

Abstract
Postsynaptic trafficking plays a key role in regulating synapse structure and function. While spiny excitatory synapses can be stable throughout adult life, their morphology and function is impaired in Alzheimer’s disease (AD). However, little is known about how AD risk genes impact synaptic function. Here we used structured superresolution illumination microscopy (SIM) to study the late-onset Alzheimer’s disease (LOAD) risk factor BIN1, and show that this protein is abundant in postsynaptic compartments, including spines. While postsynaptic Bin1 shows colocalization with clathrin, a major endocytic protein, it also colocalizes with the small GTPases Rab11 and Arf6, components of the exocytic pathway. Bin1 participates in protein complexes with Arf6 and GluA1, and manipulations of Bin1 lead to changes in spine morphology, AMPA receptor surface expression and trafficking, and AMPA receptor-mediated synaptic transmission. Our data provide new insights into the mesoscale architecture of postsynaptic trafficking compartments and their regulation by a major LOAD risk factor.
Fast excitatory neurotransmission in the mammalian brain occurs mainly at glutamatergic synapses and is mediated predominantly by the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid type of ionotropic glutamate receptor (AMPA)1. The postsynaptic side of most glutamatergic synapses is defined by dendritic spines, and spine architecture and AMPA receptor content are essential determinants of synaptic function, plasticity, behavior, and cognition2. Both spine architecture and AMPA receptor content at synapses are controlled through endocytosis and exocytic recycling3, and a proper balance of these two processes is critical for homeostatic regulation and control of dendritic spine dynamics, and therefore for normal synaptic function4. While substantial progress has been made in understanding endocytic and exocytic trafficking, the molecular mechanisms that control them, as well as the mesoscale spatial architecture of postsynaptic trafficking compartments in spines and dendrites, remain incompletely understood5.
While synapses can be stable throughout adult life, their morphology and function is severely impaired in Alzheimer’s disease (AD)2. Synapse loss is a prominent and consistent finding in postmortem brain tissue from AD patients, and cognitive decline is closely correlated with synapse loss6. Only a small fraction of patients with AD have an early-onset, familial form of the disease; the overwhelming majority of cases are late and sporadic, referred to as late-onset AD (LOAD). The genetic substrates of familial AD are fairly well understood7, but the complex genetic architecture of LOAD is only beginning to be uncovered. Genome-wide association studies (GWAS) have implicated up to ~40 genes in LOAD risk8,9. However, their roles in the pathogenesis of LOAD are poorly understood, due to incomplete knowledge about their basic neurobiological functions.
Recent large GWAS screens identified the BIN1 gene locus as an important LOAD genetic susceptibility locus, with the rs744373 single nucleotide polymorphism (SNP) risk allele having an odds ratio (OR) of 1.17 and a minor allele frequency (MAF) of 0.298. Importantly, the AlzGene analysis of epidemiological credibility of LOAD GWAS hits lists BIN1 as the second most significant LOAD risk factor after APOE (see www.alzgene.com for specifics about analysis). The BIN1 gene encodes the protein BIN1 (bridging integrator 1, also known as amphiphysin 2), implicated in several biological processes including tumor suppression, apoptosis, and trafficking10. The best known function of amphiphysins is in endocytosis, mediated by their interactions with several proteins associated with clathrin-coated pits, including clathrin, dynamin, and AP211. BIN1 has been shown to regulate intracellular trafficking through inducing membrane curvature, clustering dynamin, and binding to the actin cytoskeleton12,13. These roles have been primarily demonstrated with regards to endocytosis, but some studies suggest that BIN1 plays a role in exocytosis as well14,15.
In this study, we show that synaptic trafficking is overrepresented among LOAD risk factors. We thus investigated the basic functions of rodent Bin1 in neuronal trafficking, as a prominent representative of this subclass of LOAD risk factors. Consistent with a synaptic role of Bin1, we find that Bin1 protein is present in postsynaptic compartments, including spines and dendritic shafts, in addition to presynaptic regions. While postsynaptic Bin1 shows partial colocalization with clathrin, it shows similarly strong colocalization with the small GTPases Rab11 and Arf6. Bin1 knockdown affects the size and organization of postsynaptic Rab11 and Arf6-positive structures, but not those of clathrin. Bin1 participates in protein complexes with Arf6 as well as with GluA1, and manipulations of Bin1 lead to small but significant changes in spine morphology, AMPA receptor surface expression and trafficking, and AMPA receptor-mediated synaptic transmission. Our data provide new insights into the mesoscale architecture of postsynaptic trafficking, as well as into the basic neuronal functions of a major LOAD risk factor.
RESULTS
Network analysis of LOAD risk factors reveals a role for synaptic trafficking
In order to uncover biological processes that might be generally relevant for LOAD risk, we performed gene ontology (GO) analysis on 40 genes that have been identified in GWAS studies for LOAD risk. This analysis revealed the top 3 GO clusters of biological processes to be vesicle-mediated transport/endocytosis, cellular component assembly, and lipid transport/metabolism (Fig 1A,B). Evidence for the importance of trafficking-related LOAD risk genes functioning at synapses came when we generated a protein interaction network of the proteins encoded by the 11 LOAD risk genes present in the vesicle-mediated transport/endocytosis GO cluster. This network revealed a significant enrichment of proteins present in the postsynaptic density (PSD)16, suggesting that trafficking protein networks in the postsynaptic compartment might be generally relevant for LOAD risk (Fig 1C). When we generated a similar network around just BIN1, which has been identified multiple times in LOAD GWAS studies, we again found a highly significant enrichment of PSD proteins, as well as trafficking proteins and GTPase-related proteins (Fig 1D). Importantly, BIN1 itself was present in the PSD-expressed protein dataset. These findings support the idea that trafficking in the postsynaptic compartment, perhaps involving regulation of GTPases, might significantly contribute to the risk generated by BIN1 risk alleles, and perhaps other LOAD risk genes. We decided to further study Bin1 in rodent neurons to understand the role this protein plays in synaptic neurobiology, with the hope that our findings might more generally inform how synaptic trafficking contributes to LOAD risk.
Fig 1. Postsynaptic localization of trafficking-related LOAD risk factor Bin1.

(A) The top 3 GO biological clusters of 40 genes identified in GWAS studies of LOAD risk.
(B) Top 5 individual GO terms within the top GO cluster. The dotted line indicates significance level of P= 0.05.
(C) Protein interaction network of 11 LOAD risk genes present in the vesicle-mediated transport/endocytosis GO cluster. Network was generated using the GeneMANIA in Cytoscape. This network has a significant enrichment of proteins present in the PSD (P=0.0002).
(D) BIN1 protein interaction network reveals a significant enrichment of PSD genes (P= 4.0×10−7), trafficking genes (P= 0.0002), and GTPase-related genes (P= 5.0×10−5).
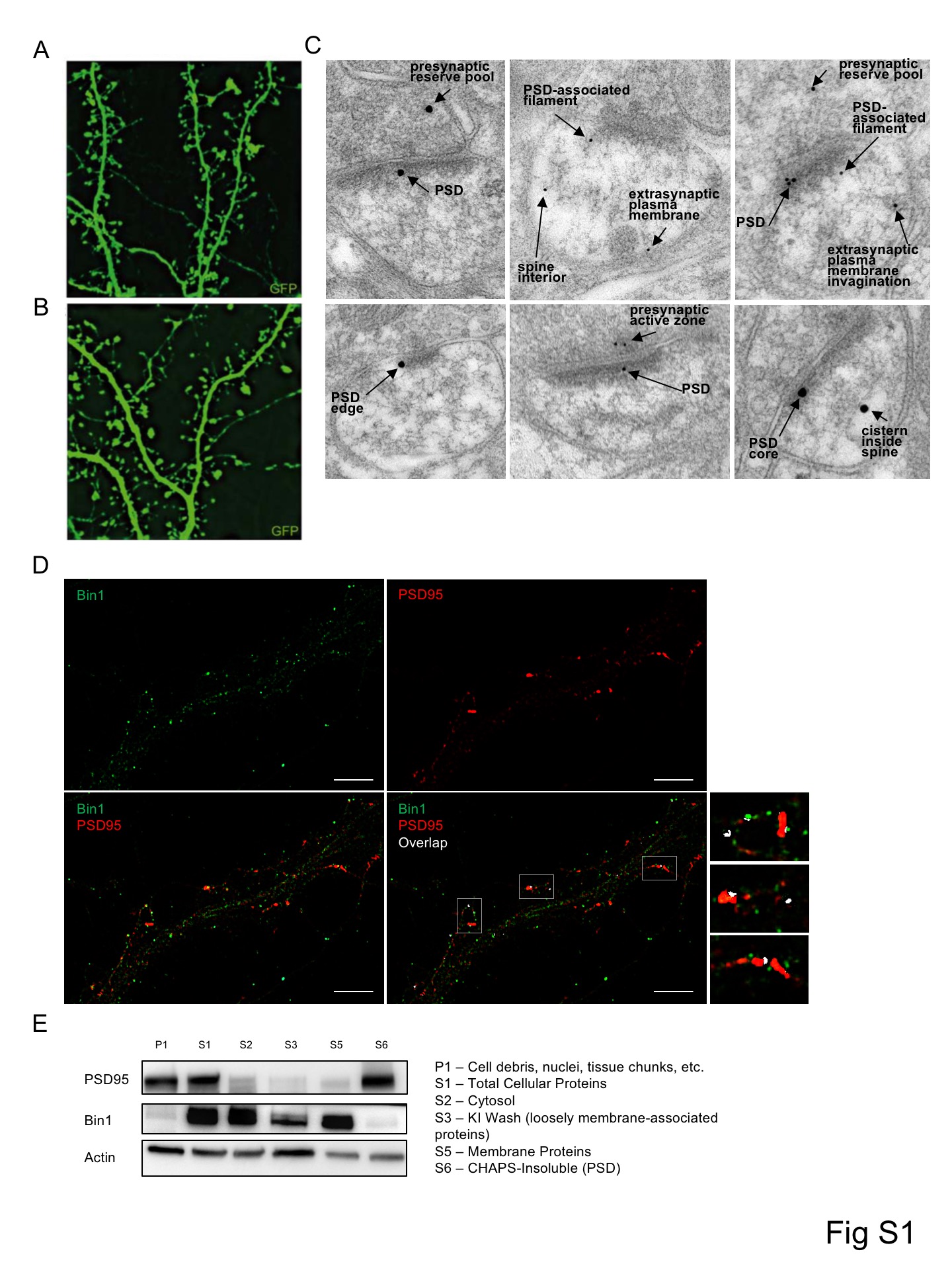
(E) Top: Single plane SIM image of a dendritic region stained for Bin1 and presynaptic marker synapsin1. GFP cell fill is outlined (see Fig S1A). Scale bar = 5 μm
Bottom: Representative SIM images of boxed spines in above image.
(F) Top: Single plane SIM image of a dendritic region stained for Bin1 and GluA1. GFP cell fill is outlined (see Fig S1B). Scale bar = 5 μm
Bottom: Representative SIM images of boxed spines above image.
(G) Representative 3D reconstruction of a stack of SIM images showing the relative localization of Bin1 and GluA1 within a spine. Scale bar = 50 nm
(H) Immuno-electron microscopy for Bin1. Left micrograph is low-magnification view of neuropil. Labeled spines have been colorized green and axon terminals pink; a thin immunopositive dendritic process (left) is colorized blue.
(I) Bin1-associated gold particles in immuno-EM images are found in the synaptic membrane/PSD, as well as near the PSD in extrasynaptic membranes.
Structured illumination microscopy (SIM) reveals subsynaptic compartmentalization of Bin1 in spines
In order to examine the distribution of Bin1 within postsynaptic microcompartments we used structured illumination microscopy (SIM) due to its ability to resolve fluorescence localization with high spatial resolution. SIM imaging revealed that neuronal Bin1 immunofluorescence defined distinct structures. Bin1 showed some colocalization with the presynaptic marker Synapsin1, but was often localized adjacent to Synapsin1 (Fig 1E, Fig S1A). In contrast, Bin1 showed substantial colocalization with GluA1, a major postsynaptic protein in glutamatergic synapses (Fig 1F, Fig S1B). 3D reconstructions revealed that Bin1 in the spine head was closely associated with GluA1 (Fig 1G).
To confirm the patterns of subsynaptic localization of Bin1 detected by SIM and to validate our SIM analysis, we used an independent method, immunoelectron microscopy (immuno-EM). Importantly, comparisons of immuno-EM labeling of Bin1 between cortex and cerebellum showed differential staining similar to what was seen in Allen Brain Atlas in situ hybridization (ISH) studies (ISH: 186% cortex vs. cerebellum; Immuno-EM: 140% cortex vs. cerebellum), supporting the specificity of the Bin1 antibody used in these studies. In the cortex, subsynaptic localization of Bin1 labeling showed immunoreactivity in spines was most often detected at or near the PSD (Fig 1H). Bin1 was almost always associated with electron-dense structures; labeling was seen both at the core and edge of the PSD, as well as near PSD-associated filaments. Bin1 was also often detected at the extrasynaptic plasma membrane, inside the spine, and in the presynaptic compartment (Fig 1H, Fig S1C). Quantification of immuno-EM labeling revealed that Bin1 concentration in spines was ~2.5 times higher than in large dendritic shafts, and was ~2 times higher in axon terminals than in dendritic shafts. The PSD compartment showed the highest concentrations, about 3.5 times higher than in shafts. SIM imaging also revealed colocalization between Bin1 and the postsynaptic density protein PSD95 (Fig S1D), further supporting Bin1 localization within the postsynaptic compartment. To better define the spatial organization of Bin1 in dendritic spine, we determined the tangential distribution of Bin1 along the plasma membrane. We found that Bin1 labeling was both at synaptic and non-synaptic membranes, concentrating just lateral to the edge of the PSD (Fig 1I). Interestingly, PSD-enriched protein fractions showed relatively low abundance of Bin1 (Fig S1E, observation without quantification), supporting the localization of Bin1 close to the PSD, but not necessarily extensive integration into this structure.
Bin1 functions in postsynaptic trafficking
To further examine possible functions of Bin1 in postsynaptic trafficking, we used SIM to map the relative localization of Bin1 with markers for major trafficking compartments, including clathrin (Chc, to identify clathrin-coated pits and vesicles), Rab5 (a marker of early endosomes), EEA1 (early endosomes), and Rab11 (recycling endosomes) (Fig 2A). Bin1 showed the highest colocalization with clathrin, and unexpectedly, also with Rab11 (further supported by Manders analysis (Fig 2B)). These findings suggested that Bin1 might regulate multiple distinct trafficking events in dendrites and spines. To explore this possibility, we focused on the endocytic and recycling compartments, where Bin1 had shown highest colocalization. We manipulated the expression of Bin1 in neurons by knockdown and overexpression (validated in Fig S2A-E), and assessed the impact of these manipulations on the area and number of Chc-labeled and Rab11-labeled structures (Fig 2C, Fig S2F-L). Surprisingly, and contrary to predictions based on interactions of Bin1 with clathrin, we found that neither total area nor number of Chc-labeled structures were affected by manipulating Bin1 expression (Fig S2G-J). Instead, knockdown of Bin1 resulted in increased area and number of Rab11-labeled structures (Fig 2C). Overexpression of Bin1, however, did not affect the total area nor number of Rab11-labeled structures (Fig S2K,L). Taken together, these data suggest that Bin1 participates in several steps of postsynaptic trafficking, including clathrin-mediated endocytosis and trafficking from recycling endosomes to the plasma membrane. Reduced Bin1 may interfere with exocytic recycling, causing a buildup of Rab11.
Fig 2. Bin1 regulates recycling endosomes in spines and interacts with Arf6.

(A) Single plane SIM images of relative localization of Bin1 and major trafficking markers in the dendritic compartment. Regions in boxes are shown below each image set in high magnification where region of co-localization are highlighted. Scale bar = 5 μm.
(B) Quantification of Bin1 co-localization with major trafficking markers based on SIM images. One-way ANOVA with Bonferroni post-hoc tests * P<0.05, ** P<0.01
(C) Left: Representative flattened confocal images of Rab11 staining in GFP filled control (scr) and Bin1 knockdown (kd) neurons. The outline indicates the GFP-cell fill (Fig S2F). Scale bar = 10 μm.
Right: Quantification of area occupied by and number of Rab11 positive puncta in scr and kd neurons (n = 12 cells). Area occupied: Mann-Whitney test; Puncta number: unpaired t-test *** P<0.001, **** P<0.0001
(D) Single plane SIM images of the relative localization of Bin1 and Arf6 in dendrites and spines. Scale bar = 5 μm.
(E) Quantification of Bin1 co-localization with trafficking markers in SIM images. One-way ANOVA with Bonferroni post-hoc tests ** P<0.01, *** P<0.001
(F) Representative 3D reconstruction of a stack of SIM images showing the relative localization of Bin1 and Arf6 in spines. Inset enlargement; scale bar = 50 nm.
(G) Montage of spine depicted in (F) to illustrate Arf6 and Bin1 contact sites (white arrowheads).
(H) Results of co-immunoprecipitation experiment that demonstrates pulldown of Arf6 with Bin1 immunoprecipitation from rat cortex homogenates.
(I) Flattened confocal image showing proximity-ligation assay (PLA) reveals an interaction of Bin1 and Arf6 in rat cortical neuron cultures, reflected by green PLA puncta. Red outline represents mCherry cell-filled pyramidal neuron. Inset: Zoomed in view of PLA puncta in dendritic spines and shaft. Scale bar = 20 μm
(J) Quantification of PLA puncta normalized to number of cell bodies in the imaging frame (identified by DAPI nuclear staining). Bin1 Ab and Arf6 Ab conditions represent negative controls where each antibody was used singly in the PLA assay. Kruskal-Wallis with Dunn’s post-hoc tests * P<0.05, *** P<0.001
Bin1 physically and functionally interacts with Arf6
The PPI network constructed around Bin1 showed an enrichment in GTPase-related genes (Fig 1D). We therefore examined the colocalization of the exocytosis-associated small GTPase Arf6 with Bin1 using SIM. Arf6 is known to regulate traffic from recycling endosomes to the plasma membrane17–19. In spines and adjacent shaft, Bin1 often colocalized with Arf6 (Fig 2D, arrowheads), with Manders analysis showing greater colocalization with Arf6 than with other trafficking markers (Fig 2E).
We used SIM to further analyze the relative localization of Bin1 with Arf6 vesicles in relationship to spine architecture. In some spines, Bin1-positive structures were completely distinct from Arf6 vesicles (Fig S3Aa). In other cases, Bin1-positive structures were in apparent contact with Arf6 vesicles (Fig S3Ab). Commonly, a gradient of colocalization could be seen at the contact site between Bin1 and Arf6 vesicles (Fig S3Ac). Sometimes one spine contained several of these localization patterns. To further visualize these spatial relationships, we generated 3D reconstructions of spines (Fig 2F, Fig S3B). The localization patterns described above could be visualized readily in such reconstructions; sites of contact between Bin1 and Arf6 were often clearly visible (Fig 2F inset), and multiple Bin1 and Arf6 vesicles could be visualized within one spine. We also examined a series of optical slices, allowing us to follow each structure in 3D across the entire spine (Fig 2G). Multiple Bin1-positive structures could extend and merge to contact a single Arf6 vesicle (open arrowhead). One Arf6 vesicle could thus contact one or several Bin1-positive structures (arrowheads). Furthermore, Arf6 and Bin1 vesicles were often enriched at opposite poles of the spine, making contact near the center of the spine.
Based on this colocalization, we hypothesized that Bin1 and Arf6 may physically interact. In support of this, co-immunoprecipitation experiments showed that Bin1 immunoprecipitation pulled down Arf6 (Fig 2H). Additionally, we used a proximity ligation assay (PLA) to investigate potential interactions between Bin1 and Arf6. This assay utilizes complimentary oligonucleotide-conjugated secondary antibodies to prime a reaction that generates fluorescent puncta only when these oligonucleotides are able to bind each other (requiring a maximum distance of 30–40 nm between probes). We observed significant generation of PLA puncta with Bin1 and Arf6 antibody co-staining of rodent cultured neurons, but few puncta when either antibody was used alone (Fig 2I,J). Together, these results strongly support a physical interaction between Bin1 and Arf6 in neurons.
To assess potential functional interactions between Bin1 and Arf6, we manipulated Bin1 expression levels and assessed the impact on the distribution of postsynaptic Arf6. Knockdown of Bin1 robustly increased the area occupied and number of Arf6-positive structures (Fig 3A,, Fig S3C), whereas overexpression of Bin1 did not significantly affect their distribution (Fig S3D,E). Reduced Bin1 levels caused accumulation of Arf6 in intracellular compartments, a pattern similar to Rab11, consistent with a role for Bin1 in modulating recycling to the plasma membrane. To test if the knockdown was specific, we rescued the effects on Arf6-positive structures by overexpressing an RNAi-resistant form of Bin1 (Fig S3F).
Fig 3. Bin1 regulates Arf6 and dendritic spine morphology.

(A) Left: Representative flattened confocal images of Arf6 staining in GFP filled control (scr) and Bin1 knockdown (kd) neurons. The outline indicates the GFP-cell fill (see Fig S3C). Scale bar = 10 μm
Right: Quantification of area occupied by and number of Arf6 positive puncta in scr and kd neurons (scr: n = 19 cells, kd = 17 cells). Area occupied: Mann-Whitney test; Puncta number: Welch’s t-test *** P<0.001
(B) Representative single plane confocal images showing total and active Arf6 staining in N2A cells with and without Bin1-GFP transfection. Scale bar = 5 μm
(C) Quantification of active/total Arf6 ratio in N2A cells transfected with GFP (n=16 cells) or Bin1-GFP (n=16 cells). All results were normalized to the average of the value of GFP transfection condition for each experiment. Mann-Whitney test *** P<0.001
(D) Representative flattened confocal images of dendrites in GFP-filled neurons expressing scrambled (scr) or Bin1 knockdown (kd) plasmids. Scale bar = 10 μm
(E) Quantification shows that knockdown of Bin1 decreases spine size without altering spine density (n = 26 cells for each condition). Spine size: Mann-Whitney test; Spine density: unpaired t-test **** P<0.0001
(F) Histogram showing frequency distribution of all individual spine areas measured in (E) (scr: n = 1049 spines, kd: n = 1120 spines).
(G) Representative flattened confocal images of dendrites in GFP-filled neurons in control (ctrl) and Bin1 overexpressing (ox) neurons. Scale bar = 10 μm
(H) Quantification shows that overexpression of Bin1 leads to decreased spine density and increased spine size (ctrl: n = 24 cells, ox: n = 20 cells). Spine size: Mann-Whitney test; Spine density: unpaired t-test * P<0.05, *** P<0.001
(I) Histogram showing frequency distribution of all individual spine areas measured in (H) (ctrl: n = 2407 spines, ox: n = 1740 spines)
(J) Representative flattened confocal images of PSD95 in scr and kd GFP-expressing neurons. Scale bar = 10 μm
(K) The size of PSD95 puncta is not altered in Bin1 knockdown neurons (scr: n = 10 cells, kd = 12 cells). Welch’s t-test.
Because we had evidence of a physical and functional interaction between Bin1 and Arf6, we next wished to test whether Bin1 may regulate Arf6 through regulation of Arf6 GTPase activity. We used a conformation-specific Arf6 antibody that only binds active, GTP-bound Arf6 (Arf6-GTP), and saw that staining with this antibody (normalized to total Arf6 staining) was increased in N2A cells with Bin1-GFP transfection compared to GFP transfection (Fig 3B,C), with no significant difference seen in total Arf6 staining (Fig S3G). Altogether, these data suggest that Bin1 participates in protein complexes with Arf6, and functions at least in part in the forward trafficking of Arf6-labeled vesicular structures, potentially through regulation of Arf6 GTPase activity.
Bin1 modulates spine morphology
Given its partial localization in dendritic spines and roles in membrane traffic, we hypothesized that Bin1 can modulate spine morphology. Indeed, Bin1 knockdown caused a small but significant reduction in spine area, but did not affect spine density (Fig 3D-F). Conversely, overexpression of Bin1 caused a small but significant increase in spine area, along with reduced spine density (Fig 3G-I). Since spine size is correlated with PSD area20, we asked whether the smaller spines caused by Bin1 knockdown also had smaller postsynaptic densities (assessed by PSD95 immunofluorescence, Fig 3J). Interestingly, we found that Bin1 knockdown did not affect PSD95 cluster areas, suggesting that its primary effect is on membrane trafficking (Fig 3K).
Based on the effects we observed on spine morphology by Bin1 knockdown/overexpression, we hypothesized that altered Bin1 expression may impact AMPA receptor-mediated synaptic transmission. To test this, we examined the impact of altered Bin1 levels on AMPAR currents (Fig 4A-D). Consistent with the reduction in spine size, knockdown of Bin1 significantly reduced the mean amplitude of AMPAR mEPSCs (Fig 4A,B). Conversely, Bin1 overexpression led to an increase in mEPSC amplitudes, reflected in the distribution of amplitude values in the cumulative probability plot (Fig 4C,D). No difference was observed in average mEPSC frequencies between experimental condition and the control condition with either knockdown or overexpression (Fig S4A). Additionally, no differences in resting membrane potential or input resistance were detected either with Bin1 knockdown or overexpression (not shown).
Fig. 4. Bin1 regulates glutamatergic neurotransmission and interacts with the AMPAR subunit GluA1.

(A) Representative traces of currents recorded from scr and kd treated cortical neurons
(B) Quantification of AMPA mEPSCs in cortical neurons reveals a significant reduction of mEPSC amplitude upon Bin1 knockdown (kd) (scr: n = 14 cells, kd = 22 cells). Unpaired t-test ** P<0.01
(C) Representative traces of currents recorded from control and Bin1 overexpressing (ox) cortical neurons
(D) Quantification of AMPA mEPSCs shows that Bin1 overexpression does not affect average mEPSC amplitude but shifts distribution of values toward more high amplitudes (scr: n = 24 cells, kd: n = 22 cells). Mann-Whitney test.
(E) Representative single plane SIM image of individual spines and line scans showing areas of colocalization between Bin1 and GluA1.
(F) Representative 3D reconstruction of a stack of SIM images, showing the relative localization of Bin1 and GluA1 in spines. Inset, higher-magnification view; scale bar = 50 nm.
(G) Montage of spine depicted in (F) to illustrate GluA1 and Bin1 contact sites (white arrowheads).
(H) 3D surface plot showing total area of GluA1 staining (y-axis) as a function of both spine area (x-axis) and total area of Bin1 staining (z-axis). Legend shows color-coding for y-axis values on graph.
(I) 3D surface plot showing total area of surface GluA1 (surfGluA1) staining (y-axis) as a function of both total area of Bin1 staining (x-axis) and spine area (z-axis). Legend shows color-coding for y-axis values on graph.
(J) Reciprocal coimmunoprecipitation of Bin1 and GluA1 from rat cortex homogenate suggests participation in common complexes.
(K) Flattened confocal image showing proximity-ligation assay (PLA) reveals an interaction of Bin1 and GluA1 in rat cortical neuron cultures, reflected by green PLA puncta. Red outline represents mCherry cell-filled pyramidal neuron. White arrowheads denote instances of interaction sites at dendritic spines. Inset: Zoomed in view of PLA puncta in dendritic spines and shaft. Scale bar = 20 μm
(L) Quantification of PLA puncta normalized to number of cell bodies in the imaging frame (identified by DAPI nuclear staining). Bin1 Ab and GluA1 Ab conditions represent negative controls where each antibody was used singly in the PLA assay. Kruskal-Wallis with Dunn’s post-hoc tests * P<0.05, ** P<0.01
Bin1 colocalizes and interacts with the GluA1 AMPA receptor subunit
We next examined the spatial relationship between Bin1 and GluA1 in spines using SIM (Fig 4E-G). GluA1 often defined a single large cluster in the spine head, though occasional smaller GluA1–positive structures could be detected in the spine head or neck. While non-colocalizing immunofluorescence could be observed (Fig 4Ea), small or large sites of colocalization were often detected (Fig 4Eb). Occasionally, Bin1-positive structures were located within a larger GluA1 cluster (Fig 4Ec). 3D reconstructions revealed more detail about the spatial relationship between Bin1 and GluA1 (Fig 4F, Fig S4B). While GluA1 immunofluorescence concentrated near the plasma membrane, extensions protruding into the interior of the spine could also be detected. Some of these contacted Bin1-positive structures located inside the spine, or near the opposite pole of the spine (Fig 4F inset). Stacks of optical slices confirmed that projections from the GluA1 cluster contacted several Bin1-positive structures (Fig 4G, arrowheads).
To gain a quantitative understanding of the relationships between Bin1, GluA1, and spine architecture, we performed 3D correlation analyses between sets of 3 parameters measured by SIM to understand how the different variables we measured might influence each other. We created surface plots in a xyz coordinate system using a distance least-square fitting approach. We first analyzed the 3D relationships of total area of GluA1 clusters with the total area of Bin1 clusters and total spine area (Fig 4H). These plots show that GluA1 levels increase as a function of both spine area and total Bin1 area, and that both of these factors influence GluA1 levels independently. This suggests that the correlation between GluA1 and Bin1 levels is not simply related to spine area. We next looked at surface GluA1 levels (surfGluA1) as a function of total Bin1 area and total spine area (Fig 4I). Interestingly, we saw that in smaller spines, surfGluA1 was not correlated with Bin1 levels, but in larger spines, there is a robust increase in surfGluA1 levels with increased Bin1. This suggests a greater role of Bin1 in influencing GluA1 trafficking in larger spines.
Based on the colocalization of Bin1 with GluA1 in spines, we hypothesized that the two proteins may physically interact. Indeed, coimmunoprecipitation and reciprocal coimmunoprecipitation (Fig 4J) from rat cortex homogenates showed that GluA1 and Bin1 participate in common protein complexes in vivo in brain. Additionally, we again performed PLA to look for interactions between Bin1 and GluA1, and found significant generation of PLA puncta with Bin1 and GluA1 antibody co-staining (Fig 4K,L). Interestingly, these PLA puncta were frequently observed in what appeared to be dendritic spines in an mCherry cell-filled pyramidal neuron (Fig 4K, arrowheads), consistent with colocalization of Bin1 and GluA1 in these compartments observed with SIM imaging discussed above. This raises the possibility that Bin1 may regulate GluA1 exocytic trafficking by forming complexes with this cargo protein.
Bin1 modulates synaptic expression of AMPA receptors
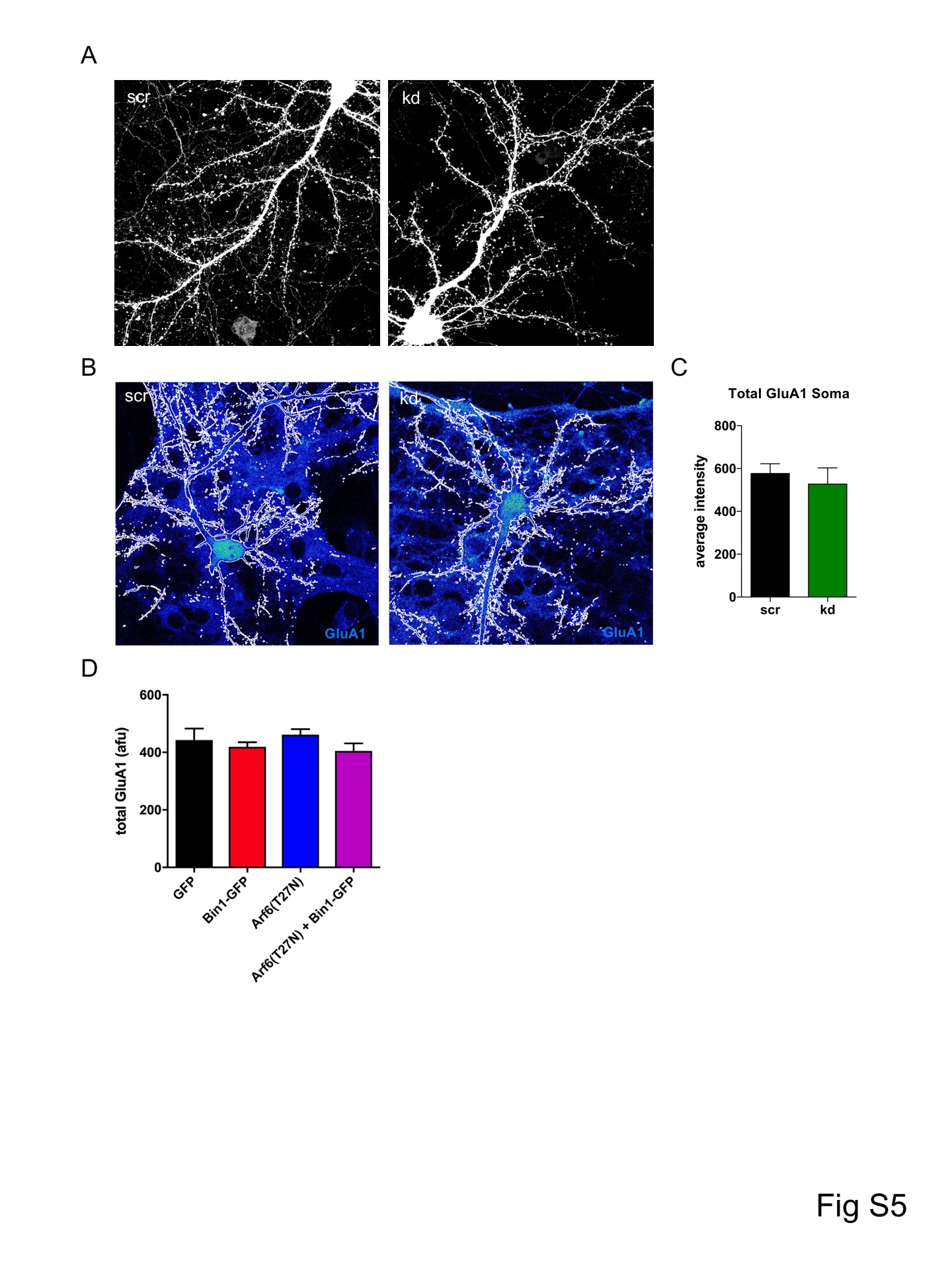
The changes in mEPSC amplitude with Bin1 knockdown/overexpression suggest that Bin1 may be required for maintaining AMPA receptors at the cell surface. To test this hypothesis, we knocked down Bin1 in neurons and examined the surface expression of GluA1 in spines and dendrites (Fig 4A,B, Fig S5A). Knockdown of Bin1 reduced surface expression of GluA1 both in spines and in the dendritic shaft (Fig 4B), whereas somatic total GluA1 content was not changed (Fig S5B,C), consistent with a role in maintaining surface receptor pools. To gain insight into the role of Bin1 in the dynamics of surface GluA1 expression, we analyzed the rate of exocytosis of GluA1 using a fluorescence recovery after photobleaching (FRAP) assay, which utilizes a pH-sensitive SEpHluorin-tagged GluA1 (SEP-GluA1) that is only fluorescent when expressed at the cell surface. We photobleached a large region of dendrite and followed the fluorescence recovery primarily driven by SEP-GluA1 trafficking to the surface (Fig 5C-F). To minimize the contribution of lateral diffusion of fluorescent SEP-GluA1 from unbleached regions to the fluorescence recovery, we analyzed a small region in the middle of the bleached dendritic region (Fig 5D). Bin1 knockdown significantly reduced the recovery of fluorescence compared to control neurons (Fig 5E,F), suggesting that loss of Bin1 impairs the ability of GluA1 to traffic to the surface, which likely underlies the reduction in AMPA currents and surface GluA1 levels seen with Bin1 knockdown.
Fig. 5. Bin1 regulates surface expression of GluA1 in an Arf6-dependent manner.

(A) Representative flattened heat map confocal images of surface GluA1 (surfGluA1) staining in scr and kd GFP cell-filled (white outline) neurons (see Fig S5A for cell fill). Scale bar = 20 μm Bottom: Zoomed in regions outlined in boxes in above images.
(B) Quantification indicates that Bin1 knockdown reduces surface expression of GluA1 in spines and shaft (n = 13 cells for each condition). Welch’s t-test ** P<0.01
(C) Schematic of FRAP experimental paradigm, illustrating fluorescence recovery by exocytosis in pH-sensitive SEP-GluA1 expressing cells.
(D) Representative single plane confocal image of the apical dendrite of a control cell demonstrating the FRAP paradigm. A 33 μm wide segment was bleached, while only the middle 2 μm section was quantified to limit the effects of lateral diffusion on fluorescence recovery. Dendrite outlines were created using the co-expressed, cell-filling mCherry.
(E) Cells in which Bin1 expression was inhibited presented with significantly slower fluorescence recoveries than scramble control cells (n = 12 cells per condition). Repeated measures ANOVA, * P<.05
(F) Single plane confocal images showing heat map time-lapse examples of a control and knockdown cell. Quantified region outlined in magenta. Scale bar = 2 μm
(G) Single plane confocal images of total and surface GluA1 staining in N2A cells with transfection conditions indicated to the left of images. Scale bar = 3 μm
(H) Quantification of surface/total GluA1 ratios with indicated transfection conditions. All results were normalized to the average of the value of GFP transfection condition for each experiment (n = 16 cells per condition). One-way ANOVA with Bonferroni post-hoc tests ** P<0.01
To determine if the interaction between Arf6 and Bin1 might be involved in the regulation of GluA1 by Bin1, we performed surface GluA1 staining experiments in N2A cells, which endogenously express GluA121 (Fig 5G). Importantly, we were able to detect GluA1 surface and total staining in these cells (Fig 5G), and Bin1-GFP transfection increased GluA1 surface/total staining compared to GFP transfected cells (Fig 5H). When we co-transfected a dominant negative Arf6 (Arf6(T272N)), this increase in surface GluA1 was blocked (Fig 5H). Importantly, there was no difference in total GluA1 between transfection conditions (Fig S5D), supporting a specific effect on GluA1 surface trafficking and not total GluA1 expression. These results suggest that Bin1 regulation of GluA1 surface levels requires Arf6 activity. We therefore propose a model where Bin1 may function with Arf6 in exocytosis from recycling endosomes, carrying cargoes such as GluA1 to the plasma membrane. In this model, when Bin1 is knocked down, this exocytosis is reduced, leading to a reduction in surface GluA1 expression and smaller spine heads.
DISCUSSION
Here we used bioinformatics to show that synaptic trafficking is overrepresented among LOAD risk factors. As a representative of this protein network, we investigated the basic synaptic functions of Bin1. We used SIM to gain insight into the spatial architecture of Bin1 localization below the diffraction limit of visible light, cross-validating the immuno-EM approach. SIM allowed us to simultaneously image multiple fluorophores relative to spine architecture in response to molecular manipulations, and to carry out detailed analysis of mesoscale architecture of trafficking events. A combination of SIM and EM allowed us to map the spatial distribution of Bin1 in synaptic regions. Bin1 is present both pre- and postsynaptically, and EM shows association of a pool of Bin1 with postsynaptic vesicular-cisternal structures and the plasma membrane, in addition to presynaptic vesicles, presynaptic active zone, the PSD, and cytoskeletal structures within the spine. Importantly, Bin1 labeling was highest in the spine and, more specifically, around the PSD, supporting an important role for this protein in the postsynaptic compartment.
Analysis of SIM images revealed that Bin1 colocalizes with markers for specific postsynaptic trafficking steps, suggesting roles in clathrin-dependent endocytosis, and recycling exocytosis. A role for Bin1 in endocytosis has been inferred from its similarity with amphiphysin1 and the interactions of Bin1/amphiphysin1 with clathrin, dynamin, AP2, synaptojanin, and endophilin, and from the regulation of membrane remodeling in vitro by Bin1’s N-BAR domains11. However, surprisingly, in our experiments Bin1 overexpression or knockdown did not affect the number and size of clathrin vesicles. Instead, knockdown of Bin1 caused an increase in the number and size of Rab11 and Arf6 vesicles within spines, perhaps due to a build-up caused by impaired recycling from recycling endosomes to the plasma membrane. Based on these findings, we propose that in the postsynaptic compartment, Rab11/Arf6-mediated recycling exocytosis may be more sensitive to changes in Bin1 levels compared to clathrin-mediated endocytosis. This would explain the observation that the net effect of Bin1 knockdown/overexpression is a reduction/enhancement of spine size and AMPA-mediated currents. Our FRAP experiment corroborates a role for Bin1 in protein delivery to the membrane in the postsynaptic compartment, as Bin1 knockdown reduced the rate of GluA1 exocytosis in dendrites measured by fluorescence recovery of SEP-GluA1 after photobleaching. Additionally, we provide evidence that this regulation of surface trafficking of GluA1 by Bin1 requires Arf6, as expression of a dominant-negative Arf6 blocks Bin1 enhancement of GluA1 surface levels in N2A cells. This proposed Bin1-Arf6-GluA1 trafficking pathway is further supported by our evidence of Bin1’s interaction with both Arf6 and GluA1.
Previous evidence for Bin1 in promoting recycling exocytosis comes from the observation that mutation of the C. elegans Bin1 ortholog AMPH-1 causes defective recycling of multiple cargoes, observed by intracellular accumulation of normally recycled membrane proteins15. This group also showed that knockdown of human BIN1 in HeLa cells impaired recycling of the transferrin receptor. AMPH-1 was shown to interact with RME-1, the C. elegans ortholog of the EH-domain containing protein EHD1, which interacts with Arf6 and regulates Arf6-dependent exocytic recycling22. EHD1 also regulates BIN1 activity during T-tubule formation in human myoblasts23. These findings suggest that a Bin1/Ehd1/Arf6 pathway may underlie the exocytic trafficking events in dendritic spines seen in our studies. Another EH-domain containing protein, EHBP1L1, also interacts with BIN1, and this interaction was shown to regulate Rab8-dependent exocytosis toward the polarized plasma membrane 14. All these studies suggest a role for BIN1 in exocytosis, but our studies are the first to demonstrate this role in dendritic spines supporting glutamatergic transmission.
The role of Arf6 in mediating Bin1’s regulation of GluA1 trafficking is interesting as Arf6 has been shown to regulate both internalization and exocytosis of AMPA receptors24,25. One study showed that overexpression of a dominant-negative Arf6 reduced surface GluA1 in primary rat neurons, suggesting exocytosis of GluA1 is more tightly regulated by Arf624. In further support of this, this same study showed that knockdown of the Arf6 GTPase-activating protein (GAP) AGAP3 (which they show increased Arf6 activity) led to an increase in surface GluA1, demonstrating a bidirectional effect of manipulating Arf6 activity. Our studies show that overexpression of Bin1 increases Arf6 activity, similar to what results from knockdown of AGAP3, and indeed we see similar increases in GluA1 surface levels with these two manipulations. Future studies might reveal whether Bin1 directly regulates Arf6 activity, or might indirectly act through regulation of proteins like AGAP3.
Our network analysis implicated synaptic trafficking in the neurobiology of LOAD risk, supporting a possible role for these processes in the spine loss observed in postmortem AD brain26. A role for abnormal trafficking in LOAD has been proposed based on putative functions of several LOAD risk genes 8. Among these is the BIN1 gene, which has been identified in GWAS studies that found associations between LOAD risk and SNPs near the BIN1 gene27. While the functional impact of these SNPs is not known, it is thought that they may affect BIN1 protein expression. Indeed, human postmortem studies have reported decreased BIN1 protein expression28,29, decreased BIN1 levels in the neuropil, and increased levels in the cytosol, suggesting reduced BIN1 at synapses29, or altered isoform ratio30 in subjects with AD. Interestingly, higher BIN1 expression has been associated with a delayed age at onset and shorter disease duration, suggesting a protective role27. In our studies, knockdown of Bin1 reduces Bin1 synaptic protein levels/function like what has been suggested for LOAD brains, as well as what might result from expression of LOAD-associated BIN1 SNPs. The effects we see on spine size and glutamatergic neurotransmission provides a potential mechanism for how alterations in synaptic BIN1 could contribute to the symptoms and progression of LOAD. Besides addressing the basic postsynaptic functions of Bin1, our studies provide a first step toward understanding the role of BIN1 and other GWAS risk factors in the pathogenesis of LOAD. The effects of Bin1 manipulations we report are small but significant, and could lead to major outcomes over the long term, consistent with a late disease onset and with the expected functional impact of a common variant risk factor. Additionally, these studies reveal a trafficking pathway that may be generally relevant for LOAD pathology, providing a potential therapeutic target for interventions in this disease.
EXPERIMENTAL PROCEDURES
Bioinformatics analysis
In order to uncover biological processes that might be generally relevant for LOAD risk, we performed GO analysis on 40 genes identified in GWAS studies of LOAD risk. These genes were present in the NHGRI-EBI Catalog of published genome-wide association studies database and all had highly significant LOAD association (P< 1×10−8) and we only included genes from studies with a replication sample. We used the GeneMANIA plugin in Cytoscape to generate a human protein interaction network from the proteins encoded by the genes in the top GO cluster (11 genes). The network was built using GO biological process-based weighting and by retrieving the top 50 related proteins to the input query and at most 20 attributes. We used the same strategy to generate the protein interaction for BIN1, except with the top 40 related proteins to the input query and at most 20 attributes.
Plasmids and antibodies
Bin1-mCherry (EX-Mm06819-M56) and Bin1 RNAi constructs were purchased from GeneCopoeia RNAi constructs expressed GFP to enable identification of transfected cells. The target sequence used was CCAGAACTTCAATAAGCAG. The RNAi resistant Bin1 mutant construct was generated using QuickChange Site-Directed Mutagenesis Kit (Stratagene). Three non-coding mutations were incorporated into the RNAi target sequence to generate the RNAi-resistant Bin1 construct. Bin1-GFP constructs were generated by replacing mCherry with GFP using In-Fusion HD cloning (Clontech). All constructs were sequence verified. pGex-nAmph232 was a gift from Pietro DeCamilli. pEGFP-N2 and pmCherry-C1 (Clontech) were overexpressed in cultured neurons for morphometric analysis experiments. The following primary antibodies were used (mAb: monoclonal antibody, pAb: polyclonal antibody) : Bin1 mAb (Millipore Cat# 05–449), Map2 mAb (Millipore Cat# MAB3418), GluA1 N-term mAb (Millipore Cat# MAB2263), GluA1 C-term pAb (Millipore Cat# ABN241), Synapsin mAb (CellSignaling Cat# 5297), PSD95 mAb (Neuromab Cat# 75–028), CHC pAb (CellSignaling Cat# 2410), Rab5 pAb (CellSignaling Cat# 2143), EEA1 pAb (CellSignaling Cat# 2411), Rab11 pAb (Sigma Cat# R5903), Arf6 pAb (Abcam Cat# ab77581), Active Arf6 mAb (New East Biosciences, Cat# 26918), CaMKIIα mAb (Millipore Cat# 05–532), DsRed pAb (to identify mCherry expression, Clontech Cat# 632496), GFP chicken pAb GFP (to identify GFP expression, Abcam Cat# ab13970).
Neuronal and Neuro-2A (N2A) cell culture and transfection
Dissociated cultures of primary cortical neurons were prepared from E18 Sprague-Dawley rat embryos or P0 C57BL6 mouse embryos. Brains were dissected in ice-cold Hank’s buffered salt solution, and cortical tissue isolated, digested with papain (Sigma; diluted in Neurobasal with EDTA (0.5 mM) and DNaseI (2 units/mL), activated with L-cysteine (1 mM) at 37°C), and mechanically dissociated in neuronal feeding media (Neurobasal + B27 supplement, Invitrogen + 0.5 mM glutamine + penicillin/streptomycin). 1hr after feeding, media was replaced. Neuronal cultures were maintained at 37°C in 5% CO2. Neuronal feeding media was supplemented with 200 μM D,L-amino-phosphonovalerate (D,L-APV, Ascent Scientific) beginning on DIV 4. Neurons were transfected at DIV21 with Lipofectamine 2000, providing a transfection efficiency of 0.5–1%. Plasmids (1–10 μg total DNA) and Lipofectamine 2000 (Invitrogen) were diluted in Dulbecco’s Modified Eagle Medium (DMEM) + HEPES (10 mM), mixed thoroughly together, and incubated for 20–30 minutes at 37°C before adding to cultured cells. Following transfection, neurons were supplanted in antibiotic-containing feeding media containing half conditioned and half fresh media, and allowed to express constructs for 3 days or as indicated.
Neuro-2A (N2A) cells were obtained from ATCC and grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum + penicillin/streptomycin. Cells were transfected with Lipofectamine 2000 1 day after plating on poly-D-lysine coated coverslips, and transgene was allowed to express for 2 days prior to fixing and staining of cells.
Immunocytochemistry
Cells were fixed for 10 min in 4% formaldehyde/4% sucrose in PBS. Fixed cells were permeabilized and blocked simultaneously in PBS containing 2%−4% normal goat serum and 0.2% Triton-X-100 for 1 hr at room temperature. Primary antibodies were added in PBS containing 2% normal goat serum overnight at 4 °C, followed by 3 × 10 min washes in PBS. Secondary antibodies were incubated for 1 hr at room temp, also in 2%−4% normal goat serum in PBS. Three further washes (15 min each) were performed before coverslips were mounted using ProLong antifade reagent (Life Technologies).
To label surface GluA1 in neurons, we incubated live cells with antibody to the extracellular N-terminus of GluA1 at 4 °C for 30 min. Cells were fixed for 10 min in 4% formaldehyde/4% sucrose in PBS and were then processed for immunostaining as described above. For surface and total labeling of GluA1 in N2A cells, cells were first fixed in 4%paraformaldehyde in PBS for 5 min and blocked in PBS containing 4% normal goat serum (without permeabilization). Cells were incubated with N-terminal GluA1 antibody for 1 hour in PBS + 4% normal goat serum and washed three times before being permeabilized as above and further stained for total GluA1 with C-terminal GluA1 antibody overnight at 4 °C. Cells were then processed using the secondary staining and mounting described above.
Confocal microscopy
Confocal images of immunostained neurons were obtained with a Zeiss LSM5 Pascal confocal microscope for two channel images and with a C2+ Nikon confocal microscope for three channel images. Images of neurons were taken using the 63x (Zeiss) or 60x (Nikon) oil-immersion objective (NA = 1.4) as z-series of 5–10 images, averaged 2 times, taken at 0.37 μm intervals, with 1024×1024 pixel resolution at ~140–150 nm/pixel. Detector gain and offset were adjusted in the channel of cell fill (GFP or mCherry) to include all spines and enhance edge detection.
For confocal imaging of N2A cells, images were taken using a 60x (Nikon) oil-immersion objective (NA = 1.4) as z-series of 5–7 images, summed 2 times, taken at 1 μm intervals with 512×512 pixel resolution at ~140nm/pixel.
Confocal image analysis
The amount of Rab11, CHC and Arf6 staining was quantified using Fiji (http://fiji.sc/Fiji). To determine spine protein content, spine regions generated by spine dimension analysis in MetaMorph were overlaid onto the appropriate channel and intensities within the spine regions were recorded. Dendritic shaft values were calculated as the mean intensity of 3 regions of shaft. Linescans were performed in Fiji. Experimenter was blinded to treatment for each coverslip using coded relabeling of coverslips prior to imaging. For active Arf6 and surface GluA1 experiments in N2A cells, transfected cells were identified by positive GFP fluorescence, and analyzed area was generated based on thresholding of either total Arf6 or total GluA1 in a single slice in roughly the middle of the cell. Intensity of active Arf6 or surface GluA1 was normalized to total Arf6 and total GluA1, respectively. For each condition, 4 cells were imaged on each of two coverslips per experiment from two independent experiments for a total of 16 cells per condition.
Membrane Fractionation
Cortices from 6-week-old mice were homogenized in sucrose buffer (20 mm HEPES, pH 7.4, 320 mm sucrose, and 5 mm EDTA) supplemented with protease inhibitor cocktail (Roche). Homogenates were centrifuged at low speed to pellet nuclei and cell debris (3000 × g for 20 min at 4°C), and the supernatant (S1) was then centrifuged at high speed (38,000 × g for 30 min at 4°C) to obtain a membrane pellet (P2). P2 was resuspended in potassium iodide buffer (20 mm HEPES, pH 7.4, 1 m KI, and 5 mm EDTA) to remove membrane-associated proteins (S3), and membranes were again collected by centrifugation (38,000 × g for 20 min at 4°C). Membranes were washed (20 mm HEPES, pH 7.4, and 5 mm EDTA) and pelleted once more (S4) before solubilizing in CHAPS buffer supplemented with protease inhibitors (20 mm HEPES, pH 7.4, 100 mm NaCl, 5 mm EDTA, and 1% CHAPS) for 2 h at 4°C. Solubilized membranes were clarified by centrifugation for 30 min at 100,000 × g at 4°C (S5). The final CHAPS-insoluble pellet was resuspended in SDS buffer (50 mm Tris, pH 7.4, 150 mm NaCl, and 1% SDS) supplemented with protease inhibitors, solubilized at 37°C for 20 min, and clarified by centrifugation (S6).
Coimmunoprecipitation assays
Coimmunoprecipitation assays were performed as33 with modifications. Briefly, adult rat cortex was dissected and homogenized in pull-down buffer (50 mM HEPES pH 7.5, 1 % Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM AEBSF with protease inhibitor cocktail (Roche) and phosphatase inhibitor (Sigma)) and solubilized for 1 hour at 4°C. Solubilized material was centrifuged at 20,000 g for 25 minutes at 4°C and the supernatant was precleared with protein A/G beads for 1 hour. Proteins were immunoprecipitated with 3 μg of antibody overnight at 4°C, followed by a 1 hour incubation with protein A/G beads. Beads were then washed extensively and bound complexes were analyzed by SDS-PAGE and western blotting.
Proximity Ligation Assay (PLA)
Neurons were prepared as described above for immunochemistry and stained overnight with indicated primary antibodies. PLA assay was then performed according to the manufacturer’s protocol using Duolink® PLA reagents from Sigma-Aldrich (Duolink® In Situ PLA® Probe Anti-Mouse PLUS Affinity purified Donkey anti-Mouse IgG (H+L), Duolink® In Situ PLA® Probe Anti-Rabbit PLUS Affinity purified Donkey anti-Rabbit IgG (H+L), Duolink® In Situ Detection Reagents Green). Negative controls using each primary antibody singly were used to determine non-specific PLA signal from each antibody. Neurons were imaged with a C2+ Nikon confocal microscope as described above, and PLA puncta were counted in ImageJ and normalized to number of cell bodies in each imaging field based on DAPI staining.
Electrophysiology
Cultured cortical neurons were used 3–4 days post-transfection (DIV24–25); they were visualized using a Zeiss Axioskop 2FS (Zeiss, Jena, Germany) upright microscope with infrared differential interference contrast video microscopy and a water-immersion 40X objective. Whole-cell recordings were performed at 21–23°C at a holding potential of −50 mV. The extracellular solution contained (in mM) 138 NaCl, 2.5 KCl, 10 HEPES, 25 glucose, 2 CaCl2, 1 MgCl2 (pH to 7.3 with NaOH). Patch pipettes were pulled from borosilicate glass (WPI) and had a resistance of 5–10 MΩ in working solution. The intracellular solution contained (in mM) 140 K-gluconate, 2 MgCl2, 2 Na2-ATP, 0.2 Na-GTP, 10 EGTA, 10 HEPES, 2 NaCl (pH to 7.3 with KOH). Traces were acquired with an Axopatch 200B amplifier (Molecular Devices) using pClamp9 software. Recordings were filtered at 2 kHz and digitized at 10 kHz. Data were analyzed with custom written MATLAB event detection software. To isolate mEPSCs, 50 μM Picrotoxin and 500 nM tetrodotoxin were added to the extracellular solution. In two cells kynurenic acid was added at the end of the recordings. This treatment completely abolished the mEPSCS confirming the glutamatergic nature of these currents.
SIM imaging and analysis
Imaging and reconstruction parameters were empirically determined with the assistance of the Nikon Imaging Center at Northwestern, with the best image in mind. Acquisition was set to 10 MHz, 14 bit with EM gain and no binning. Auto exposure was kept between 100–300 ms and the EM gain multiplier restrained below 300. Conversion gain was held at 1x unless necessary to increase signal with 2.4x. Laser power was adjusted to keep LUTs within the first quarter of the scale (<4000). Three reconstruction parameters (Illumination Modulation Contrast, High Resolution Noise Suppression and Out of Focus Blur Suppression) were extensively tested to generate consistent images across experiments without abnormal features or artifacts and producing the best Fourier transforms. Reconstruction parameters (0.96, 1.19, and 0.17) were kept consistent across experiments and imaging sessions. Resolution of images was confirmed with full-width half maximum (FWHM) measurements of a small structure within the image34. For each spine analyzed, the single plane in which the spine head was in focus, based on the cell fill, was chosen for analysis. Using the Nikon Elements software, each spine head and outlined manually in the channel of the cell fill to detect the area. Images were thresholded to allow unbiased detection of to Bin1, Arf6, and GluA1 puncta within the spine head and the area recorded. Visual assessment of fluorescence intensity was used to delineate separate or connected puncta. This approach was confirmed and kept consistent throughout all SIM analyses. The number of puncta within the spine head was quantified manually and recorded. Colocalization highlighter images and Manders’ colocalization coefficients were determined in ImageJ, after thresholding, with the use of the MBF set of plugins. Three-dimensional reconstructions of dendritic spines were created in the Nikon Elements Software by merging a dimmed maximum projection of the GFP signal with shaded volume reconstructions of the two other channels, to allow for better visualization. In order to explore multidimensional relationships between we created surface plots in a xyz coordinate system using a distance least square fitting approach. This provides a sensitive method to reveal non-salient oval data patterns. All plots were generated using the STATISTICA software.
Immuno-electron microscopy (immuno-EM)
The basic approach is as described in35. Deeply -anesthetized adult C57/BL6 mice were perfused with mixed aldehyde fixative; brains were removed, post-fixed overnight in the same fixative, and stored in pH 7.2 phosphate buffer. Small chunks of cerebral cortex were cut from 100 μm Vibratome sections, cryoprotected in 30% glycerol, and plunge-frozen in methanol cooled with dry ice. After dehydration, treatment with uranyl acetate, and infiltratration with Lowicryl HM-20 in a Leica AFS machine, blocks were polymerized with UV at 0°C, and further polymerized at room temperature. Thin sections (~80 nm) were cut and collected on nickel mesh grids, and immunoprocessed for Bin1. Secondary antibodies were conjugated to 10 nm or 20 nm-diameter gold particles, for EM visualization. Grids were examined in a Philips Tecnai transmission electron microscope at 80 KV, and images collected with a 1024 × 1024 cooled CCD (Gatan). Because antibody is restricted to the surface of the thin section, postembedding immunogold is insensitive to differences in the composition of different tissue compartments, and is therefore expected to label across the section in an unbiased matter.
Dendritic spine quantification
Two-dimensional, background-subtracted maximum projection reconstructions of images for dendritic spine morphometric analysis (area, length, and width), and quantification of spine linear density (# of spines/10 μm dendritic length) were performed using MetaMorph software (Molecular Devices). A threshold was applied to the maximum projection images to include all detectable spines, and regions along dendrites containing dendritic spines were manually traced to enclose spines but not dendritic shaft or other structures. Dendritic spine “objects”, restricted to objects with areas greater than 0.1 μm2, were automatically detected by MetaMorph, and the area, maximum length and head width of each spine was measured. Two dendritic branches (approximately 100 μm) of each neuron were analyzed. Only spines on secondary and tertiary apical dendrites were measured to reduce variability. Cultures that were directly compared were stained simultaneously and imaged with the same acquisition parameters. For each condition, 3–10 neurons each from 2–5 separate experiments were used. Experiments were performed blind to conditions and on sister cultures.
Fluorescence recovery after photobleaching (FRAP)
Mouse cortical neurons were cultured in 35mm glass bottom dishes (Poly-D-Lysine Coated, No. 0, MATEK, #P35GC-0–14C) and transfected with knockdown and scramble control plasmids as described previously. All dishes were transfected with the pH sensitive, green fluorescent SEP-GluA1 plasmid36. Cells were imaged on a Nikon A1R+ confocal with GaAsP detectors and a resonant scanner. High expressing pyramidal neurons were selected (as indicated by high levels of the co-expressed mCherry plasmid) on low magnification. Then, the primary apical dendrite was imaged starting ~50 μm from the cell body. A 512×128 pixel image was taken using a 100X objective with 110 nm /pixel, with a 1.5 multiplying lens and a 1.5 digital zoom in multiple Z planes to fully image the dendrite (9.6 μm, 48 200 nm steps). 16 red (mCherry excited with 561 nm laser) and green (SEP-GluA1 GFP excited with 488 nm laser) were averaged using the resonant scanner. After taking the pre-bleach image, bleaching was performed on a 33 μm by 13 μm section (488 laser full power, ½ scan speed, pixel dwell 4.6 for 512×512, 10 pulses for a total duration of 19.19 sec). Immediately subsequent, time-lapse imaging was performed every minute for 13 minutes (14 total post-bleach images taken). Maximum projections were analyzed using Fiji/ImageJ software. The cell-filling mCherry was thresholded to create a dendrite outline, and spines were erased to analyze just the dendritic shaft. Then, the mean pixel intensity of the middle 2 μm of the bleached region was quantified at every timepoint. Post-bleach data was background subtracted and normalized to the pre-bleach value by division. The T0 value (immediately post-bleach) was then subtracted from every timepoint, starting the recovery at a value of 0 for each cell. A total of 12 cells were analyzed for each condition on two separate days using two different neuronal culture preparations.
Statistical analysis
All statistical tests were performed with GraphPad Prism. Data were tested for normality with D’Agostino and Pearson to determine use of non-parametric (Mann-Whitney, Kruskal-Wallis), or parametric (unpaired t-test, Welch’s t-test, ANOVA) tests. For comparison between two groups with normal distributions, we employed an F test for equality of variances to determine whether to use an unpaired t-test (equal variances) or Welch’s t-test (unequal variances). Post-hoc tests were included in analyses with multiple comparisons. Tests used are indicated in figure legends, and bar graphs are displayed as mean ± SEM, unless otherwise noted. All tests were two-sided and P values were considered significant if < 0.05. N numbers refer to number of cells per condition unless otherwise stated. Required sample sizes to observe a significant effect were estimated based on previous studies in the lab.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HIGHLIGHTS.
Bioinformatics reveals a prominent role for synaptic trafficking in LOAD
SIM provides quantitative insight into the mesoscale spatial architecture of postsynaptic trafficking
Novel functions identified for the LOAD risk factor Bin1 in regulating postsynaptic endo- and exocytosis
Bin1 complexes with the small GTPase Arf6 and the AMPAR subunit GluA1 and maintains normal spine morphology and glutamatergic transmission
Acknowledgements
This work was supported by: R01s MH097216, MH107182 and MH097216 and R56 AG063433 to PP, German Research Foundation (DFG) Postdoctoral Research Fellowship SCHU2710/1-1 to BS, NS064091 MM, and NS039444 to RJW. Imaging work was partly performed at the Northwestern University Center for Advanced Microscopy generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center. Structured illumination microscopy was performed on a Nikon N-SIM system, purchased through the support of NIH 1S10OD016342-01. We thank Joshua Rappoport, Constadina Arvanitis, and Teng Leong Chew for assistance with imaging and analysis. All experiments involving animals were performed according to the Institutional Animal Care and Use Committee of NU.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
Supplementary information is available at MP’s website
REFERENCES
- 1.Anggono V & Huganir RL Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol 22, 461–469, doi: 10.1016/j.conb.2011.12.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Penzes P, Cahill ME, Jones KA, VanLeeuwen JE & Woolfrey KM Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14, 285–293, doi: 10.1038/nn.2741 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Sluijs P & Hoogenraad CC New insights in endosomal dynamics and AMPA receptor trafficking. Semin Cell Dev Biol 22, 499–505, doi: 10.1016/j.semcdb.2011.06.008 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Moore FB & Baleja JD Molecular remodeling mechanisms of the neural somatodendritic compartment. Biochim Biophys Acta 1823, 1720–1730, doi: 10.1016/j.bbamcr.2012.06.006 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Hsu VW, Bai M & Li J Getting active: protein sorting in endocytic recycling. Nat Rev Mol Cell Biol 13, 323–328, doi: 10.1038/nrm3332 (2012). [DOI] [PubMed] [Google Scholar]
- 6.DeKosky ST & Scheff SW Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27, 457–464, doi: 10.1002/ana.410270502 (1990). [DOI] [PubMed] [Google Scholar]
- 7.Bertram L & Tanzi RE Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci 9, 768–778, doi: 10.1038/nrn2494 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Karch CM & Goate AM Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 77, 43–51, doi: 10.1016/j.biopsych.2014.05.006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu JB, Tan CC, Tan L & Yu JT State of Play in Alzheimer’s Disease Genetics. J Alzheimers Dis 58, 631–659, doi: 10.3233/JAD-170062 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Tan MS, Yu JT & Tan L Bridging integrator 1 (BIN1): form, function, and Alzheimer’s disease. Trends Mol Med 19, 594–603, doi: 10.1016/j.molmed.2013.06.004 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Prokic I, Cowling BS & Laporte J Amphiphysin 2 (BIN1) in physiology and diseases. J Mol Med (Berl) 92, 453–463, doi: 10.1007/s00109-014-1138-1 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Picas L et al. BIN1/M-Amphiphysin2 induces clustering of phosphoinositides to recruit its downstream partner dynamin. Nat Commun 5, 5647, doi: 10.1038/ncomms6647 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Drager NM et al. Bin1 directly remodels actin dynamics through its BAR domain. EMBO Rep 18, 2051–2066, doi: 10.15252/embr.201744137 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakajo A et al. EHBP1L1 coordinates Rab8 and Bin1 to regulate apical-directed transport in polarized epithelial cells. J Cell Biol 212, 297–306, doi: 10.1083/jcb.201508086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pant S et al. AMPH-1/Amphiphysin/Bin1 functions with RME-1/Ehd1 in endocytic recycling. Nat Cell Biol 11, 1399–1410, doi: 10.1038/ncb1986 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bayes A et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci 14, 19–21, doi: 10.1038/nn.2719 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grossmann AH et al. The small GTPase ARF6 regulates protein trafficking to control cellular function during development and in disease. Small GTPases, 1–12, doi: 10.1080/21541248.2016.1259710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hongu T & Kanaho Y Activation machinery of the small GTPase Arf6. Adv Biol Regul 54, 59–66, doi: 10.1016/j.jbior.2013.09.014 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Johnson DL, Wayt J, Wilson JM & Donaldson JG Arf6 and Rab22 mediate T cell conjugate formation by regulating clathrin-independent endosomal membrane trafficking. J Cell Sci, doi: 10.1242/jcs.200477 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris KM & Weinberg RJ Ultrastructure of synapses in the mammalian brain. Cold Spring Harb Perspect Biol 4, doi: 10.1101/cshperspect.a005587 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhar SS, Liang HL & Wong-Riley MT Nuclear respiratory factor 1 co-regulates AMPA glutamate receptor subunit 2 and cytochrome c oxidase: tight coupling of glutamatergic transmission and energy metabolism in neurons. J Neurochem 108, 1595–1606, doi: 10.1111/j.1471-4159.2009.05929.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caplan S et al. A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. EMBO J 21, 2557–2567, doi: 10.1093/emboj/21.11.2557 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Posey AD Jr. et al. EHD1 mediates vesicle trafficking required for normal muscle growth and transverse tubule development. Dev Biol 387, 179–190, doi: 10.1016/j.ydbio.2014.01.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oku Y & Huganir RL AGAP3 and Arf6 regulate trafficking of AMPA receptors and synaptic plasticity. J Neurosci 33, 12586–12598, doi: 10.1523/JNEUROSCI.0341-13.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholz R et al. AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron 66, 768–780, doi: 10.1016/j.neuron.2010.05.003 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Penzes P & Vanleeuwen JE Impaired regulation of synaptic actin cytoskeleton in Alzheimer’s disease. Brain Res Rev 67, 184–192, doi: 10.1016/j.brainresrev.2011.01.003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karch CM, Cruchaga C & Goate AM Alzheimer’s disease genetics: from the bench to the clinic. Neuron 83, 11–26, doi: 10.1016/j.neuron.2014.05.041 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glennon EB et al. BIN1 is decreased in sporadic but not familial Alzheimer’s disease or in aging. PLoS One 8, e78806, doi: 10.1371/journal.pone.0078806 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams SL, Tilton K, Kozubek JA, Seshadri S & Delalle I Subcellular Changes in Bridging Integrator 1 Protein Expression in the Cerebral Cortex During the Progression of Alzheimer Disease Pathology. J Neuropathol Exp Neurol 75, 779–790, doi: 10.1093/jnen/nlw056 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holler CJ et al. Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer’s disease brain and correlates with neurofibrillary tangle pathology. J Alzheimers Dis 42, 1221–1227, doi: 10.3233/JAD-132450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chapuis J et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry 18, 1225–1234, doi: 10.1038/mp.2013.1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee E et al. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297, 1193–1196, doi: 10.1126/science.1071362 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Copits BA & Swanson GT Kainate receptor post-translational modifications differentially regulate association with 4.1N to control activity-dependent receptor endocytosis. J Biol Chem 288, 8952–8965, doi: 10.1074/jbc.M112.440719 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith KR et al. Psychiatric risk factor ANK3/ankyrin-G nanodomains regulate the structure and function of glutamatergic synapses. Neuron 84, 399–415, doi: 10.1016/j.neuron.2014.10.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burette AC et al. Organization of TNIK in dendritic spines. J Comp Neurol 523, 1913–1924, doi: 10.1002/cne.23770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kopec CD, Li B, Wei W, Boehm J & Malinow R Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci 26, 2000–2009, doi: 10.1523/JNEUROSCI.3918-05.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.