

Abstract
Background
Profiling the entire genome at base pair resolution in a single test offers novel insights into disease by means of dissection of genetic contributors to phenotypic features.
Methods
We performed genome sequencing for a patient who presented with atypical hereditary sensory and autonomic neuropathy, severe epileptic encephalopathy, global developmental delay, and growth hormone deficiency.
Results
Assessment of the variants detected by mapped sequencing reads followed by Sanger confirmation revealed that the proband is a compound heterozygote for rare variants within RETREG1 (FAM134B), a gene associated with a recessive form of hereditary sensory and autonomic neuropathy, but not with epileptic encephalopathy or global developmental delay. Further analysis of the data also revealed a heterozygous missense variant in DNM1L, a gene previously implicated in an autosomal dominant encephalopathy, epilepsy, and global developmental delay and confirmed by Sanger sequencing to be a de novo variant not present in parental genomes.
Conclusions
Our findings emphasize the importance of genome‐wide sequencing in patients with a well‐characterized genetic disease with atypical presentation. This approach reduces the potential for misdiagnoses.
Keywords: DNM1L, epileptic encephalopathy, HSAN, intradermal histamine test, self‐injury, whole genome sequencing
1. INTRODUCTION
High throughput sequencing offers an opportunity to analyze the genome of an individual (whole genome sequencing [WGS]) or a protein‐coding subset of a genome in a single test (whole exome sequencing [WES]). The provision of genome‐wide data from a single screen has played an important role in diagnosing rare disease patients with complex phenotypes (Posey et al., 2017; Tarailo‐Graovac et al., 2016). An emerging challenge is interpreting the genetic results and clarifying if the atypical phenotypes are an expanded clinical presentation of an already suspected monogenic disease, an unsuspected monogenic disease, or do they result from two or more distinct genetic conditions with overlapping (blended) or discrete (composite) clinical features? Population databases of untargeted populations (e.g. gnomAD) play an important role in interpretation of genomic findings (Lek et al., 2016) where thorough review of the findings is essential (Tarailo‐Graovac, Zhu, Matthews, Karnebeek, & Wasserman, 2017).
Hereditary sensory and autonomic neuropathy (HSAN) are a heterogeneous group of disorders characterized by progressive degeneration of the peripheral nervous system and presenting with prominent sensory and autonomic symptoms. Here, we report a proband presenting with a phenotype resembling an atypical form of HSAN. In addition to severe sensory and autonomic neuropathy, the patient also presented with progressive neurodegeneration, profound global developmental delay, and epileptic encephalopathy, CNS features that are previously not reported in HSAN patients. Using a singleton‐WGS analysis, we identified rare biallelic variants in RETREG1 (also known as FAM134B [MIM 613114]) and later, a rare de novo heterozygous variant in DNM1L (MIM 603850). After careful review of the variants and other patients with DNM1L deficiency, we conclude that the pathogenic de novo variant is the sole cause of the phenotypic features in our patient (including HSAN‐like phenotype), rather than a composite effect of two rare disorders.
2. METHODS
2.1. Subjects
The patient and the family were initially enrolled in to the Department of Medical Genetics GARD study for rare disease gene discovery (UBC IRB approval H09‐01228) and then subsequently enrolled into TIDEX gene discovery study (UBC IRB approval H12‐00067). They provided informed and written consent for sample collection, WGS, data analysis, and publication of the current case report. Detailed clinical presentation of the proband is available in the Data S1. In brief, the patient presented with developmental delay, decreased pain sensitivity, self‐mutilation, decreased tear production, and autonomic instability. He later developed dystonia, spasticity, and severe epileptic encephalopathy. He had an abnormal intradermal histamine test. About 0.5 ml of histamine phosphate 1:1,000 was injected intradermally in the flexor surface of the forearm, and the reaction read in 5 min. A normal response to injection is the appearance of a wheal usually 1 cm in diameter, with a zone of erythema (flare) around it, approximately 3 cm in diameter. In patients with hereditary sensory neuropathies, the wheal is not surrounded by a flare response.
2.2. WGS and confirmation
Genomic DNA was isolated using standard protocols and singleton WGS (proband only) was sequenced on an Illumina HiSeq 2500 (University of British Columbia). Our default semiautomated bioinformatics pipeline was used to analyze the data (Tarailo‐Graovac et al., 2016). Confirmation of the variants identified using WGS, as well as segregation with the disease was validated using Sanger sequencing at the CMMT/BCCHRI DNA Sequencing Core Facility (for detailed description of our WGS protocols please see Data S1).
3. RESULTS
The WGS analysis of the proband (Figure 1) revealed rare variants in only one known HSAN‐associated gene, RETREG1 (HSAN type IIB, an autosomal recessive condition [MIM 613115]) (Kurth et al., 2009). The proband is a compound heterozygote for two missense variants, validated by Sanger resequencing as compound heterozygous (Figure 1), of RETREG1 (NM_001034850; NP_001030022): (c.607G>A; p.(Val203Met)) and (c.379C>T; p.(Arg127Cys)). Both of the variants are rare according to dbSNP (v.150), NHLBI ESP, our in‐house database and gnomAD (Tarailo‐Graovac, Zhu, et al., 2017). Both of these variants affect conserved amino acids; however, while most of the tools predict the effects of the variants to be deleterious [e.g., high Combined Annotation Dependent Depletion (CADD, v1.3) (Kircher et al., 2014) scores (24.8 for p.(Val203Met); 26.0 for p.(Arg127Cys)), and probably damaging scores by PolyPhen2 (Adzhubei, Jordan, & Sunyaev, 2013)], other tools like SIFT (Kumar, Henikoff, & Ng, 2009) predict the variants to be tolerated. The evidence regarding the clinical significance of these variants, however, suggests that these are likely/benign. One of the variants is classified as likely benign by at least one clinical lab and the other is recorded as benign by all the submitting labs in the ClinVar (Landrum, Lee, & Benson, 2016). The p.(Arg127Cys) variant has been observed in eight homozygous individuals in gnomAD and the p.(Val203Met) variant in one homozygous individual. These observations combined with the fact that, to date only loss of function RETREG1 variants have been described in HSAN type IIB, lead to “likely benign” classification of both variants using the ACMG (American College of Medical Genetics and Genomics) guidelines (Richards, Aziz, & Bale, 2015).
Figure 1.
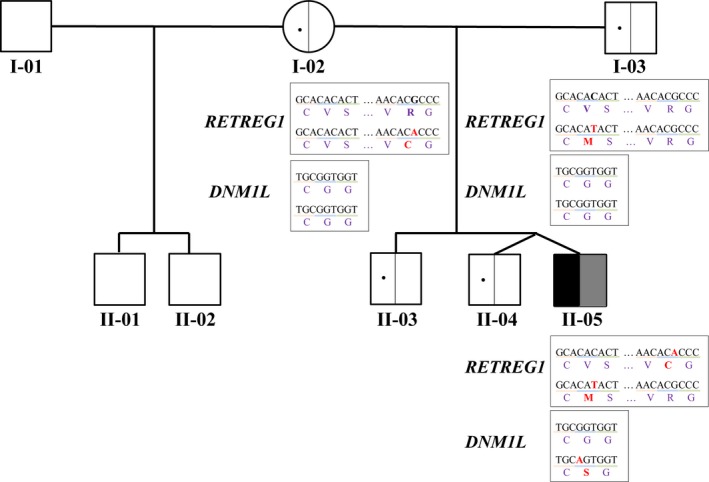
A family with an isolated case of an atypical sensory and autonomic neuropathy combined with severe epileptic encephalopathy and global developmental delay. For the simplicity, the neuropathy phenotype is denoted in black, while the CNS phenotype in gray in the affected proband (II‐05). RETREG1 variant carrier status in unaffected parents as confirmed by Sanger resequencing is depicted in left half of the symbol. The DNA (reference sequence from Human Genome GRCh37/hg19 assembly) and amino acid changes are depicted as well
Next, we also identified a heterozygous variant, confirmed de novo variant, in DNM1L (Figure 1). A dominant negative mutation in DNM1L encoding the dynamin‐like protein 1 was reported in 2007 by Waterham et al. in a patient with a lethal defect of mitochondrial and peroxisomal fission (Waterham et al., 2007). Recent publications further report developmental delay, pain insensitivity, and mitochondrial respiratory chain complex IV deficiency phenotypes (Sheffer et al., 2016); refractory epilepsy (Vanstone et al., 2016); slowly progressive mild neurological impairment (Nasca et al., 2016), and childhood onset epileptic encephalopathy (Fahrner, Liu, Perry, Klein, & Chan, 2016) in patients with DNM1L variants.
The heterozygous DNM1L (NM_012062; NP_036192) variant (c.1084G>A; p.Gly362Ser) identified here is predicted to be damaging by all the tested tools (Adzhubei et al., 2013; Kircher et al., 2014; Kumar et al., 2009), it affects a conserved amino acid, has not been observed in the NHLBI ESP, our in‐house database, or gnomAD (Tarailo‐Graovac, Zhu, et al., 2017) and according to the ACMG guidelines is classified as pathogenic (Richards et al., 2015). It has been recently reported as a pathogenic variant, identified in a patient with chronic neurological disorder, postnatal microcephaly, developmental delay, decreased respiratory chain complex IV activity, and pain insensitivity (Sheffer et al., 2016). This patient was also suspected initially to have HSAN and at the age of 1 year was examined by a Familial Dysautonomia expert. Intradermal histamine test was pathologically similar to our patient and corneal reflexes were reduced. His sequencing data, however, unlike the genome data of our patient revealed no rare variants in RETREG1 or other known HSAN‐associated genes (Sheffer personal communication). Furthermore, a heterozygous de novo variant affecting the same amino acid (c.1085G>A; p.(Gly362Asp)) has also been reported in a 7‐year‐old boy with encephalopathy (Vanstone et al., 2016). The conserved p.Gly362 amino acid is located in the domain that is important for homo‐oligomerization and was suggested to result in a dominant negative effect (Vanstone et al., 2016).
4. DISCUSSION
The diagnosis of HSAN of unknown type was entertained in this patient from very early on, based on an abnormal intradermal histamine test and the phenotype consistent with painless automutilation. Mutations in RETREG1 were identified first in 2009 to cause HSAN (Kurth et al., 2009), and spectrum of clinical presentations described since (Data S1), but none of the reported patients presented with CNS features akin to our patient.
Thus, after we identified the missense variants in RETREG1, a gene that fits well with the patient's HSAN phenotype, we continued the search for genome changes that could explain the CNS phenotype in our patient. During the prolonged ICU admission of our patient, the diagnosis of mitochondrial disease was entertained but the patient was too unstable for a muscle biopsy under general anesthesia. Identification of a de novo DNM1L variant from genome data was considered an excellent fit and was further reinforced with reports on the same (p.Gly362Ser) (Sheffer et al., 2016) or similar (p.Gly362Asp) (Vanstone et al., 2016) de novo DNM1L variants in patients presenting with refractory epilepsy published shortly after DNM1L discovery in our patient. Recurrent observation of a de novo variation in the Gly362 amino acid in DNM1L deficiency patients suggests an important role of this particular amino acid in the pathophysiology of the disease.
The case presented here is an excellent example of the emerging challenges in clinical genetics with respect to interpretation of complex phenotypes that may be due to either the occurrence of more than one genetic disorder in an individual, or new phenotypic expansion, or complete misdiagnosis in the first place. What initially seemed to be a case of an expanded phenotypic spectrum of RETREG1 deficiency with the identification of the pathogenic DNM1L and additional information on the other DNM1L patient became in fact an expansion of the DNM1L spectrum. We conclude that the identified rare missense RETREG1 variants do not contribute to the HSAN phenotype. The pain insensitivity has been observed in one patient with the same DNM1L variant (Sheffer et al., 2016), however abnormal intradermal histamine test has never been described, although it was later confirmed by the authors (Sheffer personal communication). However growth hormone deficiency has not been reported before in patients with DNM1L variants. Neither of these two patients had evidence of peroxisomal dysfunction. Our findings further illustrate the importance of genome‐wide sequencing, WGS in this case, in accurate diagnosis (Tarailo‐Graovac, Wasserman, & Van Karnebeek, 2017). Had we only applied a gene‐centric approach focused on HSAN‐implicated genes, we might either have interpreted this patient as undiagnosed, consider erroneously that the RETREG1 variants are disease causing, and suggest an expansion of the phenotypic spectrum associated with biallelic RETREG1 mutations. On the contrary, we have now expanded the phenotypic spectrum of conditions associated with this DNM1L variant as having abnormal intradermal histamine testing, which has only been associated so far with different forms of HSAN. We would like to highlight the importance of detailed and accurate description of clinical phenotyping of newly discovered genetic conditions, to have the best chance of matching a patient's clinical presentation with candidate genes found on genomic analysis.
CONFLICT OF INTEREST
All authors declare that they have no conflict of interest or financial relationships that could be considered conflict of interest.
Supporting information
ACKNOWLEDGMENTS
The authors thank the patient and the family for participating in this research study, and acknowledge Ms. A. Ghani for research coordination, Ms. X. Han, Ms. M. Higginson, and Mr. G Tran for sample handling and Sanger analysis and Ms. E. Lomba for research administration. F.R.Z was supported by a CIHR Post‐Doctoral Scholarship, NeuroDevNet post‐fellowship, and UBC Bluma Tischler fellowship. CN is a CRC Tier 1 Chair in translational Genomics. CvK is the recipient of a Michael Smith Foundation for Health Research Scholar award.
Tarailo‐Graovac M, Zahir FR, Zivkovic I, et al. De novo pathogenic DNM1L variant in a patient diagnosed with atypical hereditary sensory and autonomic neuropathy. Mol Genet Genomic Med. 2019;7:e961 10.1002/mgg3.961
Funding information
This research was supported by: the BCCH Foundation as “1st Collaborative Area of Innovation” (www.tidebc.org), Genome BC (SOF‐195 grant), the CIHR (#301221 grant), and Alberta Children's Hospital Research Institute Foundation. WGS on this project was supported by the Canadian Institute of Health Research (CIHR) [grant # MOP‐102600].
Contributor Information
Maja Tarailo‐Graovac, Email: maja.tarailograovac@ucalgary.ca.
Gabriella A. Horvath, Email: ghorvath@cw.bc.ca.
REFERENCES
- Adzhubei, I. , Jordan, D. M. , & Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen‐2. Current Protocols in Human Genetics, 76(1), 7.20.1–7.20.41. 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrner, J. A. , Liu, R. , Perry, M. S. , Klein, J. , & Chan, D. C. (2016). A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. American Journal of Medical Genetics Part A, 170, 2002–2011. 10.1002/ajmg.a.37721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher, M. , Witten, D. M. , Jain, P. , O'Roak, B. J. , Cooper, G. M. , & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46, 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4, 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Kurth, I. , Pamminger, T. , Hennings, J. C. , Soehendra, D. , Huebner, A. K. , Rotthier, A. , … Hübner, C. A. (2009). Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nature Genetics, 41, 1179–1181. 10.1038/ng.464 [DOI] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Benson, M. , et al. (2016). ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Research, 44, D862–D868. 10.3410/f.725948565.793531076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … Exome Aggregation Consortium . (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536, 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasca, A. , Legati, A. , Baruffini, E. , Nolli, C. , Moroni, I. , Ardissone, A. , … Ghezzi, D. (2016). Biallelic mutations in DNM1L are associated with a slowly progressive infantile encephalopathy. Human Mutation, 37, 898–903. 10.1002/humu.23033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey, J. E. , Harel, T. , Liu, P. , Rosenfeld, J. A. , James, R. A. , Coban Akdemir, Z. H. , … Lupski, J. R. (2017). Resolution of disease phenotypes resulting from multilocus genomic variation. New England Journal of Medicine, 376, 21–31. 10.1056/nejmoa1516767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier-Foster, J. , … ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffer, R. , Douiev, L. , Edvardson, S. , Shaag, A. , Tamimi, K. , Soiferman, D. , … Saada, A. (2016). Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. American Journal of Medical Genetics. Part A, 170, 1603–1607. 10.1002/ajmg.a.37624 [DOI] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. , Shyr, C. , Ross, C. J. , Horvath, G. A. , Salvarinova, R. , Ye, X. C. , … van Karnebeek C. D. (2016). Exome sequencing and the management of neurometabolic disorders. New England Journal of Medicine, 374, 2246–2255. 10.1056/NEJMoa1515792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. , Wasserman, W. W. , & Van Karnebeek, C. D. M. (2017). Impact of next‐generation sequencing on diagnosis and management of neurometabolic disorders: Current advances and future perspectives. Expert Review of Molecular Diagnostics, 17, 307–309. 10.1080/14737159.2017.1293527 [DOI] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. , Zhu, J. Y. A. , Matthews, A. , van Karnebeek, C. D. M. , & Wasserman, W. W. (2017). Assessment of the ExAC data set for the presence of individuals with pathogenic genotypes implicated in severe Mendelian pediatric disorders. Genetics in Medicine, 19(12), 1300–1308. 10.1038/gim.2017.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanstone, J. R. , Smith, A. M. , McBride, S. , Naas, T. , Holcik, M. , Antoun, G. , … Lines, M. A. (2016). DNM1L‐related mitochondrial fission defect presenting as refractory epilepsy. European Journal of Human Genetics, 24, 1084–1088. 10.1038/ejhg.2015.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham, H. R. , Koster, J. , van Roermund, C. W. T. , Mooyer, P. A. W. , Wanders, R. J. A. , & Leonard, J. V. (2007). A lethal defect of mitochondrial and peroxisomal fission. New England Journal of Medicine, 356, 1736–1741. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials