

Epigenetic initiation of the Th17 differentiation program is promoted by Cxxc1 via influence IL-6/STAT3 signaling.
Abstract
IL-6/STAT3 signaling is known to initiate the TH17 differentiation program, but the upstream regulatory mechanisms remain minimally explored. Here, we show that Cxxc finger protein 1 (Cxxc1) promoted the generation of TH17 cells as an epigenetic regulator and prevented their differentiation into Treg cells. Mice with a T cell–specific deletion of Cxxc1 were protected from experimental autoimmune encephalomyelitis and were more susceptible to Citrobacter rodentium infection. Cxxc1 deficiency decreased IL-6Rα expression and impeded IL-6/STAT3 signaling, whereas the overexpression of IL-6Rα could partially reverse the defects in Cxxc1-deficient TH17 cells in vitro and in vivo. Genome-wide occupancy analysis revealed that Cxxc1 bound to Il6rα gene loci by maintaining the appropriate H3K4me3 modification of its promoter. Therefore, these data highlight that Cxxc1 as a key regulator governs the balance between TH17 and Treg cells by controlling the expression of IL-6Rα, which affects IL-6/STAT3 signaling and has an impact on TH17-related autoimmune diseases.
INTRODUCTION
T helper 17 (TH17) cells, a subset of CD4+ T cells, are characterized by the secretion of interleukin-17A (IL-17A), IL-17F, IL-21, IL-22, and the transcription factors (TFs) retinoic acid receptor–related orphan receptor γt (RORγt) and RORα (1–3). RORγt and RORα are critical drivers of autoimmune tissue inflammation in human autoimmune diseases (multiple sclerosis), mouse models [experimental autoimmune encephalomyelitis (EAE)], and other autoimmune conditions (4). TH17 cells also maintain mucosal tissue homeostasis and contribute to the host defense against bacterial and fungal infections (2, 5). The activation of naive TH cells in the presence of transforming growth factor–β1 (TGF-β1) and IL-6 leads to the development of TH17 cells (6). Other cytokines such as IL-21, IL-1β, and IL-23 are crucial for the expansion, stability, and functional maturation of TH17 cells (4, 6, 7). RORγt and RORα are the master regulators of TH17 cells, and other TFs, c-Maf, IRF4 (interferon regulatory factor 4), BATF (basic leucine zipper transcription factor, ATF like), and IkBζ, are required for the induction of RORγt, IL-17, IL-21, and IL-22 in vivo and in vitro (8). IL-6, IL-21, and IL-23 can all activate signal transducers and activators of transcription 3 (STAT3), and the activation of STAT3 is crucial for their effects on TH17 cell differentiation (1). The impairment of STAT3 skews TH17 differentiation toward anti-inflammatory Treg cells, and TAZ [transcriptional coactivator with postsynaptic density 65–discs large–zonula occludens 1–binding (PDZ) motif]/TEAD1 (TEA-ATTS DNA–binding domain 1) regulates reciprocal RORγt/Foxp3 expression downstream of STAT3 (9).
Epigenetic mechanisms such as DNA methylation and histone acetylation/methylation have been reported as key players in different T cell subsets (10). In TH1 cells, permissive histone modifications and DNA demethylation in interferon-γ (IFN-γ) and Tbx21 could promote IFN-γ expression and help TH1 lineage differentiation and stability (10, 11). The TH2-specific genes (IL4, IL5, and IL13) are repressed by suppressive histone modifications and DNA methylation in the process of TH1 cell differentiation (12). However, TH2 cells contain activated histone modifications and undergo DNA demethylation at the TH2-specific gene cluster, as well as contain repressive histone modifications and undergo DNA methylation in TH1-specific genes (IFN-γ and Tbx21) (13). Foxp3, the master TF for Treg cells, was marked with permissive H3K4me3 modification and underwent DNA demethylation at its gene locus, which contributes to the determination and commitment of the Treg cell lineage (14). Similarly, genome-wide epigenetic analysis of TH17 cells uncovered the enrichment of permissive histone modifications such as H3K4me3 and DNA demethylation in the promoters of cytokine and lineage-specific genes such as IL17a, IL17f, IL21, IL23r, and Rorc (15).
CXXC finger protein 1 (Cxxc1), defined as an epigenetic regulator, binds to DNA using its CXXC finger domain and recruits SETD1 to most CpG islands (CGIs) through its Setd1-interacting domain (16, 17). Clouaire et al. (16) showed that Cxxc1 is required for H3K4me3 modification in embryonic stem cell, and Thomson et al. (18) found a concordance of Cxxc1 binding with H3K4me3 and nonmethylated CGIs in mouse brain. Our recent studies also demonstrated that Cxxc1-dependent H3K4me3 plays a critical role in thymocyte development, phagocytosis, and the bactericidal activity of macrophages (19, 20). However, the role of Cxxc1-mediated H3K4 trimethylation and DNA methylation in TH cells remains unclear.
To explore the role of Cxxc1 in TH cell differentiation, we took advantage of T cell–specific Cxxc1 knockout (KO) mice. Here, we demonstrate that Cxxc1-deficient TH17 cells exhibited impaired differentiation and stability, which led to susceptibility to bacterial infection and protected against EAE in vivo. Fluorescence-activated cell sorting (FACS) and RNA sequencing (RNA-seq) analyses identified that Cxxc1 deficiency skews TH17 differentiation toward anti-inflammatory Treg cells both in vivo and in vitro.
Via chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) analysis, we revealed genome-wide Cxxc1-binding sites in TH17 cells and H3K4me3 modification changes in Cxxc1-deficient TH17 cells compared with the wild-type (WT) control. We found that Cxxc1 bound to IL-6Rα and other TFs (IRF4 and BATF) by maintaining the appropriate H3K4me3 modification of their promoter regions. Moreover, IL-6Rα, a direct target of Cxxc1, could partially rescue the differentiation and stability defects seen in Cxxc1-deficient TH17 cells. Cxxc1 thus reciprocally regulates the balance between TH17 and Treg cells by regulating IL-6/STAT3 signaling. This suggests that the Cxxc1-mediated epigenetic program is required for T cell differentiation and TH17-related autoimmune diseases.
RESULTS
Cxxc1 promotes the differentiation of TH17 cells
To investigate the potential role of Cxxc1 in TH cell function, we generated conditional KO mice by crossing mice with loxP-flanked Cxxc1 alleles to mice with transgenic Cre driven by the distal Lck promoter (dLck-Cre mice), which mediated the deletion of genes on peripheral CD4+ and CD8+ T cells. dLckcreCxxc1fl/fl mice developed normally with no obvious difference in their T cell development in the thymus (fig. S1A). Further analysis of the peripheral T cells showed a decrease in CD8+ T cell numbers, especially in its effector/memory population, while CD4+ T cell numbers and phenotypes were normal (fig. S1, B to D). CellTrace dilution showed little impairment of the CD8+ T cell proliferation capacity of Cxxc1-deficient T cells cultured under T cell receptor (TCR) stimulation, while CD4+ T cells showed no influence of TCR stimulation (fig. S1E).
We then isolated naive CD4+ T cells from dLckcreCxxc1wt/wt and dLckcreCxxc1fl/fl mice and conducted T cell differentiation in vitro. We found that Cxxc1 ablation led to severely defective TH17 differentiation characterized by reduced IL-17A and IL-17F in two TH17 cell–polarizing conditions: (i) TGF-β1 and IL-6 and (ii) IL-1β, IL-6, and IL-23, while Foxp3 expression increased (Fig. 1, A and B). However, Cxxc1-deficient T cells exhibited no obvious difference in TH1 or TH2 differentiation and a moderate increase in induced T-regulatory cell (iTreg) differentiation (fig. S2, A to C), while the expression levels of Cxxc1 protein were consistent among these T helper subsets and in different stimulation time of TH17 cells (fig. S2, G and H). Furthermore, we also used CD4+ T cells from CreERT2+ Cxxc1fl/fl mice to test Cxxc1 function in an in vitro culture system and found a similar requirement for Cxxc1 in TH17 differentiation in the presence or absence of 4-hydroxytamoxifen treatment (which initiated the access of Cre recombinase to the nucleus and led to the deletion of Cxxc1 in vitro, as shown in fig. S2I) (Fig. 1, C and D) for 4 days, and we observed no obvious difference in TH1, TH2, or iTreg differentiation (fig. S2, D to F).
Fig. 1. TH17 cell differentiation was severely impaired in Cxxc1-deficient mice.
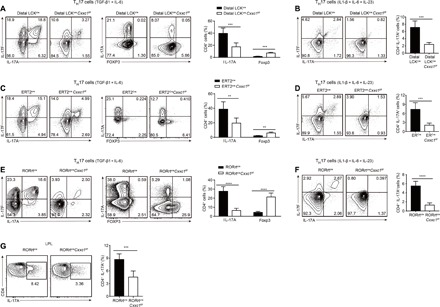
(A and B) Naive CD4+ T cells (CD4+CD25-CD62LhiCD44lo) from dLckcreCxxc1fl/fl or WT mice were differentiated into TH17 cells with (A) IL-6 and TGF-β1 or (B) IL-1β, IL-6, and IL-23 for 96 hours and then restimulated for intracellular cytokine staining. One of five to seven experiments is shown. (C and D) Naive CD4+ T cells (CD4+CD25-CD62LhiCD44lo) from ERT2creCxxc1fl/fl mice were differentiated into TH17 cells with (C) IL-6 and TGF-β1 or (D) IL-1β, IL-6, and IL-23 for 96 hours in the presence or absence of 4-OHT (4-Hydroxytamoxifen) and then restimulated for intracellular cytokine staining. One of five experiments is shown. (E and F) Naive CD4+ T cells from RORγtcreCxxc1fl/fl or WT mice were differentiated into TH17 cells with (E) IL-6 and TGF-β1 or (F) IL-1β, IL-6, and IL-23 for 96 hours and then restimulated for intracellular cytokine staining. One of seven experiments is shown. (G) Intracellular staining of IL-17A in lipoprotein lipase (LPL) CD4+ T cells in the small intestines of RORγtcreCxxc1fl/fl and WT mice. One of four experiments is shown. Error bars show the means ± SD. **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001 using the Student’s t test.
Next, we tested the function of Cxxc1 in TH17 cells by generating RORγtcre Cxxc1fl/fl mice. Similar to the dLck-mediated deletion of Cxxc1, RORγtcre Cxxc1fl/fl mice developed normally in terms of T cell development in the thymus (fig. S3A) and exhibited decreased CD8+ T cell numbers in the periphery (fig. S3, B and D), while CD4+ T cell numbers were normal (fig. S3, B and C). Then, we isolated naive CD4+ T cells from RORγtcreCxxc1wt/wt and RORγtcre Cxxc1fl/fl mice and conducted TH17 cell differentiation (TGF-β1 and IL-6 or IL-1β, IL-6, and IL-23) in vitro. Compared with the dLck-mediated deletion of Cxxc1, T cells in RORγtcreCxxc1fl/fl mice differentiated under TH17 cell–polarizing conditions showed much less IL-17A and IL-17F production than Cxxc1-competent RORγtcreCxxc1wt/wt cells and markedly increased Foxp3 production (Fig. 1, E and F). This is due to the sufficient deletion of Cxxc1 protein in TH17 cells in the RORγtcre system compared with dLckcre system (fig. S2I). Under steady-state conditions, TH17 cells are preferentially located in the lamina propria (LP) of the small intestine (3). We also showed that CD4+ T cells in the LP of RORγtcre Cxxc1fl/fl mice showed notably reduced IL-17 production in vivo (Fig. 1G), although we found a little increased frequency of Treg cells in LP and normal frequency of Treg cells in lymph nodes and spleen (fig. S3E).
To characterize IL-17A expression more specifically, we introduced IL-17AeGFP reporter mice into dLckcreCxxc1fl/fl and RORγtcre Cxxc1fl/fl backgrounds separately, and the results also confirm defective TH17 differentiation in Cxxc1-deficient cells with enhanced green fluorescent protein expression (fig. S3, F and G). These results indicate that Cxxc1 is essential for TH17 cell differentiation in vitro.
The loss of Cxxc1 affects TH17 cell function in vivo
To further evaluate whether defects in TH17 cells caused by Cxxc1 deficiency affect the development of TH17-dependent inflammatory diseases in vivo, we used a TH17-dependent autoimmune disease model, EAE, that mimics the human neuroinflammatory disease multiple sclerosis.
As we found decreased CD8+ T cell numbers in the periphery (fig. S3B), to exclude the effect of CD8+ T cell (21), we sorted naive CD4+ T cells (CD4+CD44loCD62L+) from RORγtcreCxxc1fl/fl and RORγtcreCxxc1wt/wt mice, transferred these cells into Rag1−/− mice, and then monitored them for the induction of EAE. In agreement with the results showing the in vitro defects, transfer of RORγtcreCxxc1fl/fl-naive CD4+T cells alleviated EAE, and significantly less mononuclear cell infiltration and demyelination of the spinal cord were observed (Fig. 2, A and B). Within the central nervous system (CNS)–infiltrating draining lymph nodes and spleen CD4+ T cell population, the production of IL-17A+ T helper cells was reduced in the hosts that had received RORγtcreCxxc1fl/fl cells, whereas the number of Treg cells increased in the CNS-infiltrating CD4+ T cells (Fig. 2, C and D). Cxxc1-deficient splenocytes isolated from myelin oligodendrocyte glycoprotein (MOG)–immunized mice showed impaired production of IL-17 but normal production of IFN-γ upon restimulation with the MOG peptide compared with Cxxc1-sufficient splenocytes (Fig. 2E). We found similar results when the EAE model was induced in the Rag1−/− hosts with dLckcreCxxc1fl/fl cells compared with dLckcreCxxc1wt/wt cells (fig. S4, A to C). We can find that Cxxc1 deficiency alleviated symptoms of autoimmunity in dLckcreCxxc1fl/fl (fig. S4, D to G) and RORγtcreCxxc1fl/fl (fig. S4, H to K) mice compared with appropriate control mice as well. These data suggested that the Cxxc1-deficient cells were less susceptible to the EAE disease model than Cxxc1-sufficient cells, which was in accordance with the in vitro results showing defective IL-17 expression.
Fig. 2. Cxxc1 deficiency restricts T cell–mediated autoimmunity and increases sensitivity to C. rodentium infection.
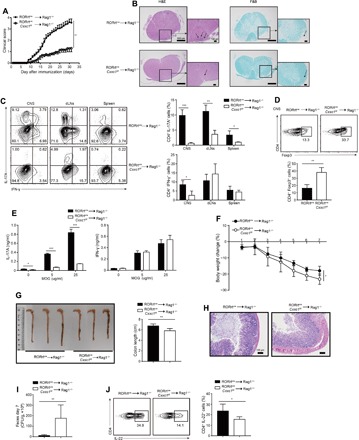
(A) Mean clinical scores for EAE in Rag1−/− recipients of RORγtcreCxxc1fl/fl (n = 13) or WT (n = 11) naive CD4+ T cells after being immunized with MOG35–55, complete Freund’s adjuvant (CFA), and pertussis toxin. Data are summed from three independent experiments. (B) Representative histology of the spinal cord of Rag1−/− mice after EAE induction (day 25). Hematoxylin and eosin (H&E) staining (left), Luxol fast blue (F&B) staining (right). (C) On day 20 after the induction of EAE in Rag1−/− hosts, CD4+ T cells were analyzed from leukocytes isolated from the CNS, draining lymph nodes (dLNs), and spleen and further analyzed for the frequency of IL-17A+ and IFN-γ+ T cells (left). Summary of CNS IL-17A+CD4+ and IFN-γ+CD4+ T cells in Rag1−/− hosts (right). One representative of three experiments is depicted. (D) The frequency of Foxp3+ cells from CNS-infiltrating lymphocytes in Rag1−/− EAE mice was determined at day 20 after immunization (top). Summary of CNS Foxp3+CD4+ cells in Rag1−/− hosts (bottom). One representative of three experiments is depicted. (E) Splenocytes were rechallenged with the MOG peptide (0, 5, and 25 μg/ml) for 3 days, and then, cytokine production was measured by enzyme-linked immunosorbent assay (ELISA). (F) Body weight changes of Rag1−/− recipients of naive CD4+ T cells from RORγtcreCxxc1fl/fl (n = 12) or WT (n = 11) mice after oral inoculation with C. rodentium at the indicated time points. Data are summed from three independent experiments. (G) Colon length for Rag1−/− recipients of naive CD4+ T cells from RORγtcreCxxc1fl/fl or WT mice after oral inoculation with C. rodentium at day 7. Summary of colon lengths in Rag1−/− hosts (right). (H) Histological analysis of representative colons from Rag1−/− hosts 7 days after inoculation. (Photo credit: Feng Lin, Institute of Immunology, Zhejiang University School of Medicine). (I) C. rodentium colony-forming units (CFUs) in the colon 7 days after inoculation. Data are summed from three independent experiments. (J) FACS analysis of IL-22 expression from isolated LPLs in Rag1−/− hosts at day 7 after inoculation. One of five experiments is shown. Error bars show the means ± SD. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 using the Student’s t test.
IL-22 is produced by leukocytes, particularly TH17 cells, and has a crucial role in host defense against bacterial infections. Ouyang and his team (5) found that IL-22 has a crucial role in the early phase of host defense against Citrobacter rodentium. We sorted naive CD4+ T cells from RORγtcreCxxc1fl/fl and RORγtcreCxxc1wt/wt mice, transferred these cells into Rag1−/− mice, and then inoculated them with C. rodentium. On day 7 after inoculation, mice that had undergone transfer of RORγtcreCxxc1fl/fl-naive CD4+ T cells developed a more aggravated infection than mice that had undergone transfer of RORγtcreCxxc1wt/wt-naive CD4+ T cells. The mice showed a significantly greater loss of body weight and shorter colon length than the WT mice (Fig. 2, F and G). Histological analysis of colons from C. rodentium–infected RORγtcreCxxc1fl/fl mice showed increased mucosal hyperplasia and submucosal inflammation compared to the RORγtcreCxxc1wt/wt mice, suggesting that Cxxc1 deficiency leads to compromised epithelial barrier function (Fig. 2H). In addition, we found that the bacterial burdens in the feces of RORγtcreCxxc1fl/fl mice were increased compared to those in the feces of RORγtcreCxxc1wt/wt mice (Fig. 2I). Cytokine analysis showed reduced IL-22 production in CD4+ T cells and group 3 innate lymphoid cell (ILC3) from the LP of hosts with Cxxc1-deficient cells transfer (Fig. 2J and fig. S5A), and ILC3’s IL-22 production impairment is possibly due to increased Treg cells (fig. S5B) (22). We found that IL-17 production was unaffected, which imply that cytokines other than IL-6 may regulate IL-17 in this model, and we found normal STAT3 phosphorylation by IL-21 stimulation of RORγtcreCxxc1fl/fl cells (fig. S5, C and D). In addition, we found similar results when we performed C. rodentium model in RORγtcreCxxc1fl/fl mice (fig. S5, E to H). These findings further support the conclusion that Cxxc1 contributes to TH17 differentiation and function in vivo.
Cxxc1-deficient TH17 cells exhibit a Treg cell–like expression profile
To further analyze the genes regulated by Cxxc1, we performed RNA-seq analysis of RORγtcreCxxc1fl/fl and RORγtcreCxxc1wt/wt TH17 cells generated in vitro in the presence of TGF-β and IL-6 for 72 hours. The RNA-seq results showed that Cxxc1-deficient TH17 cells almost completely lost their features and exhibited a Treg cell–like expression profile. Key TH17 cell–related cytokines and transcripts (e.g., Il17a, Il17f, Il21, Il22, Il6ra, Irf4, Cxcr4, Maf, and Satb1) were significantly down-regulated in RORγtcreCxxc1fl/fl TH17 cells, and the master TFs Rorc and Rora also showed a decrease in expression (Fig. 3, A and B). Key Treg cell–related transcripts (e.g., Foxp3, Ccl3, Itgae, Gpr83, Mgat5, Ikzf2, Ikzf4, Tigit, and Tnfsf10) were significantly up-regulated in RORγtcreCxxc1fl/fl TH17 cells (Fig. 3, A and B). Using a fold change of 2 and P < 0.05 to threshold parameters, we found that 782 genes were down-regulated by Cxxc1 knockdown in Cxxc1-defcient cells, and 1411 genes were up-regulated. By pathway analysis, we found the significant enrichment of many genes associated with the inflammatory response and immune-related signaling pathways (e.g., the Janus kinase–STAT cascade and tyrosine phosphorylation of STAT3 protein) (Fig. 3C). We then measured the expression of TH17 and Treg signature genes, including Il17a, Il17f, Il21, Il22, Rorc, Il6ra, Foxp3, Mgat5, Itgae, Gpr83, Tgfbr1, and Ccl3, from Cxxc1-deficient cells and Cxxc1-sufficient cells by quantitative polymerase chain reaction (qPCR) and obtained the same results as those from 72-hour RNA-seq analysis (Fig. 3D). We also verified these genes in dLCK-Cre–induced Cxxc1-deficient TH17 cells by qPCR and got the same results (fig. S5I), which indicates that Cxxc1-deficient TH17 cells exhibit a Treg cell–like expression profile.
Fig. 3. Cxxc1-deficient TH17 cells exhibit a Treg cell–like expression profile.
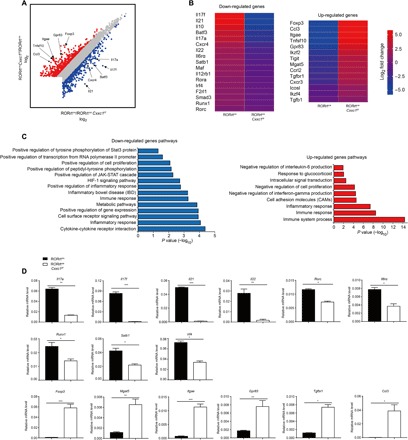
(A) Naive CD4+ T cells (CD4+CD25−CD62LhiCD44lo) from WT and RORγtcreCxxc1fl/fl mice were differentiated in the presence of TGF-β1 and IL-6 (TH17) for 72 hours, and the total RNA from the cells was analyzed by RNA-seq [STAR (structured transparent accessible reporting) method]. Scatter diagram showing changes in gene expression in WT and Cxxc1-deficient TH17 cells. Down-regulated genes are indicated in blue; up-regulated genes are indicated in red. (B) Heatmap of the fold change (log2) for differentially expressed genes (false discovery rate < 0.05 in TH17 cells is shown). (C) Pathway analysis of the down-regulated genes (left) and up-regulated genes (right). (D) The expression of the selected transcripts was quantified in TH17 cell samples differentiated from naive CD4+ T cells with TGF-β1 and IL-6 for 72 hours by real-time qPCR. One of five experiments is shown. Error bars show the means ± SD. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 using the Student’s t test.
Cxxc1 binding and H3K4me3 modification profiles of Th17 cells
Cxxc1 has two main functional domains. One domain is the N-terminal domain, which interacts with unmethylated CpG DNA to mediate its interaction with DNA methyltransferases 1 (DNMT1) and stabilizes the DNMT1 protein to regulate DNA methylation (17, 18, 23). The other is the C-terminal domain, which interacts with the Setd1 H3K4 methyltransferase complex through the Smad interaction domain to regulate histone methylation. An N-terminal fragment of Cxxc1 (residues 1 to 367) and Cxxc1 containing the point mutation C375A retain their interaction with DNMT1, but the Setd1-interacting activity of Cxxc1 is interrupted. While the C-terminal fragment of Cxxc1 (residues 361 to 656) and Cxxc1 containing the point mutation C169A still have Setd1-interacting activity and can methylate H3K4, the DNA binding activity of Cxxc1 is interrupted. To explore which functional domains within Cxxc1 were necessary for its role in TH17 differentiation, we used an in vitro TH17 differentiation system, and different vectors expressing mutated Cxxc1 proteins were overexpressed in Cxxc1-deficient TH17 cells. The overexpression of full-length Cxxc1 notably rescued the TH17 differentiation defects seen in Cxxc1-deficient cells (fig. S6A). The overexpression of the two Cxxc1 fragments (residues 1 to 367 and residues 361 to 656) and Cxxc1 containing two point mutations (C169A and C375A) showed that the C-terminal domain of Cxxc1 (residues 361 to 656, C169A) rather than the N-terminal domain of Cxxc1 (residues 1 to 367, C375A) was able to rescue TH17 differentiation in Cxxc1-deficient TH17 cells (fig. S6, A and B). Similarly, the overexpression of the C-terminal domain of Cxxc1 reduced Foxp3 expression in Cxxc1-deficient TH17 cells, but the overexpression of the N-terminal domain of Cxxc1 did not (fig. S6C). We also checked gene expression changes by real-time PCR, such as TH17 and Treg signature genes, including Il17a, Il17f, Il21, Il22, Rorc, Il6ra, Foxp3, Mgat5, Itgae, Gpr83, Tgfbr1, and Ccl3, further elucidating the role of two CXXC1 functional domains (fig. S6D). These data showed that the Setd1-interacting domain but not the DNA binding domain in Cxxc1 is crucial for TH17 differentiation, indicating that Cxxc1 may function through regulating H3K4me3 rather than through DNA methylation in TH17 cells.
To investigate the direct targets of Cxxc1 in TH17 cells, we performed ChIP-seq to map genome-wide Cxxc1-binding sites in WT TH17 cells, as well as in Cxxc1-deficient TH17 cells serving as a negative control. Compared with Cxxc1-binding sites in the mouse genome, an obvious enrichment of Cxxc1-binding sites was found in gene promoters [5-kb upstream and downstream of the transcription start site (TSS), 25.56% of Cxxc1-binding sites versus 2% of the mouse genome], exons (3.52% versus 2%), introns (32.67% verus 20%), and intergenic regions (38.25% versus 76%) (Fig. 4A). This represented a relatively high degree of enrichment at promoter regions compared with the distribution of Cxxc1-binding sites in the mouse genome. Analysis of the average binding location also showed that Cxxc1 showed high binding activity at TSS (Fig. 4B).
Fig. 4. Genome-wide maps of Cxxc1 binding and H3K4me3 modifications in TH17 cells differentiated from naive CD4+ T cells with TGF-β1 and IL-6 for 24 hours.
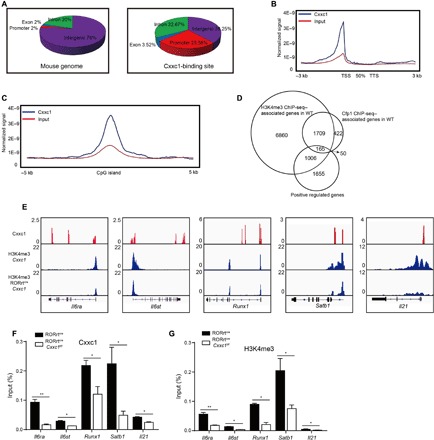
(A) Naive CD4+ T cells (CD4+CD25-CD62LhiCD44lo) from WT and RORγtcreCxxc1fl/fl mice were differentiated in the presence of TGF-β1 and IL-6 (TH17) for 24 hours, and ChIP-seq analysis was conducted to map genome-wide Cxxc1-binding sites in WT TH17 cells. Distribution of the genetic features across the whole mouse genome (mm10) (left) and the distribution of Cxxc1-binding peaks in TH17 cells (right). (B) Distribution of Cxxc1-binding peaks across extended gene bodies in TH17 cells. The tag density of Cxxc1 binding to gene bodies [between the transcription start site (TSS) and the transcription termination site (TTS)], as well as 3-kb upstream of the TSS and 3-kb downstream of the TTS regions of all RefSeq (mm10) genes, was calculated. (C) Enrichment of Cxxc1-binding peaks on CGIs. The tag density of Cxxc1 binding to CGIs and 5-kb flanking regions was calculated. (D) Overlapped regions between Cxxc1-binding sites, H3K4me3 sites, and RNA-seq down-regulated genes in WT and Cxxc1-deficient TH17 cells. (E) Integrative Genomics Viewer browser view of Cxxc1-binding peaks (red) in WT TH17 cells and H3K4me3 markers (blue) in WT and Cxxc1-deficient TH17 cells. (F) Naive WT CD4+ T cells were sorted and cultured under TH17 differentiation conditions (TGF-β and IL-6) for 24 hours, and ChIP-qPCR analysis of Cxxc1 binding at the indicated gene loci was performed. (G) Naive CD4+ T cells from WT and RORγtcreCxxc1fl/fl mice were differentiated into TH17 cells in the presence of TGF-β1 and IL-6 for 24 hours, and H3K4me3 modifications at the indicated gene loci were detected by ChIP-qPCR. The statistical significance was determined by Student’s t test. Error bars show the means ± SD. *P ≤ 0.05, **P ≤ 0.01.
As shown in Fig. 4C, Cxxc1-binding sites were enriched at the center of CGIs, and about 31% of the Cxxc1-binding sites were found to colocalize with CGIs in TH17 cells (fig. S7A). We found that Cxxc1-dependent H3K4me3 modifications might be indispensable during TH17 differentiation (fig. S6); therefore, we carried out ChIP-seq analysis to map the genome-wide H3K4me3 modifications in TH17 cells. Similar to the locations of Cxxc1-binding sites, peaks indicating H3K4me3 modifications were also found mainly in gene promoters, and a reduction in the number of H3K4me3 peaks was found in Cxxc1-deficient TH17 cells (fig. S7B).
We mapped the genes with direct CXXC1 binding, genes with at least twofold difference of H3K4me3 modifications between WT and CXXC1 KO cells, and genes positively regulated by CXXC1 (at least twofold difference in gene expression between WT and KO cells). As shown in the Venn diagram (Fig. 4D), the loci of 1874 of 2346 (80%) genes with direct CXXC1 binding were associated with H3K4me3 changes, suggesting an important role of CXXC1 in mediating the histone modification of H3K4me3 in TH cells. Our data also identified 165 genes positively regulated by CXXC1 through positively changing the H3K4me3 modifications on their loci directly (Fig. 4D and table S1). On the other hand, we were able to identify more than 4000 genes whose expression was negatively regulated by CXXC1, although, in most of the cases, through indirect signaling pathways without directly affecting the H3K4me3 on their loci (fig. S7C). Together, our data suggest that CXXC1 plays a key role in regulating gene expressions through recruitment of H3K4me3 in TH cells.
ChIP-seq data showed that Cxxc1 bound upstream of or bound to the gene body of the Il6rα, Il6st, Runx1, Satb1, Il21, Irf4, Rorc, Rora, and Batf gene loci was associated with a significant decrease in the H3K4me3 modification of the promoter regions of these genes in Cxxc1-deficient TH17 cells (Fig. 4E and fig. S7D). The direct binding of Cxxc1 in WT TH17 cells and the reduction in H3K4me3 modification in Cxxc1-deficient cells at these gene loci were confirmed by ChIP-PCR (Fig. 4, F and G, and fig. S7, E and F).
We then conducted 24-hour RNA-seq to determine potential key genes that are regulated by Cxxc1 at the early stage of differentiation and further confirmed the ChIP-seq results. We found that, even at the early stage, most of the key TH17 cell–related transcripts (e.g., Il17a, Il17f, Il21, Il22, Il6ra, Il6st, Runx1, and Satb1) were significantly down-regulated in RORγtcreCxxc1fl/fl TH17 cells and that the expression of key Treg cell–related transcripts increased (e.g., Foxp3, Ccl3, Mgat5, Itgae, and Gpr83) (fig. S7G). We measured the expression of those genes by qPCR and obtained the same results as those obtained by 24-hour RNA-seq analysis (fig. S7H). In contrast to the 72-hour RNA-seq and qPCR results, we did not find a remarkable change in Rorc, Rora, or Irf4 expression at 24 hours. Although there are interactions between CXXC1 and loci of Rorc, Rora, and Irf4, these bindings only indicate enhanced accessibility of these loci. It still requires upstream TFs to promote the expression of Rorc, Rora, and IRF4. It may take 24 hours to open the gene locus, while it may take a longer time for essential TFs to promote their expression. Similar to the ChIP-seq results, we found a decline in Il6ra and Il6st (gp130), which organize the functional receptor for IL-6. The IL-6Rα subunit binds to IL-6 and the IL-6ST subunit, which are involved in signal transduction and play a vital role in TH17 cell differentiation (24).
Cxxc1 controls TH17 cell differentiation by regulating IL-6 signaling.
We found that Il6rα showed a significant decline in H3K4me3 modification levels and mRNA expression. Then, we detected the protein levels of IL-6Rα by flow cytometry and found a significant reduction in IL-6Rα in Cxxc1-deficient cells under TH17 cell–polarizing conditions (TGF-β1 and IL-6) in different stages of differentiation (Fig. 5A). In addition, we found the same phenomenon in pathogenic TH17 cells (IL-1β, IL-6, and IL-23), indicating that the reduction of IL-6Rα is independent of TGF-β signaling (Fig. 5B). Moreover, we also detected a significant decline in IL-6Rα in dLckcre and ERT2cre deletion mice compared with that in WT mice (fig. S8A). Although IL-6ST expression was reduced at 24 hours, we found a moderate increase in IL-6ST expression at 72 hours (fig. S8B). IL-6Rα may be secreted in its soluble form, sIL-6R, which mediates the response by forming a complex with IL-6ST in a mechanism named trans-signaling (25). We therefore measured the level of sIL-6R in the supernatant by enzyme-linked immunosorbent assay (ELISA) and detected significantly lower levels of sIL-6R in Cxxc1-deficient cells than those in WT mice at all time points under TH17 cell–polarizing conditions (TGF-β1 and IL-6) (Fig. 5C). These results indicated that IL-6Rα expression was significantly reduced in Cxxc1-deficient TH17 cells both in its membrane-bound and soluble forms.
Fig. 5. The IL-6/STAT3 signaling pathway was defective after Cxxc1 deletion.
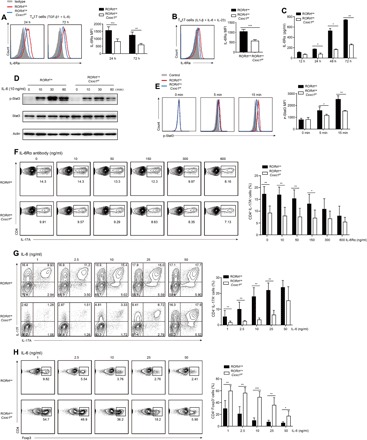
(A and B) Naive CD4+ T cells (CD4+CD25-CD62LhiCD44lo) from RORγtcreCxxc1fl/fl and WT mice were differentiated into TH17 cells with IL-6 and TGF-β1 (A) or IL-1β, IL-6, and IL-23 (B). The expression of IL-6Rα was measured by flow cytometry (left), and the mean fluorescence intensity (MFI) of IL-6Rα at different time points was measured (right). One of six experiments is shown. (C) Naive CD4+ T cells from RORγtcreCxxc1fl/fl and WT mice were differentiated into TH17 cells with IL-6 and TGF-β1, and the supernatants from cell cultures were collected at indicated time points. The amounts of IL-6Rα were then measured by ELISA. One of four experiments is shown. (D and E) Purified naive CD4+ T cells were stimulated for the indicated times with IL-6 (10 ng/ml). Phosphorylated and total STAT3 proteins were detected by Western blot assays (D) or flow cytometry (E). One of five experiments is shown. (F) Naive CD4+ T cells from WT and RORγtcreCxxc1fl/fl mice were polarized into TH17 cells in the presence of TGF-β and IL-6, and varying concentrations of IL-6Rα antibody were added. The expression levels of IL-17A and IL-17F were then analyzed by intracellular staining after 72 hours. One of six experiments is shown. (G and H) Naive CD4+ T cells from WT and RORγtcreCxxc1fl/fl mice were cultured in the presence of TGF-β1 and varying concentrations of IL-6 for 72 hours, and then, the expression levels of IL-17A, IL-17F, and Foxp3 were analyzed by intracellular staining after restimulation. One of seven experiments is shown. The statistical significance was determined by Student’s t test. Error bars show the means ± SD. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
IL-6 signaling is required for the differentiation of TH17 cells, and the activation of STAT3 is a vital component of the TH17 cell induction mechanism (26). The significant reduction of IL-6Rα in RORγtcreCxxc1fl/fl TH17 cells indicated that IL-6 signaling was possibly affected by the loss of Cxxc1 during TH17 differentiation. To assess this, we sorted RORγtcreCxxc1wt/wt and RORγtcreCxxc1fl/fl naive CD4+ T cells and stimulated them with IL-6 for different time periods, and then, we detected the activation of the downstream signaling protein STAT3. Both Western blot analysis and flow cytometry results showed a significant reduction in STAT3 activation in Cxxc1-deficient cells compared to that in WT cells stimulated with IL-6 (Fig. 5, D and E).
To further confirm the role of IL-6Rα defects in Cxxc1-deficient TH17 cells, we added different concentrations of IL-6Rα blocking antibody to TH17 culture medium in vitro. WT TH17 cells showed a marked reduction in differentiation when IL-6Rα blocking antibody was added, and the higher the concentration of IL-6Rα blocking antibody was, the lower the level of WT TH17 cell differentiation was. However, there was only a slight impact in Cxxc1-deficient TH17 cells compared with WT cells when IL-6Rα blocking antibody was added (Fig. 5F), further indicating the defects of IL-6Rα in Cxxc1-deficient TH17 cells.
IL-6 binds to IL-6Rα and is required for TH17 differentiation by activating STAT3 and inhibiting TGF-β–driven Foxp3 expression. The defects in IL-6Rα expression in TH17 cells may be compensated if the level of IL-6 is increased. To assess this, we polarized naive RORγtcreCxxc1wt/wt or RORγtcreCxxc1fl/fl CD4+ T cells into TH17 cells with varying levels of IL-6. Although IL-17 expression was only slightly restored with low and moderate levels of IL-6, it was restored with a high level of IL-6 (Fig. 5G). Moreover, IL-6 inhibited Foxp3 expression in RORγtcreCxxc1fl/fl cells in a dose-dependent manner (Fig. 5H). These data suggest that Cxxc1 may regulate TH17 differentiation dependent on the IL-6/STAT3 pathway in the modulation of early signaling events downstream of the IL-6 receptor.
IL-21 or IL-6 alone or in combination with TGF-β resulted in the up-regulation of the IL-23 receptor (IL-23R), RORγt, and the TH17 cytokines. Our flow cytometry results showed no change in IL-21R expression, while the IL-23R expression was decreased in RORγtcreCxxc1fl/fl TH17 cells (fig. S8C). The IL-6–induced expression of IL-21, a process that is dependent on STAT3 and IL-21, serves as an autocrine factor that promotes and sustains TH17 lineage commitment (1, 7). IL-21, in synergy with TGF-β, induced IL-17 expression independent of IL-6 and induced naive IL-6−/− T cells into TH17 cells (6, 27). We then cultured naive CD4+ T cells in vitro with varying concentrations of IL-21 along with TGF-β. Similar to the results observed upon the addition of varying levels of IL-6, IL-21 restored inconspicuous IL-17A and IL-17F expression at low and moderate levels, while it fully restored IL-17A and IL-17F expression in Cxxc1-deficient cells at high levels (fig. S8D). IL-23 promotes maintenance of the TH17 lineage and maintains the IL-17–secreting phenotype, but it does not promote commitment to an IL-17–secreting lineage. IL-23 could also induce IL-17A and IL-17F expression independent of IL-6 in conjunction with TGF-β in naive CD4+ T cells (6). When naive CD4+ T cells were cultured in vitro with TGF-β and varying concentrations of IL-23, defective IL-17A and IL-17F expression could not be restored, even at the highest level of IL-23, in Cxxc1-deficient cells (fig. S8E). In addition, to eliminate the residue effect of IL-6–dependent signaling, we added IL-6Rα blocking antibody in these cultures and found consistent results (fig. S8, F and G).
As TGF-β receptors are important for both TH17 and Treg cell differentiation, we detected the expression of TGF-β receptors I and II. Protein levels of TGF-β receptors I and II detected by flow cytometry analysis did not show a significant change in naive Cxxc1-deficient CD4+ T cells but showed an increase in Cxxc1-deficient TH17 cells compared with WT cells (fig. S9, A and B). Smad3 and Smad2 are downstream of TGF-β signaling, and Smad2 positively regulates the generation of TH17 cells (28), while Smad3 promotes iTreg and inhibits TH17 cell differentiation (29). Western blot results showed no significant change in the Smad2 phosphorylation level (fig. S9C), while Smad3 showed a slight increase in phosphorylation (fig. S9D). Furthermore, the TCR activation–induced phosphorylation of both ERK and JNK proteins was also normal in Cxxc1-deficient cells (fig. S9E). These results indicated that Cxxc1-deficient TH17 cells transdifferentiated into Treg cells mainly due to IL-6/STAT3 signaling defects.
IL-6Rα rescues the Cxxc1 deficiency–mediated effect on TH17 cell differentiation
To further confirm IL-6Rα defects in Cxxc1-deficient TH17 cells, we overexpressed IL-6Rα in Cxxc1-deficient TH17 cells and carried out assays. We infected Cxxc1-deficient TH17 cells with retroviruses IL-6Rα or Cxxc1 complementary DNA as a positive control. The results showed that the overexpression of IL-6Rα potently increased the production of IL-17A and IL-17F relative to that in mock-transfected control Cxxc1-deficient TH17 cells (Fig. 6A). In addition, the overexpression of IL-6Rα could also efficiently reduce Foxp3 expression in Cxxc1-deficient TH17 cells (fig. S9F). Moreover, we found that the overexpression of Cxxc1 and the C-terminal domain of Cxxc1 (residues 361 to 656, C169A) increased the expression of IL-6Rα compared to that in mock-transfected Cxxc1-deficient TH17 cells, further confirming the regulation of IL-6Rα by Cxxc1 (Fig. 6B). We also overexpressed IL-6ST in Cxxc1-deficient TH17 cells and did not find the restoration of IL-17A and IL-17F production or the inhibition of Foxp3 expression compared with controls (Fig. 6A and fig. S9F).
Fig. 6. Cxxc1 mediates TH17 cell differentiation by mediating IL-6Rα expression.
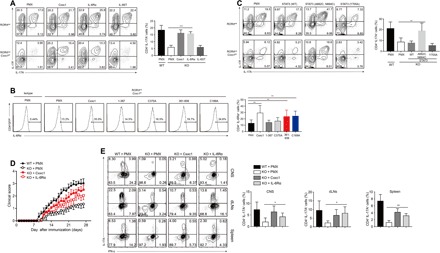
(A) Naive CD4+ T cells from WT and RORγtcreCxxc1fl/fl mice were differentiated into TH17 cells in the presence of TGF-β1 and IL-6, and 20 to 24 hours, later the cells were transfected with the indicated retrovirus (Mock, Cxxc1, IL-6Rα, and IL-6ST). IL-17A and IL-17F levels were then measured by gated CD4+GFP+ cells after retrovirus infection for 72 hours. One of six experiments is shown. (B) Sorted naive CD4+ T cells were differentiated into TH17 cells in the presence of TGF-β1 and IL-6, and 20 to 24 hours later, the cells were transfected with the indicated retrovirus. IL-6Rα levels were then measured by gated CD4+GFP+ cells after retrovirus infection for 72 hours. One of five experiments is shown. (C) Naive CD4+ T cells from WT and RORγtcreCxxc1fl/fl mice were differentiated into TH17 cells in the presence of TGF-β1 and IL-6, and 20 to 24 hours later, the cells were transfected with the indicated retrovirus [Mock, STAT3 (WT), STAT3 (A662C, N664C), and STAT3 (Y705A)]. IL-17A and IL-17F levels were then measured by gated CD4+GFP+ cells after retrovirus infection for 72 hours. One of five experiments is shown. (D) Naive CD4+ T cells from WT and RORγtcreCxxc1fl/fl mice were differentiated into TH17 cells in the presence of TGF-β1 and IL-6, and 20 to 24 hours later, the cells were transfected with the indicated retrovirus. CD4+GFP+ cells were then sorted after retrovirus infection for 72 hours and transferred into RAG1−/− hosts. Two days later, the recipient mice were immunized with MOG35–55 and FCA plus pertussis toxin to induce EAE. Clinical scores were recorded and calculated each day for the indicated times. Data are summed from three independent experiments. (E) IL-17A and IFN-γ production by CD4+ T cells isolated from CNS, draining lymph nodes, and spleens of Rag1−/− mice at the peak of disease. One representative of three experiments is depicted. Error bars show the means ± SD. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 using the Student’s t test.
To further determine defects in the activation of STAT3, which is downstream of IL-6, we overexpressed STAT3 in Cxxc1-deficient TH17 cells. Our results showed that the overexpression of the active form of STAT3 (A662C, N664C) (30) strongly increased the production of IL-17A and IL-17F and inhibited Foxp3 expression relative to that in mock-transfected control Cxxc1-deficient TH17 cells, while the overexpression of WT STAT3 and the inactive form of STAT3 had almost no apparent effect (Fig. 6C and fig. S9G). However, we did not detect a change in IL-6Rα expression when the active form of STAT3 was overexpressed, indicating that IL-6Rα is upstream of STAT3 (fig. S9H). We also detected the reduced expression of RORγt in Cxxc1-deficient TH17 cells, and RORγt overexpression in Cxxc1-deficient TH17 cells partially rescued the TH17 differentiation defect under TH0 conditions and TH17-polarizing conditions (fig. S9I). Therefore, defects in STAT3 activation impaired RORγt function at the Il17-Il17f locus.
Last, to better understand whether the overexpression of IL-6Rα in Cxxc1-deficient TH17 cells would affect the development of EAE in vivo, we sorted CD4+GFP+ T cells, transferred them into RAG1−/− hosts, and then induced EAE with MOG35–55. Mice that received WT TH17 cells developed the most severe disease, and mice that received Cxxc1-deficient TH17 cells overexpressing Cxxc1 or IL-6Rα developed more severe disease than those that received control Cxxc1-deficient TH17 cells (Fig. 6D). In addition, both the percentage and the number of TH17 cells in the CNS/draining lymph nodes/spleens of Rag1−/− mice that received IL-6Rα–overexpressing TH17 cells were notably higher than those in the control mice (Fig. 6E). These data demonstrated that IL-6Rα could potently rescue the production of IL-17A in Cxxc1-deficient TH17 cells in vitro and in vivo and that Cxxc1 could restore IL-6Rα expression in Cxxc1-deficient TH17 cells.
DISCUSSION
Epigenetic regulation is an essential mechanism to coordinate T cell differentiation. Here, we report a crucial role of Cxxc1, which directly regulates promoter-associated H3K4me3 modification and the expression of genes such as Il6rα that are essential for TH17 cell lineage specification. The transition of naive CD4 cells to TH17 cells in Cxxc1-deficient T cells was almost completely blocked, and the cells instead developed a Treg cell–like transcriptional profile.
TH17 and Treg lineage differentiation is not only controlled by a combination of their specific cytokine milieus and TFs but also subjected to epigenetic control through various mechanisms. Previous work has shown that histones maintain TH17 cell differentiation and function by inducing histone modifications at the Il17a locus (31). The H3K4me3 methyltransferase MLL/menin/TrxG complex plays a critical role in the regulation of the TH2 cell program in murine and human systems (32). In addition, deletion of the H3K9me2 methyltransferase G9a also produces both TH17 and TH2 cell response defects (33). The conditional deletion of the H3K27me3 demethylases Jmjd3 and tripartite motif-containing 28 has been reported to have an inconsistent effect on the TH17 program, possibly due to different cellular environments (34–37). However, our data showed that the loss of Cxxc1 led to a marked CD4+ T cell lineage switch from TH17 cells to Treg cells without affecting TH1 and TH2 cell differentiation. These data suggest that Cxxc1 is essential for TH17 cell differentiation and stability.
The functional receptor for IL-6 is composed of an IL-6Rα subunit that binds IL-6 and a gp130 subunit involved in signal transduction (38). The binding of IL-6 family of cytokines to their receptors activates STAT3, which is required for TH17 cell differentiation (26, 38). IL-6Rα is highly expressed in naive T cells and in the early phase of T cell activation, while IL-6Rα expression decreases in activated T cells. IL-6/gp130/STAT3 signaling is dominant in inhibiting the conversion of conventional T cells into Foxp3+ Treg cells in vivo, and in the absence of IL-6 signaling, almost no other cytokine can inhibit the conversion of T cells to Treg cells effectively (26). Our results showed a significant reduction in the IL-6Rα protein level in Cxxc1-deficient cells under TH17 cell–polarizing conditions (TGF-β1 and IL-6) at different time points. Meanwhile, Cxxc1-deficient TH17 cells exhibited significantly defective TH17 differentiation and the strong expression of Foxp3 both in vitro and in vivo. ChIP-seq analysis revealed that Cxxc1 bound to and enhanced IL-6Rα by maintaining the appropriate H3K4me3 modification of its promoter regions. The cofactor that is bound by Cxxc1 and specifically enhances IL-6Rα expression in the early phase of TH17 cell differentiation remains to be elucidated in future studies.
Both the DNA methylation and H3K4me3 domains of Cxxc1 function in different cell types and tissues (16–18). However, our overexpression assay indicated that the histone modification of Cxxc1 mostly functions in TH17 cell differentiation and stability. Our ChIP-seq data showed that Cxxc1 bound to TSS or gene body of several key genes involved in TH17 cell differentiation, including the Runx1, Satb1, IL21, Irf4, Rorc, and Rora gene loci, was associated with a significant decrease in the H3K4me3 modification of the promoter regions of these genes in Cxxc1-deficient TH17 cells. Although we suggested that IL-6Rα was the main target of Cxxc1 in TH17 cell differentiation, there are still some other target genes regulated by Cxxc1 that provide assistance in the TH17 cell differentiation process. In addition, our TH17 data illustrate that the cell type–specific binding profile of Cxxc1 may determine its primary function, which is consistent with our former analysis in thymocyte development and macrophage function.
In summary, we identified Cxxc1 to be a critical positive regulator of TH17 development at the early stage of differentiation, in which it positively regulates autoimmune disease and bactericidal activity mainly through promoting IL-6Rα expression and the subsequent activation of downstream pathways. Our findings provide insight into the association of epigenetic regulators with TH17 development and supply important clues for therapeutic approaches for the treatment of TH17-related inflammatory and autoimmune diseases.
MATERIALS AND METHODS
Experimental animals
The Cxxc1fl/fl mouse strain has been described previously (19). The ERT2cre mice were gifts from Y. W. He (Duke University Medical Center). The dLCKcre mice (JAX:012837) and IL-17AeGFP mice (018472 C57BL/6-IL-17atm1Bcgen/J) were from The Jackson Laboratories. The RORγtcre mice (JAX: 022791) were gifts from J. Qiu (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences). Rag1−/− mice were purchased from The Jackson Laboratories. All experiments were performed with 6- to 10-week-old mice unless specified. All mice were kept in the Zhejiang University Laboratory Animal Center, and all animal experimental procedures were approved by the Animal Review Committee at Zhejiang University School of Medicine.
Experimental autoimmune encephalomyelitis
To induce EAE in Rag1−/− mice, naive CD4+ T cells (CD4+CD25-CD62LhiCD44lo) from WT and RORγtcreCxxc1fl/fl or dLckcreCxxc1fl/fl mice were sorted by the Mouse CD4 Naïve T cell Enrichment Kit (no. 8804-6824-74, Invitrogen) and intravenously transferred into Rag1−/− mice at 2 × 106 cells per mouse. Two days later, the recipient mice were subjected to EAE induction.
Female age-matched Rag1−/− mice (8 to 10 weeks old) were immunized with an emulsion containing the MOG peptide MOG35–55 (200 μg per mouse; MEVGWYRSPFSRVVHLYRNGK; Sangon) in an equal amount of complete Freund’s adjuvant (200 μl per mouse; no. 7027, Chondrex Inc.). Pertussis toxin (200 ng per mouse; no. 181, List Biological Laboratories) was administered intravenously 0 and 2 days after induction. Clinical evaluation was performed daily using a five-point scale: 0, no clinical signs; 1, limp tail; 2, paraparesis (weakness, incomplete paralysis of one or two hind limbs); 3, paraplegia (complete paralysis of two hind limbs); 4, hind limb and fore limb paralysis; and 5, moribund or death.
Isolation of CNS-infiltrating mononuclear cells
Mice were intracardially perfused with 50 ml of phosphate-buffered saline (PBS). The forebrain and cerebellum were dissected, and spinal cords were collected from the spinal canal. CNS tissue was cut into pieces and digested with collagenase D (2 μg/ml; Roche Diagnostics) and deoxyribonuclease I (DNase I; 1 μg/ml; Sigma-Aldrich) at 37°C for 20 to 30 min while rotating. Mononuclear cells were isolated by passing the tissue through a 200-mesh cell filter membrane, followed by 80%/40% Percoll gradient centrifugation. Mononuclear cells were carefully removed from the interface, washed with PBS, and resuspended in culture medium for further analysis. For cytokine analysis, mononuclear cells were stimulated for 5 hours with phorbol 12-myristate 13-acetate and ionomycin (both from Sigma-Aldrich) in the presence of brefeldin A (eBioscience) and then subjected to flow cytometry analysis to detect intracellular IL-17A, IFN-γ, and Foxp3.
Infection of mice with C. rodentium and colony-forming unit counts
RAG−/− mice were provided with autoclaved water supplemented with antibiotics [ampicillin (1 g/liter), metronidazole (1 g/liter), neomycin (1 g/liter), and vancomycin (0.5 g/liter)] for 6 days and then provided with autoclaved water for 1 day. Then, naive CD4+ T cells (CD4+CD25-CD62LhiCD44lo) from WT and RORγtcreCxxc1fl/fl mice were sorted and intravenously transferred into Rag1−/− mice at 2 × 106 cells per mouse. Two days later, the recipient mice were subjected to C. rodentium infection as described (39). Briefly, mice were gavaged with 5 × 108 C. rodentium cells in 250 μl of PBS per mouse. Bacteria were prepared by shaking at 37°C overnight in LB broth, and then, the cultures were serially diluted and plated to measure the colony-forming units. Body weight was measured daily. Fecal pellets were collected, weighed, and then homogenized in sterile PBS, and C. rodentium colonies were identified on the basis of morphology after 18 to 24 hours of incubation at 37°C on MacConkey agar plates.
Histological analyses
To analyze CNS histology, mice were euthanized 22 days after EAE induction, and spinal cords were fixed in 4% paraformaldehyde and embedded in paraffin. Sections were cut and stained with Luxol fast blue and hematoxylin and eosin (H&E). To analyze colon histology, the colons from Rag1−/− hosts 7 days after inoculation with C. rodentium were collected, treated as described above, and stained with H&E.
Isolation of LP lymphocytes
Mouse small intestines were dissected, and fat tissues and Peyer’s patches were removed. The intestines were cut open longitudinally and washed with Dulbecco’s modified Eagle’s medium (DMEM) until no fecal pellets were observed. The intestines were then cut into approximately 5-mm-long pieces. The intestinal pieces were incubated in 37°C prewarmed DMEM containing 3% fetal bovine serum (FBS), 20 mM Hepes, 5 mM EDTA, and dithiothreitol (0.15 mg/ml) for 10 min with constant agitation by droppers in a 37°C water bath. The digested cells that were collected were intraepithelial lymphocytes. Then, the left small intestine was incubated in a solution of 3% FBS, 20 mM Hepes, DNase I (0.125 mg/ml), and collagenase II (0.5 mg/ml) in 37°C prewarmed DMEM for 5 min with constant agitation by droppers in a 37°C water bath, and the dissociated cells that were collected were LP lymphocytes. Last, the collected cells were isolated by passing the tissue through a 200-mesh cell filter membrane, followed by 80%/40% Percoll (GE Healthcare) gradient centrifugation. Cells were carefully removed from the interface, washed with PBS, and resuspended in culture medium for further analysis.
Flow cytometry and related reagents
All flow cytometric data were collected on a FACS Calibur or FACS LSR II system (both from BD Biosciences) and analyzed using FlowJo analysis software v7.6.1. For intracellular cytokine staining, cells were stimulated for 5 hours at 37°C with phorbol 12-myristate 13-acetate (50 ng/ml; Sigma), ionomycin (1 mg/ml; Sigma-Aldrich), and brefeldin A (eBioscience). After staining for surface markers, cells were fixed and permeabilized according to the manufacturer’s instructions (eBioscience). Intracellular staining was processed using intracellular fixation buffer (eBioscience), and a TF staining buffer set (eBioscience) was used for RORγt and Foxp3 staining. For the detection of phosphorylated STAT3 by flow cytometry, BD Phosflow Fix Buffer I and Perm/Wash Buffer I were used.
The following antibodies (clone names are in parentheses) with different fluorochrome labels were purchased from eBioscience: CD4 (RM4-5), CD8a (53-6.7), TCRβ (H57-597), CD44 (IM7), CD62L (MEL-14), IFN-γ (XMG1.2), IL-17A (TC11-18H10.1), IL-4 (11B11), and RORγt (B2D). The following reagents were purchased from BioLegend: IL-23R (12B2B64), IL-21R (4A9), CD126 (D7715A7), Foxp3 (MF-14), and IL-17F (9D3.1C8).
For Western blot and ChIP, anti-Cxxc1 (1:1000 dilution for Western blot; 6 μg for each immunoprecipitation and ChIP reaction; ab56035) was purchased from Abcam. H3K4me3 (4 μg for each ChIP reaction; 39915) was purchased from Active Motif. Anti–pC-SMAD2 (3101), anti-SMAD2 (3103), anti-SMAD3 (9523), anti–pC-SMAD3 (9520), anti-STAT3 (Tyr705) (9131), anti-STAT3 (9132), anti-Erk (Thr202/Tyr204) (4370), anti-Erk (4695), anti-JNK (T183/Y185) (9251), and anti-JNK (9258) were obtained from Cell Signaling Technology.
Isolation and differentiation of naive CD4+ T cells
Naive CD4+ T cells (CD4+CD25−CD62LhiCD44lo) were purified by a FACS Aria II flow cytometer or sorted by the Mouse CD4 Naïve T cell Enrichment Kit (no. 8804-6824-74, Invitrogen). Naive CD4+T cells were cultured with irradiated (30 Gy) anaphase-promoting complex sorted from spleen at a ratio of 1:3 and were activated with anti-CD3 (2 μg/ml) and anti-CD28 (3 μg/ml) in a 48-well plate (5 × 105 T cells per well). T cells were cultured in RPMI 1640 medium supplemented with 10% FBS, sodium pyruvate, penicillin-streptomycin, and 2-mercaptoethanol.
For nonpathogenic TH17 cell differentiation, culture medium was supplemented with IL-6 (20 ng/ml), TGF-β1 (5 ng/ml), anti–IL-4 (10 ng/ml), anti–IL-12 (10 ng/ml), and anti–IFN-γ (10 ng/ml). For pathogenic TH17 cells differentiation, culture medium was supplemented with IL-1β (20 ng/ml), IL-6 (20 ng/ml), and IL-23 (20 ng/ml), anti–IL-4 (10 ng/ml), anti–IL-12 (10 ng/ml), and anti–IFN-γ (10 ng/ml). Other T cell differentiation were performed: TH1, IL-12 (20 ng/ml) and anti–IL-4 (10 mg/ml); TH2, IL-4 (50 ng/ml), anti–IFN-γ (10 ng/ml), and anti–IL-12 (10 mg/ml); iTreg cells, TGF-β1 (5 ng/ml), anti–IL-4 (10 ng/ml), anti–IL-12 (10 ng/ml), and anti–IFN-γ (10 ng/ml). Neutralizing anti–IFN-γ (XMG1.2), anti–IL-4 (11B11), and anti–IL-12 (C17.8) were from BioLegend.
Retrovirus production and cell transfection
Retroviruses were produced in Plat-E cells. Plat-E cells were transfected with pMX-IRES-GFP plasmids containing the indicated genes, and the medium was replaced twice with 3 ml of fresh medium every 10 hours after transfection. The retrovirus-containing supernatant was collected 72 hours after the medium was replaced for the second time and used to infect T cells.
Sorted naive CD4+ T cells were differentiated into TH17 cells in the presence of TGF-β1 and IL-6 (48-well plate, 0.5 × 106 cells per well); 20 to 24 hours later, the cells were transfected with 1 ml of the indicated retrovirus in the presence of polybrene (10 μg/ml) and 10 mM Hepes and infected for 2 hours at 1500g at 32°C. After transfection, the cells were resuspended in TH17 differentiation medium and cultured for 3 days. The indicated cytokines (e.g., IL-17A and IL-17F) and other TFs (e.g., Foxp3 and RORγt) were measured by gated CD4+GFP+ cells after retrovirus infection for 72 hours.
RNA-seq analyses
For RNA-seq, total RNA was extracted from naive CD4+ T cells differentiated in the presence of TGF-β1 (5 ng/ml) and IL-6 (20 ng/ml) for 24 or 72 hours using the RNeasy kit (Qiagen). Library construction and sequencing were performed on a BGISEQ-500 platform by the Wuhan Genomic Institution (www.genomics.org.cn; BGI, Shenzhen, China). All reads were mapped to the mm10 mouse genome, and the uniquely mapped reads were subjected to RNA-seq data analysis using the Hierarchical Indexing for Spliced Alignment of Transcripts system (40).
ChIP and data analyses
ChIP assays were performed according to the manufacturer’s instructions with modifications using the ChIP-IT kit (Active Motif, USA). Briefly, the TH17 cells were fixed with 1% formaldehyde, and then, the cross-linked chromatin was sonicated in a 4°C water bath using a Bioruptor Pico sonicator (Diagenode) to obtain DNA fragments between 150 and 500 base pairs (bp) in size. For Cxxc1 ChIP-seq, 5 × 106 TH17 of cells and 6 μg of Cxxc1 antibody were used for each sample. For H3K4me3 ChIP-seq, 3 × 106 of TH17 cells and 4 μg of H3K4me3 antibody were used for each sample.
The immunoprecipitated DNA was purified and subjected to sequencing library preparation using a VAHTSTM Universal DNA Library Prep Kit for Illumina V2 (Vazyme Biotech Co. Ltd.) according to the manufacturer’s protocol. The DNA libraries were then sequenced with an Illumina HiSeq X Ten system at Veritas Genetics in Hangzhou.
Sequenced reads of 150 bp were obtained using the CASAVA 1.8.2 package (Illumina). All reads were mapped to the mm10 mouse genome, and uniquely mapped reads were subjected to a further peak identification process. MACS2_V2.1.1 was used to identify significant peaks (q = 0.05) with both input DNA and ChIP DNA in Cxxc1-deficient cells as controls. The output of the peak files was converted by IGV browser. To calculate the tag density for Cxxc1-binding sites or H3K4me3 modifications around the TSS or at the centers of CGIs, uniquely mapped tags were summarized in 100-bp windows, and all window tag counts were normalized by the total number of bases in the windows and the total read number for the given sample.
Statistical analyses
Statistical analyses were performed using GraphPad Prism (GraphPad Software). The statistical significance was determined by Student’s t test. All error bars shown in this article represent SDs. Significance levels (P values) are presented in the figures.
Supplementary Material
Acknowledgments
We thank X. L. Liu (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences) for gifts of cell lines; J. Qiu (Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences) for providing C. rodentium and mice; L. Shen (Zhejiang University) for helping with ChIP-seq data analysis; H. Y. Fang (Zhejiang University) for providing reagents; and L. R. Lu and D. Wang for their helpful discussion. We thank Y. Y. Huang, Y. W. Li, and J. J. Wang (Zhejiang University) for helping with the cell sorting; Y. Zhang and R. Ma (Zhejiang University) for feeding the mice. Funding: This work was supported, in part, by grants from the National Basic Research Program of China 973 Program (2015CB943301), the National Natural Science Foundation of China (81830006, 31670887, 31870874, and 31800734), Zhejiang Provincial Key Project of Research and Development (2019C03043), the Zhejiang Natural Science Foundation (LQ16H030003), and the Zhejiang Science and Technology Program(2017C37117 and 2017C37170). Author contributions: L.Wa. and F.L. designed the research. F.L., X.M., Y.G., W.C., Q.X., Z.H., W.L., J.C., S.H., and X.Z. performed the experiments and data analysis. L.Wa. and F.L. wrote the manuscript. L.L., C.W., J.W., W.Q., L.We., and D.W. provided expertise and advice. L.We. and L.Wa. supervised the project. Competing interests: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The ChIP-seq and RNA-seq datasets were deposited in the GSE accession:GSE132208 and the SRA accession: PRJNA545626. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax1608/DC1
Supplementary Materials and Methods
Fig. S1. Phenotype of Cxxc1 conditional KO mice.
Fig. S2. Analysis of T cell differentiation in vitro from WT and Cxxc1-deficient mice.
Fig. S3. Phenotype of RORγtcreCxxc1wt/wt and RORγtcreCxxc1fl/fl mice.
Fig. S4. Cxxc1 deficiency restricts T cell–mediated autoimmunity.
Fig. S5. Cxxc1 deficiency increased sensitivity to C. rodentium infection.
Fig. S6. Cxxc1 regulates TH17 differentiation with its H3K4me3 function.
Fig. S7. Genome-wide maps of Cxxc1 binding and H3K4me3 modifications in TH17 cells differentiated from naive CD4+ T cells with TGF-β1 and IL-6 for 24 hours.
Fig. S8. The IL-6–Stat3 signaling pathway was defective after Cxxc1 deletion.
Fig. S9. Cxxc1 mediates TH17 cell differentiation by mediating IL-6Rα expression.
Table S1. Down regulated genes in Cfp1 deficient Th17 cells.
REFERENCES AND NOTES
- 1.Wei L., Laurence A., Elias K. M., O’Shea J. J., IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J. Biol. Chem. 282, 34605–34610 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korn T., Bettelli E., Oukka M., Kuchroo V. K., IL-17 and Th17 cells. Annu. Rev. Immunol. 27, 485–517 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Ivanov I. I., McKenzie B. S., Zhou L., Tadokoro C. E., Lepelley A., Lafaille J. J., Cua D. J., Littman D. R., The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Langrish C. L., Chen Y., Blumenschein W. M., Mattson J., Basham B., Sedgwick J. D., McClanahan T., Kastelein R. A., Cua D. J., IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201, 233–240 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng Y., Valdez P. A., Danilenko D. M., Hu Y., Sa S. M., Gong Q., Abbas A. R., Modrusan Z., Ghilardi N., de Sauvage F. J., Ouyang W., Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Zhou L., Ivanov I. I., Spolski R., Min R., Shenderov K., Egawa T., Levy D. E., Leonard W. J., Littman D. R., IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 8, 967–974 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Nurieva R., Yang X. O., Martinez G., Zhang Y., Panopoulos A. D., Ma L., Schluns K., Tian Q., Watowich S. S., Jetten A. M., Dong C., Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 448, 480–483 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Okamoto K., Iwai Y., Oh-Hora M., Yamamoto M., Morio T., Aoki K., Ohya K., Jetten A. M., Akira S., Muta T., Takayanagi H., IκBζ regulates TH17 development by cooperating with ROR nuclear receptors. Nature 464, 1381–1385 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Geng J., Yu S., Zhao H., Sun X., Li X., Wang P., Xiong X., Hong L., Xie C., Gao J., Shi Y., Peng J., Johnson R. L., Xiao N., Lu L., Han J., Zhou D., Chen L., The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat. Immunol. 18, 800–812 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Wei G., Wei L., Zhu J., Zang C., Hu-Li J., Yao Z., Cui K., Kanno Y., Roh T.-Y., Watford W. T., Schones D. E., Peng W., Sun H.-w., Paul W. E., O’Shea J. J., Zhao K., Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoenborn J. R., Dorschner M. O., Sekimata M., Santer D. M., Shnyreva M., Fitzpatrick D. R., Stamatoyonnapoulos J. A., Wilson C. B., Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-γ. Nat. Immunol. 8, 732–742 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makar K. W., Wilson C. B., DNA methylation is a nonredundant repressor of the Th2 effector program. J. Immunol. 173, 4402–4406 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Fields P. E., Lee G. R., Kim S. T., Bartsevich V. V., Flavell R. A., Th2-specific chromatin remodeling and enhancer activity in the Th2 cytokine locus control region. Immunity 21, 865–876 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Kitagawa Y., Wing J. B., Sakaguchi S., Transcriptional and epigenetic control of regulatory T cell development. Prog. Mol. Biol. Transl. Sci. 136, 1–33 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Akimzhanov A. M., Yang X. X. O., Dong C., Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J. Biol. Chem. 282, 5969–5972 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Clouaire T., Webb S., Skene P., Illingworth R., Kerr A., Andrews R., Lee J.-H., Skalnik D., Bird A., Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 26, 1714–1728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown D. A., Di Cerbo V., Feldmann A., Ahn J., Ito S., Blackledge N. P., Nakayama M., McClellan M., Dimitrova E., Turberfield A. H., Long H. K., King H. W., Kriaucionis S., Schermelleh L., Kutateladze T. G., Koseki H., Klose R. J., The SET1 complex selects actively transcribed target genes via multivalent interaction with CpG island chromatin. Cell Rep. 20, 2313–2327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomson J. P., Skene P. J., Selfridge J., Clouaire T., Guy J., Webb S., Kerr A. R. W., Deaton A., Andrews R., James K. D., Turner D. J., Illingworth R., Bird A., CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 464, 1082–1086 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao W., Guo J., Wen X., Miao L., Lin F., Xu G., Ma R., Yin S., Hui Z., Chen T., Guo S., Chen W., Huang Y., Liu Y., Wang J., Wei L., Wang L., CXXC finger protein 1 is critical for T-cell intrathymic development through regulating H3K4 trimethylation. Nat. Commun. 7, 11687 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hui Z., Zhou L., Xue Z., Zhou L., Luo Y., Lin F., Liu X., Hong S., Li W., Wang D., Lu L., Wang J., Wang L., Cxxc finger protein 1 positively regulates GM-CSF-derived macrophage phagocytosis through Csf2rα-mediated signaling. Front. Immunol. 9, 1885 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sinha S., Boyden A. W., Itani F. R., Crawford M. P., Karandikar N. J., CD8+ T-cells as immune regulators of multiple sclerosis. Front. Immunol. 6, 619 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauche D., Joyce-Shaikh B., Jain R., Grein J., Ku K. S., Blumenschein W. M., Ganal-Vonarburg S. C., Wilson D. C., McClanahan T. K., Malefyt R. D. W., Macpherson A. J., Annamalai L., Yearley J. H., Cua D. J., LAG3+ regulatory T cells restrain interleukin-23 producing CX3CR1+ gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity 49, 342–352.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Tate C. M., Lee J.-H., Skalnik D. G., CXXC finger protein 1 contains redundant functional domains that support embryonic stem cell cytosine methylation, histone methylation, and differentiation. Mol. Cell. Biol. 29, 3817–3831 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boulanger M. J., Chow D.-C., Brevnova E. E., Garcia K. C., Hexameric structure and assembly of the interleukin-6/IL-6 α-receptor/gp130 complex. Science 300, 2101–2104 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Jones G. W., McLoughlin R. M., Hammond V. J., Parker C. R., Williams J. D., Malhotra R., Scheller J., Williams A. S., Rose-John S., Topley N., Jones S. A., Loss of CD4+ T cell IL-6R expression during inflammation underlines a role for IL-6 trans signaling in the local maintenance of Th17 cells. J. Immunol. 184, 2130–2139 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Korn T., Mitsdoerffer M., Croxford A. L., Awasthi A., Dardalhon V. A., Galileos G., Vollmar P., Stritesky G. L., Kaplan M. H., Waisman A., Kuchroo V. K., Oukka M., IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. U.S.A. 105, 18460–18465 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korn T., Bettelli E., Gao W., Awasthi A., Jager A., Strom T. B., Oukka M., Kuchroo V. K., IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature 448, 484–487 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malhotra N., Robertson E., Kang J., SMAD2 is essential for TGFβ-mediated Th17 cell generation. J. Biol. Chem. 285, 29044–29048 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez G. J., Zhang Z., Chung Y., Reynolds J. M., Lin X., Jetten A. M., Feng X. H., Dong C., Smad3 differentially regulates the induction of regulatory and inflammatory T cell differentiation. J. Biol. Chem. 284, 35283–35286 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pernet V., Joly S., Jordi N., Dalkara D., Guzik-Kornacka A., Flannery J. G., Schwab M. E., Misguidance and modulation of axonal regeneration by Stat3 and Rho/ROCK signaling in the transparent optic nerve. Cell Death Dis. 4, e734 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe Y., Onodera A., Kanai U., Ichikawa T., Obata-Ninomiya K., Wada T., Kiuchi M., Iwamura C., Tumes D. J., Shinoda K., Yagi R., Motohashi S., Hirahara K., Nakayama T., Trithorax complex component Menin controls differentiation and maintenance of T helper 17 cells. Proc. Natl. Acad. Sci. U.S.A. 111, 12829–12834 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamashita M., Hirahara K., Shinnakasu R., Hosokawa H., Norikane S., Kimura M. Y., Hasegawa A., Nakayama T., Crucial role of MLL for the maintenance of memory T helper type 2 cell responses. Immunity 24, 611–622 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Antignano F., Burrows K., Hughes M. R., Han J. M., Kron K. J., Penrod N. M., Oudhoff M. J. S., Wang S. K., Min P. H., Gold M. J., Chenery A. L., Braam M. J., Fung T. C., Rossi F. M., McNagny K. M., Arrowsmith C. H., Lupien M., Levings M. K., Zaph C., Methyltransferase G9A regulates T cell differentiation during murine intestinal inflammation. J. Clin. Invest. 124, 1945–1955 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z., Cao W., Xu L., Chen X., Zhan Y., Yang Q., Liu S., Chen P., Jiang Y., Sun X., Tao Y., Hu Y., Li C., Wang Q., Wang Y., Chen C. D., Shi Y., Zhang X., The histone H3 lysine-27 demethylase Jmjd3 plays a critical role in specific regulation of Th17 cell differentiation. J. Mol. Cell Biol. 7, 505–516 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Li Q., Zou J., Wang M., Ding X., Chepelev I., Zhou X., Zhao W., Wei G., Cui J., Zhao K., Wang H. Y., Wang R.-F., Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T-cell differentiation. Nat. Commun. 5, 5780 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chikuma S., Suita N., Okazaki I. M., Shibayama S., Honjo T., TRIM28 prevents autoinflammatory T cell development in vivo. Nat. Immunol. 13, 596–603 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Jiang Y., Liu Y., Lu H., Sun S.-C., Jin W., Wang X., Dong C., Epigenetic activation during T helper 17 cell differentiation is mediated by Tripartite motif containing 28. Nat. Commun. 9, 1424 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quintana F. J., Old dog, new tricks: IL-6 cluster signaling promotes pathogenic TH17 cell differentiation. Nat. Immunol. 18, 8–10 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Qiu J., Heller J. J., Guo X., Chen Z.-m., Fish K., Fu Y.-X., Zhou L., The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 36, 92–104 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax1608/DC1
Supplementary Materials and Methods
Fig. S1. Phenotype of Cxxc1 conditional KO mice.
Fig. S2. Analysis of T cell differentiation in vitro from WT and Cxxc1-deficient mice.
Fig. S3. Phenotype of RORγtcreCxxc1wt/wt and RORγtcreCxxc1fl/fl mice.
Fig. S4. Cxxc1 deficiency restricts T cell–mediated autoimmunity.
Fig. S5. Cxxc1 deficiency increased sensitivity to C. rodentium infection.
Fig. S6. Cxxc1 regulates TH17 differentiation with its H3K4me3 function.
Fig. S7. Genome-wide maps of Cxxc1 binding and H3K4me3 modifications in TH17 cells differentiated from naive CD4+ T cells with TGF-β1 and IL-6 for 24 hours.
Fig. S8. The IL-6–Stat3 signaling pathway was defective after Cxxc1 deletion.
Fig. S9. Cxxc1 mediates TH17 cell differentiation by mediating IL-6Rα expression.
Table S1. Down regulated genes in Cfp1 deficient Th17 cells.