the induction and remission of a depression-related behavioral state.
Both CORT and ketamine are well positioned to regulate dendritic spine remodeling at a molecular level. Circadian oscillations in glucocorticoid activity have been implicated in balancing spine formation and elimination through both transcription-dependent processes and rapid, nongenomic mechanisms converging on the regulation of actin cytoskeletal remodeling by LIM kinase 1 and its substrate cofilin (80). Chronic excessive CORT exposure disrupts this balance, leading to widespread spine loss (80, 81). Stress effects on microglial activation may also be involved (82–86). Ketamine is well positioned to regulate spine density through the activation of brain-derived neurotrophic factor (BDNF)-TrkB and mammalian target of rapamycin complex 1 (mTORC1) signaling (4, 5, 50). BDNF-dependent signaling may also be involved in facilitating clustered spine restoration after ketamine treatment. Activity-dependent BDNF signaling at existing spines can “prime” neighboring dendritic sites for structural plasticity by activating a tripartite signaling mechanism involving Cdc42, Rac1, and RhoA (87).
Our results also define a specific causal role for PFC spinogenesis in mediating transitions among a depression-related behavioral state, behavioral remission, and spontaneous recurrence. Ketamine’s acute effects on depression-related behavior and circuit function occur rapidly and precede the onset of spine formation, which in turn suggests that spine remodeling may be an activity-dependent adaptation to changes in circuit function (83, 88) and is consistent with theoretical models implicating synaptic homeostasis mechanisms in depression and the stress response (89, 90). Although not required for inducing ketamine’s effects acutely, these newly formed spines are critical for sustaining the anti-depressant effect over time. Of note, interfering with ketamine-induced spine formation blocked its effects on motivated escape behavior but not on sucrose preference, indicating a specific role for prefrontal spinogenesis in sustaining some but not all antidepressant behavioral changes. Spine remodeling in the hippocampus, amygdala, nucleus accumbens, and other structures may support other behaviors, and some behavioral changes may be sustained by other mechanisms (5, 89, 91–95).
By monitoring spine remodeling longitudinally, our experiments also reveal that although ketamine reverses chronic CORT effects on spine density and behavior, this apparent normalization belies the facts that most lost spines are not restored in their original positions and the baseline configuration of prefrontal spines is persistently altered. Whereas ~45% of new spines persisted for at least 4 days after ketamine treatment—indicating a high likelihood that they contain functional synapses—the remaining ~55% were quickly lost. Together, the failure to fully restore the baseline configuration of PFC synapses immediately after ketamine treatment and the subsequent loss of at least half of these newly formed spines may act as substrates for the spontaneous recurrence of depression-related behaviors—a common finding in clinical studies 1 week after treatment (1–3). Pharmacological and neurostimulatory interventions for enhancing and preserving the rescue of lost synapses may therefore be useful for promoting sustained remission.
Materials and methods summary
For additional details on each section below, as well as information on stress paradigms, ketamine treatment, stereotaxic coordinates, behavioral assays, immunohistochemistry, histological verification, and statistical analyses and other information, see the supplementary materials.
Animals
Young adult (aged 9 to 20 weeks) male Thyl/YFP-H transgenic mice and C57BL/6J mice were used for spine imaging and other experiments, respectively. Animals were group-housed under a 12-hour-12-hour light-dark cycle and provided ad libitum access to food and water (except during restraint stress). Sample sizes for each experiment were determined by using G*Power analysis software, on the basis of anticipated effect sizes that were estimated from previously published reports whenever they were available, and were powered to detect moderate, biologically meaningful effect sizes. AH studies were approved by the Weill Cornell Institutional Animal Care and Use Committee.
Surgery
Stereotactic injections were performed under anesthesia to introduce viral vectors. AAVl-hSyn-GCaMP6s was injected into the PFC for calcium imaging and fiber photometry experiments. AAV1-SARE-Arc-PSDelta1.2-mVenus-PARac1 was expressed in the mPFC for spine deletion experiments. For prism implantations, 4-mm-diameter circular craniotomies were centered over the midline, and 1.5-mm right-angle boro-silicate glass prisms were implanted in the mPFC contralateral to the viral injection site by using a digital micromanipulator. For fiber photometry experiments, a smaller craniotomy was opened, and 400-um-diameter multimodal optical fibers or custom dual-core implantable fiber optic cannulas were implanted above the injection sites.
Data acquisition summary
All in vivo images were acquired by using a 2P laser-scanning microscope (Olympus RS) with a Mai Tai DeepSee laser tuned to 920 nm and a 25×, 1.0NA water-immersion objective (4-mm working distance). For calcium imaging experiments, time-lapse images were acquired at ~1 frame per second in awake mice. Z-stacks through up to 250-μm depths were acquired for spine and AS-PaRac1 experiments. Fiber photometry was conducted during tail suspension behavior. GCaMP6s was excited with a 470-nm light-emitting diode (LED) through an optical fiber patch cord (modulated at a frequency of 521 Hz). Calcium signals were aligned to behavioral measures via transistor-to-transistor logic (TTL) pulses.
Data analysis summary
All data were analyzed by investigators who were blind to the experimental condition. Dendritic spine remodeling was assessed in ImageJ in Thy1/YFP mice on randomly selected dendritic branch segments containing clearly distinguishable spines. Formed and eliminated spines were identified in pairs of images obtained from the same areas over given intervals, and rates were quantified as a proportion of the total number of spines evaluated. Calcium imaging data preprocessing and cell segmentation were performed by using established methods (90–92) instantiated in MATLAB scripts. Fluorescence signal time series [the change in fluorescence divided by the baseline fluorescence (ΔF/F)] were calculated for each neuron relative to a 40-s sliding window baseline. Photometry data were acquired continuously during tail suspension behavior and analyzed by using MATLAB scripts to normalize fluorescence signals and identify the onset and termination of calcium transients. For details on the analysis of clustered spine remodeling, calcium imaging (correlated activity and ensemble events), and photometry data (predicting TST behavior), see the supplementary materials.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the Liston lab for helpful discussions.
Funding: This work was supported by funds from the Whitehall Foundation, Hartwell Foundation, One Mind Institute, Rita Allen Foundation, and National Institute of Mental Health (R00 MH097822, R01 MH109685, and R01 MH118451) and by a Klingenstein-Simons Fellowship in Brain Science. R.N.M.-S. was supported by a fellowship from the NSF. H.B. was supported by KAKENHI grant 17H06312. H.K. was supported by CREST (JPMJCR1652) from JST, SRPBS (17dm0107120h002) from AMED, and a grant-in-aid (26221001) and WPI from MEXT of Japan. D.C.S. and R.N.F. were supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the National Institutes of Health under award T32GM007739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program. R.N.F. was supported by a fellowship from the NIH (F30 MH115622). L.G. was supported by Simons Foundation award 429965. T.A.M. was supported by NIH grants R01 DA08259 and HL136520.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: AAV1-SARE-Arc-PSDdelta1.2-mVenus-PARac1 is available upon request from H.K. (hkasai@m.u-tokyo.ac.jp) and H.B. (hbito@m.u-tokyo.ac.jp) under a material transfer agreement with the University of Tokyo. All other materials are commercially available. All data needed to evaluate the conclusions in the paper are present in the paper or the supplementary materials.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Berman RM et al. , Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351–354 (2000). doi: 10.1016/S0006-3223(99)00230-9; [DOI] [PubMed] [Google Scholar]
- 2.Zarate CA Jr., et al. , A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63, 856–864 (2006). doi: 10.1001/archpsyc.63.8.856; [DOI] [PubMed] [Google Scholar]
- 3.Ibrahim L et al. , Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: Results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology 37, 1526–1533 (2012). doi: 10.1038/npp.2011.338; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li N et al. , mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964 (2010). doi: 10.1126/science.1190287; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Autry AE et al. , NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95 (2011). doi: 10.1038/nature10130; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zanos P et al. , NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486 (2016). doi: 10.1038/nature17998; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phoumthipphavong V, Barthas F, Hassett S, Kwan AC, Longitudinal effects of ketamine on dendritic architecture in vivo in the mouse medial frontal cortex. eNeuro 3, ENEURO.0133–15.2016 (2016). doi: 10.1523/ENEURO.0133-15.2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duman RS, Malberg J, Thome J, Neural plasticity to stress and antidepressant treatment. Biol. Psychiatry 46, 1181–1191 (1999). doi: 10.1016/S0006-3223(99)00177-8; [DOI] [PubMed] [Google Scholar]
- 9.Radley JJ et al. , Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb. Cortex 16, 313–320 (2006). doi: 10.1093/cercor/bhi104; [DOI] [PubMed] [Google Scholar]
- 10.Cook SC, Wellman CL, Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J. Neurobiol 60, 236–248 (2004). doi: 10.1002/neu.20025; [DOI] [PubMed] [Google Scholar]
- 11.Liston C et al. , Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J. Neurosci 26, 7870–7874 (2006). doi: 10.1523/JNEUROSCI.1184-06.2006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gourley SL, Swanson AM, Koleske AJ, Corticosteroid-induced neural remodeling predicts behavioral vulnerability and resilience. J. Neurosci 33, 3107–3112 (2013). doi: 10.1523/JNEUROSCI.2138-12.2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pattwell SS et al. , Dynamic changes in neural circuitry during adolescence are associated with persistent attenuation of fear memories. Nat. Commun 7, 11475 (2016). doi: 10.1038/ncomms11475; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chia TH, Levene MJ, Microprisms for in vivo multilayer cortical imaging. J. Neurophysiol 102, 1310–1314 (2009). doi: 10.1152/jn.91208.2008; [DOI] [PubMed] [Google Scholar]
- 15.Andermann ML et al. , Chronic cellular imaging of entire cortical columns in awake mice using microprisms. Neuron 80, 900–913 (2013). doi: 10.1016/j.neuron.2013.07.052; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Low RJ, Gu Y, Tank DW, Cellular resolution optical access to brain regions in fissures: Imaging medial prefrontal cortex and grid cells in entorhinal cortex. Proc. Natl. Acad. Sci. U.S.A 111, 18739–18744 (2014). doi: 10.1073/pnas.1421753111; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen T-W et al. , Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013). doi: 10.1038/nature12354; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunaydin LA et al. , Natural neural projection dynamics underlying social behavior. Cell 157, 1535–1551 (2014). doi: 10.1016/j.cell.2014.05.017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui G et al. , Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 494, 238–242 (2013). doi: 10.1038/nature11846; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayashi-Takagi A et al. , Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525, 333–338 (2015). doi: 10.1038/nature15257; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karatsoreos IN et al. , Endocrine and physiological changes in response to chronic corticosterone: A potential model of the metabolic syndrome in mouse. Endocrinology 151, 2117–2127 (2010). doi: 10.1210/en.2009-1436; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bodnoff SR et al. , Enduring effects of chronic corticosterone treatment on spatial learning, synaptic plasticity, and hippocampal neuropathology in young and mid-aged rats. J. Neurosci 15, 61–69 (1995). doi: 10.1523/JNEUROSCI.15-01-00061.1995; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luine VN, Spencer RL, McEwen BS, Effects of chronic corticosterone ingestion on spatial memory performance and hippocampal serotonergic function. Brain Res. 616, 65–70 (1993). doi: 10.1016/0006-8993(93)90193-Q; [DOI] [PubMed] [Google Scholar]
- 24.McLay RN, Freeman SM, Zadina JE, Chronic corticosterone impairs memory performance in the Barnes maze. Physiol. Behav 63, 933–937 (1998). doi: 10.1016/S0031-9384(97)00529-5; [DOI] [PubMed] [Google Scholar]
- 25.Radley J, Morilak D, Viau V, Campeau S, Chronic stress and brain plasticity: Mechanisms underlying adaptive and maladaptive changes and implications for stress-related CNS disorders. Neurosci. Biobehav. Rev 58, 79–91 (2015). doi: 10.1016/j.neubiorev.2015.06.018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karatsoreos IN, Bhagat S, Bloss EB, Morrison JH, McEwen BS, Disruption of circadian clocks has ramifications for metabolism, brain, and behavior. Proc. Natl. Acad. Sci. U.S.A 108, 1657–1662 (2011). doi: 10.1073/pnas.1018375108; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Y et al. , A mouse model of depression induced by repeated corticosterone injections. Eur. J. Pharmacol 581, 113–120 (2008). doi: 10.1016/j.ejphar.2007.12.005; [DOI] [PubMed] [Google Scholar]
- 28.Gourley SL et al. , Regionally specific regulation of ERK MAP kinase in a model of antidepressant-sensitive chronic depression. Biol. Psychiatry 63, 353–359 (2008). doi: 10.1016/j.biopsych.2007.07.016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitra R, Sapolsky RM, Acute corticosterone treatment is sufficient to induce anxiety and amygdaloid dendritic hypertrophy. Proc. Natl. Acad. Sci. U.S.A 105, 5573–5578 (2008). doi: 10.1073/pnas.0705615105; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strekalova T, Spanagel R, Bartsch D, Henn FA, Gass P, Stress-induced anhedonia in mice is associated with deficits in forced swimming and exploration. Neuropsychopharmacology 29, 2007–2017 (2004). doi: 10.1038/sj.npp.1300532; [DOI] [PubMed] [Google Scholar]
- 31.Gourley SL, Taylor JR, Recapitulation and reversal of a persistent depression-like syndrome in rodents. Curr. Protoc. Neurosci. Chapter 9, 32 (2009). doi: 10.1002/0471142301.ns0932s49; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shansky RM, Hamo C, Hof PR, McEwen BS, Morrison JH, Stress-induced dendritic remodeling in the prefrontal cortex is circuit specific. Cereb. Cortex 19, 2479–2484 (2009). doi: 10.1093/cercor/bhp003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J, Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33, 121–129 (2010). doi: 10.1016/j.tins.2010.01.001; [DOI] [PubMed] [Google Scholar]
- 34.Gökçe O, Bonhoeffer T, Scheuss V, Clusters of synaptic inputs on dendrites of layer 5 pyramidal cells in mouse visual cortex. eLife 5, e09222 (2016). doi: 10.7554/eLife.09222; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleindienst T, Winnubst J, Roth-Alpermann C, Bonhoeffer T, Lohmann C, Activity-dependent clustering of functional synaptic inputs on developing hippocampal dendrites. Neuron 72, 1012–1024 (2011). doi: 10.1016/j.neuron.2011.10.015; [DOI] [PubMed] [Google Scholar]
- 36.Takahashi N et al. , Locally synchronized synaptic inputs. Science 335, 353–356 (2012). doi: 10.1126/science.1210362; [DOI] [PubMed] [Google Scholar]
- 37.Makino H, Malinow R, Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron 72, 1001–1011 (2011). doi: 10.1016/j.neuron.2011.09.036; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McBride TJ, Rodriguez-Contreras A, Trinh A, Bailey R, Debello WM, Learning drives differential clustering of axodendritic contacts in the barn owl auditory system. J. Neurosci 28, 6960–6973 (2008). doi: 10.1523/JNEUR0SCI.1352-08.2008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Druckmann S et al. , Structured synaptic connectivity between hippocampal regions. Neuron 81, 629–640 (2014). doi: 10.1016/j.neuron.2013.11.026; [DOI] [PubMed] [Google Scholar]
- 40.Rah J-C et al. , Thalamocortical input onto layer 5 pyramidal neurons measured using quantitative large-scale array tomography. Front. Neural Circuits 7,177 (2013). doi: 10.3389/fncir.2013.00177; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K, Spine growth precedes synapse formation in the adult neocortex in vivo. Nat. Neurosci 9, 1117–1124 (2006). doi: 10.1038/nn1747; [DOI] [PubMed] [Google Scholar]
- 42.Lohmann C, Bonhoeffer T, A role for local calcium signaling in rapid synaptic partner selection by dendritic filopodia. Neuron 59, 253–260 (2008). doi: 10.1016/j.neuron.2008.05.025; [DOI] [PubMed] [Google Scholar]
- 43.Carrillo-Reid L, Yang W, Bando Y, Peterka DS, Yuste R, Imprinting and recalling cortical ensembles. Science 353, 691–694 (2016). doi: 10.1126/science.aaf7560; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller JE, Ayzenshtat I, Carrillo-Reid L, Yuste R, Visual stimuli recruit intrinsically generated cortical ensembles. Proc. Natl. Acad. Sci. U.S.A 111, E4053–E4061 (2014). doi: 10.1073/pnas.1406077111; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamm JP, Peterka DS, Gogos JA, Yuste R, Altered cortical ensembles in mouse models of schizophrenia. Neuron 94, 153–167.e8 (2017). doi: 10.1016/j.neuron.2017.03.019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warden MR et al. , A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 492, 428–432 (2012). doi: 10.1038/nature11617; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cryan JF, Mombereau C, Vassout A, The tail suspension test as a model for assessing antidepressant activity: Review of pharmacological and genetic studies in mice. Neurosci. Biobehav. Rev 29, 571–625 (2005). doi: 10.1016/j.neubiorev.2005.03.009; [DOI] [PubMed] [Google Scholar]
- 48.Ma X-C et al. , Long-lasting antidepressant action of ketamine, but not glycogen synthase kinase-3 inhibitor SB216763, in the chronic mild stress model of mice. PLOS ONE 8, e56053 (2013). doi: 10.1371/journal.pone.0056053; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang C et al. , R-ketamine: A rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl. Psychiatry 5, e632 (2015). doi: 10.1038/tp.2015.136; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duman RS, Aghajanian GK, Sanacora G, Krystal JH, Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med 22, 238–249 (2016). doi: 10.1038/nm.4050; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krishnan V et al. , Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131, 391–404 (2007). doi: 10.1016/j.cell.2007.09.018; [DOI] [PubMed] [Google Scholar]
- 52.Castren E, Hen R, Neuronal plasticity and antidepressant actions. Trends Neurosci. 36, 259–267 (2013). doi: 10.1016/j.tins.2012.12.010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Popoli M, Yan Z, McEwen BS, Sanacora G, The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci 13, 22–37 (2011). doi: 10.1038/nrn3138; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ota KT et al. , REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat. Med 20, 531–535 (2014). doi: 10.1038/nm.3513; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duman RS, Monteggia LM, A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127 (2006). doi: 10.1016/j.biopsych.2006.02.013; [DOI] [PubMed] [Google Scholar]
- 56.Berton O et al. , Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311, 864–868 (2006). doi: 10.1126/science.1120972; [DOI] [PubMed] [Google Scholar]
- 57.Hodes GE et al. , Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc. Natl. Acad. Sci. U.S.A 111, 16136–16141 (2014). doi: 10.1073/pnas.1415191111; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hodes GE, Kana V, Menard C, Merad M, Russo SJ, Neuroimmune mechanisms of depression. Nat. Neurosci 18, 1386–1393 (2015). doi: 10.1038/nn.4113; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miller AH, Maletic V, Raison CL, Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 65, 732–741 (2009). doi: 10.1016/j.biopsych.2008.11.029; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AFT, Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat. Neurosci 10, 376–384 (2007). doi: 10.1038/nn1846; [DOI] [PubMed] [Google Scholar]
- 61.Arnsten AFT, Stress signalling pathways that impair prefrontal cortex structure and function. Nat. Rev. Neurosci 10, 410–422 (2009). doi: 10.1038/nrn2648; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Birnbaum SG et al. , Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science 306, 882–884 (2004). doi: 10.1126/science.1100021; [DOI] [PubMed] [Google Scholar]
- 63.Joëls M, Baram TZ, The neuro-symphony of stress. Nat. Rev. Neurosci 10, 459–466 (2009). doi: 10.1038/nrn2632; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Y et al. , Correlated memory defects and hippocampal dendritic spine loss after acute stress involve corticotropin-releasing hormone signaling. Proc. Natl. Acad. Sci. U.S.A 107, 13123–13128 (2010). doi: 10.1073/pnas.1003825107; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yuen EY et al. , Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 73, 962–977 (2012). doi: 10.1016/j.neuron.2011.12.033; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yuen EY et al. , Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc. Natl. Acad. Sci. U.S.A 106, 14075–14079 (2009). doi: 10.1073/pnas.0906791106; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dias-Ferreira E et al. , Chronic stress causes frontostriatal reorganization and affects decision-making. Science 325, 621–625 (2009). doi: 10.1126/science.1171203; [DOI] [PubMed] [Google Scholar]
- 68.Chaudhury D et al. , Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 493, 532–536 (2013). doi: 10.1038/nature11713; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Friedman A, Jump-starting natural resilience reverses stress susceptibility. Science 346, 555 (2014). doi: 10.1126/science.1260781; [DOI] [PubMed] [Google Scholar]
- 70.Li B et al. , Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature 470, 535–539 (2011). doi: 10.1038/nature09742; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hikosaka O, The habenula: From stress evasion to value-based decision-making. Nat. Rev. Neurosci 11, 503–513 (2010). doi: 10.1038/nrn2866; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Proulx CD, Hikosaka O, Malinow R, Reward processing by the lateral habenula in normal and depressive behaviors. Nat. Neurosci 17, 1146–1152 (2014). doi: 10.1038/nn.3779; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lammel S et al. , Input-specific control of reward and aversion in the ventral tegmental area. Nature 491, 212–217 (2012). doi: 10.1038/nature11527; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC, Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature 487, 183–189 (2012). doi: 10.1038/nature11160; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McGirr A, LeDue J, Chan AW, Xie Y, Murphy TH, Cortical functional hyperconnectivity in a mouse model of depression and selective network effects of ketamine. Brain 140, 2210–2225 (2017). doi: 10.1093/brain/awx142; [DOI] [PubMed] [Google Scholar]
- 76.Hultman R et al. , Brain-wide electrical spatiotemporal dynamics encode depression vulnerability. Cell 173, 166–180. e14 (2018). doi: 10.1016/j.cell.2018.02.012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou T et al. , History of winning remodels thalamo-PFC circuit to reinforce social dominance. Science 357, 162–168 (2017). doi: 10.1126/science.aak9726; [DOI] [PubMed] [Google Scholar]
- 78.Wang F et al. , Bidirectional control of social hierarchy by synaptic efficacy in medial prefrontal cortex. Science 334, 693–697 (2011). doi: 10.1126/science.1209951; [DOI] [PubMed] [Google Scholar]
- 79.Bagot RC et al. , Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat. Commun 6, 7062 (2015). doi: 10.1038/ncomms8062; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liston C et al. , Circadian glucocorticoid oscillations promote learning-dependent synapse formation and maintenance. Nat. Neurosci 16, 698–705 (2013). doi: 10.1038/nn.3387; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liston C, Gan W-B, Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. Proc. Natl. Acad. Sci. U.S.A 108, 16074–16079 (2011). doi: 10.1073/pnas.1110444108; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stephan AH, Barres BA, Stevens B, The complement system: An unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci 35, 369–389 (2012). doi: 10.1146/annurev-neuro-061010-113810; [DOI] [PubMed] [Google Scholar]
- 83.Stellwagen D, Malenka RC, Synaptic scaling mediated by glial TNF-alpha. Nature 440, 1054–1059 (2006). doi: 10.1038/nature04671; [DOI] [PubMed] [Google Scholar]
- 84.Paolicelli RC et al. , Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458 (2011). doi: 10.1126/science.1202529; [DOI] [PubMed] [Google Scholar]
- 85.Schafer DP et al. , Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012). doi: 10.1016/j.neuron.2012.03.026; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wohleb ES, Franklin T, Iwata M, Duman RS, Integrating neuroimmune systems in the neurobiology of depression. Nat. Rev. Neurosci 17, 497–511 (2016). doi: 10.1038/nrn.2016.69; [DOI] [PubMed] [Google Scholar]
- 87.Hedrick NG et al. , Rho GTPase complementation underlies BDNF-dependent homo-and heterosynaptic plasticity. Nature 538, 104–108 (2016). doi: 10.1038/nature19784; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Turrigiano GG, The self-tuning neuron: Synaptic scaling of excitatory synapses. Cell 135, 422–435 (2008). doi: 10.1016/j.cell.2008.10.008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Duman RS, Aghajanian GK, Synaptic dysfunction in depression: Potential therapeutic targets. Science 338, 68–72 (2012). doi: 10.1126/science.1222939; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kavalali ET, Monteggia LM, Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am. J. Psychiatry 169, 1150–1156 (2012). doi: 10.1176/appi.ajp.2012.12040531; [DOI] [PubMed] [Google Scholar]
- 91.Russo SJ, Nestler EJ, The brain reward circuitry in mood disorders. Nat. Rev. Neurosci 14, 609–625 (2013). doi: 10.1038/nrn3381; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.LaPlant Q et al. , Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci 13, 1137–1143 (2010). doi: 10.1038/nn.2619; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mitra R, Jadhav S, McEwen BS, Vyas A, Chattarji S, Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proc. Natl. Acad. Sci. U.S.A 102, 9371–9376 (2005). doi: 10.1073/pnas.0504011102; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang Y et al. , Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 554, 317–322 (2018). doi: 10.1038/nature25509; [DOI] [PubMed] [Google Scholar]
- 95.LeGates TA et al. , Reward behaviour is regulated by the strength of hippocampus-nucleus accumbens synapses. Nature 564, 258–262 (2018). doi: 10.1038/s41586-018-0740-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.