

Two distinct antagonist-bound structures of CysLT1R reveal unique ligand-binding modes and signaling mechanisms.
Abstract
The G protein–coupled cysteinyl leukotriene receptor CysLT1R mediates inflammatory processes and plays a major role in numerous disorders, including asthma, allergic rhinitis, cardiovascular disease, and cancer. Selective CysLT1R antagonists are widely prescribed as antiasthmatic drugs; however, these drugs demonstrate low effectiveness in some patients and exhibit a variety of side effects. To gain deeper understanding into the functional mechanisms of CysLTRs, we determined the crystal structures of CysLT1R bound to two chemically distinct antagonists, zafirlukast and pranlukast. The structures reveal unique ligand-binding modes and signaling mechanisms, including lateral ligand access to the orthosteric pocket between transmembrane helices TM4 and TM5, an atypical pattern of microswitches, and a distinct four-residue–coordinated sodium site. These results provide important insights and structural templates for rational discovery of safer and more effective drugs.
INTRODUCTION
Cysteinyl leukotriene receptor type 1 (CysLT1R) along with CysLT2R are two G protein (heterotrimeric GTP-binding protein)–coupled receptors (GPCRs) activated by the endogenous leukotrienes LTC4, LTD4, and LTE4, produced from arachidonic acid (1). CysLT1R is a nanomolar affinity receptor for LTD4, with lower affinities for LTC4 and LTE4 (2). It is broadly expressed in most types of leukocytes, lung, spleen, intestines, pancreas, prostate, and smooth muscle, predominately activating Gq/11, while also signaling through Gi/o and through G protein–independent pathways (1). CysLT1R is a key player in allergic and inflammatory disorders, such as asthma, allergic rhinitis, atopic dermatitis, and urticaria, and is involved in cardiovascular diseases and several types of cancer (3–8). The selective CysLT1R antagonists montelukast, zafirlukast, and pranlukast are often used for the treatment of asthma, allergic rhinitis, and rhinosinusitis (9). Despite their bronchodilator and anti-inflammatory properties, they have shown low effectiveness in some patients and gastrointestinal symptoms and neuropsychiatric side effects have been reported (8, 10–12), although no causal relationship has yet been established. To gain deeper understanding into the functional mechanisms of CysLTRs, we determined the crystal structures of CysLT1R in complexes with zafirlukast and pranlukast at 2.5- and 2.7-Å resolution, respectively.
RESULTS
CysLT1R structure determination using synchrotron and x-ray free-electron laser (XFEL) sources
Human CysLT1R was engineered for crystallization by fusion of a thermostabilized apocytochrome b562RIL (13) into the third intracellular loop (ICL3) and a truncation of the C terminus. This minimally modified construct was crystallized in lipidic cubic phase (LCP) (14) as a complex with zafirlukast or pranlukast. Crystals of CysLT1R bound to pranlukast (CysLT1R-pran) reached an average size of 250 μm by 15 μm by 15 μm, and the structure was obtained using four cryocooled crystals at a synchrotron source. Crystals of CysLT1R bound to zafirlukast (CysLT1R-zafir), however, could not be optimized to grow larger than 10 μm, and therefore, the structure was obtained with an x-ray free-electron laser (15) at room temperature (RT) using 5 μm by 2 μm by 2 μm microcrystals. CysLT1R-pran structure was solved in a monoclinic P21 space group with one monomer per asymmetric unit, while CysLT1R-zafir structure was obtained in a triclinic P1 space group with two monomers per asymmetric unit, forming a parallel dimer through the TM4 (transmembrane helix 4)–TM6 interface (figs. S1 to S3 and table S1).
Overall receptor architecture
Overall, CysLT1R-zafir and CysLT1R-pran structures share the same canonical heptahelical transmembrane domain (7TM) fold and similar conformations [Cα root mean square deviation (RMSD), 1.3 Å] (Fig. 1, A and B); however, the chemically distinct ligands demonstrate different binding modes, resulting in major deviations observed in the extracellular parts of TM5 and the second extracellular loop (ECL2), as discussed below. As expected, we captured both CysLT1R antagonist structures in an inactive state, with the TM6 conformation resembling that in other inactive structures, e.g., β2-adrenergic receptor (β2AR) and P2Y1R (Fig. 2C). The structure of CysLT1R-pran reveals a new disulfide bond variant between residues C141.23 and C2677.25 [superscript refers to the Ballesteros-Weinstein GPCR residue numbering scheme (16)], connecting the extracellular tips of the unusually long TM1 and TM7 helices (Fig. 1A). While disulfide bonds between an extended N terminus and the TM7-ECL3 junction have been observed in many GPCRs, a disulfide bond directly linking and stabilizing two TMs has not been previously reported. Supporting the importance of this disulfide bond for ligand binding and signaling, alanine mutations of either of these cysteines resulted in a two- to sixfold drop in the LTD4 potency (table S2).
Fig. 1. Overall structure of CysLT1R and its comparison with other receptors.
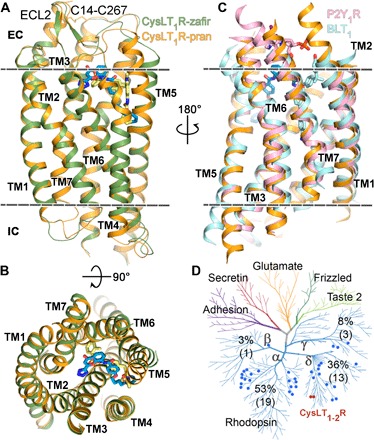
(A) Side and (B) top views of CysLT1R-pran (receptor, orange; ligand, blue) and CysLT1R-zafir (receptor, green; ligand, yellow). (C) Superposition of CysLT1R-pran with BLT1-BIIL260 [Protein Data Bank (PDB) ID 5X33, light blue] and P2Y1R-BPTU (PDB ID 4XNV, pink). (D) Distribution of lipid receptors on the human GPCR sequence homology tree. CysLT1R and CysLT2R are marked as red dots, and other lipid receptors are marked as blue dots. The percentage of the lipid receptors located on each branch is shown, with the exact numbers given in parenthesis. The membrane boundaries are shown as dashed lines in (A) and (C).
Fig. 2. Functional motifs of CysLT1R show unusual inactive-state features.
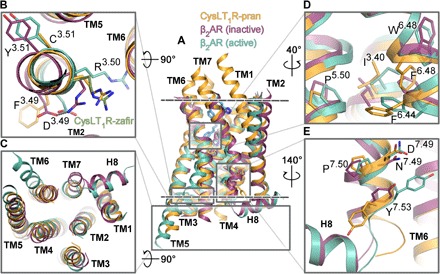
(A) Superposition of CysLT1R-pran (orange) with β2AR in inactive (PDB ID 2RH1; violet) and active (PDB ID 3SN6; teal) conformations. The membrane boundaries are shown as dashed lines. Loops are removed for clarity. (B to E) Zoom in on functional elements: DRY motif (B), intracellular region (C), P-I-F motif (D), and NPxxY motif (E). A different conformation of R1213.50 in CysLT1R-zafir (chain A) is shown as green sticks in (B).
In both CysLT1R structures, electron density terminates at a flexible SGG3008.48 motif, and the intracellular helix 8 (H8) is not resolved, despite the presence of an amphipathic α-helical F8.50xxxLxxF motif. While a glycine in the position 8.47 occurs in many GPCRs, the GG8.48 pair is unique for CysLT1R (with an exception of BLT1 that has a GG8.48G motif), introducing an additional flexibility in the TM7-H8 junction. A natural single-nucleotide variant G3008.48S was linked to a strong atopic phenotype in the Tristan da Cunha population, markedly increasing their predisposition to asthma (17). In our IP1 assays, the mutation G3008.48S showed improved signal transduction in agreement with previous studies (18), while the mutation G3008.48E (as in CysLT2R) abolished cell surface expression, and the C-terminal truncation at G3008.48 rendered the receptor nonresponsive to LTD4 (table S2), supporting the importance of H8 and its conformational stability in signaling.
Unusual pattern of microswitches
Specific features that set CysLT1R apart from other class A receptors are evident in its unique combination of functional motifs or “microswitches” (Fig. 2) (19). Thus, the highly conserved DR3.50Y motif is replaced by FR3.50C in CysLT1R (Fig. 2B), eliminating the common for inactive-state structures salt bridge between D3.49 and R3.50 and conferring more flexibility to R3.50. This residue adopts two different conformations in CysLT1R-pran and CysLT1R-zafir structures (Fig. 2B). Restoring the motif by mutating F1203.49D substantially decreases receptor expression and renders the protein nonresponsive to LTD4, while C1223.51Y mutation does not change the potency of LTD4 in IP1 assays (table S2).
A conserved P5.50-I3.40-F6.44 motif has been characterized in many class A GPCRs as a key microswitch, coupling conformational changes in the orthosteric ligand pocket with transitions in the cytoplasmic G protein– or β-arrestin–binding site (20). While the P-I-F residues are conserved in CysLT1R, they are found in a different conformation than in most other antagonist-bound GPCR structures, e.g., in β2AR bound to carazolol [Protein Data Bank (PDB) ID 2RH1]. In both CysLT1R structures, concerted rearrangements of I3.40 and F6.44 rotamers result in a “switched-on” conformation of this motif, reminiscent of an active-like state (Fig. 2D). This change is apparently related to the replacement of the neighboring “toggle switch” W6.48 by F6.48. Unlike W6.48 in other GPCRs, the side chain of F6.48 in CysLT1R adopts a “downward” conformation, where it would clash with the conformation of F6.44 from the P-I-F motif found in inactive receptors. Notably, analysis of other known δ-branch GPCR structures (fig. S4) reveals that all of them also have F6.48 instead of W6.48, and all of them have a P-I-F motif in the same switched-on conformation as in CysLT1R, despite representing antagonist-bound inactive states. On the intracellular side (Fig. 2C), the TM6 of CysLT1R is well aligned with the inactive conformation of this helix in other receptors. However, the TM7 in the NPxxY7.53 motif region is shifted (~3 Å) inward compared to the inactive-state β2AR and most other GPCRs. This TM7 shift also appears to be common for inactive δ-branch class A GPCR structures (Fig. 2E).
Unique sodium-binding site
The sodium-binding site in both CysLT1R structures has a well-defined electron density for a Na+ ion (fig. S3, E to G) and is distinct from previously observed sites, representing a new variant of this key functional element of class A GPCRs (21, 22). Na+ ion in CysLT1R is coordinated by four side chains (D692.50, S1103.39, N2877.45, and D2917.49), two of which are acidic, and one water molecule, with all distances within a typical for Na+ coordination 2.4- to 2.6-Å range (Fig. 3). In contrast, in adenosine A2A receptor (A2AAR) and β1AR, Na+ is directly coordinated by only two side chains (D2.50 and S3.39) and three waters, and in proteinase-activated receptor 1 (PAR1) and delta opioid receptor (DOR) by three side chains and two waters. Moreover, the Na+ position in CysLT1R is shifted by ~1.5 Å toward the intracellular side compared to its location in α- and γ-branch receptors, resembling another δ-branch receptor, PAR1 (PDB ID 3VW7). The difference is that in CysLT1R, the N7.45 side chain directly coordinates Na+ instead of making an indirect water-mediated contact with S7.45 in PAR1. Such a tight and well-defined coordination enabled reliable assignment of Na+ at 2.5-Å resolution, which otherwise may not be sufficient for this assessment. The tight binding of Na+ is also corroborated by a relatively high Na+ affinity of 39 ± 11 mM, estimated from its effect on receptor thermostability (fig. S5A).
Fig. 3. Sodium-binding pocket in CysLT1R.
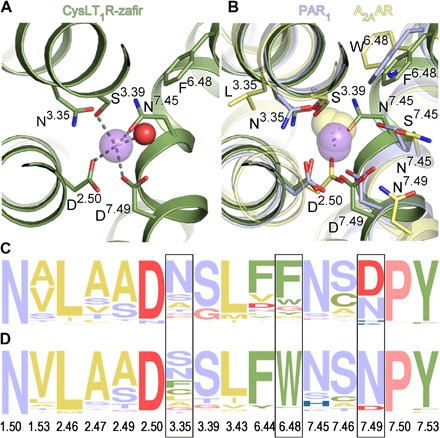
(A) Details of Na+ (purple sphere) coordination. Water molecule is shown as a red sphere. (B) Comparison of all high-resolution GPCR structures with resolved Na+. Sodium ions are shown as purple spheres for the δ-branch receptors: CysLT1R-zafir (green), PAR1 (light purple; PDB ID 3VW7), PAR2 (PDB ID 5NDD), and as yellow spheres for receptors from the α-branch: A2AAR (yellow; PDB ID 4EIY) and β1AR (turkey; PDB ID 4BVN), and the γ-branch: DOR (PDB ID 4N6H). (C and D) Frequency analysis of amino acid occurrence in the sodium pocket of the δ-branch class A GPCRs (C) and other class A receptors excluding the δ-branch (D). Yellow color marks amino acids with hydrophobic side chains; green, aromatic; red, negatively charged; blue, positively charged; purple, polar uncharged; pink, Gly and Pro. Frames indicate positions with the largest differences. The frequency analysis was performed using the weblogo.berkeley.edu server.
In agreement with the crystal structures, we observed a pronounced specific sodium effect on CysLT1R thermostability, with melting temperature increased as much as 8°C upon addition of Na+ at a physiological (150 mM) concentration (fig. S5). This effect was reproducible in the Apo state and in the presence of zafirlukast. It was specific for sodium and was reduced in D692.50N and D2917.49N mutants, with an overall receptor stability decreased in these mutants. In IP1 assays at physiological sodium concentrations, three of the sodium site mutations (N1063.35A, S1103.39A, and D2917.49N) improved agonist and decreased antagonist potencies, likely by weakening sodium binding, while N1063.35A and S1103.39A also increased constitutive activity of the receptor and reduced LTD4 efficacy (fig. S6). The latter effect of reduced signaling response was also observed for sodium site mutations in A2AAR (23). For D692.50N and N2877.45A mutations, we observed decreased LTD4 potency and/or efficacy (table S2).
CysLT1R ligand–binding pocket
The ligand-binding pocket in both CysLT1R-zafir and CysLT1R-pran structures stretches from ECL2 all the way across the receptor toward a gap between TM4 and TM5, deep in the middle section of the 7TM bundle (Fig. 4), which is different from any previously observed pockets in GPCR structures (fig. S4). Zafirlukast and pranlukast are very distinct chemically and, despite overall similar binding positions, show substantial variation in contacts within the pocket (Fig. 4, G and H). The extracellular (EC) part of the binding pocket is lined up with polar and charged residues that interact with the ligands. Thus, Y1043.33 makes extensive polar interactions with the tetrazol and benzopyran moieties of pranlukast, as well as with sulfonamide of zafirlukast, while Y2496.51 directly interacts only with zafirlukast (Fig. 4, A and D). The side chain of R792.60 anchors pranlukast via a salt bridge to its tetrazole moiety. In contrast, zafirlukast does not directly contact R792.60, and its toluene group points toward TM6 and TM7, making hydrophobic interactions. Docking of LTD4 into the CysLT1R structures suggests that the endogenous agonist follows a similar path and shares several interactions with pranlukast (fig. S7). The glycine carboxyl of LTD4 forms a salt bridge with R792.60 and a hydrogen bond with Y1043.33, mimicking the tetrazol group of pranlukast, while the cysteinyl carboxyl engages in an additional salt bridge and hydrogen bonds with R221.31, Y261.35, Y832.64, and Q2747.32. Other key LTD4 interactions include a hydrogen bond to Y2496.51, stacking with R2536.55, and hydrophobic interactions with V1865.35 and V2777.35. Mutations in most of these residues markedly reduce or completely abolish LTD4 signaling (table S2).
Fig. 4. Orthosteric ligand-binding pocket in CysLT1R.
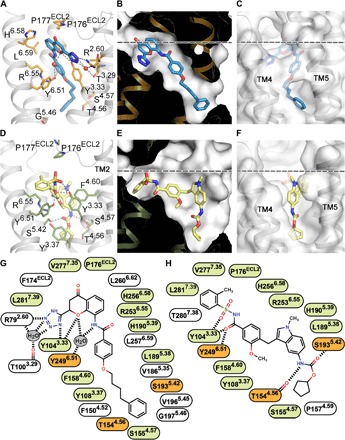
(A and D) Details of ligand-receptor interactions for pranlukast (A) and zafirlukast (D). (B and E) Pocket shapes for pranlukast (B) and zafirlukast (E). (C and F) Pocket entrance for pranlukast, closed “gate” (C), and zafirlukast, open “gate” (F). (G and H) 2D representations of receptor-ligand interactions for pranlukast (G) and zafirlukast (H). Water molecules are shown as red spheres in (A). Residues engaged in the same type of interactions with both zafirlukast and pranlukast are colored in light green, and those engaged in different types are colored in orange (G and H). The membrane boundary is shown as a dashed line in (B, C, E, and F).
Lateral entrance of ligands directly from the membrane
While the binding locations of the benzamide group of zafirlukast and pranlukast are very similar, both the ligand binding and the receptor conformation in the vicinity of the TM4-TM5 gap diverge markedly (Fig. 4, B and E). In CysLT1R-pran, the extended phenylbutyl chain of the ligand is enclosed entirely within the 7TM bundle, with only a small opening between T1544.56 and V1925.41 toward the lipid bilayer. In CysLT1R-zafir, a large-scale (~7 Å) movement of the extracellular tip of TM5 and a corresponding rearrangement in ECL2 create a wide sideways opening in the 7TM bundle, which accommodates the indole group of the ligand and is sufficient for passing the whole ligand into the pocket laterally (Fig. 4, C and F). The tilting motion of the extracellular part of TM5 is apparently facilitated by the highly conserved P2015.50 hinge, which is made even more flexible by G1975.46 as found in CysLTRs, LPA4,5,6, and BLT1. This TM5 plasticity suggests that the opening between TM4 and TM5 can serve as an important gate for lateral ligand entry into the orthosteric pocket.
Molecular dynamics reveals flexibility of the ligand access gate
Molecular dynamics simulations demonstrate that the open-gate conformation of the pocket as found in the CysLT1R-zafir complex is metastable: After zafirlukast is removed, the open pocket collapses typically within 200 ns into a closed conformation, resembling the one observed in the CysLT1R-pran structure (fig. S8 and movie S1). In some cases, however, the open-gate conformation lingers much longer due to a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid molecule entering the pocket from the membrane (movie S2). Moreover, we observed that the gate can close and open spontaneously within a 1-μs simulation (movie S3), indicating a relatively low barrier for transitions between the open and close states, supporting the lateral ligand access route into the orthosteric pocket in CysLT1R.
DISCUSSION
As described in this study, two CysLT1R structures in complex with antiasthmatic drugs pranlukast and zafirlukast shed light on the mechanism of ligand entry in lipid receptors and provide an important example of receptor plasticity in binding of two chemically distinct antagonists. The structures suggest a new GPCR activation mechanism in which the P-I-F microswitch is preactivated while the receptor is stabilized in the inactive state by a tightly bound sodium ion coordinated by four residues, including two acids. In addition, these structures suggest a potential binding mode for native cysteinyl leukotrienes and offer high-quality templates for the rational design of tool compounds and novel lead compounds for drug discovery and validation.
Despite the fact that dihydroxy and cysteinyl leukotrienes are related through arachidonic acid synthetic pathways, CysLT1R shares little sequence identity with leukotriene B4 receptors (BLT1/BLT2, 26/20% identity in 7TM domain, respectively) located on the γ-branch of class A GPCRs (19, 24). Instead, CysLT1R evolutionary belongs to the δ-branch together with purinergic (29% identity with P2Y1R), proteinase-activated (29% identity with PAR1), and platelet-activating factor (29% identity with PAFR) receptors. This observation is corroborated by a close structural similarity to P2Y1R (25) (Cα RMSD, 1.1 Å on 90% of residues in 7TM) compared with BLT1 (26) (Cα RMSD, 2.3 Å) (fig. S4). Such a high level of structural similarity as that between CysLT1R and P2Y1R is typically observed within members of GPCR subfamilies, which is especially noteworthy because CysLT1R and P2Y1R bind completely different types of ligands (lipids versus nucleotides) and share only 3 identical out of 30 residues in their respective orthosteric binding pockets.
Since the first observation of sodium effect distinguishing between antagonist and agonist binding in opioid receptors over 45 years ago (27) and the more recent discovery of an allosteric sodium-binding site in the A2AAR (28), sodium ion is emerging as an essential modulator of signaling in many class A GPCRs (21, 22). CysLT1R structure reveals a new type of the sodium-binding site coordinated by four residues and one water. The exact combination of the four sodium–coordinating residues, D2.50, D7.49, N7.45, and S3.39, occurs in sequences of 39 (about 6%) of nonolfactory class A GPCRs, suggesting that tight coordination of Na+ may be important for the function of these receptors. Notably, PAR1 (29) and PAR2 (30) have been so far the only two other structures solved with D2.50 and D7.49 intact and with a Na+ ion observed in the sodium-binding pocket, while the other two GPCRs with these naturally occurring residues [P2Y1R (25) and P2Y12R (31)] had D7.49N mutations introduced to improve receptor yield and/or stability.
Cysteinyl leukotrienes are lipid signaling molecules, and in general, ligand-access routes into the orthosteric pocket directly from the membrane have been hypothesized for several lipid receptors. Thus, a small opening was observed between TM3 and TM4 in FFAR1 (free fatty acid receptor 1) (PDB ID 4PHU) (32), and between TM1 and TM7 in S1P1 (sphingosine-1-phosphate receptor 1) (PDB ID 3V2Y) (33), while access to the orthosteric pocket between TM4 and TM5 was proposed for two other δ-branch lipid receptors, PAFR (PDB ID 5ZKP) (34) and LPAR6 (lysophosphatidic acid receptor 6) (PDB ID 5XSZ) (35), based on docking in PAFR and the positioning of an isolated alkyl chain found in the LPAR6 crystal structure. The CysLT1R-zafir structure, however, directly captures the TM5 entry gate, jammed in its open state by the ligand, revealing details of the ligand entry and suggesting possibility for this dynamic lateral gating mechanism in other lipid receptors.
CysLTRs control bronchodilation and inflammation in the lungs, and therefore, they have been considered as prominent targets for the treatment of asthma. According to the Centers for Disease Control and Prevention, asthma is a highly debilitating chronic condition, widespread among children, affecting quality of life and productivity of almost 10% of the population of our modern society and may result in death (36). This work should contribute toward a better understanding of the disease and development of more efficient and safe treatments against asthma and associated disorders, directly improving lives of millions of people.
MATERIALS AND METHODS
Protein engineering for structural studies
The human wild-type CysLT1R DNA was codon optimized for insect cell expression and cloned into a modified pFastBac1 vector (Invitrogen) containing an expression cassette with a hemagglutinin (HA) signal sequence, followed by a Flag tag and a 10× His tag at the N terminus. Tags were separated from the receptor sequence by the tobacco etch virus (TEV) protease cleavage site. To facilitate crystallization, a thermostabilized apocytochrome b562RIL (BRIL; PDB ID 1M6T) was fused into ICL3 of CysLT1R (K222–K223 with S and SG linkers, respectively) with the intact N terminus and the C terminus truncated after K311. A complete DNA sequence of the crystallized CysLT1R construct is provided in Supplementary Materials and Methods.
Insect cell expression and purification of the CysLT1R construct for crystallization
High-titer recombinant baculovirus (109 viral particles per milliliter) was obtained using the Bac-to-Bac Baculovirus Expression System (Invitrogen). Sf9 cells at a cell density of (2–3) × 106 cells ml−1 were infected with the virus at a multiplicity of infection of 10 with the addition of 8 μM zafirlukast (Cayman Chemical). Cells were harvested by centrifugation at 48 hours after infection and stored at −80°C until use.
Insect cell membranes were disrupted by thawing frozen cell pellets in a hypotonic buffer containing 10 mM Hepes (pH 7.5), 10 mM MgCl2, 20 mM KCl, and protease inhibitor cocktail [PIC; 500 μM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (Gold Biotechnology), 1 μM E-64 (Cayman Chemical), 1 μM leupeptin (Cayman Chemical), 150 nM aprotinin (AG Scientific)] with the ratio of 50 μl per 100 ml of lysis buffer. Extensive washing of raw membranes was performed by repeated centrifugation for 30 min at 220,000g at 4°C and resuspension in the same buffer and then in a high-salt buffer containing 10 mM Hepes (pH 7.5), 10 mM MgCl2, 20 mM KCl, 1 M NaCl, and PIC (50 μl per 200 ml of lysis buffer) (two and three times, respectively). Purified membranes were then resuspended in the presence of 25 μM zafirlukast or pranlukast, iodoacetamide (2 mg ml−1), and PIC (50 μl per 50 ml of resuspension buffer) and incubated at 4°C for 30 min before solubilization. Receptor was extracted from the membrane using 1% (w/v) n-dodecyl-β-d-maltopyranoside (DDM; Avanti Polar Lipids) and 0.2% (w/v) cholesteryl hemisuccinate (CHS; Sigma-Aldrich) with continuous stirring at 4°C for 3.5 hours. The supernatant was isolated by centrifugation at 460,000g for 45 min at 4°C and incubated with TALON IMAC (immobilized metal affinity chromatography) resin (Clontech) overnight at 4°C in the presence of 10 mM imidazole.
The resin was then washed at 4°C with six column volumes (CVs) of 100 mM Hepes (pH 7.5), 250 mM NaCl, 10% (v/v) glycerol, 0.1% (w/v) DDM, 0.02% (w/v) CHS, 15 mM imidazole, PIC (50 μl per 100 ml of buffer), 10 mM MgCl2, and 8 mM adenosine 5′-triphosphate, and with six CVs of 50 mM Hepes (pH 7.5), 250 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 30 mM imidazole, and PIC (50 μl per 100 ml of buffer). Then, the buffer was replaced with 50 mM Hepes (pH 7.5), 250 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, and 10 mM imidazole, and CysLT1R was treated with PNGase F (Sigma-Aldrich) for 4.5 hours to deglycosylate the receptor. The protein was then eluted with 5 CVs of 50 mM Hepes (pH 7.5), 250 mM NaCl, 10% (v/v) glycerol, 0.015% (w/v) DDM, 0.003% (w/v) CHS, and 300 mM imidazole. A PD-10 desalting column (GE Healthcare) was used to remove imidazole. The protein was then treated overnight at 4°C with His-tagged TEV protease (home-made) to remove the N-terminal Flag and His tags. The TEV protease and the cleaved 10× His tag were removed by incubating the sample for 1.5 hours with TALON IMAC resin. The receptor was then concentrated to 50 to 60 mg ml−1 with a 100-kDa molecular weight cutoff concentrator (Millipore). In the case of crystallization with zafirlukast, 50 μM zafirlukast (Sigma-Aldrich) was added to the elution buffer, 200 μM after the desalt procedure, and 10 μM into the washing buffers. In the case of crystallization with pranlukast, 50 μM pranlukast (Sigma-Aldrich) was added to the elution buffer and after the desalt procedure and 10 μM into the washing buffers. Protein purity and monodispersity were tested by SDS–polyacrylamide gel electrophoresis and analytical size exclusion chromatography (aSEC). Typically, protein purity exceeded 95%, and aSEC profiles showed a single peak with less than 10% aggregation level (fig. S1A). Protein stability was assessed by a microscale thermoshift assay using a 10 μM CPM [7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin, Invitrogen; λex = 384 nm; λem = 470 nm] dye on a Rotorgene-Q (QIAGEN) instrument (37). Typical melting temperatures were higher than 70°C (fig. S1B).
LCP crystallization of CysLT1R
The CysLT1R-BRIL in complex with pranlukast was crystallized using the LCP method by mixing 40% of protein (50 mg ml−1) with 60% of lipid (monoolein and cholesterol, 9:1, w/w) using a syringe lipid mixer (15). After a clear LCP formed, the mixture was dispensed onto 96-well glass sandwich plates (Marienfeld) as 40-nl drops and overlaid with 800 nl of precipitant solutions using an NT8-LCP robot (FORMULATRIX). Crystals appeared after 2 to 10 days and reached their full size within 2 weeks using 100 mM sodium citrate (pH 6), 200 to 600 mM lithium nitrate, and 30 to 38% (v/v) polyethylene glycol 400 (PEG400) as a precipitant solution. Typical crystal size was 250 μm by 15 μm by 15 μm, and crystals had a twiggy shape (fig. S1D). Crystals were harvested directly from LCP using 100- to 200-μm MicroMount loops (MiTeGen) and flash frozen in liquid nitrogen. For the XFEL data collection, CysLT1R-BRIL purified with zafirlukast was crystallized in syringes as described (38), using 75 to 175 mM sodium phosphate, 24 to 34% (v/v) PEG400, 100 mM Hepes (рН 7), and 1 μM zafirlukast as a precipitant. Typical crystal size was 5 μm by 2 μm by 2 μm (fig. S1C).
Data collection using synchrotron radiation
Crystallographic data collection of CysLT1R-pran crystals was performed at the ID30b beamline of the European Synchrotron Radiation Facility (ESRF), Grenoble, France. Overall, nine CysLT1R-pran datasets (two 90° and seven 10° wedges) were collected from four needle-shaped crystals using a PILATUS 6M detector and an unattenuated 20-μm beam (wavelength, 0.97717 Å). Data were collected using 0.1° to 0.2° oscillations per frame with an exposure calculated by BEST (39), so that the overall dose per dataset was 20 MGy. Data were processed using XDS (x-ray detector software) and scaled with XSCALE (40). Rejection of outliers was performed using correlation coefficients produced by XSCALE, as described (41). Drop of Rmeas in the lowest-resolution shell after an individual dataset exclusion was used as a supporting criterion of its nonisomorphism.
Data collection using x-ray free-electron laser
XFEL data of CysLT1R-zafir crystals were collected at the CXI (coherent x-ray imaging) instrument of the Linac Coherent Light Source (LCLS) at SLAC National Accelerator Laboratory, Menlo Park, California. The LCLS was operated at a wavelength of 1.302 Å (9.52 keV) delivering individual x-ray pulses of 43-fs duration focused into a spot size of ~1.5 μm in diameter using a pair of Kirkpatrick-Baez mirrors. LCP laden with protein microcrystals was injected at RT inside a vacuum chamber into the beam focus region using an LCP injector (42) with a 50-μm-diameter capillary at a flow rate of 0.3 μl min−1. Microcrystals ranged in size from 2 to 10 μm. Diffraction data were collected at a pulse repetition rate of 120 Hz with the 2.3-megapixel Cornell-SLAC Pixel Array Detector. The beam was attenuated to 6.3% of full intensity (1.9 × 1010 photons per pulse) to avoid detector saturation. A total number of 315,374 detector images were collected, of which 40,720 were identified as potential crystal hits with more than 20 Bragg peaks using Cheetah (43), corresponding to an average hit rate of 13%. Autoindexing and structure factor integration of the crystal hits were performed using the Monte Carlo integration routine with the “pushres 2.4” option implemented in CrystFEL (version 0.6.3 + 23ea03c7) (44). Peak detection parameters were extensively optimized for Cheetah, and experimental geometry was refined for CrystFEL with geoptimizer (45). The overall time of data collection from a sample with a total volume of about 15 μl was approximately 45 min and yielded 30,129 indexed patterns (indexing rate 90%).
Structure determination
The structure of CysLT1R-pran was initially solved by molecular replacement using the program Phaser (46) with two independent search models of the P2Y1R 7TM domain (PDB ID 4XNW) and BRIL from the high-resolution A2AAR structure (PDB ID 4EIY). Then, CysLT1R-zafir structure was determined by molecular replacement using 7TM domain and BRIL from the final CysLT1R-pran structure.
Model building for both CysLT1R-pran and CysLT1R-zafir structures was performed by cycling between manual inspection with Coot (47) using both 2mFo-DFc and mFo-DFc maps and automatic refinement with autoBUSTER (48) or phenix.refine (49). Restraints for pranlukast and zafirlukast were generated using the web server GRADE (version 1.2.9, available at www.globalphasing.com) and phenix.elbow. The correct orientation of the methylbenzene ring in zafirlukast (fig. S3B) was selected on the basis of the B factor and density fit comparison between two possible conformations.
In addition to zafirlukast modeled in the orthosteric binding site, an additional zafirlukast molecule was found in molecule B only, located mostly in the lipid bilayer just outside of the receptor-binding site (fig. S2, C and D). This zafirlukast molecule was likely trapped in this location due to a relatively high ligand concentration used during crystallization.
The CysLT1R-zafir structures from two molecules A and B in the asymmetric unit show very high similarity (RMSD, 0.7 Å within 7TM; RMSD, 1.1 Å for the whole structure). The main differences include a 4-Å deviation of a flexible part of ECL2 and conformations of several side chains exposed to the lipid bilayer and solvent. The final data collection and refinement statistics are shown in table S1.
Molecular docking
To prepare LTD4 for molecular docking, we extracted its chemical structure from the PubChem web database (https://pubchem.ncbi.nlm.nih.gov/compound/5280878), assigned charges at pH 7.0, and generated three-dimensional (3D) ligand structure from its 2D representation, using the Monte Carlo optimization and the MMFF-94 force field. As for the receptor, we preprocessed the CysLT1R-pran structure by adding missing residues, optimizing side-chain rotamers, and removing water molecules. Rectangular box enclosing the ligand-binding site of pranlukast in CysLT1R was used as the sampling space for docking. We kept the receptor rigid during the docking simulations and sampled ligand conformations in the internal coordinate space using biased probability Monte Carlo optimization with the sampling parameter (docking effort) set to 50. We performed three independent docking runs and selected the binding pose with the lowest docking score. All simulations were performed using the ICM-Pro software package (Molsoft, San Diego).
IP1 production assay
For the CysLT1R functional assays, the initial pcDNA3.1(+)-CYSLTR1 plasmid coding for CysLT1 wild-type receptor, cMyc tag for labeling, and β-globin intron for stability (19) was received as a gift from J. Stankova (Université de Sherbrooke, Canada). The 3× HA tag was then inserted by overlapping polymerase chain reaction (PCR) instead of cMyc for better antibody detection, and desired gene modifications (point mutations, truncations, or partner protein fusion) were introduced by overlapping PCR.
The Cisbio IP-One kit was used according to the manufacturer’s instructions. Human embryonic kidney (HEK) 293 cells were seeded onto poly-l-lysine–coated 384-well plates at 20,000 cells per well and transfected with 40 ng of DNA coding for the wild-type CysLT1R or for the CysLT1R mutants using the X-tremeGENE HP (Roche) agent. At 48 hours after transfection, the medium was removed, and the cells were washed with fresh Hanks’ balanced salt solution. Cells were either stimulated directly with a range of LTD4 concentrations (10−12 to 10−6 M) prepared in IP1 stimulation buffer, or sequentially stimulated with a range of zafirlukast and pranlukast concentrations (10−11 to 10−5 M), and LTD4 concentrations corresponding to the EC80 (80% of effective concentration) for each mutant. After equilibration for 30 min at 37°C, the cells were lysed with IP1-D2 and Ab-Crypt reagents in lysis buffer and then incubated at RT for 1 hour. The plate was read on a Tecan Genios Pro plate reader using a HTRF (homogeneous time-resolved fluorescence) filter set (λex = 320 nm, λem = 620 and 655 nm). Data were plotted using the three-parameter EC50/IC50 fit of GraphPad Prism 7 (San Diego) and represent the means ± SD of at least two independent experiments performed in quadruplicate.
Cell surface expression determined by enzyme-linked immunosorbent assay
HEK293 cells were seeded in 24-well plates coated with poly-l-lysine (Sigma-Aldrich) at 100,000 cells per well and transfected with 375 ng of plasmid coding for the wild-type or mutant CysLT1R using X-tremeGENE HP (Roche). Forty-eight hours after transfection, cells were fixed with 3.7% (v/v) formaldehyde in tris-buffered saline [TBS; 20 mM tris-HCl (pH 7.5) and 150 mM NaCl] for 5 min at RT. Cells were washed three times with TBS and incubated for 1 hour in TBS supplemented with 3% fat-free milk (w/v) to block nonspecific binding sites. A mouse monoclonal anti-HA antibody coupled to horseradish peroxidase (Roche) was added at 1:1000 dilution in TBS with 3% fat-free dry milk for 3 hours at RT. Following incubation, cells were washed twice with TBS before the addition of 250 μl of 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich). Plates were incubated at RT for 15 min, and the reaction was stopped by the addition of 250 μl of 2N HCl. The reagent (200 μl) was transferred into a 96-well plate, and the absorbance was read at 450 nm on a Tecan Genios Pro plate reader. Cells transfected with the empty pcDNA3.1(+) vector were used to determine the background. Data were plotted using GraphPad Prism 7 and represent the means ± SD of at least two independent experiments performed in quadruplicate.
Effects of monovalent ions on receptor stability
Effects of monovalent ions were assayed using the CysLT1R crystallization construct and two mutants D2.50N and D7.49N based on it. Each receptor construct was purified, as described above for the crystallization setup, using KCl instead of NaCl in hypotonic and solubilization buffers. KCl was replaced with choline chloride during the washing step on the Talon resin. After elution, the protein solution was desalted from imidazole, concentrated to 1 mg ml−1, and diluted 50 times with the assay buffer: 10 μM CPM dye, 25 mM Hepes (pH 7.5), 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, containing one of the ions (Na+, K+, or Rb+) at concentrations between 0 and 150 mM. The ionic strength in all samples was compensated by choline chloride to an overall ion concentration of 150 mM. The samples were then incubated at 4°C in the dark for 15 min, and their thermal stability was analyzed using a microscale fluorescence assay as previously described (37). Briefly, fluorescence from the CPM dye (see the purification methods part) was recorded during a temperature ramp from 25° to 80°C with a 1.5°C min−1 rate using a Rotor-Gene Q real-time PCR machine (QIAGEN). Melting curves were collected for n = 3 independent experiments in triplicates. For all constructs, experiments were carried out in the presence of 50 μM zafirlukast. In the case of the CysLT1R crystallization construct, additional experiments without ligand (CysLT1R-apo) were conducted. All melting curves were fitted in GraphPad Prism 7 using the Boltzmann sigmoidal function to obtain the melting temperatures. Plots of ΔTm versus ion concentration revealed nonspecific linear effects for K+ and Rb+ in the case of CysLT1R, and for all ions in the case of D2.50N and D7.49N mutants. To estimate sodium affinity for CysLT1R crystallization construct, ΔTm versus [Na+] curves were fitted in GraphPad Prism 7 using the “One site—Total and nonspecific binding” function, with nonspecific effect taken from K+ data. In all the other cases, the ion effects on thermostability were fitted to linear functions, and the ion effect at 150 mM concentration was calculated. The results were expressed as means ± SD for three independent experiments performed in triplicates.
Molecular dynamics simulations
The CysLT1R-pran and CysLT1R-zafir structures were preprocessed to assign their protonation states and to model missing side chains using the energy-based optimization protocols available in ICM-Pro (v3.8-6) (50). Missing loops were modeled using Loop modeling and regularization protocols in ICM-Pro. The preprocessed structures were used for molecular dynamics simulation as previously described (51), using input files generated by the CHARMM-GUI web server (52). The initial membrane coordinates were assigned by aligning the receptor models to CB1 receptor coordinates retrieved from the OPM (Orientations of Proteins in Membranes) database (53). The structure was simulated in a periodic box containing 178 POPC lipids, 11,898 water molecules, 31 sodium, and 44 chloride ions. After the initial energy minimization, the system was equilibrated for 10 ns, followed by production runs of up to 1 μs for CysLT1R-zafir and 650 ns for CysLT1R-pran apo-state structures. The simulations were performed using the Gromacs (v.2018) simulation package (54), and the plots were generated using the Matplotlib plotting package available in Python. The simulations were performed either on NVIDIA P100 GPU enabled nodes made available by the Google Cloud Platform or with NVIDIA K-80 or P100 GPU enabled clusters at the High-Performance Computing Center at the University of Southern California.
Supplementary Material
Acknowledgments
We thank S. Ustinova, A. Awawdeh, P. Utrobin, J. Velasquez, and Y. Kovalev for technical assistance; P. Buslaev and I. Maslov for help with the data analysis; R. C. Stevens and members of his group, M. Audet, C. Hanson, and M. Chu, for advice and help with the GPCR expression; and J. Stankova for providing CysLT1R plasmid. We acknowledge the Structural Biology Group of the ESRF and especially A. N. Popov for assistance with crystallographic data collection. The crystallographic synchrotron data collection experiments were performed on beamlines ID29, ID30a-3, and ID30b at the ESRF, Grenoble, France. Funding: This work was supported in part by the Russian Science Foundation project no. 19-14-00261 (A.L., A.G., E.M., A.M., E.L., and N.S.), the NIH grants R35 GM127086 (V.C.), R21 DA042298 (W.L.), and R01 GM124152 (W.L.), and the NSF Science and Technology Center grant 1231306 (U.W., W.L., and V.C.). V.G. was supported by the special agreement CEA (IBS)–HGF (FZJ) STC 5.1, the Grenoble Instruct Centre (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL), the French Infrastructure for Integrated Structural Biology (FRISBI; ANR-10-INSB-05-02), and the New Generation of Drugs for Alzheimer’s Disease project (GRAL; ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). É.B.-O. was supported by a research fellowship from the Institut de Pharmacologie de Sherbrooke (IPS) and Centre d’excellence en neurosciences de l’Université de Sherbrooke (CNS). R.B. was supported by a research fellowship from the Canadian Institutes of Health Research (CIHR) and from the Fonds de Recherche en Santé du Québec (FRQ-S). P.S. holds a Canada Research Chair in Neurophysiopharmacology of Chronic Pain, and part of this work was supported by a CIHR grant (FDN-148413). V.P. acknowledges support from the European Regional Development Fund ELIBIO (project “Structural dynamics of biomolecular systems” CZ.02.1.01/0.0/0.0/15_003/0000447). Sample preparation for XFEL was supported by the grant 18-02-40020 from the Russian Foundation for Basic Research (RFBR). Use of the LCLS, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. We acknowledge NIH Commons Credits CCREQ-2016-03-00010, Google Cloud Platform credits, and USC High-Performance Computing Center for computer resources used in MD simulations. Author contributions: A.L. and A.G. optimized the constructs, developed the expression and purification procedure, expressed and purified the proteins, screened the ligands, crystallized the protein-ligand complexes, collected synchrotron data, and prepared the initial draft. A.G., E.M., V.B., A.L., A.Bo., V.P., and A.M. collected x-ray diffraction data at ESRF. W.L., A.Ba., A.I., and V.C. collected x-ray diffraction data at LCLS. H.H. and U.W. operated the sample injector during XFEL data collection. É.B.-O., R.B., J.-M.L., and P.S. performed and analyzed the cell signaling and cell surface expression assays. E.M., V.B., and G.W.H. determined and refined the structures. V.C., V.K., A.L., A.G., V.B., P.P., E.M., and A.M. performed the data analysis/interpretation. E.L., A.S., E.E., and N.S. helped with construct optimization, protein expression, and purification. P.P. and V.K. performed molecular docking and structure analysis. N.P. and V.K. performed molecular dynamics simulations. V.K. provided initial construct design and supervised computational analysis and modeling. A.Ba. helped with XFEL data analysis. A.L., A.G., V.K., P.P., V.B., E.M., A.M., and V.C. wrote the manuscript with help from the other authors. V.C., V.K., V.G., A.M., and V.B. initiated the project. A.M. and V.B. organized the project implementation, were responsible for the overall project management, and cosupervised the research. V.C. supervised the whole project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Coordinates and structure factors have been deposited in the PDB under the accession codes 6RZ4 (CysLT1R-pran) and 6RZ5 (CysLT1R-zafir). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax2518/DC1
Supplementary Materials and Methods
Fig. S1. Protein purification and crystallization.
Fig. S2. Crystal packing.
Fig. S3. Examples of electron density.
Fig. S4. Comparison of CysLT1R-pran structure and ligand binding pose with other lipid and δ-branch class A GPCRs.
Fig. S5. Effect of monovalent cations on CysLT1R stability.
Fig. S6. Effects of LTD4 and CysLT1R antagonists on IP1 production.
Fig. S7. Docking of LTD4 in CysLT1R.
Fig. S8. MD simulations of the TM4-TM5 gate closure.
Table S1. Crystallographic data collection and refinement statistics.
Table S2. Signaling and cell surface expression data for CysLT1R.
Movie S1. Rapid closure of the ligand access gate.
Movie S2. Lipid molecule enters the ligand access gate.
Movie S3. Spontaneous opening and closing of the ligand access gate.
Reference (55)
REFERENCES AND NOTES
- 1.Bäck M., Dahlén S.-E., Drazen J. M., Evans J. F., Serhan C. N., Shimizu T., Yokomizo T., Rovati G. E., International Union of Basic and Clinical Pharmacology. LXXXIV: Leukotriene receptor nomenclature, distribution, and pathophysiological functions. Pharmacol. Rev. 63, 539–584 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Lynch K. R., O'Neill G. P., Liu Q., Im D.-S., Sawyer N., Metters K. M., Coulombe N., Abramovitz M., Figueroa D. J., Zeng Z., Connolly B. M., Bai C., Austin C. P., Chateauneuf A., Stocco R., Greig G. M., Kargman S., Hooks S. B., Hosfield E., Williams D. L., Ford-Hutchinson A. W., Caskey C. T., Evans J. F., Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature 399, 789–793 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Bäck M., Powell W. S., Dahlén S. E., Drazen J. M., Evans J. F., Serhan C. N., Shimizu T., Yokomizo T., Rovati G. E., Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR review 7. Br. J. Pharmacol. 171, 3551–3574 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burke L., Butler C. T., Murphy A., Moran B., Gallagher W. M., O’Sullivan J., Kennedy B. N., Evaluation of cysteinyl leukotriene signaling as a therapeutic target for colorectal cancer. Front. Cell Dev. Biol. 4, 103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ingelsson E., Yin L., Bäck M., Nationwide cohort study of the leukotriene receptor antagonist montelukast and incident or recurrent cardiovascular disease. J. Allergy Clin. Immunol. 129, 702–707.e2 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Laidlaw T. M., Boyce J. A., Cysteinyl leukotriene receptors, old and new; implications for asthma. Clin. Exp. Allergy 42, 1313–1320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuyama M., Yoshimura R., Cysteinyl-leukotriene1 receptor is a potent target for the prevention and treatment of human urological cancer. Mol. Med. Rep. 3, 245–251 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Capra V., Thompson M. D., Sala A., Cole D. E., Folco G., Rovati G. E., Cysteinyl-leukotrienes and their receptors in asthma and other inflammatory diseases: Critical update and emerging trends. Med. Res. Rev. 27, 469–527 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Capra V., Ambrosio M., Riccioni G., Rovati G. E., Cysteinyl-leukotriene receptor antagonists: Present situation and future opportunities. Curr. Med. Chem. 13, 3213–3226 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Asano K., Shiomi T., Hasegawa N., Nakamura H., Kudo H., Matsuzaki T., Hakuno H., Fukunaga K., Suzuki Y., Kanazawa M., Yamaguchi K., Leukotriene C4 synthase gene A(−444)C polymorphism and clinical response to a CYS-LT1 antagonist, pranlukast, in Japanese patients with moderate asthma. Pharmacogenetics 12, 565–570 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Law S. W. Y., Wong A. Y. S., Anand S., Wong I. C. K., Chan E. W., Neuropsychiatric events associated with leukotriene-modifying agents: A systematic review. Drug Safety 41, 253–265 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Shimbo J., Onodera O., Tanaka K., Tsuji S., Churg-Strauss syndrome and the leukotriene receptor antagonist pranlukast. Clin. Rheumatol. 24, 661–662 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Chun E., Thompson A. A., Liu W., Roth C. B., Griffith M. T., Katritch V., Kunken J., Xu F., Cherezov V., Hanson M. A., Stevens R. C., Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caffrey M., Cherezov V., Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 4, 706–731 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stauch B., Cherezov V., Serial femtosecond crystallography of G protein–coupled receptors. Annu. Rev. Biophys. 47, 377–397 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ballesteros J. A., Weinstein H., Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428 (1995). [Google Scholar]
- 17.Thompson M. D., Capra V., Takasaki J., Maresca G., Rovati G. E., Slutsky A. S., Lilly C., Zamel N., McIntyre Burnham W., Cole D. E., Siminovitch K. A., A functional G300S variant of the cysteinyl leukotriene 1 receptor is associated with atopy in a Tristan da Cunha isolate. Pharmacogenet. Genomics 17, 539–549 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Yaddaden L., Véronneau S., Thompson M. D., Rola-Pleszczynski M., Stankova J., Cellular signalling of cysteinyl leukotriene type 1 receptor variants CysLT1-G300S and CysLT1-I206S. Prostaglandins Leukot. Essent. Fatty Acids. 105, 1–8 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Katritch V., Cherezov V., Stevens R. C., Structure-function of the G protein–coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schönegge A.-M., Gallion J., Picard L.-P., Wilkins A. D., Le Gouill C., Audet M., Stallaert W., Lohse M. J., Kimmel M., Lichtarge O., Bouvier M., Evolutionary action and structural basis of the allosteric switch controlling β2AR functional selectivity. Nat. Commun. 8, 2169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katritch V., Fenalti G., Abola E. E., Roth B. L., Cherezov V., Stevens R. C., Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 39, 233–244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massink A., Gutiérrez-de-Terán H., Lenselink E. B., Ortiz Zacarías N. V., Xia L., Heitman L. H., Katritch V., Stevens R. C., IJzerman A. P., Sodium ion binding pocket mutations and adenosine A2A receptor function. Mol. Pharmacol. 87, 305–313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zarzycka B., Zaidi S. A., Roth B. L., Katritch V., Harnessing ion-binding sites for gpcr pharmacology. Pharmacol. Rev. 71, 571–595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fredriksson R., Lagerström M. C., Lundin L.-G., Schiöth H. B., The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63, 1256–1272 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Zhang D., Gao Z.-G., Zhang K., Kiselev E., Crane S., Wang J., Paoletta S., Yi C., Ma L., Zhang W., Han G. W., Liu H., Cherezov V., Katritch V., Jiang H., Stevens R. C., Jacobson K. A., Zhao Q., Wu B., Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 520, 317–321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hori T., Okuno T., Hirata K., Yamashita K., Kawano Y., Yamamoto M., Hato M., Nakamura M., Shimizu T., Yokomizo T., Miyano M., Yokoyama S., Na+−mimicking ligands stabilize the inactive state of leukotriene B4 receptor BLT1. Nat. Chem. Biol. 14, 262–269 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Pert C. B., Pasternak G., Snyder S. H., Opiate agonists and antagonists discriminated by receptor binding in brain. Science 182, 1359–1361 (1973). [DOI] [PubMed] [Google Scholar]
- 28.Liu W., Chun E., Thompson A. A., Chubukov P., Xu F., Katritch V., Han G. W., Roth C. B., Heitman L. H., IJzerman A. P., Cherezov V., Stevens R. C., Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang C., Srinivasan Y., Arlow D. H., Fung J. J., Palmer D., Zheng Y., Green H. F., Pandey A., Dror R. O., Shaw D. E., Weis W. I., Coughlin S. R., Kobilka B. K., High-resolution crystal structure of human protease-activated receptor 1. Nature 492, 387–392 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng R. K. Y., Fiez-Vandal C., Schlenker O., Edman K., Aggeler B., Brown D. G., Brown G. A., Cooke R. M., Dumelin C. E., Doré A. S., Geschwindner S., Grebner C., Hermansson N.-O., Jazayeri A., Johansson P., Leong L., Prihandoko R., Rappas M., Soutter H., Snijder A., Sundström L., Tehan B., Thornton P., Troast D., Wiggin G., Zhukov A., Marshall F. H., Dekker N., Structural insight into allosteric modulation of protease-activated receptor 2. Nature 545, 112–115 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Zhang J., Zhang K., Gao Z.-G., Paoletta S., Zhang D., Han G. W., Li T., Ma L., Zhang W., Müller C. E., Yang H., Jiang H., Cherezov V., Katritch V., Jacobson K. A., Stevens R. C., Wu B., Zhao Q., Agonist-bound structure of the human P2Y12 receptor. Nature 509, 119–122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srivastava A., Yano J., Hirozane Y., Kefala G., Gruswitz F., Snell G., Lane W., Ivetac A., Aertgeerts K., Nguyen J., Jennings A., Okada K., High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 513, 124–127 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Hanson M. A., Roth C. B., Jo E., Griffith M. T., Scott F. L., Reinhart G., Desale H., Clemons B., Cahalan S. M., Schuerer S. C., Sanna M. G., Han G. W., Kuhn P., Rosen H., Stevens R. C., Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao C., Tan Q., Xu C., He L., Yang L., Zhou Y., Zhou Y., Qiao A., Lu M., Yi C., Han G. W., Wang X., Li X., Yang H., Rao Z., Jiang H., Zhao Y., Liu J., Stevens R. C., Zhao Q., Zhang X. C., Wu B., Structural basis for signal recognition and transduction by platelet-activating-factor receptor. Nat. Struct. Mol. Biol. 25, 488–495 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Taniguchi R., Inoue A., Sayama M., Uwamizu A., Yamashita K., Hirata K., Yoshida M., Tanaka Y., Kato H. E., Nakada-Nakura Y., Otani Y., Nishizawa T., Doi T., Ohwada T., Ishitani R., Aoki J., Nureki O., Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature 548, 356–360 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Centers for Disease Control and Prevention, 2017 National Health Interview Survey (NHIS) Data (2018); www.cdc.gov/asthma/most_recent_national_asthma_data.htm.
- 37.Alexandrov A. I., Mileni M., Chien E. Y. T., Hanson M. A., Stevens R. C., Microscale fluorescent thermal stability assay for membrane proteins. Structure 16, 351–359 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Liu W., Wacker D., Gati C., Han G. W., James D., Wang D., Nelson G., Weierstall U., Katritch V., Barty A., Zatsepin N. A., Li D., Messerschmidt M., Boutet S., Williams G. J., Koglin J. E., Seibert M. M., Wang C., Shah S. T. A., Basu S., Fromme R., Kupitz C., Rendek K. N., Grotjohann I., Fromme P., Kirian R. A., Beyerlein K. R., White T. A., Chapman H. N., Caffrey M., Spence J. C. H., Stevens R. C., Cherezov V., Serial femtosecond crystallography of G protein-coupled receptors. Science 342, 1521–1524 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bourenkov G. P., Popov A. N., A quantitative approach to data-collection strategies. Acta Crystallogr. D Biol. Crystallogr. 62, 58–64 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giordano R., Leal R. M. F., Bourenkov G. P., McSweeney S., Popov A. N., The application of hierarchical cluster analysis to the selection of isomorphous crystals. Acta Crystallogr. D Biol. Crystallogr. 68, 649–658 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Weierstall U., James D., Wang C., White T. A., Wang D., Liu W., Spence J. C. H., Bruce Doak R., Nelson G., Fromme P., Fromme R., Grotjohann I., Kupitz C., Zatsepin N. A., Liu H., Basu S., Wacker D., Won Han G., Katritch V., Boutet S., Messerschmidt M., Williams G. J., Koglin J. E., Marvin Seibert M., Klinker M., Gati C., Shoeman R. L., Barty A., Chapman H. N., Kirian R. A., Beyerlein K. R., Stevens R. C., Li D., Shah S. T. A., Howe N., Caffrey M., Cherezov V., Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 5, 3309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barty A., Kirian R. A., Maia F. R. N. C., Hantke M., Yoon C. H., White T. A., Chapman H., Cheetah: Software for high-throughput reduction and analysis of serial femtosecond x-ray diffraction data. J. Appl. Cryst. 47, 1118–1131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White T. A., Kirian R. A., Martin A. V., Aquila A., Nass K., Barty A., Chapman H. N., CrystFEL: A software suite for snapshot serial crystallography. J. Appl. Cryst. 45, 335–341 (2012). [Google Scholar]
- 45.Yefanov O., Mariani V., Gati C., White T. A., Chapman H. N., Barty A., Accurate determination of segmented x-ray detector geometry. Opt. Express 23, 28459–28470 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.G. Bricogne, BUSTER Version 2.10.3 (Global Phasing Ltd., 2011). [Google Scholar]
- 49.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abagyan R., Totrov M., Kuznetsov D., ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 15, 488–506 (1994). [Google Scholar]
- 51.Zhang H., Han G. W., Batyuk A., Ishchenko A., White K. L., Patel N., Sadybekov A., Zamlynny B., Rudd M. T., Hollenstein K., Tolstikova A., White T. A., Hunter M. S., Weierstall U., Liu W., Babaoglu K., Moore E. L., Katz R. D., Shipman J. M., Garcia-Calvo M., Sharma S., Sheth P., Soisson S. M., Stevens R. C., Katritch V., Cherezov V., Structural basis for selectivity and diversity in angiotensin II receptors. Nature 544, 327–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jo S., Kim T., Iyer V. G., Im W., CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Lomize M. A., Pogozheva I. D., Joo H., Mosberg H. I., Lomize A. L., OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 55.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax2518/DC1
Supplementary Materials and Methods
Fig. S1. Protein purification and crystallization.
Fig. S2. Crystal packing.
Fig. S3. Examples of electron density.
Fig. S4. Comparison of CysLT1R-pran structure and ligand binding pose with other lipid and δ-branch class A GPCRs.
Fig. S5. Effect of monovalent cations on CysLT1R stability.
Fig. S6. Effects of LTD4 and CysLT1R antagonists on IP1 production.
Fig. S7. Docking of LTD4 in CysLT1R.
Fig. S8. MD simulations of the TM4-TM5 gate closure.
Table S1. Crystallographic data collection and refinement statistics.
Table S2. Signaling and cell surface expression data for CysLT1R.
Movie S1. Rapid closure of the ligand access gate.
Movie S2. Lipid molecule enters the ligand access gate.
Movie S3. Spontaneous opening and closing of the ligand access gate.
Reference (55)