

Abstract
Depression is a complex disorder that takes an enormous toll on individual health. As affected individuals display a wide variation in their clinical symptoms, the precise neural mechanisms underlying the development of depression remain elusive. Although it is impossible to phenocopy every symptom of human depression in rodents, the preclinical field has had great success in modeling some of the core affective and neurovegetative depressive symptoms, including social withdrawal, anhedonia, and weight loss. Adaptations in select cell populations may underlie these individual depressive symptoms and new tools have expanded our ability to monitor and manipulate specific cell types. This review outlines some of the most recent preclinical discoveries on the molecular and neurophysiological mechanisms in reward circuitry that underlie the expression of behavioral constructs relevant to depressive symptoms.
Introduction
Millions of individuals suffer from depressive disorders worldwide and up to 40% of patients do not adequately respond to antidepressant medications [1]. Major depressive disorder (MDD) is a symptomatically heterogeneous disease, spanning cognitive, emotional, motivational, and physiological domains. Identifying the exact etiology of MDD is challenging, as MDD patients present with a constellation of symptoms that are not likely explained by a single unifying mechanism. However, human functional imaging and postmortem tissue studies have identified abnormalities in several brain regions [2–6] including nuclei within the brain’s reward pathway. Altered reward circuit function is theorized to underlie the loss of pleasure and amotivational syndrome experienced by most MDD patients, and many studies have strived to uncover the precise circuit, cellular, and molecular adaptations responsible for this anhedonia.
Preclinical rodent models for studying depression have been useful for identifying the cell-type-specific mechanisms that are otherwise inaccessible in human studies. Although rats and mice do not likely experience the complex cognitive aspects of clinical depression, rodents, similar to humans, want to work for rewards such as food, sex, and social interaction. When either species show diminished interest in these rewards, it is termed “anhedonia” and is modeled several ways preclinically. The most common models generate anhedonia with chronic stress or by using animals selectively bred for this behavior [7]. Chronically stressed rodents have reduced reward preference, as measured by a loss of preference for sucrose solutions and time spent interacting with a novel con-specific. Stressed animals also tend to spend less time struggling against inescapable stressors such as forced swimming or tail suspension. The stress-induced behavioral phenotypes are reversed by chronic, but not acute antidepressant treatment, and are thus posited to have both face and predictive validity [8]. However, it is of paramount importance to consider a range of behavioral tests when assessing depression-like behavior in rodents. Compounds that produce acute behavioral effects in a forced-swim or tail-suspension test (i.e., increase struggling time) may not translate to meaningful therapeutics when administered chronically and the meaning of “time spent struggling” has been recently called into question [9]. For the sake of brevity, we will briefly describe the common depression models mentioned in this review and refer the reader to [7, 8] for more in-depth coverage.
Chronic unpredictable stress
One of the more widely used depression models employs a battery of chronic, mild physical stressors, termed chronic mild, variable, or unpredictable stress (herein abbreviated CUS). In this paradigm, rodents are exposed to a series of stressors such as cage tilting and disrupted lights for 8–12 weeks [10], after which animals show reduced sucrose preference. Both male and female rodents can be used in CUS paradigms and the stress results in other “depression-like behaviors,” such as increased immobility in the forced-swim test. The pattern of stressors can vary between laboratories and many groups study only animals that lose sucrose preference after stress.
Learned helplessness
In “learned helplessness,” rodents are first exposed to a series of unescapable foot shocks. Animals are then given an option to escape the shock: those that fail to escape display “learned helplessness [11],” which attempts to mimic the sense of powerlessness felt by some MDD patients. Although this model also generates a cohort of “resilient” animals (those that escape the shock), it often relies solely on “escape failures” to measure depression-like behavior.
Social defeat stress
In addition to physical stressors, the preclinical field has moved toward psychosocial stressors, as the stressful experiences that precipitate MDD likely represent a combination thereof. The most widely used and standardized psychosocial stressor is chronic social defeat stress (CSDS). CSDS subjects mice to once-daily bouts of agonistic social confrontation with a resident aggressor and continuous sensory interaction with the resident for 10 days [12]. Approximately 70% of mice that undergo CSDS are termed “stress-susceptible,” displaying a robust depression-like phenotype marked by reduced social interaction, increased anhedonia, and significant body-weight changes [13]. The remaining ~30% of mice retain preference for social interaction and are termed “stress-resilient.” Historically, CSDS has been restricted to adult male mice, but recent procedural modifications allow for inclusion of female subjects [14–16] and adolescents [17]. Each model for depression affords unique advantages and disadvantages, and we refer the reader to in-depth discussion in Czéh et al. [7].
It is impossible to cover what spans at least six decades of work on the neural underpinnings of depression. Due to the great successes of measuring reward-related behavior in preclinical models, this review will focus on how two key components of reward circuitry, the ventral tegmental area (VTA) and nucleus accumbens (NAc), undergo adaptations in models for studying depression. First, we will describe recent discoveries on how cellular and molecular adaptations in the VTA drive anhedonia. Second, we will cover similar adaptations in the NAc with a focus on the contributions of different neuron subtypes. Third, we will highlight an emerging role for inflammatory and non-neuronal cells within the reward circuit in mediating behavioral constructs relevant to depression.
Ventral tegmental area
Arguably one of the most important nodes in motivation and reward circuitry is the VTA. Reward learning and connectivity between the VTA and striatum is impaired in MDD patients [18, 19]. However, this does not likely represent a weakening of solely reward-related signals. Although dopaminergic neurons in the VTA fire in response to unpredicted reward and in anticipation of impending reward [20], they also respond to aversive events [21, 22], contrary to the old idea that the VTA signals exclusively “positive” events. It is also worth mentioning the VTA contains not only dopaminergic neurons, but GABAergic and glutamatergic neurons. Finally, MDD symptoms often manifest bidirectionally between patients (e.g., increased or decreased appetite), adding additional complexity to the interpretation of altered VTA activity.
To dissect how the VTA is associated with depression symptoms, the preclinical field has invested a great deal of effort characterizing molecular and physiological changes that occur in VTA neurons from animals exhibiting depression-like behaviors. Then, to link these changes to the behavior, researchers have manipulated the cellular and molecular activity to reverse or recapitulate anhedonia. In some instances, decreased VTA activity is enough to elicit anhedonia, whereas in others, increased VTA activity is required. It is becoming increasingly evident that where the VTA dopamine neurons project is as important as the directionality of their activity. Below, we summarize the most recent studies and propose some remaining questions.
Stress-mediated increases in dopamine neuron activity
Chronic physical or psychosocial stressors increase VTA dopamine neuron activity to elicit depression-like behavior. Following CSDS, stress-susceptible animals have increased VTA dopamine neuron firing [13, 23–29] that persists for several weeks after stress termination [28]. Elevated firing appears to drive the susceptible phenotype, as optogenetic stimulation of VTA dopamine neurons recapitulates stress susceptibility in previously resilient animals [23, 24, 30]. However, increased VTA activity drives susceptibility through specific projection targets (Fig. 1, top): inhibiting VTA dopamine neurons that project to the NAc induces resilience but inhibiting VTA dopamine projections to medial prefrontal cortex (mPFC) promotes CSDS susceptibility [23]. Chronic restraint stress also increases VTA firing [31] and burst firing is increased in vivo during an encounter with an aggressive rat [32]. Dendritic spine density, a proxy for excitatory synapse density and activity, is also increased in the VTA of CSDS-susceptible mice [33]. These lasting increases in VTA dopamine neuron firing after chronic stress appear to drive the susceptible phenotype, but little is known regarding physiological adaptations to VTA non-dopaminergic neurons. Elucidating the cellular and molecular adaptations that promote increased VTA neuronal activity will be important for identifying druggable targets for treating depression symptoms.
Fig. 1.
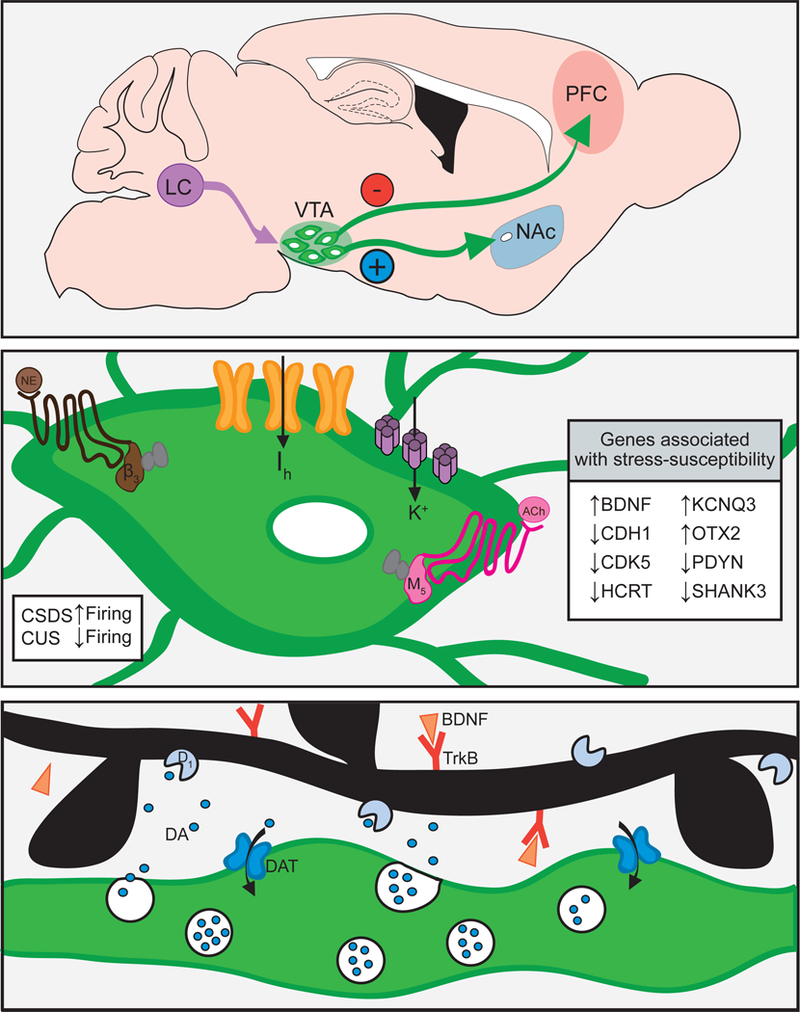
Stress alters ventral tegmental area dopamine neurons to generate depressive behavior. Top: Simplified schematic of ventral tegmental area (VTA) projections associated with stress susceptibility. Inhibition of the VTA to prefrontal cortex (PFC) projection is pro-depressant. Stimulation of the VTA to nucleus accumbens (NAc) projection is pro-depressant. Noradrenergic neurons from the locus coeruleus (LC) project to the VTA where they modulate excitability and stress-susceptibility. Middle: VTA dopamine neuron firing is increased in mice susceptible to social defeat (CSDS) and decreased after chronic unpredictable stress (CUS). Excitability of VTA dopamine neurons is modulated by norepinephrine through α1 and β3 receptors (brown), acetylcholine through muscarinic receptors (pink), and a balancing of Ih and K+ currents through HCN (orange) and KCNQ channels (purple). Inset box details genes associated with stress susceptibility in the VTA. Bottom: Dopamine varicosity (green) releasing dopamine in the NAc. Stress is associated with greater phasic dopamine release, upregulation of dopamine transporters (DAT), dopamine receptors, and BDNF-TrkB signaling
Stress-induced alterations in the intrinsic properties of VTA neurons can drive increased VTA activity through changes in neuronal excitability. One modulator of excitability and firing frequency is the hyperpolarization-activated current (Ih), or “pacemaker current,” which is conducted through hyperpolarization-activated cyclic nucleotide gated (HCN) channels [34]. The stress-mediated increases in VTA dopamine neuron firing are caused by increased Ih currents [29, 35] (Fig. 1, middle). Increased Ih currents occur specifically in NAc, but not mPFC projecting VTA dopamine neurons [30], aligning with the role of these projections in mediating stress susceptibility [23]. Optogenetic stimulations that promote stress susceptibility also increase dopamine neuron excitability [23]. Ih and intrinsic excitability are upregulated in CSDS-susceptible mice [30], but somewhat paradoxically, Ih is upregulated to a greater extent in resilient mice. Through homeostatic adaptations, resilient mice upregulate K+ channels to reduce excitability as a consequence of the enhanced Ih [13, 30]. Ih potentiation with repeated lamotrigine increases K+ currents and decreases neuronal excitability in previously susceptible mice to reverse social and sucrose preference deficits [25]. KCNQ K+ channels regulate excitability thresholds and counteract membrane depolarizations, and upregulation of this specific inhibitory driving force is thought to underlie altered excitability and resilience. In support of this, KCNQ overexpression or administration of KCNQ-openers rescues depressive behavior [25, 27] by reducing firing of NAc projecting, but not PFC projecting VTA dopamine neurons [25]. KCNQ channel Kv7.4 is an attractive target in depression, as this subtype is more selectively expressed in VTA (not substantia nigra) dopamine neurons and Kv7.4 currents are reduced in susceptible mice [27]. Sucrose preference deficits, social avoidance, and VTA excitability are reduced by opening Kv7.4 channels with fasudil [27], a drug already used in humans [36, 37].
Dopamine neuron excitability is also modulated by receptors expressed on the membrane, as well as intracellular signaling molecules that change receptor expression and trafficking (Fig. 1, middle). For example, norepinephrine released from the locus coeruleus (LC) acts at adrenergic receptors expressed on dopamine neurons to regulate neuronal excitability and CSDS-susceptibility [26]. Repeated activation of VTA projecting LC neurons promotes CSDS-resilience through α1 and β3 adrenergic receptors on VTA dopamine neurons [38]. Activation of these receptors balances Ih and K+ currents to reverse the hyperactivity of NAc projecting VTA neurons [38]. Endo-cannabinoids acting at cannabinoid receptors also alter VTA firing, including cannabinoid receptor CB2. CB2 activation reduces VTA dopamine neuron firing [39] and immobility in forced-swim and tail-suspension tests [40]. Mice over-expressing CB2 have reduced vulnerability to CUS and depression-like behaviors at baseline [41, 42], whereas CB2 knockout in dopaminergic cells enhances forced-swim and tail-suspension immobility [43]. Acetylcholine through cholinergic receptors further modulates VTA activity, particularly the M5-type muscarinic acetylcholine receptor (mAChR), which enhances tonic excitability of dopamine neurons [44]. Enhanced VTA cholinergic tone increases forced-swim immobility and anhedonia through mAChRs [45], and mAChR or nicotinic acetylcholine receptor (nAChR) antagonism reduces forced-swim immobility [46]. Downstream changes in intracellular signaling cascades can also influence excitability by altering receptor function. For example, chronic stress increases extracellular signal-regulated kinase 2 (ERK2) activity in the VTA and inhibiting VTA ERK2 rescues CSDS-susceptibility and reduces forced-swim and tail-suspension immobility by reducing dopamine neuron excitability [47]. As ERK2 reduces inhibitory currents through GABAA receptors [48], ERK2 inhibition may suppress dopamine neuron hyperactivity. There are likely additional molecular players that regulate dopamine neuron excitability and compounds targeting these regulators may prove useful therapeutic agents.
Stress-mediated decreases in dopamine neuron activity
As alluded to above (“Stress-mediated increases in dopamine neuron activity”), adaptations to dopaminergic neurons depend on the stressor or depression model. VTA burst firing increases during an agonistic encounter [32] and after repeated social defeat [13], as well as during acute stressors such as restraint or foot-shock [49, 50]. However, chronic mild or chronic cold stress instead reduce VTA population activity [50, 51]. Rats exposed to chronic mild stress have fewer spontaneously active dopamine neurons [52, 53], an effect more pronounced in females [53]. VTA firing is also reduced in mice exposed to CUS and optogenetic stimulation in this context reverses, instead of promotes, stress-induced behavioral deficits [54]. Decreased activity of VTA dopamine neurons may not reflect a weakening of solely reward-related signals, and the time-course and intensity of the stress is likely to generate a different set of molecular and physiological adaptations. It is important to understand the mechanisms underlying opposite physiological changes in models that result in the same behavioral outcome.
Naturally, the mechanisms driving decreased VTA activity after CUS differ from those that cause CSDS-mediated increases in activity. One such mechanism is the L-type voltage gated calcium channel Cav1.3, which decreases neuronal excitability [55]. Increased VTA Cav1.3 channel activity enhances anhedonia and social-behavior deficits [56], in agreement with the idea that reduced VTA activity drives specific depressive behaviors [54]. Acetylcholine signaling through β2-nAChRs also modulates VTA activity, whereby β2-nAChR signaling switches dopamine neurons to an excited state [57]. Peroxisome proliferator-activated receptor α (PPARα) signaling increases β2-nAChR phosphorylation, which subsequently decreases VTA dopamine neuron firing [58]. Somewhat paradoxically, long-term activation of PPARα decreases β2-nAChR phosphorylation, increases dopamine neuron bursting, and rescues sucrose self-administration after stress [59]. Apart from excitability, reduced VTA activity can also arise from a loss of excitatory input. Mice that develop anhedonia after systemic lipopolysaccharide have reduced plasma membrane expression of AMPA receptor subunit GluR1 [60], indicative of decreased excitatory input onto dopamine neurons.
Although the bidirectional changes in VTA activity reported in CUS and CSDS studies may reflect methodological differences, it is possible that decreased VTA activity following CUS and increased activity following CSDS reflect subpopulations of dopamine neurons with different projection targets. Those that project to mPFC may have decreased activity, whereas those that project to NAc may have increased activity. Careful dissection of individual cell types is needed to understand this discrepancy and the differing molecular and receptor-level mechanisms changing dopamine neuron activity and excitability.
Classical antidepressant actions on VTA dopamine neuron activity
Similar to stressors, classical antidepressant treatments either increase or decrease VTA dopamine neuron activity. Two weeks of fluoxetine normalizes Ih currents in dopaminergic neurons after CSDS [29], indicating one mechanism of classic antidepressant action may be restoration of baseline dopamine neuron activity. Indeed, both CSDS and CUS-induced firing changes are reversed by Ih current normalization after HCN2 overexpression [30, 61]. Two weeks of escitalopram reduces VTA firing rate and bursting activity [62], whereas other antidepressants or electroconvulsive therapy increase burst firing [63, 64]. However, these disparate findings are difficult to interpret, as both behavioral and serotoninergic effects of anti-depressant citalopram are affected by housing conditions (i.e., group or single housing) [65]. The rats used in ref. [63] were group-housed during treatment, whereas the housing conditions used in ref. [62] are unclear. Perhaps classical antidepressants fail in some individuals because the VTA firing rate is changed incorrectly, i.e., they increase activity when decreased activity would be more therapeutic. Future work could compare how classic and new rapid acting antidepressants alter VTA firing rates in several different stress paradigms or treatment-resistant patients to address this possibility.
Molecular adaptations in the VTA
Numerous molecular changes accompany the electro-physiological and behavioral changes, including brain-derived neurotrophic factor (BDNF) and signaling molecules downstream of BDNF (Fig. 1, bottom). The appearance of depressive-like behavior following CSDS is mediated in part by increased BDNF signaling in the VTA to NAc circuit, reviewed elsewhere in detail [66]. Optogenetic stimulation of NAc projecting VTA neurons exacerbates susceptibility to social stress in a BDNF-TrkB receptor-dependent manner [24]. However this is circuit specific as BDNF has antidepressant actions in other brain regions that are required for the antidepressant effects of ketamine [67].
Protein kinase B (AKT), downstream of BDNF signaling, is a molecular link between depressive behavior and altered dopamine neuron activity. Decreased AKT is associated with increased VTA dopaminergic neuron excitability and reduced AKT tone leads to reductions in membrane GABAA receptor expression and GABA release. Activated phospho-AKT is decreased in the VTA of susceptible mice, an effect reversed by fluoxetine and recapitulated by expression of a dominant negative AKT mutant. In rats subject to forced-swim, downregulated AKT in the VTA enhances immobility and decreases sucrose preference [68]. Other downstream kinases, such as Cyclin dependent kinase 5 (Cdk5) also regulate depressive behavior by altering dopamine release. Cdk5 phosphorylates the rate-limiting enzyme for dopamine synthesis, ultimately influencing dopamine release in the terminal field. VTA Cdk5 knockout decreases sucrose preference and increases immobility in the forced-swim test, an effect reversed by elevating cAMP in dopamine neurons [69].
Psychosocial [13] and unpredictable stress [70, 71] profoundly alter gene expression beyond the BDNF pathway in total VTA tissue (Fig. 1, middle). When differentially expressed genes are classified by Gene Ontology and grouped by gene function, stress alters the expression of genes that regulate actin cytoskeleton, calcium signaling, cholinergic synapses, and dopaminergic synapses. A major goal for the field is to identify the upstream regulators of these transcriptional changes. Recent work has identified the transcription factor Otx2 as one such upstream regulator responsible for “stress-priming” the VTA [72]. Stress in a late postnatal period primes the VTA to be in a depression-like state by suppressing expression of Otx2. Transient VTA Otx2 overexpression rescues the enhanced CSDS-susceptibility and downregulation of Otx2-regulated genes caused by late postnatal stress [72]. Identifying common transcriptional regulators of the genes altered in depression may aid in identifying new therapeutics.
Remaining questions
Together, these recent studies provide insight into how stress exposure alters the physiology of VTA dopamine neurons and provides clues into the underlying molecular mechanisms controlling depressive behavior. However, several questions remain. Namely, are the bidirectional changes in VTA activity after CSDS or CUS a consequence of stress paradigm, such that each stressor causes unique cellular and molecular adaptations? Or can the changes in VTA activity be viewed along a continuum, whereby “too much” or “too little” Ih current disrupts baseline firing? Do the firing changes instead reflect different symptoms—i.e., social withdrawal vs anhedonia? Or do they reflect the activity of separate cell populations within the VTA [73]? Specific VTA afferents regulate reward and aversion [74]. Is there a role for specific inputs onto VTA dopamine neurons in depression? What are the contributions of non-dopaminergic cells in the VTA that receive similar inputs to dopaminergic cells [75]? Finally, what are the molecular mechanisms driving the changes within subpopulations, do they vary within dopaminergic and non-dopaminergic cells, and how can we target them to ameliorate depressive symptoms?
Nucleus accumbens
One of the major projection targets of VTA dopamine neurons is the NAc. The primary projection neurons of the NAc are medium-spiny neurons (MSNs), which are divided into two subtypes based on the expression of dopamine D1 or D2 receptors (D1-MSNs and D2-MSNs, respectively) [76]. There is little anatomical overlap between MSN subtypes or their projection targets [77, 78]. NAc D1-MSNs send projections back to the VTA, as well as the substantia nigra and ventral pallidum (VP), whereas D2-MSNs project exclusively to the VP [77, 78]. Under normal conditions, the actions of D1-and D2-MSNs generate balanced behavioral output [79, 80]. However, biased activity of one subtype over another is hypothesized to lead to depression [81].
NAc function is altered in stressed or anhedonic animals. NAc MSNs undergo structural and physiological changes after chronic stress dependent on MSN subtype. Glutamatergic, dopaminergic, and peptidergic transmission are also altered by chronic stress in the NAc. Increasing glutamatergic transmission into NAc can either promote or rescue stress-related behaviors dependent on the afferent projection. Similarly, alterations to NAc dopamine release and uptake are contingent on the stress paradigm. An array of transcriptional changes accompanies altered NAc function. These include up-or downregulation of cellular morphology molecules, glucocorticoid and glutamate receptors, and transcription factors. Epigenetic changes are one potential mechanism by which such a vast array of genes exhibit altered expression; however, much of this work has not differentiated between MSN subtype. Given the often oppositional changes in D1-and D2-MSNs, continued effort to identify the physiological and molecular changes in specific subtypes will be key to understanding how the NAc contributes to depression. Below, we summarize the most recent studies and propose some remaining questions.
Bidirectional changes in D1-and D2-MSN activity
Chronic stress alters MSN activity by changing excitatory input and intrinsic excitability. Excitatory input is weakened onto D1-MSNs and strengthened onto D2-MSNs of chronically stressed mice [82, 83] (Fig. 2). Optogenetic D1-MSN stimulation reverses social and sucrose preference deficits, whereas D2-MSN stimulation enhances stress susceptibility [82]. At the level of individual dendritic spines, there is evidence for synaptic strengthening of the larger, mushroom spines on D1-MSNs and weakening of mushroom spines on D2-MSNs in stress-resilient mice [84]. Long-term potentiation correlates with spine enlargement [85], suggesting one mechanism of stress resilience may be a strengthening of glutamatergic input onto D1-MSNs. At baseline, calcium transients in D1-, but not D2-MSN are associated with social interaction [86], consistent with D1-MSN activation being sufficient to drive social behavior [87]. D1-MSN peak calcium transient amplitude is larger in mice that will later become resilient, suggesting increased D1-MSN baseline activity; however, this difference is abolished after the first aggressive encounter [86]. D1-MSN calcium transient frequency is reduced during social interaction in stress-susceptible mice [88]. Thus, reductions in D1-MSN, but not D2-MSN activity, drive social avoidance after CSDS.
Fig. 2.
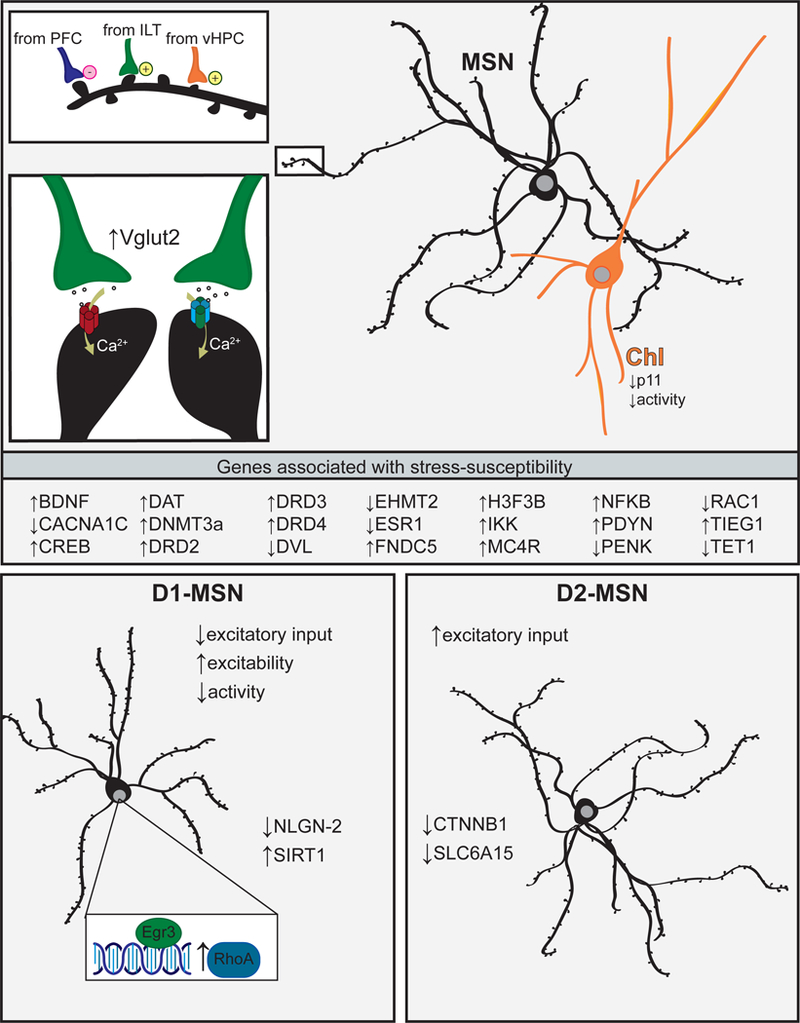
Depression is associated with cell-type-specific adaptations to nucleus accumbens medium-spiny neurons. Top, upper left: Susceptibility to social defeat stress is associated with a weakening of gluta-matergic inputs from prefrontal cortex (PFC) and strengthening of inputs from intralaminar thalamus (ILT) and ventral hippocampus (vHPC) to nucleus accumbens medium-spiny neurons (NAc MSNs). Top, lower left: Stress-susceptible mice have increased expression of glutamate transporter Vglut2, increased calcium-permeable AMPA receptors (red), and increased GluN2B containing NMDA receptors (blue). Top, right: Cholinergic interneurons (ChI) have reduced protein p11 and ChI inhibition induces depressive behavior. Middle: Genes associated with stress susceptibility in the NAc. Bottom, left: In stress-susceptible mice, dopamine D1 receptor expressing MSNs undergo dendritic atrophy. D1-MSNs also have decreased excitatory input, increased intrinsic excitability, and reduced activity in vivo. Neuroligin-2 (NLGN-2) expression is decreased selectively in D1-MSNs. Sirtuin-1 (SIRT1), Early Growth Response 3 (Egr3), and RhoA expression are increased selectively in D1-MSNs. Inset: Egr3 transcriptionally regulates expression of RhoA in stress-susceptible mice. Bottom, right: In stress-susceptible mice, dopamine D2 receptor expressing MSNs do not undergo dendritic atrophy, but may increase spine density. D2-MSNs have increased excitatory input. β-catenin (CTNNB1) and transporter Slc6a15 expression are reduced selectively in D2-MSNs
Despite reductions in excitatory input, D1-MSNs of susceptible mice have increased excitability [82, 89] and calcium transient amplitudes [88]. Increased D1-MSN excitability may arise as a homeostatic adaptation to the loss of excitatory input through altered structural morphology (see “Structural plasticity” below). One potential mechanism for enhanced D1-MSN excitability is through increased glucocorticoid receptor signaling. Glucocorticoids can enhance excitability [90] and D1-MSN glucocorticoid receptor knockout promotes stress resilience [91]. However, glucocorticoid-mediated plasticity may be NAc subregion-dependent. After cold water forced-swim, synaptic strength is increased in MSNs only from the NAc shell, an effect blocked by pre-administration of a glucocorticoid receptor antagonist [92]. Voltage-gated calcium channels also regulate neuronal excitability. Cacna1c, which codes for the voltage-dependent calcium channel-α1C subunit (Cav1.2), is decreased in the NAc of CSDS-susceptible mice. NAc Cacna1c knockdown enhances stress susceptibility and increases anhedonia [93], indicating a possible role for calcium channels in depressive behavior. However, it is unclear which MSN subtype undergoes synaptic plasticity in this study. It will be important to characterize the mechanisms underlying altered synaptic strength in specific cell types due to the bidirectional change in D1-and D2-MSN activity.
Changes to glutamatergic function in NAc
The NAc receives glutamatergic inputs from several afferents and stress alters excitatory transmission arising from the thalamus, mPFC, and hippocampus (Fig. 2, top) [94–96]. Increased glutamatergic transmission from intralaminar thalamus to NAc synapses promotes CSDS-susceptibility, whereas blocking transmission at these synapses blocks social avoidance [95]. NAc glutamate transporter Vglut2 is increased in female mice after subchronic variable stress [94], similar to CSDS-susceptible male mice [95]. Vglut2 is expressed predominately by the thalamus and brainstem neurons [97], lending further support to enhanced thalamo-accumbal transmission in stress susceptibility. Conversely, glutamate release at mPFC to NAc synapses is decreased in CSDS-susceptible mice and stimulation of inputs from mPFC or amygdala promotes CSDS-resilience [96]. Some evidence suggests ventral hippocampus (vHip) to NAc synapses are strengthened in CSDS-susceptible mice [96]. Stimulation of vHip to NAc synapses is pro-depressive in an acute forced-swim stress but does not impair social behavior in the absence of stress [96]. In contrast, vHip to NAc synapses are weakened following chronic multimodal stress; however, this appears to occur selectively onto D1-MSNs [98]. NAc afferents may differentially target D1-or D2-MSNs [99], begging the question if the changes to glutamatergic inputs are MSN subtype specific.
Glutamate signals through a variety of postsynaptic receptors including ionotropic AMPA and NMDA receptors. Increased NAc AMPA receptor function (increased GluR1:GluR2 ratio) is associated with CSDS-susceptibility [100]. Arc protein regulates AMPA receptor trafficking and promotes endocytosis [101]. Social defeat upregulates NAc Arc protein in rats who exhibit proactive (“fighting back”) coping behavior [102]. It is tempting to speculate that decreased AMPA receptor function, via Arc upregulation, contributes to the more resilient behavioral outcome in proactive-coping rats. Interestingly, mice expressing a G2019S mutation in leucine-rich repeat kinase 2 (LRRK2) are unusually resilient to CSDS and fail to accumulate inward rectifying AMPA currents typical of susceptibility. NAc glutamatergic synapses in LRRK2 mutant mice lack functional inwardly rectifying calcium-permeable AMPARs [103]. Blocking NAc calcium-permeable AMPARs reverses social-behavior deficits [56]. Together, these studies reveal an important role for specific AMPARs in mediating CSDS-susceptibility.
Glutamate signaling through NMDA receptors is key for depressive behavior in D2-MSNs. Anhedonia in chronic pain models is associated with prolonged NMDA receptor signaling in D2-MSNs due to an increase in the proportion of GluN2B containing NMDA receptors [104]. In agreement, rendering NMDA receptors non-functional in D2-MSNs by GluN1 knockout results in a depression-resistant phenotype, marked by prolonged latency to immobility in forced-swim and tail suspension [105]. Further, the antidepressant effects of fluoxetine require downregulation of the NMDA receptor signaling partner CaMKII [106, 107]. As excitatory input is enhanced onto D2-MSNs, it is probably enhanced calcium influx through calcium-permeable AMPA and NMDA receptors on D2-MSNs that drive CSDS-susceptibility.
Dopaminergic function
The NAc receives dense dopaminergic input from the VTA, and for an excellent review on how stress impacts extra-cellular dopamine, we refer the reader to ref. [108]. Acutely, NAc dopamine receptor blockade is pro-depressive during tail suspension [54] and global dopamine receptor antagonism reduces sucrose preference [109]. However, dopamine receptor antagonism during CSDS does not block susceptibility [24]. At both expression and protein level, NAc dopamine receptors and dopamine transporter (DAT) are upregulated in stressed rats (Fig. 1, bottom) and increased D2-family receptor expression is linked to decreased sucrose preference [70]. Work from the Jones lab also supports a role for altered DAT in stress. Rats reared in social isolation have increased NAc dopamine release and faster uptake compared with those reared in group housing but reduced basal dopamine levels [110]. In slice preparations, evoked NAc dopamine release and uptake rates are also increased in socially defeated rats [111]. This contrasts with unaltered dopamine release in socially defeated mice [24]. It is still unclear how altered (or unaltered) NAc dopamine release drives social avoidance and other depression-like behaviors due to the dearth of real-time dopamine measurements in behaving animals.
Neuropeptides
Neuropeptides exhibit diverse, often long-lasting effects on gene expression and excitability. Stress promotes the release of neuropeptide dynorphin and activation of endogenous opioid signaling [112]. Dynorphin mRNA is increased in the NAc shell of CSDS-susceptible mice [113] and expression of structurally similar nociceptin/orphanin FQ is increased in both shell and core of socially defeated rats [114]. NAc dynorphin concentration is decreased in rats reared in social isolation, but this is thought to be a consequence of increased release, as κ-opioid receptors are functionally hyperactive [115]. Although dynorphin is associated with stress susceptibility, enkephalin is associated with stress resiliency [116]. NAc enkephalin mRNA is downregulated following chronic restraint [117] but upregulated in rats resilient to the stress [118]. These small opioid peptides signal through inhibitory G-protein-coupled receptors that modulate MSN excitability. Chronic restraint increases NAc melanocortin receptor MC4R expression and MC4R knockdown prevents stress-induced weight loss and decreased sucrose preference [83]. The ligand for MC4R, α-melanocyte-stimulating hormone, exerts its prodepressive effects through D1-MSNs by altering excitability and increasing calcium-permeable AMPA receptors [119]. Neuropeptides can influence several targets due to volume transmission, but how they influence MSN-subtype excitability seems to be key for their role in depression-like behaviors.
Gene expression
There are several genes implicated in response to stress and depressive behavior (Fig. 2, middle). For example, transcription factor CREB is stimulated in the NAc following stress and its activation drives anhedonia (recently reviewed in ref. [120]). ΔFosB, a truncated isoform of FosB, is induced by stress or antidepressant exposure and is associated with resilience (recently reviewed in refs. [120, 121]), particularly in D1-MSNs [100, 122]. Recent work suggests the protective effect of voluntary wheel running on stress-induced depression is through a CREB-dependent induction of ΔFosB [123] and Fosb targeted histone acetylation drives resilience in D1-MSNs, but susceptibility in D2-MSNs [124].
Depressive behavior is mediated in part by increased NAc BDNF signaling (reviewed in ref. [66]). FosB and CREB can regulate BDNF expression, and BDNF upregulation induced by synaptic activity depends on Wnt secretion [125]. Wnt leads to activation of the kinase disheveled (DVL). Activated DVL inhibits glycogen synthase kinase-3β (GSK3β) and regulates several downstream targets including β-catenin (β-cat). DVL is downregulated in the NAc of postmortem MDD patients and GSK3β inhibition promotes CSDS-resilience [126]. β-Cat signaling is down-regulated specifically in D2-MSNs of susceptible mice and excising β-cat from the NAc increases stress susceptibly [127]. β-Cat regulates Dicer1, a microRNA regulator that, in turn, modulates other known pro-susceptibility genes such as Arc and Npas4 [127].
Many studies have focused on signaling pathways downstream of BDNF signaling. Indeed, phospho-extracellular signaling regulated kinase is increased in the NAc shell of D1-MSNs in susceptible mice [24]. Transcription factors downstream of BDNF, such as transforming growth factor β-inducible early gene-1 [102], and Early Growth Response 3 (Egr3) [88] are also upregulated after social defeat (Fig. 2, middle). Egr3 is upregulated specifically in D1-MSNs of susceptible mice and its knockdown can prevent altered synaptic activity and stress susceptibility [88]. Genes that are under the control of these upregulated transcription factors also exhibit increased expression. For example, the mitochondrial biogenesis regulator PPAR-γ coactivator 1-α (PGC1α) is regulated by both CREB [128] and Egr3 [129]. Social defeat increases NAc PGC1α and its downstream myokine FNDC5, and FNDC5 is upregulated more in resilient mice [130]. FNDC5 induces thermogenesis and elevates mitochondrial gene expression [131], and resilient mice have increased NAc metabolism [132]. These mitochondrial and metabolic changes may allow for increased expression of other resilience-associated genes by providing more ATP and are worth further investigation.
Beyond CREB and Egr3, other transcription factors regulate gene expression to contribute to stress-related behaviors. Estrogen receptor-α (ERα), a ligand-gated transcription factor, upregulates the expression of genes associated with nervous system development and downregulates genes associated with immune system processes. In female mice subject to chronic variable stress, NAc ERα is decreased in the nuclear fraction of both D1-and D2-MSNs. In both male and female mice, NAc ERα overexpression increases sucrose preference and promotes CSDS-resilience in males [133]. NAc ERα overexpression in male mice also induces a transcriptional profile similar to that of resilient mice [133]. Ligand-activated transcription factors may be useful targets for pharmacotherapy, as expression of resilience-related genes could be turned off and on.
Modern genetic approaches allow for transcriptional profiling of select cell types [134] and this approach has afforded new insights into genes changed exclusively in D1-or D2-MSNs (Fig. 2, bottom). For example, the MDD risk gene SLC6A15 [135] is decreased in postmortem MDD NAc. In mice susceptible to CSDS, Slc6a15 is decreased selectively in D2-MSNs and stress susceptibility is recapitulated by decreasing Slc6a15 specifically in D2-MSNs [136]. Similar to opposing electrophysiological changes in D1-and D2-MSNs, genes important for synapse maintenance and dendritic architecture are differentially expressed between the cell types. Synaptic cell-adhesion molecule Neurologin 2 (Nlgn2) is decreased specifically in D1-MSNs of susceptible mice. D1-MSN Nlgn2 knockdown leads to susceptibility, whereas Nlgn2 knockdown in D2-MSNs confers resilience [137]. The GTPase RhoA, which negatively regulates dendritic complexity, is increased specifically in D1-MSNs of susceptible mice [138]. RhoA is transcriptionally regulated by Egr3 [88] and in stress-naive mice, increased RhoA in D1-MSNs decreases sucrose preference in males, grooming behavior in females, and forced-swim immobility. Upregulation of RhoA in D1-MSNs either by Egr3 overexpression or RhoA over-expression promotes stress susceptibility [88, 138]. After CSDS, inhibiting downstream effector Rho-kinase reverses social withdrawal and forced-swim immobility [138]. These cell-type-specific changes in gene expression are candidate mechanisms for bidirectional changes in activity (discussed above in “Bidirectional changes in D1-and D2-MSN activity”) and structure (discussed below in “Structural plasticity”). Future work using this approach is likely to uncover additional candidate mechanisms within the cell types.
Epigenetic mechanisms
Epigenetic modifications are a candidate mechanism for how stress alters gene expression to convey the risk for depression. DNA methyltransferases (DNMTs), histone methyltransferases, and histone deacetylases (HDAC) alter the structure and function of chromatin to regulate gene expression [139]. Repressive histone modifications are both up-and downregulated after social defeat stress, and these changes are linked to genes with known roles in CSDS-susceptibility. The repressive histone lysine methyl-transferase G9a decreases TrkB-CREB signaling and G9a expression is decreased in the NAc of CSDS-susceptible mice [140]. G9a overexpression promotes resilience, thought to occur by normalization of TrkB-CREB signaling [140]. Permissive acetylation on the Rac1 promotor, a GTPase downregulated by CSDS, is significantly reduced in susceptible mice, and susceptible mice have enhanced methylation directly upstream of the promotor [141]. Similarly, there is increased repressive trimethylation along the promoter region of Rac1 after social isolation stress [142]. Intra-NAc HDAC inhibition reverses social avoidance induced by CSDS, likely caused by increased Rac1 expression [141]. CSDS upregulates transcriptional repressor Dnmt3a in the NAc and chronic intra-NAc DNMT inhibition restores social interaction [143]. Subchronic stress also upregulates NAc Dnmt3a, but to a greater extent in stressed females. Dnmt3aoverexpression makes both sexes susceptible to 3 days of subchronic variable stress [144], indicating transcriptional repression is a shared mechanism of stress susceptibility between the sexes.
Apart from DNMTs, epigenetic modifications such as hydroxymethylation can also repress gene expression. The hydroxymethylase 10–11 translocation methylcytosine dioxygenase 1 (Tet1) catalyzes the conversion of 5-methycytosine to 5-hydroxymethlcytosine. Tet1 is decreased in the NAc of susceptible mice. Paradoxically, NAc Tet1 knockout increases sucrose preference and social interaction in stressed mice. However, Tet1 overexpression induces a resilience-like NAc gene expression profile [145]. The replacement of canonical histones with histone variants (“histone turnover”) can also alter DNA binding and subsequent gene expression. Expression of activity-dependent histone variant H3.3 (H3f3b) is increased in the NAc in postmortem MDD tissue, after early life stress, and in CSDS-susceptible mice, an effect prevented in mice by environmental enrichment [146]. Stalling histone turnover with viral knockdown of H3.3 produces resilience after CSDS partly due to normalization of transcriptional dysregulation of genes associated with morphology and plasticity [146]. Although stressors clearly regulate gene expression through epigenetic mechanisms, we have little insight into how they function in cell subtypes. Indeed, the deacetylase SIRT1 promotes CSDS-susceptibility through D1-MSNs [147]. Histone acetylation on the Fosb promotor drives resilience in D1-MSNs, but susceptibility in D2-MSNs [124]. Thus, manipulating the epigenetic mechanisms in specific MSN subtypes might allow for more precise control over gene expression and novel therapeutics.
Structural plasticity
Morphological changes are a consequence of altered gene expression and excitatory input after CSDS. Indeed, social avoidance correlates with reductions in volume of the cingulate cortex, NAc, thalamus, raphe, and bed nucleus of the stria terminalis, suggesting the neurons in these regions may undergo structural atrophy [148]. In the NAc, dendritic complexity of D1-, but not D2-MSNs, is reduced in CSDS-susceptible mice. Reduced D1-MSN complexity is sufficient to drive CSDS-susceptibility [88, 138]. NAc MSN spine density, dendritic length, and branch points are decreased after stress hormone corticosterone [149] or gestational stress [150]. Reduced dendritic complexity largely agrees with changes in other brain regions [151, 152] and is associated with reduced excitatory input but increased intrinsic excitability [88, 89], which may reflect homeostatic self-tuning [153]. The mechanism underlying structural plasticity of MSNs in CSDS is altered expression and activity of Rho GTPases [88, 89, 138, 141].
In contrast, CUS increases dendritic length of NAc core MSNs, which is reversed by imipramine or fluoxetine treatment [154]. MSN spine density is also increased in rats susceptible to learned helplessness [155]. However, many of these studies did not examine dendritic complexity or spine density in specific MSN subtypes. Spine density is unchanged in D1-MSNs [88, 89], yet there is evidence MSNs from susceptible mice have more stubby spines [156]. Pro-susceptibility Dnmt3a overexpression [143] or constitutively active IκB kinase (IKK) [156, 157] both increase MSN spine density. Thus, we believe the stress-related changes to spine density occur on D2-MSNs, as D2-MSNs do not undergo dendritic atrophy but have enhanced excitatory input [82]. Future work should confirm increased NAc spine density occurs specifically on D2-MSNs and identify the mechanisms causing increased dendritic length after CUS.
Cholinergic interneurons
Approximately 95% of NAc neurons are MSNs; however, the remaining cells, namely interneurons, also contribute to depression-like behaviors. Silencing NAc cholinergic interneurons (ChIs) results in anhedonia and increases immobility in the forced-swim and tail-suspension tests [158]. Decreased ChI activity may modulate local MSN activity to cause depressive behaviors, as optogenetic inhibition of ChI enhances MSN spiking [159]. The role of ChIs is also interesting to consider in the context of NAc dopamine. The dopamine release thought to be important for driving anti-depressive behavior [54] may arise from synchronized NAc ChI activity and be independent of altered VTA activity [160, 161], although this remains untested. One molecular mechanism that contributes to NAc ChI involvement in anhedonia is the calcium binding protein p11. Mice lacking p11 [162], p11 reductions in NAc [163], or p11 knockout in NAc ChIs [158] is sufficient to reduce sucrose preference and increase forced-swim immobility (Fig. 2, top). In the PFC, reduced p11 is associated with increased repressive methylation on the p11 promotor [164], but it is unclear if this also occurs in NAc ChIs. Continued effort to identify the molecular and physiological changes to all cell types in the NAc will be critical for understanding how the NAc encodes depressive behaviors.
Remaining questions
Together, these studies provide insight into how stress exposure alters NAc MSN physiology to elicit depressive behavior. We now have many clues as to the underlying mechanisms, but several questions remain. Why do many divergent transcriptional alterations in the NAc result in the same behavioral outcome and are they a cause or a consequence of anhedonia? How do cellular and molecular changes in NAc projection neurons lead to stress adaptations in target regions including the VTA and VP, the latter which displays distinct circuit alterations after stress [165]? Although hypothesized, is the BDNF critical for CSDS-induced depression actually released from dopaminergic terminals in the NAc? TrkB, but not dopamine receptor antagonism blocks the induction of social avoidance. However, BDNF may be modulating local dopamine release [166, 167] to drive other aspects of anhedonia. Do BDNF and dopamine act on specific MSN subtypes to drive depression? What are the upstream mechanisms governing MSN-subtype-specific atrophy after stress? Both BDNF [168] and dopamine [169] mediate dendritic growth. Dopamine denervation drives atrophy of striatal MSNs, but to a greater extent in D1-MSNs [170], suggesting there may yet be a dopaminergic component to CSDS-susceptibility. Further work is needed to determine the precise mechanisms by which these signaling molecules cause transcriptional changes to alter the morphology and physiology of the two MSN subtypes.
Non-neuronal cell types in reward circuitry
At least 40% of cells in the rodent [171] and human [172] brain are non-neuronal, but how non-neuronal cells interact with reward circuitry in MDD is unknown. There is particular interest in how inflammatory cell types in the brain cause depressive symptoms, as anti-inflammatory treatments reduce depressive symptoms in humans [173] and chronic stress enhances inflammation in animal models [174]. In the periphery, chronically stressed mice upregulate the pro-inflammatory cytokine interleukin 6 (IL-6) [175–177]. IL-6 drives production of inflammatory T-helper 17 (Th17) cells, which promote learned helplessness and social avoidance, and mice unable to produce Th17 are resilient to learned helplessness [178]. CSDS-susceptible mice upregulate cytokines IL-1β and CXCL1, and have more circulating leukocytes [175]. Further, prolonged restraint stress decreases anti-inflammatory IL-4 and IL-10 [176]. In the brain, cytokines such as interferon-α promote depressive behaviors [179] and serotonin reuptake inhibitors have anti-inflammatory properties [180]. CSDS promotes recruitment of circulating monocytes and macrophages into the brain perivascular space via complement 3 receptor and CX3C chemokine receptor 1 signaling [181–183]. Chronic stress also increases the leakiness of the blood-brain barrier to permit infiltration of peripheral immune cells and cytokines such as IL-6 [184].
Microglia
As microglia are the resident immune cells of the central nervous system (CNS) and microglial transcripts are upregulated after stress [185], they are an attractive linker between inflammation and depression in the brain. Microglia can be neuroprotective or neurotoxic dependent on the release of pro-inflammatory or anti-inflammatory factors. Their dichotomous function is typically described by the M1/M2 paradigm (reviewed in ref. [186]). M1 microglia respond to injury or infection by releasing pro-inflammatory cytokines such as IL-1β. M2 microglia dampen pro-inflammatory immune responses with anti-inflammatory cytokines and can enhance neurotrophic factors. Microglia play a key role in altering synaptic landscape and for an excellent review we refer the reader to ref. [187].
Chronic stress alters microglia density, morphology, and function dependent on stress duration and brain region [188]. Anhedonia caused by high-fat diet or restraint stress is associated with increased ionized calcium-binding adaptor molecule 1-positive microglia in several brain regions including the NAc [189, 190]. Microglia from socially defeated mice have an increased inflammatory profile and produce more IL-6 after lipopolysaccharide (LPS) stimulation [191], similar to enhanced IL-6 release from the peripheral leukocytes of stressed mice [175]. Microglia in CUS mice have enhanced colony stimulating factor 1 receptor expression and enhanced phagocytotic activity, paralleled by CSF1 elevations in postmortem MDD tissue [185]. Viral knockdown of neuronal Csf1 attenuates dendritic spine pruning on mPFC neurons and prevents stress-induced anhedonia and forced-swim immobility [185]. As phagocytotic microglia contribute to dendritic remodeling in the mPFC, it is tempting to speculate microglia may also contribute to dendritic spine changes on NAc MSNs.
Microglia release pro-inflammatory cytokines such as tumor necrosis factor-α and IL-1β, which go on to activate other signaling molecules such as IKK and downstream transcription factor nuclear factor (NF)-κB. CSDS increases IKK and NF-κB, along with the number of immature dendritic spines in the NAc of susceptible mice [156, 157]. In the absence of stress, constitutively active IKK increases thin spine density, increases forced-swim immobility, and decreases sucrose preference after acute stress [156, 157]. Viral inhibition of IKK and NF-κB in NAc also prevents neuroinflammation and forced-swim immobility caused by high-fat diet [190]. It remains to be determined which specific cytokines are released by microglia in the NAc to activate IKK and drive changes to dendritic spines and behavior. Cytokines can also be released by neurons and in a spinal nerve ligation model of anhedonia, cytokine CCL2 is upregulated in both NAc D1-and D2-MSNs. Ccl2 reduction with short hairpin RNA restores sucrose preference and increases swimming in the forced-swim test [192]. As CCL2 recruits monocytes to the site of inflammation, this molecule may serve as another link between inflammation and dendritic remodeling. Further work is needed to identify the precise cytokines that contribute to maladaptive neuroinflammation in stress and depression.
The function of microglia in stress and depression is not limited to neurotoxic signaling but may also involve a degree of neuroprotection. When lymphocytes from socially defeated mice are transplanted into Rag2−/− lymphopoenic mice, recipient mice exhibit more social interaction and less tail-suspension immobility [193]. The “stress programmed” lymphocytes alter the microglia in Rag2−/− mice, causing them to skew toward a more neuroprotective, M2-like profile [193]. Similarly, intraperitoneal injection of Gram-negative bacterial endotoxin LPS typically elicits a rapid inflammatory response, globally activates microglia, and produces depressive behavior. However, in mice previously exposed to CUS, LPS decreases forced-swim immobility [188]. Does microglial activation instead help prune the immature dendritic spines generated by stress in NAc or other brain regions [156, 157]? Support for this idea comes from mice with autophagy-deficient microglia. Mice that lack microglial autophagy-related protein 7 exhibit impaired social interaction and increased immature synapses [194]. Further, severe combined immunodeficiency mice have impaired social preference [195]. Thus, although M1 microglia and infiltrating macrophages may contribute to depressive behavior via increased inflammatory signaling, there may be a neuroprotective role of M2 microglia or beneficial synaptic pruning by M1. Microglia found in healthy reward circuitry are incredibly diverse [196] and more work is needed to profile their function in pathologic states. Further, how microglia either respond or contribute to the altered neurochemical environment referenced above remains unknown.
Non-inflammatory cell types
Bacteria in the gastrointestinal tract directly influence neural activity through sensory neurons, or by modulating the function of the hypothalamic–pituitary–adrenal axis (reviewed in ref. [197]). Recent work indicates gut microbiota can also influence neural metabolism [198]. The gut microbiome controls gut permeability and inflammation, and may thus participate in the neuroimmune mechanisms of depression [197]. More work is needed to determine the precise mechanisms by which microbiota interact with both neuronal and non-neuronal cell types in reward circuitry or other brain regions to produce depressive behavior.
It is important to note other non-inflammatory glial cells may contribute to depression. Indeed, CUS can downregulate myelin and oligodendrocyte-specific transcripts in the NAc [199]. Mice with reduced glutamate transporter GLT-1 expression in habenular astrocytes are more stress-susceptible and exhibit increased tail-suspension immobility at baseline [200], in agreement with reduced glutamate transporter expression in learned helpless rats [201]. As glutamate clearance modulates neuronal excitability [200] and glutamatergic synapse strength contributes to depression [202], how astrocytic glutamate clearance contributes to excitatory synapse strength and subsequent depressive behavior is also worth further investigation in the reward circuit.
Remaining questions
Together, these studies provide compelling evidence for reward circuit non-neuronal and neuroimmune mechanisms of depression. It will be important to profile how interactions between glial cells, macrophages, and neurons shift from a normal to pathologic state and how this leads to anhedonia. How do microglia remodel synapses in depression? Do they prune specific spines on specific cell types, or participate in the formation of new spines [203]? What causes the blood–brain barrier to degrade in some brain regions but not others [184, 204]? Are these changes a consequence of physical stressors or would they also extend to emotional stressors [205]? How does the gut microbiome influence anhedonia and how is this altered by changes in food intake during depressive episodes? Neuroscientists must not discount the contribution of the periphery to the changes in CNS function that drive MDD.
Concluding remarks
Dysregulation of the brain’s reward circuitry is heavily implicated in the symptomology of depression and the use of genetic and viral tools has enabled us to define the precise cell types that contribute to anhedonia. These studies have revealed important adaptations in both dopaminergic and glutamatergic function within the VTA and NAc, and notably these adaptations can differ in directionality based on cell type or stress paradigm. However, much remains to be investigated, especially with respect to the contribution of non-neuronal and inflammatory cell types in depressive behavior. Further, we must define the precise molecular mechanisms that explain the discrepancies involving the contribution of dopamine release and changes to burst firing and excitability of VTA neurons. It is difficult to understand how different stress paradigms yield similar behavioral outcomes but divergent cellular adaptations. Although frustrating for preclinical scientists, the unique adaptations after differing stressors is perhaps unsurprising and may afford an opportunity to address the heterogeneity of MDD in humans.
Although rapid tests such as forced swim and sucrose preference have been important for screening new antidepressant compounds due to their high-throughput nature, these tests should be combined with more complex assessments of motivated behavior and assessed across stress paradigms. It will be important to include behaviors that separate rodents based on response to classical antidepressants [206]. The “treatment-resistant” population may undergo adaptations yet undiscovered and might be more helpful in identifying compounds for treatment-resistant depression. Similarly, although we know changes to NAc D1-and D2-MSN function are bidirectional, most of this work was done solely in male rodents. Moving forward, we must determine the contribution of specific cell types in anhedonic females and the extent to which circulating gonadal hormones, among other unidentified sex differences, alter the development of depression. This is especially important to consider in the context of post-partum depression, which is both uniquely female and inadequately addressed in preclinical models.
Finally, despite significant progress made in elucidating the cellular and molecular mechanisms driving anhedonia and depressive behavior in rodents, new antidepressant treatments have not been approved for use in humans. Much of the validation of the molecular changes found in rodents relies on tissue collected from postmortem MDD brains. It is possible we are ruling out valid molecular targets by predominately measuring changes in gene expression from MDD patients who have completed suicide. This subset of patients certainly does not represent the entirety of individuals suffering from MDD and we must try to identify biomarkers that are more readily accessible in living humans with MDD. By improving our understanding of the specific cellular and molecular mechanisms of depression, we have the potential to uncover new treatments that may ameliorate symptoms in otherwise antidepressant resistant populations.
Acknowledgements
We acknowledge Drs CA Calarco, JF Cheer, and DP Covey for instructive comments on this manuscript. MKL is supported by NIH R01DA038613, R01MH106500, and R01DA047843. MEF is supported by NIH F32MH116574.
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Holtzheimer PE, Mayberg HS. Stuck in a rut: rethinking depression and its treatment. Trends Neurosci. 2011;34:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drevets WC. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr Opin Neurobiol. 2001;11:240–9. [DOI] [PubMed] [Google Scholar]
- 3.Liotti M, Mayberg HS. The role of functional neuroimaging in the neuropsychology of depression. J Clin Exp Neuropsychol. 2001;23:121–36. [DOI] [PubMed] [Google Scholar]
- 4.Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. [DOI] [PubMed] [Google Scholar]
- 5.Zhu M-Y, Klimek V, Dilley GE, Haycock JW, Stockmeier C, Overholser JC, et al. Elevated levels of tyrosine hydroxylase in the locus coeruleus in major depression. Biol Psychiatry. 1999;46:1275–86. [DOI] [PubMed] [Google Scholar]
- 6.Drysdale AT, Grosenick L, Downar J, Dunlop K, Mansouri F, Meng Y, et al. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat Med. 2017;23:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Czéh B, Fuchs E, Wiborg O, Simon M. Animal models of major depression and their clinical implications. Prog Neuropsycho-pharmacol Biol Psychiatry. 2016;64:293–310. [DOI] [PubMed] [Google Scholar]
- 8.Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13:1161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anyan J, Amir S. Too depressed to swim or too afraid to stop? A reinterpretation of the forced swim test as a measure of anxiety-like behavior. Neuropsychopharmacology. 2018;43:931–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willner P The chronic mild stress (CMS) model of depression: history, evaluation and usage. Neurobiol Stress. 2017;6:78–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vollmayr B, Gass P. Learned helplessness: unique features and translational value of a cognitive depression model. Cell Tissue Res. 2013;354:171–8. [DOI] [PubMed] [Google Scholar]
- 12.Golden SA, Covington HE, Berton O, Russo SJ. A standardized protocol for repeated social defeat stress in mice. Nat Protoc. 2011;6:1183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krishnan V, Han M-HH, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi A, Chung J-R, Zhang S, Zhang H, Grossman Y, Aleyasin H, et al. Establishment of a repeated social defeat stress model in female mice. Sci Rep. 2017;7:12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris AZ, Atsak P, Bretton ZH, Holt ES, Alam R, Morton MP, et al. A novel method for chronic social defeat stress in female mice. Neuropsychopharmacology. 2018;43:1276–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iñiguez SD, Flores-Ramirez FJ, Riggs LM, Alipio JB, Garcia-Carachure I, Hernandez MA, et al. Vicarious social defeat stress induces depression-related outcomes in female mice. Biol Psychiatry. 2018;83:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iñiguez SD, Riggs LM, Nieto SJ, Dayrit G, Zamora NN, Shawhan KL, et al. Social defeat stress induces a depression-like phenotype in adolescent male c57BL/6 mice. Stress. 2014;17:247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar P, Goer F, Murray L, Dillon DG, Beltzer ML, Cohen AL, et al. Impaired reward prediction error encoding and striatal-midbrain connectivity in depression. Neuropsychopharmacology. 2018;43:1581–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blood AJ, Iosifescu DV, Makris N, Perlis RH, Kennedy DN, Dougherty DD, et al. Microstructural abnormalities in subcortical reward circuitry of subjects with major depressive disorder. PLoS ONE. 2010;5:e13945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schultz W Reward prediction error. Curr Biol. 2017;27: R369–R371. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto M, Hikosaka O. Two types of dopamine neuron distinctly convey positive and negative motivational signals. Nature. 2009;459:837–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen JY, Haesler S, Vong L, Lowell BB, Uchida N. Neuron-type-specific signals for reward and punishment in the ventral tegmental area. Nature. 2012;482:85–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493:532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wook Koo J, Labonte B, Engmann O, Calipari ES, Juarez B, Lorsch Z, et al. Essential role of mesolimbic brain-derived neurotrophic factor in chronic social stress–induced depressive behaviors. Biol Psychiatry. 2016;80:469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedman AK, Juarez B, Ku SM, Zhang H, Calizo RC, Walsh JJ, et al. KCNQ channel openers reverse depressive symptoms via an active resilience mechanism. Nat Commun. 2016;7:11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isingrini E, Perret L, Rainer Q, Amilhon B, Guma E, Tanti A, et al. Resilience to chronic stress is mediated by noradrenergic regulation of dopamine neurons. Nat Neurosci. 2016;19:560–3. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Sun H, Ding J, Niu C, Su M, Zhang L, et al. Selective targeting of M-type potassium Kv 7.4 channels demonstrates their key role in the regulation of dopaminergic neuronal excitability and depression-like behaviour. Br J Pharmacol. 2017;174:4277–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Razzoli M, Andreoli M, Michielin F, Quarta D, Sokal DM. Increased phasic activity of VTA dopamine neurons in mice 3 weeks after repeated social defeat. Behav Brain Res. 2011; 218:253–7. [DOI] [PubMed] [Google Scholar]
- 29.Cao J-LJ-L, Covington HE, Friedman AK, Wilkinson MB, Walsh JJ, Cooper DC, et al. Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci. 2010;30:16453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman AK, Walsh JJ, Juarez B, Ku SM, Chaudhury D, Wang J, et al. Enhancing depression mechanisms in midbrain dopamine neurons achieves homeostatic resilience. Science. 2014;344:313–9. 10.1126/science.1249240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anstrom KK, Woodward DJ. Restraint increases dopaminergic burst firing in awake rats. Neuropsychopharmacology. 2005; 30:1832. [DOI] [PubMed] [Google Scholar]
- 32.Anstrom KK, Miczek KA, Budygin EA. Increased phasic dopamine signaling in the mesolimbic pathway during social defeat in rats. Neuroscience. 2009;161:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qu Y, Yang C, Ren Q, Ma M, Dong C, Hashimoto K. Regional differences in dendritic spine density confer resilience to chronic social defeat stress. Acta Neuropsychiatr. 2018;30:117–22. [DOI] [PubMed] [Google Scholar]
- 34.Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–85. [DOI] [PubMed] [Google Scholar]
- 35.Wanat MJ, Hopf FW, Stuber GD, Phillips PEM, Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol. 2008;586:2157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao J, Zhu X, Wang Q, Zhang D, Cui C-S, Zhang P, et al. Acute effects of Rho-kinase inhibitor fasudil on pulmonary arterial hypertension in patients with congenital heart defects. Circ J. 2015;79:1342–8. [DOI] [PubMed] [Google Scholar]
- 37.Fukumoto Y, Yamada N, Matsubara H, Mizoguchi M, Uchino K, Yao A, et al. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ J. 2013;77:2619–25. [DOI] [PubMed] [Google Scholar]
- 38.Zhang H, Chaudhury D, Nectow AR, Friedman AK, Zhang S, Juarez B, et al. Alpha1 and beta3 adrenergic receptor-mediated mesolimbic homeostatic plasticity confers resilience to social stress in susceptible mice. Biol Psychiatry. 2018;85:226–36. https://doi.org/10.10167J.BI0PSYCH.2018.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H-Y, Gao M, Liu Q-R, Bi G-H, Li X, Yang H-J, et al. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc Natl Acad Sci USA. 2014;111:E5007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bahi A, Al Mansouri S, Al Memari E, Al Ameri M, Nurulain SM, Ojha S. β-Caryophyllene, a CB2 receptor agonist produces multiple behavioral changes relevant to anxiety and depression in mice. Physiol Behav. 2014;135:119–24. [DOI] [PubMed] [Google Scholar]
- 41.García-Gutiérrez M, Pérez-Ortiz J, Gutiérrez-Adán A, Manzanares J. Depression-resistant endophenotype in mice overexpressing cannabinoid CB2 receptors. Br J Pharmacol. 2010;160:1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.García-Gutiérrez MS, Manzanares J. Overexpression of CB2 cannabinoid receptors decreased vulnerability to anxiety and impaired anxiolytic action of alprazolam in mice. J Psycho-pharmacol. 2011;25:111–20. [DOI] [PubMed] [Google Scholar]
- 43.Liu Q-R, Canseco-Alba A, Zhang H-Y, Tagliaferro P, Chung M, Dennis E, et al. Cannabinoid type 2 receptors in dopamine neurons inhibits psychomotor behaviors, alters anxiety, depression and alcohol preference. Sci Rep. 2017;7:17410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 2012;76:116–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Small KM, Nunes E, Hughley S, Addy NA. Ventral tegmental area muscarinic receptors modulate depression and anxiety-related behaviors in rats. Neurosci Lett. 2016;616:80–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Addy NA, Nunes EJ, Wickham RJ. Ventral tegmental area cholinergic mechanisms mediate behavioral responses in the forced swim test. Behav Brain Res. 2015;288:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iñiguez SD, Vialou V, Warren BL, Cao J-L, Alcantara LF, Davis LC, et al. Extracellular signal-regulated kinase-2 within the ventral tegmental area regulates responses to stress. J Neurosci. 2010;30:7652–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bell-Horner CL, Dohi A, Nguyen Q, Dillon GH, Singh M. ERK/MAPK pathway regulates GABAA receptors. J Neurobiol. 2006;66:1467–74. [DOI] [PubMed] [Google Scholar]
- 49.Valenti O, Lodge DJ, Grace AA. Aversive stimuli alter ventral tegmental area dopamine neuron activity via a common action in the ventral hippocampus. J Neurosci. 2011;31:4280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valenti O, Gill KM, Grace AA. Different stressors produce excitation or inhibition of mesolimbic dopamine neuron activity: response alteration by stress pre-exposure. Eur J Neurosci. 2012;35:1312–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang C, Grace AA. Amygdala-ventral pallidum pathway decreases dopamine activity after chronic mild stress in rats. Biol Psychiatry. 2014;76:223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreines JL, Owrutsky ZL, Gagnon KG, Grace AA. Divergent effects of acute and repeated quetiapine treatment on dopamine neuron activity in normal vs. chronic mild stress induced hypodopaminergic states. Transl Psychiatry. 2017;7:1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rincón-Cortés M, Grace AA. Sex-dependent effects of stress on immobility behavior and VTA dopamine neuron activity: modulation by ketamine. Int J Neuropsychopharmacol. 2017;20:823–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai H-C, Finkelstein J, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. 2013; 493:537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McKinney BC, Sze W, Lee B, Murphy GG. Impaired long-term potentiation and enhanced neuronal excitability in the amygdala of Ca(V)1.3 knockout mice. Neurobiol Learn Mem. 2009;92:519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martínez-Rivera A, Hao J, Tropea TF, Giordano TP, Kosovsky M, Rice RC, et al. Enhancing VTA Cav1.3 L-type Ca2+ channel activity promotes cocaine and mood-related behaviors via overlapping AMPA receptor mechanisms in the nucleus accumbens. Mol Psychiatry. 2017;22:1735–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mameli-Engvall M, Evrard A, Pons S, Maskos U, Svensson TH, Changeux J-P, et al. Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron. 2006;50:911–21. [DOI] [PubMed] [Google Scholar]
- 58.Melis M, Carta S, Fattore L, Tolu S, Yasar S, Goldberg SR, et al. Peroxisome proliferator-activated receptors-alpha modulate dopamine cell activity through nicotinic receptors. Biol Psychiatry. 2010;68:256–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheggi S, Melis M, De Felice M, Aroni S, Muntoni AL, Pelliccia T, et al. PPARα modulation of mesolimbic dopamine transmission rescues depression-related behaviors. Neuropharmacology. 2016;110:251–9. [DOI] [PubMed] [Google Scholar]
- 60.Sekio M, Seki K. Lipopolysaccharide-induced depressive-like behavior is associated with α1-adrenoceptor dependent down-regulation of the membrane GluR1 subunit in the mouse medial prefrontal cortex and ventral tegmental area. Int J Neuropsychopharmacol. 2015;18:pyu005. 10.1093/ijnp/pyu005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhong P, Vickstrom CR, Liu X, Hu Y, Yu L, Yu H-G, et al. HCN2 channels in the ventral tegmental area regulate behavioral responses to chronic stress. Elife. 2018;7:pii: e32420 10.7554/eLife.32420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dremencov E, El Mansari M, Blier P . Effects of sustained serotonin reuptake inhibition on the firing of dopamine neurons in the rat ventral tegmental area. J Psychiatry Neurosci. 2009;34:223–9. [PMC free article] [PubMed] [Google Scholar]
- 63.West CHK, Weiss JM. Effects of chronic antidepressant drug administration and electroconvulsive shock on activity of dopaminergic neurons in the ventral tegmentum. Int J Neuropsychopharmacol. 2011;14:201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sekine Y, Suzuki K, Ramachandran PV, Blackburn TP, Ashby CR. Acute and repeated administration of fluoxetine, citalopram, and paroxetine significantly alters the activity of midbrain dopamine neurons in rats: an in vivo electrophysiological study. Synapse. 2007;61:72–7. [DOI] [PubMed] [Google Scholar]
- 65.Dankoski EC, Agster KL, Fox ME, Moy SS, Wightman RM. Facilitation of serotonin signaling by SSRIs is attenuated by social isolation. Neuropsychopharmacology. 2014;39:2928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14:609–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Björkholm C, Monteggia LM. BDNF – a key transducer of antidepressant effects. Neuropharmacology. 2016;102:72–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krishnan V, Han M-H, Mazei-Robison M, Iñiguez SD, Ables JL, Vialou V, et al. AKT signaling within the ventral tegmental area regulates cellular and behavioral responses to stressful stimuli. Biol Psychiatry. 2008;64:691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhong P, Liu X, Zhang Z, Hu Y, Liu SJ, Lezama-Ruiz M, et al. Cyclin-dependent kinase 5 in the ventral tegmental area regulates depression-related behaviors. J Neurosci. 2014;34:6352–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bai M, Zhu X, Zhang L, Zhang Y, Xue L, Wang Y, et al. Divergent anomaly in mesocorticolimbic dopaminergic circuits might be associated with different depressive behaviors, an animal study. Brain Behav. 2017;7:e00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun X, Song Z, Si Y, Wang JH. microRNA and mRNA profiles in ventral tegmental area relevant to stress-induced depression and resilience. Prog Neuropsychopharmacol Biol Psychiatry. 2018;86:150–65. [DOI] [PubMed] [Google Scholar]
- 72.Peña CJ, Kronman HG, Walker DM, Cates HM, Bagot RC, Purushothaman I, et al. Early life stress confers lifelong stress susceptibility in mice via ventral tegmental area OTX2. Science (80-). 2017;356:1185–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang H, de Jong JW, Tak Y, Peck J, Bateup HS, Lammel S. Nucleus accumbens subnuclei regulate motivated behavior via direct inhibition and disinhibition of VTA dopamine subpopulations. Neuron, 2018;97:434–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, et al. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 2012;491:212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beier KT, Steinberg EE, DeLoach KE, Xie S, Miyamichi K, Schwarz L, et al. Circuit architecture of VTA dopamine neurons revealed by systematic input-output mapping. Cell. 2015;162:622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kupchik YM, Brown RM, Heinsbroek JA, Lobo MK, Schwartz DJ, Kalivas PW. Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat Neurosci. 2015;18:1230–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith RJ, Lobo MK, Spencer S, Kalivas PW. Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways). Curr Opin Neurobiol. 2013;23:546–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gerfen CR. The neostriatal mosaic: multiple levels of compart-mental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320. [DOI] [PubMed] [Google Scholar]
- 80.Nicola SM. The nucleus accumbens as part of a basal ganglia action selection circuit. Psychopharmacology (Berl). 2007;191:521–50. [DOI] [PubMed] [Google Scholar]
- 81.Maia TV, Frank MJ. From reinforcement learning models to psychiatric and neurological disorders. Nat Neurosci. 2011; 14:154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Francis TC, Chandra R, Friend DM, Finkel E, Dayrit G, Miranda J, et al. Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol Psychiatry. 2015;77:212–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature. 2012;487:183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Khibnik LA, Beaumont M, Doyle M, Heshmati M, Slesinger PA, Nestler EJ, et al. Stress and cocaine trigger divergent and cell Ttype-specific regulation of synaptic transmission at single spines in nucleus accumbens. Biol Psychiatry. 2016;79:898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Muir J, Lorsch ZS, Ramakrishnan C, Deisseroth K, Nestler EJ, Calipari ES, et al. In vivo fiber photometry reveals signature of future stress susceptibility in nucleus accumbens. Neuropsychopharmacology. 2018;43:255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, et al. Natural neural projection dynamics underlying social behavior. Cell. 2014;157:1535–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Francis TC, Chandra R, Gaynor A, Konkalmatt P, Metzbower SR, Evans B, et al. Molecular basis of dendritic atrophy and activity in stress susceptibility. Mol Psychiatry. 2017;22:1512–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Francis TC, Gaynor A, Chandra R, Fox ME, Lobo MK. The selective RhoA inhibitor Rhosin promotes stress resiliency through enhancing D1-MSN plasticity and reducing hyper-excitability. Biol Psychiatry. 2019. 10.1016/j.biopsych.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Duvarci S, Paré D. Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J Neurosci. 2007; 27:4482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barik J, Marti F, Morel C, Fernandez SP, Lanteri C, Godeheu G, et al. Chronic stress triggers social aversion via glucocorticoid receptor in dopaminoceptive neurons. Science. 2013;339:332–5. [DOI] [PubMed] [Google Scholar]
- 92.Campioni MR, Xu M, McGehee DS. Stress-induced changes in nucleus accumbens glutamate synaptic plasticity. J Neurophysiol. 2009;101:3192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Terrillion CE, Francis TC, Puche AC, Lobo MK, Gould TD. Decreased nucleus accumbens expression of psychiatric disorder risk gene Cacna1c promotes susceptibility to social stress. Int J Neuropsychopharmacol. 2017;20:428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brancato A, Bregman D, Ahn HF, Pfau ML, Menard C, Cannizzaro C, et al. Sub-chronic variable stress induces sex-specific effects on glutamatergic synapses in the nucleus accumbens. Neuroscience. 2017;350:180–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Christoffel DJ, Golden SA, Walsh JJ, Guise KG, Heshmati M, Friedman AK, et al. Excitatory transmission at thalamo-striatal synapses mediates susceptibility to social stress. Nat Neurosci. 2015;18:962–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bagot RC, Parise EM, Peña CJ, Zhang H-X, Maze I, Chaudhury D, et al. Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat Commun. 2015;6:7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fremeau RT, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, et al. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–60. [DOI] [PubMed] [Google Scholar]
- 98.LeGates TA, Kvarta MD, Tooley JR, Francis TC, Lobo MK, Creed MC, et al. Reward behaviour is regulated by the strength of hippocampus–nucleus accumbens synapses. Nature. 2018;564:258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Barrientos C, Knowland D, Wu MMJ, Lilascharoen V, Huang KW, Malenka RC, et al. Cocaine-induced structural plasticity in input regions to distinct cell types in nucleus accumbens. Biol Psychiatry. 2018;84:893–904. 10.1016/j.biopsych.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vialou V, Robison AJ, Laplant QC, Covington HE, Dietz DM, Ohnishi YN, et al. DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat Neurosci. 2010;13:745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, et al. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52:445–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Coppens CM, Siripornmongcolchai T, Wibrand K, Alme MN, Buwalda B, de Boer SF, et al. Social defeat during adolescence and adulthood differentially induce BDNF-regulated immediate early genes. Front Behav Neurosci. 2011;5:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Matikainen-Ankney BA, Kezunovic N, Menard C, Flanigan M, Zhong Y, Russo SJ, et al. Parkinson’s Disease-linked LRRK2-G2019S mutation alters synaptic plasticity and promotes resilience to chronic social stress in young adulthood. J Neurosci. 2018;38:9700–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schwartz N, Temkin P, Jurado S, Lim BK, Heifets BD, Polepalli JS, et al. Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science. 2014;345:535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Joffe ME, Vitter SR, Grueter BA. GluN1 deletions in D1-and A2A-expressing cell types reveal distinct modes of behavioral regulation. Neuropharmacology. 2017;112:172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Robison AJ, Bartlett RK, Bass MA, Colbran RJ. Differential modulation of Ca2+ /calmodulin-dependent protein kinase II activity by regulated interactions with N -methyl-D-aspartate receptor NR2B subunits and α-actinin. J Biol Chem. 2005;280:39316–23. [DOI] [PubMed] [Google Scholar]
- 107.Robison AJ, Vialou V, Sun H-S, Labonte B, Golden SA, Dias C, et al. Fluoxetine epigenetically alters the CaMKIIα promoter in nucleus accumbens to regulate ΔFosB binding and antidepressant effects. Neuropsychopharmacology. 2014;39:1178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Holly EN, Miczek KA. Ventral tegmental area dopamine revisited: effects of acute and repeated stress. Psychopharmacology (Berl). 2016;233:163–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Muscat R, Willner P. Effects of dopamine receptor antagonists on sucrose consumption and preference. Psychopharmacology (Berl). 1989;99:98–102. [DOI] [PubMed] [Google Scholar]
- 110.Yorgason JT, Calipari ES, Ferris MJ, Karkhanis AN, Fordahl SC, Weiner JL, et al. Social isolation rearing increases dopamine uptake and psychostimulant potency in the striatum. Neuropharmacology. 2016;101:471–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Deal AL, Konstantopoulos JK, Weiner JL, Budygin EA. Exploring the consequences of social defeat stress and intermittent ethanol drinking on dopamine dynamics in the rat nucleus accumbens. Sci Rep. 2018;8:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Knoll AT, Carlezon WA. Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bérubé P, Laforest S, Bhatnagar S, Drolet G. Enkephalin and dynorphin mRNA expression are associated with resilience or vulnerability to chronic social defeat stress. Physiol Behav. 2013;122:237–45. [DOI] [PubMed] [Google Scholar]
- 114.Der-Avakian A, D’Souza MS, Potter DN, Chartoff EH, Carlezon WA, Pizzagalli DA, et al. Social defeat disrupts reward learning and potentiates striatal nociceptin/orphanin FQ mRNA in rats. Psychopharmacology (Berl). 2017;234:1603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Karkhanis AN, Rose JH, Weiner JL, Jones SR. Early-life social isolation stress increases kappa opioid receptor responsiveness and downregulates the dopamine system. Neuropsychopharmacology. 2016;41:2263–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Henry MS, Gendron L, Tremblay M-E, Drolet G. Enkephalins: endogenous analgesics with an emerging role in stress resilience. Neural Plast. 2017;2017:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Poulin JF, Laforest S, Drolet G. Enkephalin downregulation in the nucleus accumbens underlies chronic stress-induced anhedonia. Stress. 2014;17:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sweis BM, Veverka KK, Dhillon ES, Urban JH, Lucas LR. Individual differences in the effects of chronic stress on memory: behavioral and neurochemical correlates of resiliency. Neuroscience. 2013;246:142–59. [DOI] [PubMed] [Google Scholar]
- 119.Chuang J-C, Krishnan V, Yu HG, Mason B, Cui H, Wilkinson MB, et al. A β3-Adrenergic-leptin-melanocortin circuit regulates behavioral and metabolic changes induced by chronic stress. Biol Psychiatry. 2010;67:1075–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Manning CE, Williams ES, Robison AJ. Reward network immediate early gene expression in mood disorders. Front Behav Neurosci. 2017;11:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nestler EJ. ΔFosB: a transcriptional regulator of stress and antidepressant responses. Eur J Pharmacol. 2015;753:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lobo MK, Zaman S, Damez-Werno DM, Koo JW, Bagot RC, DiNieri JA, et al. FosB induction in striatal medium spiny neuron subtypes in response to chronic pharmacological, emotional, and optogenetic stimuli. J Neurosci. 2013;33:18381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mul JD, Soto M, Cahill ME, Ryan RE, Takahashi H, So K, et al. Voluntary wheel running promotes resilience to chronic social defeat stress in mice: a role for nucleus accumbens ΔFosB. Neuropsychopharmacology. 2018;43:1934–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hamilton PJ, Burek DJ, Lombroso SI, Neve RL, Robison AJ, Nestler EJ, et al. Cell-type-specific epigenetic editing at the Fosb gene controls susceptibility to social defeat stress. Neuropsychopharmacology. 2018;43:272–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang W, Shi Y, Peng Y, Zhong L, Zhu S, Zhang W, et al. Neuron activity–induced Wnt signaling up-regulates expression of brain-derived neurotrophic factor in the pain neural circuit. J Biol Chem. 2018;293:15641–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wilkinson MB, Dias C, Magida J, Mazei-Robison M, Lobo M, Kennedy P, et al. A novel role of the WNT-dishevelled-GSK3 signaling cascade in the mouse nucleus accumbens in a social defeat model of depression. J Neurosci. 2011;31:9084–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dias C, Feng J, Sun H, Shao NY, Mazei-Robison MS, Damez-Werno D, et al. β-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature. 2014;516:51–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–83. [DOI] [PubMed] [Google Scholar]
- 129.Chandra R, Engeln M, Francis TC, Konkalmatt P, Patel D, Lobo MK. A role for peroxisome proliferator-activated receptor gamma coactivator-1? in nucleus accumbens neuron subtypes in cocaine action. Biol Psychiatry. 2017;81:564–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhan G, Huang N, Li S, Hua D, Zhang J, Fang X, et al. PGC-1α–FNDC5–BDNF signaling pathway in skeletal muscle confers resilience to stress in mice subjected to chronic social defeat. Psychopharmacology (Berl). 2018;235:3351–8. [DOI] [PubMed] [Google Scholar]
- 131.Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012; 481:463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Larrieu T, Cherix A, Duque A, Rodrigues J, Lei H, Gruetter R, et al. Hierarchical status predicts behavioral vulnerability and nucleus accumbens metabolic profile following chronic social defeat stress. Curr Biol. 2017;27:2202–10. [DOI] [PubMed] [Google Scholar]
- 133.Lorsch ZS, Loh Y-HE, Purushothaman I, Walker DM, Parise EM, Salery M, et al. Estrogen receptor α drives pro-resilient transcription in mouse models of depression. Nat Commun. 2018;9:1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dougherty JD. The expanding toolkit of translating ribosome affinity purification. J Neurosci. 2017;37:12079–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kohli MA, Lucae S, Saemann PG, Schmidt MV, Demirkan A, Hek K, et al. The neuronal transporter gene SLC6A15 confers risk to major depression. Neuron. 2011;70:252–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chandra R, Francis TC, Nam H, Riggs LM, Engeln M, Rudzinskas S, et al. Reduced Slc6a15 in nucleus accumbens D2-neurons underlies stress susceptibility. J Neurosci. 2017;37:6527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Heshmati M, Aleyasin H, Menard C, Christoffel DJ, Flanigan ME, Pfau ML, et al. Cell-type-specific role for nucleus accumbens neuroligin-2 in depression and stress susceptibility. Proc Natl Acad Sci USA. 2018;115:1111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fox ME, Chandra R, Menken MS, Larkin EJ, Nam H, Engeln M, et al. Dendritic remodeling of D1 neurons by RhoA/Rho-kinase mediates depression-like behavior. Mol Psychiatry. 2018. 10.1038/s41380-018-0211-5. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Klengel T, Pape J, Binder EB, Mehta D. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology. 2014;80:115–32. [DOI] [PubMed] [Google Scholar]
- 140.Covington HE, Maze I, Sun H, Bomze HM, DeMaio KD, Wu EY, et al. A role for repressive histone methylation in cocaine-Iinduced vulnerability to stress. Neuron. 2011;71:656–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Golden SA, Christoffel DJ, Heshmati M, Hodes GE, Magida J, Davis K, et al. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat Med. 2013;19:337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wilkinson MB, Xiao G, Kumar A, LaPlant Q, Renthal W, Sikder D, et al. Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. J Neurosci. 2009;29:7820–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.LaPlant Q, Vialou V, Covington HE, Dumitriu D, Feng J, Warren BL, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hodes GE, Pfau ML, Purushothaman I, Ahn HF, Golden SA, Christoffel DJ, et al. Sex differences in nucleus accumbens transcriptome profiles associated with susceptibility versus resilience to subchronic variable stress. J Neurosci. 2015;35:16362–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Feng J, Pena CJ, Purushothaman I, Engmann O, Walker D, Brown AN, et al. Tet1 in nucleus accumbens opposes depression-and anxiety-like behaviors. Neuropsychopharmacology. 2017;42:1657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lepack AE, Bagot RC, Peña CJ, Loh Y-HE, Farrelly LA, Lu Y, et al. Aberrant H3.3 dynamics in NAc promote vulnerability to depressive-like behavior. Proc Natl Acad Sci USA. 2016;113:12562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kim H-D, Hesterman J, Call T, Magazu S, Keeley E, Armenta K, et al. SIRT1 mediates depression-like behaviors in the nucleus accumbens. J Neurosci. 2016;36:8441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Anacker C, Scholz J, O’Donnell KJ, Allemang-Grand R, Diorio J, Bagot RC, et al. Neuroanatomic differences associated with stress susceptibility and resilience. Biol Psychiatry. 2016;79:840–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Morales-Medina JC, Sanchez F, Flores G, Dumont Y, Quirion R. Morphological reorganization after repeated corticosterone administration in the hippocampus, nucleus accumbens and amygdala in the rat. J Chem Neuroanat. 2009;38:266–72. [DOI] [PubMed] [Google Scholar]
- 150.Haim A, Sherer M, Leuner B. Gestational stress induces persistent depressive-like behavior and structural modifications within the postpartum nucleus accumbens. Eur J Neurosci. 2014;40:3766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shansky RM, Hamo C, Hof PR, McEwen BS, Morrison JH. Stress-induced dendritic remodeling in the prefrontal cortex is circuit specific. Cereb Cortex. 2009;19:2479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–22. [DOI] [PubMed] [Google Scholar]
- 153.Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bessa JM, Morais M, Marques F, Pinto L, Palha JA, Almeida OFX, et al. Stress-induced anhedonia is associated with hypertrophy of medium spiny neurons of the nucleus accumbens. Transl Psychiatry. 2013;3:e266–e266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yang C, Shirayama Y, Zhang J-c, Ren Q, Hashimoto K Regional differences in brain-derived neurotrophic factor levels and dendritic spine density confer resilience to inescapable stress. Int J Neuropsychopharmacol. 2015;18:pyu121–pyu121. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 156.Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, Ahn HF, et al. IκB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci. 2011;31:314–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Christoffel DJ, Golden SA, Heshmati M, Graham A, Birnbaum S, Neve RL, et al. Effects of inhibitor of κB kinase activity in the nucleus accumbens on emotional behavior. Neuropsychopharmacology. 2012;37:2615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Warner-Schmidt JL, Schmidt EF, Marshall JJ, Rubin AJ, Arango-Lievano M, Kaplitt MG, et al. Cholinergic interneurons in the nucleus accumbens regulate depression-like behavior. Proc Natl Acad Sci USA. 2012;109:11360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Witten IB, Lin S-C, Brodsky M, Prakash R, Diester I, Anikeeva P, et al. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science (80-). 2010;330:1677–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cachope R, Mateo Y, Mathur BN, Irving J, Wang H-L, Morales M, et al. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2012;2:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. [DOI] [PubMed] [Google Scholar]
- 162.Svenningsson P, Chergui K, Rachleff I, Flajolet M, Zhang X, El Yacoubi M, et al. Alterations in 5-HT1B receptor function by p11 in depression-like states. Science. 2006;311:77–80. [DOI] [PubMed] [Google Scholar]
- 163.Alexander B, Warner-Schmidt J, Eriksson T, Tamminga C, Arango-Llievano M, Ghose S, et al. Reversal of depressed behaviors in mice by p11 gene therapy in the nucleus accumbens. Sci Transl Med. 2010;2:54ra76–54ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Melas PA, Rogdaki M, Lennartsson A, Björk K, Qi H, Witasp A, et al. Antidepressant treatment is associated with epigenetic alterations in the promoter of P11 in a genetic model of depression. Int J Neuropsychopharmacol. 2012;15:669–79. [DOI] [PubMed] [Google Scholar]
- 165.Knowland D, Lilascharoen V, Pacia CP, Shin S, Wang EH-J, Lim BK. Distinct ventral pallidal neural populations mediate separate symptoms of depression. Cell. 2017;170:284–.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bosse KE, Maina FK, Birbeck JA, France MM, Roberts JJP, Colombo ML, et al. Aberrant striatal dopamine transmitter dynamics in brain-derived neurotrophic factor-deficient mice. J Neurochem. 2012;120:385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Martin-Iverson MT, Todd KG, Altar CA. Brain-derived neurotrophic factor and neurotrophin-3 activate striatal dopamine and serotonin metabolism and related behaviors: interactions with amphetamine. J Neurosci. 1994;14:1262–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. [DOI] [PubMed] [Google Scholar]
- 169.Shinohara R, Taniguchi M, Ehrlich AT, Yokogawa K, Deguchi Y, Cherasse Y, et al. Dopamine D1 receptor subtype mediates acute stress-induced dendritic growth in excitatory neurons of the medial prefrontal cortex and contributes to suppression of stress susceptibility in mice. Mol Psychiatry. 2018;23:1717–30. [DOI] [PubMed] [Google Scholar]
- 170.Gagnon D, Petryszyn S, Sanchez MG, Bories C, Beaulieu JM, De Koninck Y, et al. Striatal neurons expressing D1 and D2 receptors are morphologically distinct and differently affected by dopamine denervation in mice. Sci Rep. 2017;7:41432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Herculano-Houzel S, Lent R. Isotropic fractionator: a simple, rapid method for the quantification of total cell and neuron numbers in the brain. J Neurosci. 2005;25:2518–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.von Bartheld CS, Bahney J, Herculano-Houzel S. The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J Comp Neurol. 2016;524:3865–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Köhler O, Benros ME, Nordentoft M, Farkouh ME, Iyengar RL, Mors O, et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects. JAMA Psychiatry. 2014;71:1381. [DOI] [PubMed] [Google Scholar]
- 174.Kubera M, Obuchowicz E, Goehler L, Brzeszcz J, Maes M. In animal models, psychosocial stress-induced (neuro)inflammation, apoptosis and reduced neurogenesis are associated to the onset of depression. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:744–59. [DOI] [PubMed] [Google Scholar]
- 175.Hodes GE, Pfau ML, Leboeuf M, Golden SA, Christoffel DJ, Bregman D, et al. Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci USA. 2014;111:16136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Voorhees JL, Tarr AJ, Wohleb ES, Godbout JP, Mo X, Sheridan JF, et al. Prolonged restraint stress increases IL-6, reduces IL-10, and causes persistent depressive-like behavior that is reversed by recombinant IL-10. PLoS ONE. 2013;8:e58488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Monje FJ, Cabatic M, Divisch I, Kim E-J, Herkner KR, Binder BR, et al. Constant darkness induces IL-6-dependent depression-like behavior through the NF-B signaling pathway. J Neurosci. 2011;31:9075–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Beurel E, Harrington LE, Jope RS. Inflammatory T helper 17 cells promote depression-like behavior in mice. Biol Psychiatry. 2013;73:622–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hayley S, Scharf J, Anisman H. Central administration of murine interferon-α induces depressive-like behavioral, brain cytokine and neurochemical alterations in mice: A mini-review and original experiments. Brain Behav Immun. 2013;31:115–27. [DOI] [PubMed] [Google Scholar]
- 180.Walker FR. A critical review of the mechanism of action for the selective serotonin reuptake inhibitors: do these drugs possess anti-inflammatory properties and how relevant is this in the treatment of depression? Neuropharmacology. 2013;67:304–17. [DOI] [PubMed] [Google Scholar]
- 181.Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci. 2013;33:13820–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, et al. Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry. 2014;75:970–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Crider A, Feng T, Pandya CD, Davis T, Nair A, Ahmed AO, et al. Complement component 3a receptor deficiency attenuates chronic stress-induced monocyte infiltration and depressive-like behavior. Brain Behav Immun. 2018;70:246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S, et al. Social stress induces neurovascular pathology promoting depression. Nat Neurosci. 2017;20:1752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wohleb ES, Terwilliger R, Duman CH, Duman RS. Stress-induced neuronal colony stimulating factor 1 provokes microglia-mediated neuronal remodeling and depressive-like behavior. Biol Psychiatry. 2018;83:38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53:1181–94. [DOI] [PubMed] [Google Scholar]
- 187.Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77:10–8. [DOI] [PubMed] [Google Scholar]
- 188.Kreisel T, Frank MG, Licht T, Reshef R, Ben-Menachem-Zidon O, Baratta MV, et al. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry. 2014;19:699–709. [DOI] [PubMed] [Google Scholar]
- 189.Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, Pow DV, et al. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun. 2010;24:1058–68. [DOI] [PubMed] [Google Scholar]
- 190.Décarie-Spain L, Sharma S, Hryhorczuk C, Issa-Garcia V, Barker PA, Arbour N, et al. Nucleus accumbens inflammation mediates anxiodepressive behavior and compulsive sucrose seeking elicited by saturated dietary fat. Mol Metab. 2018;10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, et al. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci. 2011;31:6277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wu X-B, Jing P-B, Zhang Z-J, Cao D-L, Gao M-H, Jiang B-C, et al. Chemokine receptor CCR2 contributes to neuropathic pain and the associated depression via increasing NR2B-mediated currents in both D1 and D2 dopamine receptor-containing medium spiny neurons in the nucleus accumbens shell. Neuropsychopharmacology. 2018;43:2320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Brachman RA, Lehmann ML, Maric D, Herkenham M. Lymphocytes from chronically stressed mice confer antidepressant-like effects to naive mice. J Neurosci. 2015;35:1530–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kim H-J, Cho M-H, Shim WH, Kim JK, Jeon E-Y, Kim D-H, et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry. 2017;22:1576–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, et al. Unexpected role of interferon-y in regulating neuronal connectivity and social behaviour. Nature. 2016;535:425–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.De Biase LM, Schuebel KE, Fusfeld ZH, Jair K, Hawes IA, Cimbro R, et al. Local cues establish and maintain region-specific phenotypes of basal ganglia microglia. Neuron. 2017;95:341–.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Foster JA, McVey Neufeld K-A. Gut-brain axis: how the microbiome influences anxiety and depression. Trends Neurosci. 2013;36:305–12. [DOI] [PubMed] [Google Scholar]
- 198.Zheng P, Zeng B, Zhou C, Liu M, Fang Z, Xu X, et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol Psychiatry. 2016;21:786–96. [DOI] [PubMed] [Google Scholar]
- 199.Liu J, Dietz K, Hodes GE, Russo SJ, Casaccia P. Widespread transcriptional alternations in oligodendrocytes in the adult mouse brain following chronic stress. Dev Neurobiol. 2018;78:152–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Cui W, Mizukami H, Yanagisawa M, Aida T, Nomura M, Isomura Y, et al. Glial dysfunction in the mouse habenula causes depressive-like behaviors and sleep disturbance. J Neurosci. 2014;34:16273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zink M, Vollmayr B, Gebicke-Haerter PJ, Henn FA. Reduced expression of glutamate transporters vGluT1, EAAT2 and EAAT4 in learned helpless rats, an animal model of depression. Neuropharmacology. 2010;58:465–73. [DOI] [PubMed] [Google Scholar]
- 202.Thompson SM, Kallarackal AJ, Kvarta MD, Van Dyke AM, LeGates TA, Cai X. An excitatory synapse hypothesis of depression. Trends Neurosci. 2015;38:279–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun. 2018;9:1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lehmann ML, Weigel TK, Cooper HA, Elkahloun AG, Kigar SL, Herkenham M. Decoding microglia responses to psychosocial stress reveals blood-brain barrier breakdown that may drive stress susceptibility. Sci Rep. 2018;8:11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Warren BL, Vialou VF, Iniguez SD, Alcantara LF, Wright KN, Feng J, et al. Neurobiological sequelae of witnessing stressful events in adult mice. Biol Psychiatry. 2013;73:7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Der-Avakian A, Mazei-Robison MS, Kesby JP, Nestler EJ, Markou A. Enduring deficits in brain reward function after chronic social defeat in rats: susceptibility, resilience, and antidepressant response. Biol Psychiatry. 2014;76:542–9. [DOI] [PMC free article] [PubMed] [Google Scholar]