

We report a novel pathway where FANCD2 binds to nuclear receptors, COUP-TFII/TR4, to promote alternative lengthening of telomeres.
Abstract
Alternative lengthening of telomeres (ALT) is known to use homologous recombination (HR) to replicate telomeric DNA in a telomerase-independent manner. However, the detailed process remains largely undefined. It was reported that nuclear receptors COUP-TFII and TR4 are recruited to the enriched GGGTCA variant repeats embedded within ALT telomeres, implicating nuclear receptors in regulating ALT activity. Here, we identified a function of nuclear receptors in ALT telomere maintenance that involves a direct interaction between COUP-TFII/TR4 and FANCD2, the key protein in the Fanconi anemia (FA) DNA repair pathway. The COUP-TFII/TR4-FANCD2 complex actively induces the DNA damage response by recruiting endonuclease MUS81 and promoting the loading of the PCNA-POLD3 replication complex in ALT telomeres. Furthermore, the COUP-TFII/TR4-mediated ALT telomere pathway does not require the FA core complex or the monoubiquitylation of FANCD2, key steps in the canonical FA pathway. Thus, our findings reveal that COUP-TFII/TR4 regulates ALT telomere maintenance through a novel noncanonical FANCD2 pathway.
INTRODUCTION
Cancer cells have to maintain telomere length to overcome telomere shortening–induced cell crisis. The majority of cancer cells (85 to 90%) do so by reactivating telomerase, while the rest of the small but significant number of cancer cells relies on the alternative lengthening of telomeres (ALT) pathway to elongate telomere (1, 2). So far, multiple molecular hallmarks have been identified in ALT cells, including heterogeneous telomere length, abundance of extrachromosomal telomeric repeat DNA, elevated levels of telomere recombination, and ALT-associated promyelocytic leukemia (PML) bodies (APBs), which colocalize with telomeres and contain numerous DNA repair proteins (1, 2). These phenotypic characteristics supported the model that ALT cells may induce the DNA damage response in the telomere region and activate the homologous recombination (HR) pathway to replicate telomere repeats and repair DNA damage. However, in mammalian cells, telomere repeats are typically bound by the Shelterin protein complex and form a T-loop structure, therefore protecting chromosome ends from being recognized as double-strand break and inhibiting the DNA damage response as well as HR (3). How the DNA damage response and HR can be specifically activated in ALT telomeres remains poorly understood.
COUP-TFII (NR2F2) and TR4 (NR2C2) belong to orphan nuclear receptors, both binding to the direct repeats of A/GGGTCA DNA sequence to regulate target gene transcription (4, 5). COUP-TFII has been shown to play critical roles in cell fate specification, organogenesis, angiogenesis, and metabolism as well as in a variety of diseases (6). Accumulated evidence also reveals that COUP-TFII promotes prostate cancer progression and metastasis as well as tumor angiogenesis (7–9), highlighting COUP-TFII as a potential target for cancer therapy. On the other hand, it has been reported that TR4 is involved in neuronal development, fertility, energy homeostasis, and prostate cancer progression (10, 11).
Recent reports demonstrated that COUP-TFII and TR4 are associated with telomeres specifically in ALT cells (12–14). COUP-TFII and TR4 usually bind to A/GGGTCA direct repeats but not the classical telomere repeats, GGGTTA (4, 5). However, in ALT cells, one variant telomere repeat, GGGTCA, has been enriched markedly, which provides a platform for COUP-TFII/TR4 binding to telomeres (13, 14). At present, little is known about the functions of COUP-TFII/TR4 in the ALT pathway.
FANCD2 belongs to Fanconi anemia complementation group (FANC) proteins and plays key roles in the Fanconi anemia (FA) pathway for DNA interstrand cross-linker (ICL) repair (15, 16). In response to DNA damage, eight FANC proteins (A, B, C, E, F, G, L, and M) assemble into an FA core complex that recruits and monoubiquitylates FANCD2 and FANCI. The FANCI-D2 complex then forms nuclear foci together with other DNA damage response proteins, which, in turn, remove ICL sites and induce HR to repair DNA damage (15, 16).
To further investigate the functions of COUP-TFII, we decided to screen for COUP-TFII–interacting proteins through a high-throughput bimolecular fluorescence complementation (BiFC) assay (17). We identified FANCD2 as an interacting protein of COUP-TFII, and further analysis confirmed that FANCD2 interacts with both COUP-TFII and TR4. Because ALT is an HR-dependent process, we hypothesize that FANCD2 could be recruited to the ALT telomeres through its interaction with COUP-TFII/TR4, therefore eliciting HR in the ALT telomeres. To test this hypothesis, we focused our study on the functions of COUP-TFII/TR4 and FANCD2 in the ALT pathway and uncovered a novel mechanism by which the DNA damage response and HR are induced in ALT telomeres, leading to the noncanonical pathway to telomere lengthening and maintenance.
RESULTS
FANCD2 interacts with COUP-TFII and TR4
To identify the novel interacting proteins of COUP-TFII, we performed a BiFC screen using a hORFeome-based prey library generated previously (17). In this assay, the N terminus of Venus yellow fluorescent protein (YFPN) was fused with COUP-TFII, and the C terminus of Venus YFP (YFPC) was fused with proteins in open reading frame (ORF) libraries or positive control protein, SMAD4, which was shown to interact with COUP-TFII (8). When the YFPN and YFPC fragments are in close proximity via interactions between their partners, they fold into a functional YFP protein (Fig. 1A) (17). As shown in fig. S2A, few cells with coexpression of YFPN–COUP-TFII and negative control (YFPC) displayed YFP signals, while a large number of YFP-positive cells were seen with coexpression of YFPN–COUP-TFII and positive control, SMAD4-YFPC. Using SMAD4 as a positive control, we picked up ORFs with a percentage of YFP positivity higher than SMAD4 and identified 179 COUP-TFII–interacting candidates from ~11,880 ORFs (table S1). We performed DAVID functional annotation cluster to analyze the functions of these candidates (fig. S1). As expected, the top clusters are involved in transcriptional regulation and some functions have been reported to be regulated by COUP-TFII, such as response to oxidative stress and muscle development (18–20).
Fig. 1. FANCD2 interacts with COUP-TFII and TR4.
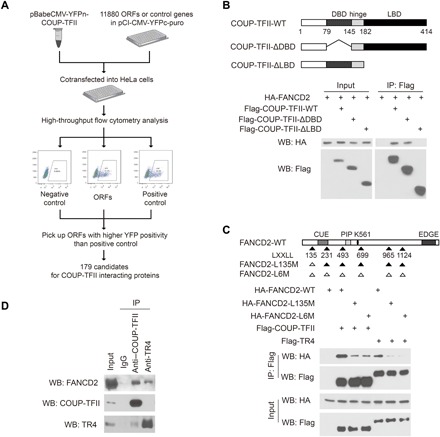
(A) Outline of BiFC screen for COUP-TFII–interacting proteins. (B) (Top) Schematic diagrams of COUP-TFII and its mutants. COUP-TFII contains the N-terminal DBD (DNA binding domain), middle hinge region, and the C-terminal LBD (ligand-binding domain). (Bottom) COUP-TFII binds to FANCD2 through its LBD. HA-FANCD2 was cotransfected with Flag-tagged COUP-TFII or its mutants into 293T cells. Lysates were subjected to IP with Flag antibody and Western blot (WB) with antibodies as indicated. (C) (Top) Schematic diagrams of FANCD2 and its mutants. FANCD2 contains six LXXLL motifs; the position of the first amino acids in each LXXLL motif is marked. Black and white triangles label LXXLL motifs and AXXAA mutants, respectively. CUE, coupling of ubiquitin conjugation to endoplasmic reticulum degradation domain; PIP, PCNA-interacting protein motif. (Bottom) LXXLL-135 motif is critical for FANCD2 binding to COUP-TFII and TR4. IP and Western blot were performed as described in (B). (D) Endogenous interactions between FANCD2 and COUP-TFII/TR4. Lysates isolated from WI38-VA13/2RA cells (2 × 107) were subjected to IP with COUP-TFII, TR4, or immunoglobulin G (IgG) antibody and analyzed by Western blot as indicated.
To our surprise, FANCD2, the key protein for DNA ICL repair, is on the list (table S1 and fig. S2A). Because the FANCD2-BiFC fragment used in the BiFC screen only contains 1 to 231 amino acids of FANCD2, we first asked whether COUP-TFII could interact with both FANCD2-BiFC and full-length FANCD2. COUP-TFII is shown to bind to FANCD2 (Fig. 1, B and C) as well as FANCD2-BiFC (fig. S2B) by co-immunoprecipitation (Co-IP) assays.
We next asked what regions of COUP-TFII are important for interacting with FANCD2. COUP-TFII consists of a DNA binding domain (DBD) in the middle and a ligand-binding domain (LBD) in the C terminus. Deletion of the LBD, but not the DBD, abolished COUP-TFII interaction with FANCD2 (Fig. 1B), suggesting that the interacting regions reside in the LBD. It has been well documented that the LBD is important for mediating interactions between nuclear receptors and other co-regulators or transcription factors by interacting with LXXLL motifs (21). FANCD2 contains six LXXLL motifs, while only one resides in the regions between amino acids 135 and 139 of the FANCD2-BiFC fragment. To test whether this LXXLL motif was critical for FANCD2 association with COUP-TFII, we generated a mutation in this motif. As shown in fig. S2B, mutation of the LXXLL-135 motif to AXXAA (L135M) eliminates the binding of FANCD2-BiFC fragment to COUP-TFII in the Co-IP assay. Mutation of this motif also markedly weakened the interactions between full-length FANCD2 and COUP-TFII (Fig. 1C). Because the FANCD2-L135M mutant still exhibits a weak interaction with COUP-TFII/TR4, we further asked whether the other five additional LXXLL motifs might also be involved in this interaction. Mutation of all five additional LXXLL motifs (FANCD2-L6M) in FANCD2-L135M did not further reduce its interaction with COUP-TFII/TR4 when compared with that of the FANCD2-L135M mutant (Fig. 1C), while FANCD2-BiFC fragment, which contains only the L135 motif, binds to COUP-TFII to a level comparable with that of wild-type FANCD2 (fig. S2C), suggesting that these five additional LXXLL motifs are unlikely involved in FANCD2 interaction with COUP-TFII/TR4. Collectively, these results indicate that the LBD of COUP-TFII and the LXXLL-135 motif of FANCD2 are critical for their interaction. To further investigate whether these interactions occur with endogenous proteins, we used Co-IP assays and illustrated that FANCD2 binds to COUP-TFII in WI38-VA13/2RA cells (Fig. 1D).
Because TR4, another member of the orphan nuclear receptor family, is also reported to be enriched in the ALT telomere (12–14), we investigated whether TR4 could also interact with FANCD2. As shown in Fig. 1C, FANCD2 associates with TR4, and this association also requires the LXXLL-135 motif of FANCD2. Moreover, endogenous binding of TR4 and FANCD2 can be detected in WI38-VA13/2RA cells (Fig. 1D). These results indicate that TR4 interacts with FANCD2 in an analogous way as COUP-TFII.
COUP-TFII and TR4 recruit FANCD2 to ALT through direct interaction
FANCD2 is a key factor in the FA pathway, where it is recruited and monoubiquitylated by the FA core complex, and further induces the DNA damage response and HR to promote ICL repair (15, 16). To ascertain whether FANCD2 can be recruited into ALT telomeres by interacting with COUP-TFII/TR4, we first checked the localization of COUP-TFII/TR4 and FANCD2 by immunofluorescence and telomere–fluorescence in situ hybridization (FISH). COUP-TFII and TR4 are colocalized with telomeres in both U2-OS (Fig. 2A) and WI38-VA13/2RA (fig. S3A) cells (ALT+), but not in HeLa cells (ALT−) (fig. S3B), a specific pattern consistent with previously published reports (12–14). Moreover, a COUP-TFII mutant lacking the DBD fails to accumulate at telomeres (fig. S3C), similar to previously published results of TR4, indicating that COUP-TFII/TR4 is recruited to telomeres through direct DNA binding. FANCD2 displays triple colocalization with COUP-TFII/TR4 and telomeres in ALT cells but not in HeLa cells (Fig. 2A and fig. S3). These colocalizations at ALT telomeres are significantly reduced (Fig. 2C and fig. S3E) upon efficient knockdown of COUP-TFII/TR4 (Fig. 2B and fig. S3D). In contrast, the FANCD2 foci outside of telomeres, which may arise from the canonical DNA damage response, remain similar upon knockdown of COUP-TFII/TR4 (Fig. 2C). These results indicate that COUP-TFII/TR4 specifically regulate the recruitment of FANCD2 to the ALT telomere.
Fig. 2. COUP-TFII/TR4 recruits FANCD2 to ALT telomeres through direct interaction.
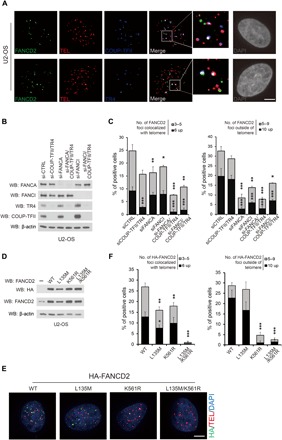
(A) FANCD2 colocalized with COUP-TFII/TR4 in ALT telomeres. FANCD2 (green) and COUP-TFII or TR4 (blue) were detected by immunofluorescence, and telomeres (red) were detected by FISH. The triple colocalized signals (arrows) are shown in white. Scale bar, 5 μm. (B and C) COUP-TFII/TR4 regulates the recruitment of FANCD2 to the ALT telomere. (B) U2-OS cells were transfected with siRNA for 72 hours. Knockdown efficiency was determined by Western blot as described. (C) Quantification of FANCD2 and telomere localization. Both FANCD2 foci that colocalized with telomeres and those not colocalized with telomeres were counted. (D to F) Colocalization of HA-FANCD2 and its mutants with ALT telomeres. (D) Inducible HA-FANCD2 and its mutants were introduced to U2-OS cells as indicated, and the expression levels were confirmed by Western blot. (E) Representative images for localization of different mutants of HA-FANCD2 and telomeres in U2-OS cells. Scale bar, 5 μm. (F) Quantification of HA-FANCD2 and telomere localization. WT, wild type. For (C) and (F), data show mean ± SD from n = 3 experiments. At least 150 cells were counted in each experiment. *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with nonparametric).
Because the FA core complex is required for the recruitment of FANCD2 to DNA damage sites (15, 16), we next tested whether it also regulates FANCD2 association with the ALT telomere. Disruption of the FA core complex by knocking down FANCA markedly decreased FANCD2 foci outside the telomere (Fig. 2C and fig. S3E), consistent with previously published reports. The FANCD2 foci that colocalized with telomeres were reduced slightly with knockdown of FANCA itself but were further reduced when both FANCA and COUP-TFII/TR4 were knocked down (Fig. 2C and fig. S3E). Consistently, depletion of FANCI also caused a significant reduction of FANCD2 signals outside of telomeres but not those associated with telomeres (Fig. 2C and fig. S3E). These results suggest that the recruitment of FANCD2 to telomeres is mainly dependent on COUP-TFII/TR4, which is different from the DNA damage response induced by the classical FA pathway.
To further investigate the mechanisms for FANCD2 recruitment to ALT telomeres, we generated inducible cell lines to overexpress wild type and mutants of FANCD2 with hemagglutinin (HA) tag (Fig. 2D) and measured the HA-FANCD2 foci using an HA-specific antibody. As expected, the FANCD2 signals outside of telomeres were abolished when a monoubiquitylation mutant, K561R, was used, confirming that monoubiquitylation of FANCD2 at K561 is critical for its recruitment to DNA damage sites (Fig. 2, E and F). However, the COUP-TFII/TR4–interacting domain mutant LXXLL-135 has no effect on FANCD2 accumulation on DNA damage sites, suggesting that COUP-TFII/TR4 is not likely to play a role in the canonical DNA damage response. Mutation of the LXXLL-135 motif greatly reduced the colocalization of FANCD2 to the telomeres in comparison to the wild type (Fig. 2, E and F), supporting the premise that the LXXLL-135 motif is required for FANCD2 association with COUP-TFII/TR4 (Fig. 1C) and its recruitment to telomeres. The K561R FANCD2 mutant, which can no longer be monoubiquitylated by the FA core complex, still exhibited a substantial colocalization albeit at a reduced level in the telomeres, and these signals were lost when both K561R and L135M were mutated in FANCD2 (Fig. 2, E and F). These findings indicate that FANCD2 can be recruited to ALT via a monoubiquitylation-independent pathway that required the interaction of FANCD2 with COUP-TFII/TR4.
The recruitment of FANCD2 to ALT telomeres by COUP-TFII/TR4 occurs mainly in G2 phase
It has been reported that the recruitment of FANCD2 by the FA core complex to DNA damage sites is cell cycle dependent, which is coupled to DNA replication in S phase (22, 23). We therefore tested whether the accumulation of FANCD2 in ALT telomeres is also cell cycle regulated. U2-OS cells were arrested in early S phase by double thymidine block and then released, and cell cycle progression and FANCD2 foci were monitored at different time points. As shown in Fig. 3 (A and B), the FANCD2 signals outside of telomeres are enriched mostly in S phase, decreased in G2 phase, and became almost undetectable in G1 phase. In contrast, the FANCD2 signals colocalizing with telomeres showed slight increase in S phase, reached a peak in G2 phase, and quickly declined to baseline in G1 phase. At the same time, the time course of FANCD2 monoubiquitylation showed that the level of monoubiquitylation of FANCD2 was very low to undetectable in G2 phase (Fig. 3B), supporting the notion that the recruitment of FANCD2 to ALT telomeres is not dependent on its monoubiquitylation.
Fig. 3. The recruitment of FANCD2 to ALT telomeres by COUP-TFII/TR4 occurs mainly in G2 phase.
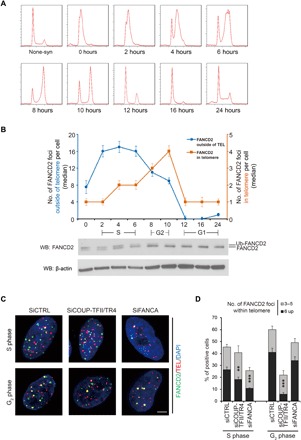
(A and B) The FANCD2 signals within the telomere and outside the telomere displayed different cell cycle–dependent patterns. (A) Cell cycle progression was determined by PI staining and flow cytometry. (B) The top graph shows the quantification of FANCD2 and telomere localization. At least 100 cells from n = 2 experiments were counted at each time point for both FANCD2 foci colocalized with telomeres (orange line) and those not colocalized with telomeres (blue line). Data show median ± SEM. Lower blots show the monoubiquitylation pattern of FANCD2 in different cell cycle stages. (C and D) COUP-TFII/TR4– and FANCA-regulated FANCD2 recruitment to ALT telomeres through different pathways. U2-OS cells were transfected with siRNAs for 48 hours, synchronized as in (A), and harvested at 4 hours (S phase) and 10 hours (G2 phase) after release. Representative images (C) and quantification (D) for localization of FANCD2 and telomeres in cells of S and G2 phases after siRNA treatment. Scale bar, 5 μm. At least 150 cells were counted in each experiment. Values are mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with nonparametric).
Moreover, results obtained from COUP-TFII/TR4 or FANCA knockdown experiments indicated that the recruitment of FANCD2 in S phase is mainly dependent on the FA core complex, while that in G2 phase is mainly dependent on COUP-TFII/TR4 (Fig. 3, C and D). These results indicate that the recruitment of FANCD2 to ALT telomeres requires the presence of COUP-TFII/TR4 and takes place mainly in G2 phase, which is distinct from the recruitment of FANCD2 to the DNA damage sites that requires the FA core complex and occurs in S phase.
Together, these results strongly suggest that there are two pathways to recruit FANCD2 to telomeres: The FA core complex–dependent pathway mediates the canonical DNA damage response during S phase; in contrast, the COUP-TFII/TR4-dependent pathway mediates noncanonical functions, such as telomere lengthening and maintenance during G2 phase.
COUP-TFII/TR4 and FANCD2 are required for telomere maintenance in ALT cells
To test whether COUP-TFII/TR4 and FANCD2 could regulate ALT activity and telomere elongation, we generated a series of knockout cells by CRISPR-Cas9. Subsequent to the introduction of Cas9 and single-guide RNA (sgRNA) targeting specifically either COUP-TFII/TR4, FANCD2, FANCA, or FANCI, a marked decrease in the expression of specific targeting protein was seen in WI38-VA13/2RA cells (Fig. 4A). As a measurement of ALT activity, we first determined the telomere sister chromatid exchange (T-SCE) rate, C-circle level, and APB formation in these knockout cell lines. As shown in Fig. 4 (B to D) and fig. S4, depletion of COUP-TFII/TR4 or FANCD2 results in significant reduction of C-circle levels (Fig. 4B), APB formation (Fig. 4C), and T-SCE rate (Fig. 4D), suggesting critical roles of these proteins in regulating ALT activity. However, knockout of FANCA or FANCI, two other components of the canonical FA pathway, had no effect on C-circle level (Fig. 4B), APB formation (Fig. 4C), or T-SCE rate (Fig. 4D), indicating that the classical FA pathway is not required for ALT induction.
Fig. 4. COUP-TFII/TR4 and FANCD2 are critical for telomere maintenance in ALT cells.
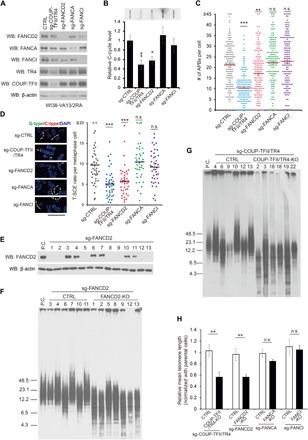
(A) Western blot denoted the knockout efficiency in WI38-VA13/2RA cells after Cas9 and sgRNA introduction. (B) C-cycle assay in different knockout cells. Values are mean ± SD from n = 3 experiments. (C) APB formation in different knockout cells. At least 150 cells from n = 3 experiments were counted for each sample. Values are mean ± SEM. n.s., not significant. (D) The T-SCE rate was measured by Co-FISH, G-strand in green and C-strand in red, and arrows point to the T-SCE signals. The T-SCE rate was calculated from the number of T-SCE/signal–positive chromosome ends for each metaphase cell. At least 40 metaphase cells from n = 3 experiments were counted for each sample. Values are mean ± SEM. Scale bar, 5 μm. For (B) to (D), *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with nonparametric). (E) Expression level of FANCD2 in single clones. P.C., parental cells. (F and G) Knockout of FANCD2 or COUP-TFII/TR4 shortened telomere length in ALT cells. The telomere length of each clone was detected by terminal restriction fragment (TRF) assay at PD ~25. (H) Quantification of relative mean telomere length in different groups of single clones. Values are mean ± SEM. *P < 0.05; **P < 0.01 (Student’s t test).
Furthermore, to elucidate the function of FANCD2 in ALT telomere maintenance, we evaluated the telomere length of FANCD2 knockout and control cells at different PDs (population doublings) by terminal restriction fragment (TRF) assay. Notably, the telomere length of FANCD2 knockout cells showed substantial shortening at PD ~25 in comparison to parental cells, while no notable changes occurred in control cells (fig. S5, B and C), indicating that FANCD2 was necessary for telomere maintenance in ALT cells. However, FANCD2 knockout cells grew much slower than wild-type cells and were quickly taken over by the wild-type cells so that we could not detect further changes in telomere length at later passages (fig. S5A). To confirm the function of FANCD2 in ALT telomere maintenance, we selected a few single clones and divided them into two groups, FANCD2-KO (FANCD2 is undetectable) and control (FANCD2 positive), and then measured the telomere length of each clone at PD ~25. When compared to parental cells, all seven FANCD2-KO clones displayed significant shortening of telomeres, with an average telomere length of about 50% of that of the parental cells (Fig. 4, E, F, and H). Although the control clones had variable telomere length, their average length was close to the parental cells, suggesting that the telomere shortening seen in the FANCD2-KO cells is not due to clonal variation. However, the telomere length of these clones did not continue to shorten over time. This is consistent with previous reports that disruption of telomere maintenance mechanisms in ALT cells resulted in a quick shortening and then stabilization of telomere length (24). Moreover, depletion of FANCD2 also shortened telomeres in U2-OS cells (fig. S6, A and B) but not in HeLa cells (fig. S6, C and D), indicating a specific effect of FANCD2 in ALT cells. Notably, telomere shortening was also observed in COUP-TFII and TR4 double-knockout cells (Fig. 4, G and H, and fig. S7A) but not in FANCA or FANCI knockout cells (Fig. 4H and fig. S7). Together, these results support the notion that COUP-TFII/TR4 and FANCD2 are required for ALT telomere maintenance, while the classical FA pathway is not involved.
FANCD2 recruits MUS81 to induce the DNA damage response and subsequently promotes the loading of POLD3 in ALT telomeres
For ICL repair, FANCD2 co-operates with multiple endonucleases, including FAN1 (25), MUS81 (26), and the SLX4 complex (XPF-ERCC1-SLX4-SLX1) (27, 28), to generate a double-strand break and subsequent removal of ICL sites, followed by recruitment of multiple DNA damage response proteins to induce HR and repair double-strand break. To test whether the recruitment of these nucleases also contributes to the FANCD2-regulated ALT pathway, we first examined the localization of these proteins in ALT cells. MUS81, FAN1, and SLX4 all displayed colocalization with telomeres in WI38-VA13/2RA cells (fig. S8A). However, the pathways leading to their recruitment appear to be quite different. The levels of MUS81 in telomeres were greatly reduced upon knockdown of COUP-TFII/TR4 or FANCD2, but not FANCA (Fig. 5A and fig. S8A), suggesting that MUS81 is recruited to ALT telomeres specifically by a COUP-TFII/TR4-FANCD2 complex, but not by the canonical FA pathway. In contrast, association of FAN1 in telomeres mainly depended on FANCD2 and FANCA, but not COUP-TFII/TR4 (Fig. 5A and fig. S8A), indicating that the recruitment of FAN1 to ALT telomeres relies on the FA pathway rather than the COUP-TFII/TR4-FANCD2 complex. COUP-TFII/TR4, FANCD2, and FANCA do not regulate the recruitment of SLX4 to telomeres, as their silencing did not affect SLX4 association with telomeres (Fig. 5A and fig. S8A).
Fig. 5. FANCD2 recruits MUS81 to induce the DNA damage response and subsequently promotes the loading of POLD3 in ALT telomeres.
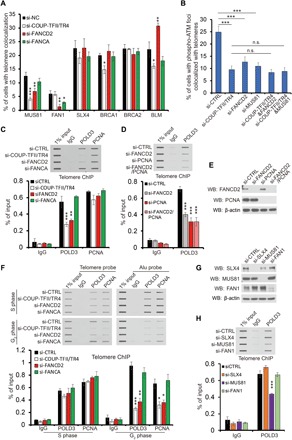
(A and B) Signals of MUS81, FAN1, SLX4, BRCA1, BRCA2, BLM, and phospho-ATM colocalized with ALT telomeres upon different siRNA treatment were quantified as indicated. Cells with three or more colocalization foci were set as positive, and at least 150 cells were counted in each experiment. Values are mean ± SD from n = 3 experiments. (C, D, and H) Telomere ChIP against POLD3 and PCNA in U2-OS cells after siRNA treatment as indicated. Knockdown efficiency was determined by Western blot in (E) and (G). (F) Telomere or Alu ChIP against POLD3 and PCNA in synchronized U2-OS cells. Cells were treated with different siRNAs and synchronized at S or G2 phase, and ChIP assay was performed as indicated. The immunoprecipitated DNA was detected by dot blot using telomere probe or Alu probe. Quantification of Alu ChIP was shown in fig. S10B. For (C), (D), (F), and (H), bar graph shows the percentage of telomere DNA pulled down in comparison with input. Data show mean ± SD from n = 3 experiments. *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with nonparametric).
To elucidate whether the recruitment of MUS81 by the COUP-TFII/TR4-FANCD2 complex affected the DNA damage response in ALT telomeres, we measured the signals for ATM Ser1981 phosphorylation (p-ATM), a surrogate for the presence of DNA double-strand break. As shown in fig. S8 (B and C), depletion of COUP-TFII/TR4, FANCD2, or MUS81, but not FAN1, FANCA, or FANCI, resulted in a significant reduction of p-ATM signals in ALT telomeres. Notably, knockdown of FANCD2 or MUS81 in COUP-TFII/TR4-depleted cells did not further reduce p-ATM accumulation in ALT telomeres (Fig. 5B), supporting the notion that there is a COUP-TFII/TR4-FANCD2-MUS81 axis that is responsible for inducing the DNA damage response in ALT telomeres. In contrast, knocking down of SLX4 resulted in an increase in p-ATM signals in telomeres (fig. S8, B and C), suggesting that SLX4 plays an opposite role and suppresses the DNA damage response in telomeres.
FANCD2 has been shown to associate with multiple HR proteins, including BRCA1 (22), BRCA2 (29), as well as BLM (30), and recruits them to promote HR for ICL repair. However, these proteins did not appear to be involved in the FANCD2-mediated ALT pathway, since knockdown of FANCD2 did not reduce the recruitment of BRCA1 or BRCA2 and even increased the signals of BLM in telomeres (Fig. 5A and fig. S8A). Depletion of COUP-TFII/TR4 resulted in a significant decrease of BRCA1 and BLM signals in telomeres (Fig. 5A and fig. S8A), suggesting that COUP-TFII/TR4 might induce different DNA repair pathways in ALT besides the FANCD2 pathway.
Recently, it has been shown that DNA double-strand breaks induce the loading of the PCNA– RFC1–Pol δ replication complex in ALT telomeres and enacts long-tract telomere synthesis (24). Because PCNA has been shown to interact with FANCD2 (31), we examined whether FANCD2 is also involved in the recruitment of the PCNA–Pol δ complex to ALT. To test this possibility, we measured the telomeric signals of POLD3, the accessory subunit of Pol δ, and PCNA by telomere chromatin IP (ChIP) assay. As shown in fig. S9, POLD3 and PCNA bind to telomere sequence more frequently in ALT(+) cells when compared with ALT(−) HeLa cells, consistent with previous report. The association of POLD3 in telomere was significantly reduced after depletion of COUP-TFII/TR4 or FANCD2 in U2-OS cells (Fig. 5C), and similar results were also observed by measuring the colocalization of POLD3 with telomeres (fig. S8D), suggesting that the loading of POLD3 requires the COUP-TFII/TR4-FANCD2 complex. Consistently, depletion of PCNA or FANCD2 resulted in similar levels of reduction of POLD3 binding in telomeres, and double knockdown of both proteins did not show a further decrease of POLD3 association, indicating that FANCD2 cooperates with PCNA to regulate the POLD3 loading in ALT telomeres (Fig. 5, D and E). However, the recruitment of PCNA to telomere does not appear to be dependent on the COUP-TFII/TR4-FANCD2 complex, as silencing of COUP-TFII/TR4 or FANCD2 had no notable effect on PCNA signals in telomeres (Fig. 5C), suggesting a complicated mechanism underlying the regulation of the PCNA-POLD3 complex loading to telomeres.
To address this puzzle, we asked whether COUP-TFII/TR4-FANCD2–dependent recruitment of PCNA may be overwhelmed by the recruitment of PCNA during DNA synthesis in S phase. We have shown earlier that the recruitment of FANCD2 by COUP-TFII/TR4 occurs mainly in G2 phase; therefore, it is possible that FANCD2 regulation of the PCNA-POLD3 complex loading to telomeres occurs in a cell cycle–dependent manner. To address this question, we determined the binding of POLD3 and PCNA to the telomere sequence as well as the Alu sequence (as a control for nontelomere region) by ChIP assay using synchronized U2-OS cells. As expected, POLD3 and PCNA associate with both telomere and Alu sequences in S phase (Fig. 5F), as part of regular genomic DNA replication before cell division, and this association is not significantly altered upon COUP-TFII/TR4, FANCD2, or FANCA knockdown (Fig. 5F and fig. S10B). However, in G2 phase, the binding of POLD3 and PCNA to telomeres is comparable with that in S phase, whereas their binding to Alu repeats was markedly reduced, indicating a specific recruitment of the POLD3-PCNA complex to telomeres in G2 phase. This G2 phase–specific recruitment was dependent on the COUP-TFII/TR4-FANCD2 complex, as depletion of COUP-TFII/TR4 or FANCD2 results in a significant reduction of both POLD3 and PCNA binding to telomeres. In contrast, knockdown of FANCA has no effect, indicating that recruitment of both PCNA and POLD3 requires COUP-TFII/TR4-FANCD2 for telomere lengthening. We also measured cell cycle progression in synchronized cells. Knockdown of COUP-TFII/TR4, FANCD2, or FANCA did not affect cell cycle progression (fig. S10A), thus excluding the possibility that the reduction of POLD3 and PCNA binding to telomere is due to changes in cell cycle progression. Because it was shown previously that DNA double-strand breaks induce the loading of the PCNA–RFC1–Pol δ replication complex to ALT telomeres, we further tested whether the endonuclease MUS81, SLX4, or FAN1 is also involved in regulating the association of POLD3 with telomeres. Binding of POLD3 to ALT telomeres was reduced with knockdown of MUS81, but not knockdown of SLX4 or FAN1 (Fig. 5, G and H), consistent with our observation that the COUP-TFII/TR4-FANCD2-MUS81 axis is required for the induction of the DNA damage response in ALT telomeres in G2 phase. Together, these results reveal a novel pathway in which the COUP-TFII/TR4-FANCD2 complex recruits MUS81 to generate double-strand break in ALT telomeres and subsequently promotes the loading of POLD3 to initiate telomere replication in cooperation with PCNA.
Monoubiquitylation of FANCD2 is not required for its function in the ALT pathway
The monoubiquitylation of FANCD2 at the K561 site by the FA core complex is an essential step for ICL repair, which allows multiple DNA damage response proteins to associate with FANCD2 through their ubiquitin-binding domain to promote DNA repair processes (15, 16). As shown earlier, recruitment of FANCD2 to telomeres is relatively normal even with a mutant defective in monoubiquitylation (K561R), raising the possibility that monoubiquitylation of FANCD2 may not be important in the DNA repair process in the ALT pathway (Fig. 2, E and F). To further investigate this hypothesis, we generated an inducible system to express different types of CRISPR-Cas9–resistant FANCD2 mutants, introduced them into WI38-VA13/2RA cells, and subsequently knocked out endogenous FANCD2. As shown in Fig. 6A, the expression levels of different FANCD2 mutants are similar as compared to endogenous FANCD2. Consistent with that observed in FANCD2 knockdown cells, knockout of FANCD2 resulted in a marked decrease of MUS81, p-ATM, and POLD3 signals in telomeres (Fig. 6, B and C). Reintroduction of wild-type FANCD2 into the knockout cells largely restored the recruitment of MUS81, p-ATM, and POLD3 to telomeres (Fig. 6, B and C). The K561R mutant was able to restore the telomeric signals of MUS81, p-ATM, and POLD3 in ALT cells to a similar extent as wild-type FANCD2 (Fig. 6, B and C), suggesting that monoubiquitylation of FANCD2 at K561 is not important for these processes (Fig. 6, B and C). FANCD2 with K561R mutation also restored the reduction of the T-SCE rate in FANCD2 knockout cells, indicating that FANCD2 could promote HR in ALT telomeres through a monoubiquitylation-independent pathway. In contrast, mutation of the LXXLL-135 motif abolished the ability of FANCD2 to complement for the loss of the recruitment of MUS81, p-ATM, and POLD3, as well as the reduction of the T-SCE rate in ALT cells (Fig. 6, B to D), thus highlighting the importance of COUP-TFII/TR4-FANCD2 interaction for regulating the DNA damage response in ALT telomeres. We also measured the time course of telomere changes in these cell lines. As shown in Fig. 6 (E and F), reintroduction of wild-type FANCD2 rescued telomere shortening caused by FANCD2-KO through 25 PDs. Unfortunately, the cells harboring FANCD2-K561R or FANCD2-L135M are rather unstable and could not be cultured long enough to test their ability to rescue telomere shortening. Together, these results indicate that the function of FANCD2 in ALT telomeres depends on its interactions with COUP-TFII/TR4, but not its status of monoubiquitylation.
Fig. 6. Monoubiquitylation of FANCD2 is not required for its function in the ALT pathway.
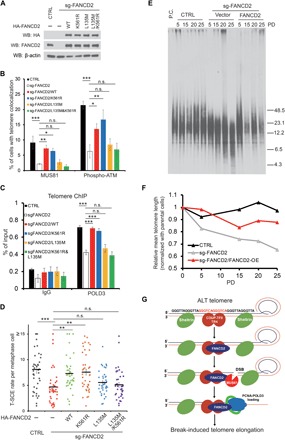
(A) Western blot confirmed the reintroduction of HA-FANCD2 and its mutants in FANCD2-KO WI38-VA13/2RA cells. (B) Quantification of MUS81 and p-ATM signals colocalized with telomeres in different cells as indicated. Cells with three or more colocalization foci were set as positive. At least 150 cells were counted in each experiment. Data show mean ± SD from n = 3 experiments. (C) Telomere ChIP against POLD3 in different cells as indicated. Bar graph, percentage of telomeric DNA pulled down. Data show mean ± SD from n = 3 experiments. (D) Quantification of T-SCE rate in different cells as indicated. At least 40 metaphase cells from n = 3 experiments were counted for each sample. Values are mean ± SEM. (E) Time course of telomere length change in different cells as indicated. (F) Quantification of data in (E). (G) Model for ALT induced by the COUP-TFII/TR4-FANCD2 complex (see text). *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with nonparametric).
DISCUSSION
ALT is a telomerase-independent pathway for telomere maintenance, which is frequently activated in sarcoma, astrocytoma, glioblastoma, and pancreatic neuroendocrine tumors (32–34). Although the mechanisms underlying the ALT pathway remain unclear, accumulating evidence supports the model that ALT cells can elicit the DNA damage response in the telomere region and activate break-induced replication or rolling cycle amplification to elongate telomeres (1, 2). However, the DNA damage response was typically inhibited by the Shelterin complex and specific structure in telomeres (3), suggesting that ALT telomeres must possess a unique but yet unknown feature to avert the protections of the Shelterin complex. In support of this notion, the absence of ATRX and/or DAXX proteins as well as the presence of heterozygous mutation of histone H3.3 and H3.1 genes were frequently found in ALT cells (33, 34). These mutations might disrupt the incorporation and modification of H3.3 in telomeres, resulting in the reduction of telomere compaction, thus rendering it more susceptible to DNA damage signals. In addition, GGGTCA variant repeats have been shown to be enriched in telomeres of ALT cells. These repeats provide the binding sites for orphan nuclear receptors COUP-TFII and TR4, which, in turn, recruit other downstream DNA remodeling factors to ALT telomeres (13, 14, 35). COUP-TFII/TR4 may compete with the Shelterin complex in binding to telomeres, therefore disrupting the inhibition of the DNA damage response in ALT telomeres. Aside from the aforementioned possibilities, there are no clear mechanisms for how the DNA damage response is induced in ALT telomeres. Here, we propose a novel pathway wherein COUP-TFII/TR4 recruits FANCD2 to ALT telomeres and actively induces the DNA damage response for ALT telomere maintenance (Fig. 6G).
FANCD2 serves as a central player in the FA pathway, which is required for DNA ICL repair in concert with DNA replication (15, 16). The FA core complex recruits and monoubiquitylates FANCD2 as well as FANCI in S phase, and then the FANCI-D2 complex recruits downstream DNA damage response proteins to remove the ICL sites and generate double-strand breaks, and further promotes HR-directed repair (15, 16). In ALT cells, abundant FANCD2 signals are found outside of telomeres during S phase, and this localization is dependent on the FA core complex. In contrast, the recruitment of FANCD2 to telomeres in ALT cells occurs mainly in G2 phase, a process that is dependent on COUP-TFII/TR4 but not the FA core complex, suggesting that this recruitment is not induced by regular DNA damage signals. Our data showed that COUP-TFII/TR4 and FANCD2 are required for telomere maintenance in ALT cells, while FANCA and FANCI are dispensable. Although the monoubiquitylation of FANCD2 by the FA core complex has been shown to play important roles for FANCD2 accumulation in DNA damage sites and recruitment of downstream DNA damage response proteins, unexpectedly, this modification is dispensable for FANCD2 recruitment and induction of the DNA damage response in the ALT telomere. Together, our results highlight the importance of interaction of COUP-TFII/TR4-FANCD2 in ALT telomeres and how ALT cells use these interactions to create a novel noncanonical process to elicit the DNA damage response for telomere maintenance, instead of the conventional DNA damage response induced by the FA pathway.
MUS81, together with FAN1 and the SLX4 complex (XPF-ERCC1-SLX1-SLX4), serves as the downstream endonucleases of the FA pathway, which incise and generate double-strand break to remove ICL sites (25, 26, 36, 37). The recruitment of the FAN1 and SLX4 complex to the ICL sites is mediated by their interactions with the monoubiquitylated FANCI-D2 complex (25, 27), while the detailed mechanisms underlying MUS81 accumulation at ICL sites remain unclear. It has been shown that MUS81 colocalized with ALT telomeres and enhanced the T-SCE rate in ALT cells (38). Our results further uncovered a novel process by which MUS81 is recruited to ALT telomeres by the COUP-TFII/TR4-FANCD2 complex and subsequently induces double-strand breaks in ALT telomeres. This process does not require monoubiquitylation of FANCD2 or the presence of FANCA and FANCI and is distinct from the conventional repair process elicited by the FA pathway. Moreover, our data also revealed that SLX4 plays an opposite role in inducing breaks in ALT telomeres, and its recruitment is not dependent on the COUP-TFII/TR4-FANCD2 complex. Our findings are consistent with previous report that SLX4 could be recruited to telomeres through interaction with TRF2 in both ALT(+) and ALT(−) cells, where it suppresses the DNA damage response and protects telomere stability (39). MUS81 is a structure-specific endonuclease and prefers to incise the 3′-flap or replication fork but not regular double-strand DNA (40). Thus, some intrinsic-specific structures might be present in ALT telomeres to facilitate incision by MUS81. Recently, it was shown that COUP-TFII/TR4 binding to ALT telomeres results in the formation of bridges between their target loci, which induce telomere clustering and relocalization (14). This raises an interesting question of whether FANCD2 and MUS81 are involved in the resolution of these bridges so that the DNA damage response in ALT telomeres can be initiated.
For ICL repair, FANCD2 typically associates with a series of HR proteins, such as BRCA1, BRCA2, and BLM, to promote RAD51-mediated HR (22, 29, 30). However, depletion of FANCD2 did not affect the recruitment of these proteins in ALT telomeres, indicating that the function of FANCD2 for ALT telomere maintenance was not dependent on such a canonical HR pathway. Recent report showed that DNA double-strand breaks induce the loading of the PCNA-RFC1-POLD3 replication complex in ALT telomeres to activate long-tract telomere synthesis (24). Here, our results revealed that the PCNA-POLD3 replication complex could be specifically recruited to ALT telomeres in G2 phase, and this recruitment depends on the COUP-TFII/TR4-FANCD2 complex. Furthermore, MUS81, the downstream endonuclease of the COUP-TFII/TR4-FANCD2 complex, is also required for POLD3 binding to telomeres, thus strongly supporting the notion that POLD3-mediated telomere replication is dependent on the COUP-TFII/TR4-FANCD2-MUS81 axis–induced DNA damage response.
Previous studies suggest a complicated role of FANCD2 in the ALT pathway. Root et al. (41) showed that knockdown of FANCD2 by small interfering RNA (siRNA) increases the telomeric DNA content and T-SCE rate in GM847 cells. However, Fan et al. (42) reported that depletion of FANCD2 results in the decreased T-SCE rate but no change in telomeric DNA content in U2-OS cells. Our studies showed that FANCD2 can be recruited to ALT telomeres through two different pathways: One pathway uses the FA core complex, is induced by DNA damage signals, and occurs primarily in S phase; the other pathway depends on COUP-TFII/TR4, is associated with GGGTCA telomeric repeats, and occurs mainly in G2 phase. Although our data showed that the association of FANCD2 with telomeres is mainly dependent on COUP-TFII/TR4 in U2-OS and WI38-VA13/2RA cells, it is possible that, in other ALT cell lines with fewer GGGTCA repeats in telomeres, the recruitment of FANCD2 may rely on the FA core complex. Whether these two different types of recruitment result in different functions of FANCD2 in the ALT pathway remains to be determined. Another interesting question is what is the physiological function of the interaction between nuclear receptors and FANCD2? Why would nature evolve such an interaction specifically for ALT cancer? COUP-TFII/TR4 interacts with FANCD2 through binding its LXXLL motif, which is a motif used in the association between nuclear receptors and co-regulators. Notably, FANCD2 has been shown to bind to the promoter region of the TAp63 gene and regulate its transcription (43), implicating a potential role of FANCD2 in transcriptional regulation in cooperation with nuclear receptors aside from the ALT pathway.
Together, our results highlight a novel noncanonical pathway for ALT telomere maintenance, where the COUP-TFII/TR4-FANCD2 complex actively elicits DNA double-strand breaks by recruiting endonuclease MUS81 and subsequently promotes the loading of the PCNA-POLD3 replication complex to mediate break-induced telomere elongation. This ALT telomere elongation pathway is distinct from DNA damage induced by the canonical FA pathway. Given that COUP-TFII and TR4 are nuclear receptors, they are excellent drug targets, as their activity can be regulated by ligands, either agonists or antagonists, which bind to their LBDs. Thus, it is feasible to find small molecules that inhibit the activities of COUP-TFII and TR4 for potential treatment of these ALT cancers.
MATERIALS AND METHODS
Cell culture
293T and HeLa cells were obtained from the Tissue Culture Core in Baylor College of Medicine and cultured in Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin. U2-OS cells were a gift from P. Zhang (Baylor College of Medicine) and cultured in McCoy’s 5A, 10% FBS, and 1% penicillin/streptomycin. WI38-VA13/2RA cells were obtained from the American Type Culture Collection and cultured in MEM, 10% FBS, and 1% penicillin/streptomycin. All cell lines were confirmed to be free of mycoplasma species by the Tissue Culture Core in Baylor College of Medicine.
Antibodies
The following primary antibodies were used in this study: anti-Flag (mouse, Sigma F1804), anti-HA [rabbit, Cell Signaling Technology (CST), 3724], anti–COUP-TFII (mouse, R&D Systems, PP-7174-00; rabbit, CST, 6434), anti-TR4 (mouse, R&D Systems, PP-0107B-00), anti-FANCD2 (rabbit, GeneTex, GTX-30142), anti-FANCA (mouse, EMD Millipore, MABC557), anti-FANCI (rabbit, Bethyl Laboratories, A301-254A), anti-MUS81 (mouse, Abcam, ab14387), anti-SLX4 (rabbit, Bethyl Laboratories, A302-270A), anti–p-ATM (Ser1981) (mouse, EMD Millipore, 05-740), anti-PCNA (mouse, Santa Cruz Biotechnology, sc-56), anti-POLD3 (rabbit, Bethyl Laboratories, A301-243A), and anti–β-actin (goat, Santa Cruz Biotechnology, sc-1616). The anti-FAN1 antibody was a gift from J. Huang (Zhejiang University, China). The horseradish peroxidase (HRP)–conjugated secondary antibodies (CST) were used for Western blot, and the fluorophore-conjugated secondary antibodies (Invitrogen) were used for indirect immunofluorescence analyses.
Plasmids
For YFPN–COUP-TFII and SMAD4-YFPC, COUP-TFII and SMAD4 were first cloned into pENTR1A (Invitrogen) and then cloned into pBabeCMV-YFPn-DEST and pCL-CMV-DEST-YFPc using Gateway (Invitrogen), respectively. For HA-tagged FANCD2 and its mutants, Flag-tagged COUP-TFII and its mutants, and Flag-TR4 were cloned into pcDNA3.1 by polymerase chain reaction (PCR). For inducible HA-FANCD2, CRISPR-Cas9–resistant FANCD2 and their mutants were cloned into pENTR1A first and then cloned into pInducer20 (Addgene #44012) by Gateway. For sgRNA expression, DNA oligos were synthesized in Sigma and cloned into lentiGuide-puro (Addgene #52963).
Bimolecular fluorescence complementation assay
The ORF library for BiFC was generated as described previously (17). HeLa cells were seeded in 96-well plates, and YFPN–COUP-TFII was cotransfected individually with YFPc-ORF or either negative control (YFPc) or positive control (SMAD4-YFPc) to each well using Biomek 3000 Laboratory Automation Workstation (Beckman Coulter). After 24 hours, cells were harvested for high-throughput flow cytometric analysis using an LSRII flow cytometer equipped with a HTS sampler (BD Biosciences). The percentage of YFP-positive cells (Score) was counted for each well, and ORFs with Score higher than positive control in the same plate were considered as candidates for COUP-TFII–interacting proteins.
IP and Western blot analysis
Cells were incubated in lysis buffer [20 mM Hepes (pH 7.9), 150 mM NaCl, 1 mM EDTA, 5% glycerol, 0.5% NP-40, and protease inhibitor] at 4°C for 30 min and centrifuged at 16,000 rpm for 15 min. The lysates were precleared by protein A/G beads (Santa Cruz Biotechnology) for 1 hour, reacted with appropriate antibody for 1 hour, and incubated with protein A/G beads overnight at 4°C. After extensive washes, immunoprecipitated proteins were eluted in SDS sample loading buffer (Bio-Rad Laboratories), separated by SDS–polyacrylamide gel electrophoresis, transferred to nitrocellulose (Bio-Rad Laboratories), and detected in Western blots with appropriate primary antibodies coupled with HRP-conjugated secondary antibody by chemiluminescence (Thermo Fisher Scientific).
RNA interference
Cells were reverse-transfected using Lipofectamine RNAiMAX (Life Technologies) with an siRNA concentration of 25 μM per the manufacturer’s instructions. The following siRNAs, synthesized by Sigma unless otherwise indicated, were used in this study: si-COUP-TFII, 5′-GUACCUGUCCGGAUAUAUU-dTdT-3′; si-TR4, AM51333 (Fisher Scientific); si-FANCA, 5′-GCAGGUCACGGUUGAUGUA-dTdT-3′; si-FANCD2, 5′-CAACAUACCUCGACUCAUU-dTdT-3′; si-FANCI, 5′-GCAGAAAGAAAUAGCGUCU-dTdT-3′; si-PCNA, 5′-GGAGGAAGCUGUUACCAUAUU-dTdT-3′; si-MUS81, 5′-GAGUUGGUACUGGAUCACAUU-dTdT-3′; si-SLX4, 5′-AAACGUGAAUGAAGCAGAA-dTdT-3′; si-FAN1, 5′-AAACCGUACUUGAGAAUGA-dTdT-3′; and negative control siRNA, Universal Negative control #1 (Sigma SIC001).
Immunofluorescence-FISH
Cells were cultured on coverslips for 24 to 48 hours, preextracted with permeabilization solution [20 mM Hepes (pH 7.9), 20 mM NaCl, 5 mM MgCl2, 300 mM sucrose, and 0.5% NP-40] for 10 min, and fixed in 4% paraformaldehyde for 10 min at room temperature. Coverslips were blocked with PBG buffer [1× phosphate-buffered saline (PBS) with 0.5% bovine serum albumin (BSA) and 0.2% gelatin] for 30 min at room temperature and incubated with primary antibody in PBG overnight at 4°C. After washing with PBST (1× PBS with 0.1% Tween 20), the coverslips were incubated with secondary antibody in PBG for 1 hour at room temperature. For FISH, the samples were refixed with 4% paraformaldehyde for 10 min and treated with 10 mM glycine for 30 min at room temperature. After washing with PBS, coverslips were denatured and incubated with TelG-Cy3 peptide nucleic acid (PNA) probe (PANAGENE F-1006) in hybridization buffer [70% deionized formamide, 10 mM tris (pH 7.4), and 0.5% Roche blocking solution] for 2 hours at room temperature, stained with 4′,6-diamidino-2-phenylindole (DAPI), and mounted with Dako fluorescent mounting medium. High-resolution imaging was performed on a GE Healthcare DVLive epifluorescence image restoration microscope using an Olympus PlanApo 60× or 40× [1.42 numerical aperture (NA)] objective and a 1.9k × 1.9k sCMOS (scientific complementary metal-oxide semiconductor) camera. Z stacks (0.3 μm) covering the whole nucleus (~5 μm) were acquired before applying a conservative restorative algorithm for quantitative image deconvolution. Maximum intensity projections were generated and used for image analysis.
Double thymidine block and cell cycle analysis
Cells at 25 to 30 % confluency were treated with 2 mM thymidine for 18 hours, washed, and replenished with fresh medium. After 10 hours, cells were treated with 2 mM thymidine again for 16 hours, replaced with new medium, and analyzed at different time points. Cell cycle progression was determined by propidium iodide (PI) staining. Cells were fixed in 70% ethanol and incubated with PI (50 μg/ml) plus ribonuclease (RNase) A (10 μg/ml) in PBS for 30 min at 37°C, followed by incubation overnight at 4°C. Stained cells were analyzed by flow cytometry.
Generation of knockout and rescue cell lines by CRISPR-Cas9
Cells were first transduced with lentiCas9-Blast, selected with blasticidin (Gibco) for 10 days, transduced with lentiGuide-puro-sgRNA, and selected by puromycin (Gibco) for 5 days. To generate single clones, cells were seeded with five cells per well into a 96-well plate. Single clones were picked up and cultured to 2 × 106 cells (PD ~25) and harvested for Western blot and TRF analysis. For rescue expression in the knockout cell lines, pInducer20 with different types of CRISPR-Cas9–resistant FANCD2 were introduced to cells before lentiGuide-puro. Cells were selected with geneticin (Gibco) for 10 days, introduced to lentiGuide-puro-sgRNA, and selected by puromycin (Gibco) for 5 days. Cells were further treated with doxycycline for 2 days to induce FANCD2 expression and harvested for Western blot, immunofluorescence-FISH, and T-SCE analysis. The following CRISPR sgRNA sequences were used in this study: sg-COUP-TFII, 5′-TATATCCGGACAGGTACGAG-3′; sg-TR4, 5′-CTGCTGTTTGGGGGATCCGG-3′; sg-FANCD2, 5′-AGTTGACTGACAATGAGTCG-3′; sg-FANCA, 5′-CCGGCTGAGAGAATACCCAC-3′; and sg-FANCI, 5′-ACTAGATAAGCAGCACAATG-3′.
Chromosome orientation telomere-FISH
Cells were cultured in fresh medium supplemented with 20 μM bromodeoxyuridine (BrdU)/bromodeoxycytidine (BrdC) (3:1) for 16 to 24 hours (less than one cell cycle). Cell cultures were treated with colcemid (0.2 mg/ml) (Roche) for the last 1 to 2 hours, allowing accumulation of mitotic cells. Cells were harvested by trypsinization and incubated in 0.075 M KCI for 15 to 30 min at 37°C, fixed in cold fixative (3:1 methanol/acetic acid), spun at 1000 rpm for 5 min, and resuspended in fixative (the volume may vary depending on cell number). Concentrated cells in fixative were then dropped onto cold, wet microscope slides and washed with fixative. The slides were placed on a humidified 42°C heating block for 1 min and left to dry overnight. Next, slides with metaphase spread were treated with RNase A (0.5 mg/ml) for 10 min at 37°C, postfixed with 4% paraformaldehyde for 10 min at room temperature, and stained with Hoechst 33258 (0.5 μg/ml) (Sigma-Aldrich) in 2× SSC for 15 min at room temperature. Slides were then flooded with 80 μl of 2× SSC under coverslip and exposed to long-wave (365-nm) ultraviolet (UV) light (CL-1000 Ultraviolet Crosslinker) for 30 min. The BrdU/BrdC-substituted DNA strands were then digested in Exonuclease III solution (10 U/μl) (Promega) in buffer supplied by the manufacturer for 30 min at 37°C. Slides were rinsed in water and allowed to air dry. Subsequently, the slides were denatured and hybridized with 0.1 μM TelG-Cy3 PNA probe (PANAGENE F-1006) for 2 hours at room temperature, washed in Hybridization Wash I [10 mM tris-HCl (pH 7.2), 70% formamide, and 0.1% BSA] for 15 min, and hybridized again with 0.1 μM TelC-FITC PNA probe (PANAGENE F-1009) for 2 hours at room temperature. Last, slides were washed in Hybridization Wash I and II [0.1 M tris-HCl (pH 7.2), 0.15 M NaCl, and 0.08% Tween 20], stained with DAPI, and mounted with Dako fluorescent mounting medium. The images were captured on the same system as immunofluorescence-FISH, except using PlanApo 100× (1.42 NA) objective. Z stacks (0.3 μm) covering ~3 μm were acquired before applying a conservative restorative algorithm for quantitative image deconvolution. Maximum intensity projections were generated and used for image analysis.
C-circle assay
Genomic DNA was extracted with the DNA Blood Kit (Qiagen) according to the manufacturer’s instructions and eluted in 10 mM tris (pH 7.8). A total of 16 ng of DNA was incubated in master mix containing BSA (0.2 mg/ml); 0.1% Tween 20; 1 mM each of deoxyadenosine triphosphate, deoxycytidine triphosphate, deoxyguanosine triphosphate, and deoxythymidine triphosphate; 4 μM dithiothreitol; 1× φ29 buffer; and 7.5 U of φ29 DNA polymerase in a final volume of 20 μl at 30°C for 8 hours and then at 65°C for 20 min. Each assay was performed with φ29 DNA polymerase, and a negative control was performed without the φ29 DNA polymerase. A slot blot analysis with (TAACCC)6-Biotin probe in a nondenatured condition was carried out to quantify each C-circle assay. The results were analyzed by ImageJ.
TRF analysis
For genomic DNA extract, cells (5 × 105 to 10 × 105) were suspended in 400 μl of lysis buffer [10 mM tris-HCl (pH 7.5), 10 mM EDTA, 10 mM NaCl, 0.5% N-lauroylsarcosine, and proteinase K (1 mg/ml)] and incubated overnight at 56°C. Lysates were mixed with 75 mM NaCl in ethanol (800 μl) and incubated at room temperature until the precipitated DNA was clearly visible. After washing with 70% ethanol twice, genomic DNA was resuspended in 80 μl of CutSmart buffer with 40 U of Hin fI and 40 U of Rsa I (New England Biolabs) and incubated overnight at 37°C. The digested DNA was ethanol-precipitated again and quantified. DNA (1 to 5 μg) was run on a 1% PFGE (pulsed-field gel electrophoresis) agarose gel in 0.5× TBE (tris–boric acid–EDTA) buffer using the CHEF-DR II System (Bio-Rad Laboratories) at 6 V/cm (initial switch time, 5 s; final switch time, 5 s) for 20 hours at 14°C. DNA was transferred to Nylon membranes (Whatman), hybridized with (TAACCC)6-Biotin probe in ULTRAhyb buffer (Invitrogen) overnight at 42°C, and detected by Chemiluminescent Nucleic Acid Detection Module (Thermo Fisher Scientific). The results were analyzed by TeloTool.
Telomere ChIP
Cells were harvested by trypsinization and washed in PBS, and 1 × 107 cells were resuspended in 1 ml of cell lysis buffer [5 mM Hepes-KOH (pH 8.0), 85 mM KCl, 0.4% (v/v) NP-40, 1× cocktail protease inhibitor, and 1 mM phenylmethylsulfonyl fluoride (PMSF)]. Cells were incubated on ice for 10 min and centrifuged at 6000g for 15 s at 4°C to pellet nuclei. After removal of the supernatant, nuclei were fixed at room temperature in the same volume of cell lysis buffer lacking NP-40 and containing 1% (v/v) formaldehyde. Fixed nuclei were incubated at room temperature for 10 min with mixing once by inversion during fixation. Cross-linking was quenched by addition of 75 μl of 1.5 M glycine and incubation for 5 min at room temperature, with two inversions during incubation. The mixture was centrifuged again at 6000g for 15 s at 4°C, and the supernatant was discarded. To the nuclei, 50 μl of nuclei lysis buffer [50 mM tris-HCl (pH 8.0), 1 mM EDTA, 1% (v/v) SDS, 1× CPI (cocktail protease inhibitor), and 1 mM PMSF] was added and incubated for 10 min on ice. The sample was then diluted by the addition of 750 μl of ice-cold buffer A [20 mM Hepes-KOH (pH 8.0), 2 mM MgCl2, 300 mM KCl, 1 mM EDTA, 10% (v/v) glycerol, and 0.1% (v/v) Triton X-100]. Chromatin was fragmented with Branson Digital Sonifier D250 at 70% duty cycle and level 3 output control for 10 × 30-s pulse with 60-s pause and was centrifuged at 14,000g for 10 min at 4°C to remove the solubilized chromatin within the supernatant. For IP, 200 μl of chromatin was added to 400 μl of ice-cold IP buffer [20 mM Hepes-KOH (pH 8.0), 150 mM KCl, 1.5 mM EDTA, 1% (v/v) Triton X-100, 1× CPI, and 1 mM PMSF] on ice, and antibody (5 and 10 μg per IP) was added as required. After 1 hour of rotation at 4°C, 30 μl of protein A/G beads (Santa Cruz Biotechnology) was added, and samples were rotated at 4°C overnight. The following day, beads were collected via centrifugation of samples for 30 s at 12,000g at room temperature, washed twice with 1 ml of room temperature buffer A and subsequently with 1 ml of TE [10 mM tris-HCl and 1 mM EDTA (pH 8.0)], and then centrifuged for 30 s at 12,000g at room temperature. Immunoprecipitated DNA was eluted from the protein A/G agarose beads by digestion with 200 μg of proteinase K in 100 μl of elution buffer [50 mM NaHCO3 and 1% (v/v) SDS], incubated at 56°C for 1 hour, purified with a QIAquick PCR Purification Kit (Qiagen), and eluted in TE. The purified DNA was dot-blotted on Bio-Dot SF (Bio-Rad Laboratories) according to the manufacturer’s protocol and detected by Southern blot using (TAACCC)6-Biotin probe or Alu probe, TGGCTCACGCCTGTAATCCCAGCACTTTGGGAGGCCGA-Biotin. The amount of DNA immunoprecipitated was normalized for each sample on the basis of the amount of input telomeric DNA, as determined by signal intensity from standard curves for each sample. The results were analyzed by ImageJ.
Statistical analyses
GraphPad Prism 6 was used to perform statistical analysis with nonparametric one-way analysis of variance (ANOVA) or two-tailed Student’s t tests as described in figure legends.
Supplementary Material
Acknowledgments
We thank L.-y. Yu-Lee and J. Hebert for editorial assistance and thank J. Huang (ZJU) for the gift of the FANCD2 construct and FAN1 antibody. Funding: The work was supported by grants from NIH HL114539 and DK059820, CPRIT RP150648 (to S.Y.T. and M.-J.T.), DK45641 (to M.-J.T.), and NCI CA211653, CPRIT RP160462, and the Welch Foundation Q-1673 (to Z.S.). We would also like to acknowledge the support of the C-BASS Shared Resource (P30CA125123), the Integrated Microscopy Core [NIH (DK56338 and CA125123) and CPRIT (RP150578)] for imaging analysis, and the Cytometry and Cell Sorting Core (NCI-CA125123) for flow cytometry at Baylor College of Medicine. Author contributions: M.X., M.-J.T., and S.Y.T. conceived and designed the experimental approaches and prepared the manuscript. M.X. performed most of the experiments. J.Q., L.W., and H.-J.L. performed a subset of experiments. C.-Y.K. contributed to the statistical analysis. D.L. contributed to the BiFC screen. Z.S. and J.C. participated in some data analysis and experimental design. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax6366/DC1
Fig. S1. DAVID functional annotation cluster analysis for COUP-TFII–interacting candidates.
Fig. S2. Identification of FANCD2 as a candidate COUP-TFII–interacting protein through BiFC screen.
Fig. S3. Related to Fig. 2.
Fig. S4. Related to Fig. 4C.
Fig. S5. Depletion of FANCD2 shortens telomere length in ALT cells.
Fig. S6. Knockout of FANCD2 or COUP-TFII/TR4 shortened telomere length in U2-OS cells but not in HeLa cells.
Fig. S7. Related to Fig. 4H.
Fig. S8. Related to Fig. 5.
Fig. S9. Telomere ChIP against POLD3 and PCNA in HeLa, U2-OS, and WI38-VA13/2RA cells.
Fig. S10. Related to Fig. 5F.
Table S1. Candidate COUP-TFII–interacting proteins.
REFERENCES AND NOTES
- 1.Shay J. W., Reddel R. R., Wright W. E., Cancer. Cancer and telomeres—An ALTernative to telomerase. Science 336, 1388–1390 (2012). [DOI] [PubMed] [Google Scholar]
- 2.O’Sullivan R. J., Almouzni G., Assembly of telomeric chromatin to create ALTernative endings. Trends Cell Biol. 24, 675–685 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Palm W., de Lange T., How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Mangelsdorf D. J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P., Evans R. M., The nuclear receptor superfamily: The second decade. Cell 83, 835–839 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooney A. J., Tsai S. Y., O’Malley B. W., Tsai M. J., Chicken ovalbumin upstream promoter transcription factor (COUP-TF) dimers bind to different GGTCA response elements, allowing COUP-TF to repress hormonal induction of the vitamin D3, thyroid hormone, and retinoic acid receptors. Mol. Cell. Biol. 12, 4153–4163 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin F.-J., Qin J., Tang K., Tsai S. Y., Tsai M.-J., Coup d'Etat: An orphan takes control. Endocr. Rev. 32, 404–421 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin J., Chen X., Xie X., Tsai M.-J., Tsai S. Y., COUP-TFII regulates tumor growth and metastasis by modulating tumor angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 3687–3692 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin J., Wu S.-P., Creighton C. J., Dai F., Xie X., Cheng C.-M., Frolov A., Ayala G., Lin X., Feng X.-H., Ittmann M. M., Tsai S.-J., Tsai M.-J., Tsai S. Y., COUP-TFII inhibits TGF-β-induced growth barrier to promote prostate tumorigenesis. Nature 493, 236–240 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin S.-C., Kao C.-Y., Lee H.-J., Creighton C. J., Ittmann M. M., Tsai S.-J., Tsai S. Y., Tsai M.-J., Dysregulation of miRNAs-COUP-TFII-FOXM1-CENPF axis contributes to the metastasis of prostate cancer. Nat. Commun. 7, 11418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin S.-J., Zhang Y., Liu N.-C., Yang D. R., Li G., Chang C., Minireview: Pathophysiological roles of the TR4 nuclear receptor: Lessons learned from mice lacking TR4. Mol. Endocrinol. 28, 805–821 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee Y. F., Lee H. J., Chang C., Recent advances in the TR2 and TR4 orphan receptors of the nuclear receptor superfamily. J. Steroid Biochem. Mol. Biol. 81, 291–308 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Dejardin J., Kingston R. E., Purification of proteins associated with specific genomic Loci. Cell 136, 175–186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conomos D., Stutz M. D., Hills M., Neumann A. A., Bryan T. M., Reddel R. R., Pickett H. A., Variant repeats are interspersed throughout the telomeres and recruit nuclear receptors in ALT cells. J. Cell Biol. 199, 893–906 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marzec P., Armenise C., Pérot G., Roumelioti F. M., Basyuk E., Gagos S., Chibon F., Déjardin J., Nuclear-receptor-mediated telomere insertion leads to genome instability in ALT cancers. Cell 160, 913–927 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Su X., Huang J., The Fanconi anemia pathway and DNA interstrand cross-link repair. Protein Cell 2, 704–711 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nalepa G., Clapp D. W., Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 18, 168–185 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Lee O.-H., Kim H., He Q., Baek H. J., Yang D., Chen L.-Y., Liang J., Chae H. K., Safari A., Liu D., Songyang Z., Genome-wide YFP fluorescence complementation screen identifies new regulators for telomere signaling in human cells. Mol. Cell. Proteomics 10, M110.001628 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu S.-P., Kao C.-Y., Wang L., Creighton C. J., Yang J., Donti T. R., Harmancey R., Vasquez H. G., Graham B. H., Bellen H. J., Taegtmeyer H., Chang C.-P., Tsai M.-J., Tsai S. Y., Increased COUP-TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nat. Commun. 6, 8245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee H.-J., Kao C.-Y., Lin S.-C., Xu M., Xie X., Tsai S. Y., Tsai M.-J., Dysregulation of nuclear receptor COUP-TFII impairs skeletal muscle development. Sci. Rep. 7, 3136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie X., Tsai S. Y., Tsai M.-J., COUP-TFII regulates satellite cell function and muscular dystrophy. J. Clin. Invest. 126, 3929–3941 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plevin M. J., Mills M. M., Ikura M., The LxxLL motif: A multifunctional binding sequence in transcriptional regulation. Trends Biochem. Sci. 30, 66–69 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Taniguchi T., Garcia-Higuera I., Andreassen P. R., Gregory R. C., Grompe M., D’Andrea A. D., S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 100, 2414–2420 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Knipscheer P., Räschle M., Smogorzewska A., Enoiu M., Ho T. V., Schärer O. D., Elledge S. J., Walter J. C., The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326, 1698–1701 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dilley R. L., Verma P., Cho N. W., Winters H. D., Wondisford A. R., Greenberg R. A., Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu T., Ghosal G., Yuan J., Chen J., Huang J., FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 329, 693–696 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Hanada K., Budzowska M., Modesti M., Maas A., Wyman C., Essers J., Kanaar R., The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 25, 4921–4932 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto K. N., Kobayashi S., Tsuda M., Kurumizaka H., Takata M., Kono K., Jiricny J., Takeda S., Hirota K., Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 6492–6496 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein Douwel D., Boonen R. A. C. M., Long D. T., Szypowska A. A., Räschle M., Walter J. C., Knipscheer P., XPF-ERCC1 acts in unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell 54, 460–471 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X., Andreassen P. R., D’Andrea A. D., Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol. Cell. Biol. 24, 5850–5862 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pichierri P., Franchitto A., Rosselli F., BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 23, 3154–3163 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howlett N. G., Harney J. A., Rego M. A., Kolling F. W. IV, Glover T. W., Functional interaction between the Fanconi Anemia D2 protein and proliferating cell nuclear antigen (PCNA) via a conserved putative PCNA interaction motif. J. Biol. Chem. 284, 28935–28942 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henson J. D., Hannay J. A., McCarthy S., Royds J. A., Yeager T. R., Robinson R. A., Wharton S. B., Jellinek D. A., Arbuckle S. M., Yoo J., Robinson B. G., Learoyd D. L., Stalley P. D., Bonar S. F., Yu D., Pollock R. E., Reddel R. R., A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin. Cancer Res. 11, 217–225 (2005). [PubMed] [Google Scholar]
- 33.Schwartzentruber J., Korshunov A., Liu X.-Y., Jones D. T. W., Pfaff E., Jacob K., Sturm D., Fontebasso A. M., Quang D.-A. K., Tönjes M., Hovestadt V., Albrecht S., Kool M., Nantel A., Konermann C., Lindroth A., Jäger N., Rausch T., Ryzhova M., Korbel J. O., Hielscher T., Hauser P., Garami M., Klekner A., Bognar L., Ebinger M., Schuhmann M. U., Scheurlen W., Pekrun A., Frühwald M. C., Roggendorf W., Kramm C., Dürken M., Atkinson J., Lepage P., Montpetit A., Zakrzewska M., Zakrzewski K., Liberski P. P., Dong Z., Siegel P., Kulozik A. E., Zapatka M., Guha A., Malkin D., Felsberg J., Reifenberger G., von Deimling A., Ichimura K., Collins V. P., Witt H., Milde T., Witt O., Zhang C., Castelo-Branco P., Lichter P., Faury D., Tabori U., Plass C., Majewski J., Pfister S. M., Jabado N., Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Heaphy C. M., de Wilde R. F., Jiao Y., Klein A. P., Edil B. H., Shi C., Bettegowda C., Rodriguez F. J., Eberhart C. G., Hebbar S., Offerhaus G. J., McLendon R., Rasheed B. A., He Y., Yan H., Bigner D. D., Oba-Shinjo S. M., Marie S. K. N., Riggins G. J., Kinzler K. W., Vogelstein B., Hruban R. H., Maitra A., Papadopoulos N., Meeker A. K., Altered telomeres in tumors with ATRX and DAXX mutations. Science 333, 425 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conomos D., Reddel R. R., Pickett H. A., NuRD-ZNF827 recruitment to telomeres creates a molecular scaffold for homologous recombination. Nat. Struct. Mol. Biol. 21, 760–770 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Zhang J., Walter J. C., Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 19, 135–142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dehé P.-M., Gaillard P.-H. L., Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell Biol. 18, 315–330 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Zeng S., Xiang T., Pandita T. K., Gonzalez-Suarez I., Gonzalo S., Harris C. C., Yang Q., Telomere recombination requires the MUS81 endonuclease. Nat. Cell Biol. 11, 616–623 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson J. S. J., Tejera A. M., Castor D., Toth R., Blasco M. A., Rouse J., Localization-dependent and -independent roles of SLX4 in regulating telomeres. Cell Rep. 4, 853–860 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciccia A., McDonald N., West S. C., Structural and functional relationships of the XPF/MUS81 family of proteins. Annu. Rev. Biochem. 77, 259–287 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Root H., Larsen A., Komosa M., al-Azri F., Li R., Bazett-Jones D. P., Stephen Meyn M., FANCD2 limits BLM-dependent telomere instability in the alternative lengthening of telomeres pathway. Hum. Mol. Genet. 25, 3255–3268 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Fan Q., Zhang F., Barrett B., Ren K., Andreassen P. R., A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res. 37, 1740–1754 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park E., Kim H., Kim J. M., Primack B., Vidal-Cardenas S., Xu Y., Price B. D., Mills A. A., D’Andrea A. D., FANCD2 activates transcription of TAp63 and suppresses tumorigenesis. Mol. Cell 50, 908–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax6366/DC1
Fig. S1. DAVID functional annotation cluster analysis for COUP-TFII–interacting candidates.
Fig. S2. Identification of FANCD2 as a candidate COUP-TFII–interacting protein through BiFC screen.
Fig. S3. Related to Fig. 2.
Fig. S4. Related to Fig. 4C.
Fig. S5. Depletion of FANCD2 shortens telomere length in ALT cells.
Fig. S6. Knockout of FANCD2 or COUP-TFII/TR4 shortened telomere length in U2-OS cells but not in HeLa cells.
Fig. S7. Related to Fig. 4H.
Fig. S8. Related to Fig. 5.
Fig. S9. Telomere ChIP against POLD3 and PCNA in HeLa, U2-OS, and WI38-VA13/2RA cells.
Fig. S10. Related to Fig. 5F.
Table S1. Candidate COUP-TFII–interacting proteins.