

Abstract
Background
Dyschromatosis symmetrica hereditaria (DSH;OMIM: #127400) is a rare autosomal dominant skin disease of hyperpigmented and hypopigmented macules on the dorsal aspects of the feet and hands. The adenosine deaminase RNA‐Specific (ADAR;OMIM: *146920) gene was identified as causing DSH. Although more than 200 mutations are reported, no research has included the pedigrees of ethnic minorities in China. To investigate clinical features and genetic factors among multi‐ethnic families, seven multi‐ethnic pedigrees with DSH were collected for analysis of hereditary characteristics and ADAR mutations.
Methods
All 15 exons and exon–intron sequences of the ADAR gene were amplified and Sanger sequenced from 25 patients and 36 normal controls from seven multi‐ethnic DSH families with 100 healthy normal controls. Seven mutations were analyzed by Polyphen 2, SIFT and Provean. All mutations in ADAR with DSH were reviewed and genetic and clinical features were summarized for analysis. The ADEAMc domain may be a hot spot of ADAR mutations among patients with DSH.
Results
Seven novel mutations were identified in seven multi‐ethnic pedigrees: c.497delA(p.Arg105fs), c.3352C>T(p.Gln1058*) and c.3722delT(p.Ser1181fs) were found in three Uygur families with DSH; c.1330A>G(p.Val332Met) and c.2702A>T(p.His841Leu) were found in two Kazakh pedigrees and c.1176G>A(p.Lys326Glu) and c.2861G>A(p.Arg892His) in two Hui pedigrees. We summarized 203 different mutations of ADAR from people with DSH.
Conclusions
Seven novel mutations were identified in seven multi‐ethnic families with DSH. Our study expands the genetic spectrum of ADAR mutations in DSH.
Keywords: adenosine deaminase acting on RNA, China, dyschromatosis symmetrica hereditaria, mutation
1. INTRODUCTION
Dyschromatosis symmetrica hereditaria (DSH; OMIM: #127400) is a rare autosomal dominant skin disease with characteristic hyperpigmented and hypopigmented spots on the dorsal aspects of the hands and feet (Hayashi & Suzuki, 2013). In some patients, freckle‐like macules appear on the face. A rash usually appears in infancy or early childhood, lasting the entire life, without any changes in distribution (Tomita and Suzuki, 2004). Some factors affect DSH phenotype such as viral infection and exposure to ultraviolet (UV) light (Zhang et al., 2016). Histologically, the distribution of melanocytes in hypopigmented and pigmentation regions is different with fewer melanocytes appearing on hypopigmented spots compared with pigmentation regions (Kondo, Suzuki, Mitsuhashi, et al., 2008). Electron microscopic examination shows large intercellular spaces and vacuoles around few melanosomes in hypopigmented skin (Omura et al., 2017).
Adenosine deaminase RNA‐Specific (ADAR; OMIM: *146920) was identified as the causal gene for DSH. The gene is located on chromosome 1q21.1–21.2 and contains 15 exons (Miyamura et al., 2003; Zhang et al., 2003). It codes for ADAR, and with alternative splicing, ADAR p150, with a promoter induced by interferon and ADAR p110, with a constitutive promoter (Patterson & Samuel, 1995). In an editing‐independent manner, ADAR is important for catalyzing the conversion from adenosine to inosine and participates in gene regulation in normal mammalian development (Song, Sakurai, Shiromoto, & Nishikura, 2016).
To date, some familial and some sporadic cases of DSH are reported in East Asia, mainly in Japanese and Chinese populations (Consigli, Zanni, Ragazzini, & Danielo, 2010). More than 200 mutations in ADAR have been identified among different populations worldwide (Tang et al., 2018). Most reports focus on Japanese and Han populations in China. Ethnic background is a major influence on DSH (Liu et al., 2006), with no causal gene mutation analysis in minority populations. We collected seven DSH families to investigate ADAR mutation to better understand the basic pathogenic mechanisms of DSH.
2. MATERIALS AND METHODS
2.1. Participants and ethics statement
Data were collected from seven multi‐ethnic DSH families from the Xinjiang Uygur Autonomous Region: three Uyghur families (Figure 1a–c), two Hasakez families (Figure 1d,e) and two Hui families (Figure 1f,g) for a total of 25 patients and 36 normal participants. The 25 affected individuals agreed on peripheral blood collection, approved by the Research Ethics Committee of China Medical University. All participants gave written informed consent. All affected individuals had irregularly shaped and sized macules on the dorsal aspects of the hands and feet.
Figure 1.
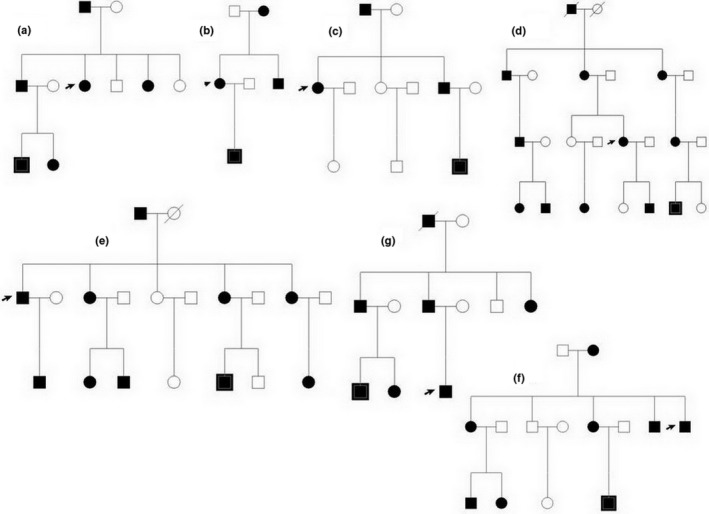
The family diagram: three Uyghur population families (a–c), two Hasakez population families (d, e) and two Hui population families (f, g)
2.2. Polymerase chain reaction (PCR)
Genomic DNA samples were extracted from peripheral blood using Universal Genomic DNA Extraction kits (TaKaRa, Dalian, China). ADAR (GenBank: NC_000001.11, GRCh38.p13)primers flanked all 15 coding exons and intron–exon boundaries (Table 1). PCR was: 1 min at 94°C, 35 cycles of 30 s at 94°C, 30 s at 58°C, 1 min at 72°C, and 5 min at 72°C. The amplification products were bi‐directional Sanger sequenced (3730xl DNA Analyzer)by the Shanghai Shenggong Company.
Table 1.
Mutations of the ADAR1 gene in multi‐ethnic pedigrees with DSH in China
| EXON | ADAR1 primer | Length | TM, °C |
|---|---|---|---|
| EXON1 (5‐3) | F(5‐3):GCGGAGGGGTTCGACTTGTA | 396 | 58 |
| R(5‐3):CGCTACGCACTGCAACACAA | |||
| Exon2‐1 (5‐3) | F(5‐3):TTCCACAGGCAGCAAAGGGA | 448 | 57 |
| R(5‐3):AGGCAATCAACACCTCTCTGTGG | |||
| Exon2‐2 (5‐3) | F(5‐3):GCCTCCAGTACCAGAGGCA | 526 | 58 |
| R(5‐3):GGTTCCAAGCCTGAGCTGAGAC | |||
| Exon2‐3 (5‐3) | F(5‐3):CCAGACGGTCATAGCCAAGGAG | 507 | 58 |
| R(5‐3):AGGGATTGCAGCTGGAGCG | |||
| Exon2‐4 (5‐3) | F(5‐3):TCTGCGACTATCTCTTCAATGTGTCTGAC | 510 | 59 |
| R(5‐3):ACTCACCTGGTGCTGCGC | |||
| Exon2‐5 (5‐3) | F(5‐3):ACCACCTGTTCATTACAATGGCCC | 407 | 58 |
| R(5‐3):GATAGGCGCCACCAAACAGC | |||
| Exon3 | F(5‐3):GGAGTTCCTTGGCCTACCCT | 348 | 58 |
| R(5‐3):CCAGATGGCAGGAGGACACC | |||
| Exon4 | F(5‐3):AACCCCTTGACAGGTGGTGG | 290 | 60 |
| R(5‐3):CAGCTGGACAGAGGACACGT | |||
| Exon5 | F(5‐3):CTGGCAGAGGCTAGGTCAGG | 296 | 61 |
| R(5‐3):TGTTGAGGGAGTCACTGGCA | |||
| Exon6 | F(5‐3):TGCCAGTGACTCCCTCAACA | 374 | 58 |
| R(5‐3):GTTTCCCTCAACTCGCCCCT | |||
| Exon7 | F(5‐3):TGTCAGGGTCTGGCACTTGT | 356 | 57 |
| R(5‐3):GCATGACAGCAAGAGCCACC | |||
| Exon8 | F(5‐3):GGTGGCTCTTGCTGTCATGC | 389 | 58 |
| R(5‐3):CGGCATGTCTCAGAGCCTCA | |||
| Exon9 | F(5‐3):TGAAAGCGGGTGCCTCTCAT | 374 | 59 |
| R(5‐3):GGGCCACAGCTCTGACCTC | |||
| Exon10 | F(5‐3):ACCTGCCTTCCTAACCAGACT | 337 | 58 |
| R(5‐3):TGGGAGACTGGAGGTGGACA | |||
| Exon11 | F(5‐3):ACTGTTTTGGAGCCCCACGA | 258 | 58 |
| R(5‐3):CCTGGACCTTGCAGAGCCTT | |||
| Exon12 | F(5‐3):AGAAACCACGCCAGGGAGTG | 281 | 57 |
| R(5‐3):CCAGTTCCAGATCCCAAGGCA | |||
| Exon13 | F(5‐3):TCCCCACATGCTTCTGCCTC | 278 | 58 |
| R(5‐3):CCCCTTGCCCACAGTGTACA | |||
| Exon14 | F(5‐3):ACCCCACACTTCCTCTCTCCT | 275 | 58 |
| R(5‐3):AAGTCAGGGCAGAGGCTTGG | |||
| Exon15‐1 | F(5‐3):gtctccactgtgagctccttatcttacag | 500 | 60 |
| R(5‐3):CTGGCCAGACCTTGCCTAGC | |||
| Exon15‐2 | F(5‐3):agcattcctcatcacatggtcagg | 580 | 58 |
| R(5‐3):GTGCAGGATGGGAGGATGGC | |||
| Exon15‐3 | F(5‐3):ctcagagggcaaagaggtgaaca | 541 | 58 |
| R(5‐3):GGTGTCACTGTCATGAGAGATATTACACCG | |||
| Exon15‐4 | F(5‐3):gccaacgggacaaatcctagagg | 640 | 61 |
| R(5‐3):CACCACGGCACCAAGTCTATGC | |||
| Exon15‐5 | F(5‐3):ctggctctctggctcctgt | 662 | 58 |
| R(5‐3):GGCTGCGCTGCCTTCTGAT | |||
| Exon15‐6 | F(5‐3):ctggagtggaagaggcctgc | 571 | 57 |
| R(5‐3):GGAATGCACAGTAGCCACAGTTCA | |||
| Exon15‐7 | F(5‐3):acacaggacagaggaggcaga | 468 | 58 |
| R(5‐3):GGCCACAGGTCCCTTTGTTC |
2.3. Bioinformation analysis
Mutations of ADAR were analyzed for evolutionary conservation and deleteriousness by the Polyphen‐2 program (http://genetics.bwh.harvard.edu/pph2) and SIFT (http://sift.jcvi.org). SWISS‐MODEL predicted the structure of ADAR gene mutations affecting the protein. UniProtKB/UniRef 100 Release 2011_12 (14 December, 2011) was used for multiple sequence alignment of ADAR among multispecies.
3. RESULTS
3.1. Mutations of the ADAR gene in multi‐ethnic pedigrees with DSH in China were seven novel mutations including four missense mutations (p.K326E, p.H841L, p.V332M and p.R892H), frameshift mutations (p.R105fs and p.S1181fs) and one nonsense mutation (p.G1058X) in seven pedigrees from different ethnic populations (Table 2, Figure 2). Mutations were not found in 100 unrelated normal controls and were not included in the NCBI SNP database, suggesting the novel mutations may be pathological mutations of DSH. Four missense mutations, p.K326E, p.H841L, p.V332M and p.R892H, may result in amino acid sequence changes and were indicated to be potentially harmful. Two frameshift mutations (p.R105fs and p.S1181fs) led to a premature termination codon (PTC). Products may include the inactive enzymes of ADAR. Nonsense mutation (p.G1058*) in exon 12 may lead to a truncated ADAR protein.
Table 2.
Primers sequence of ADAR1
| Family | Ethnic grounp | Oneset time | Leision extremities | Mutation location | Nucleotide change | Protein change | Mutation type | Report or not | Predicted Mutation effects | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Polyphen 2.0 | SIFT | Provean | |||||||||
| 1 | Uyghur | 1 year | Extremities | Exon2 | c.497delA | p.Arg105fs | Frameshift | N | — | — | Neutral |
| 2 | Uyghur | 8month | Extremities | Exon12 | c.3352C>T | p.Gln1058* | Nonsense | N | — | — | Deleterious |
| 3 | Uyghur | 2 year | Extremities and ankles | Exon15 | c.3722delT | p.Ser1181fs | Frameshift | N | — | — | Neutral |
| 4 | Kazakh | 2 year | Extremities | Exon2 | c.1330A>G | p.Val332Met | Missense | N | Damage | Damage | Neutral |
| 5 | Kazakh | 1 year | Extremities | Exon8 | c.2702A>T | p.His841Leu | Missense | N | Damage | Damage | Deleterious |
| 6 | Hui | 4 year | Extremities | Exon2 | c.1176G>A | p.Lys326Glu | Missense | N | Damage | Damage | Neutral |
| 7 | Hui | 10 month | Extremities | Exon9 | c.2861G>A | p.Arg892His | Missense | N | Damage | Damage | Deleterious |
ADAR (GenBank: NC_000001.11, GRCh38.p13).
Figure 2.
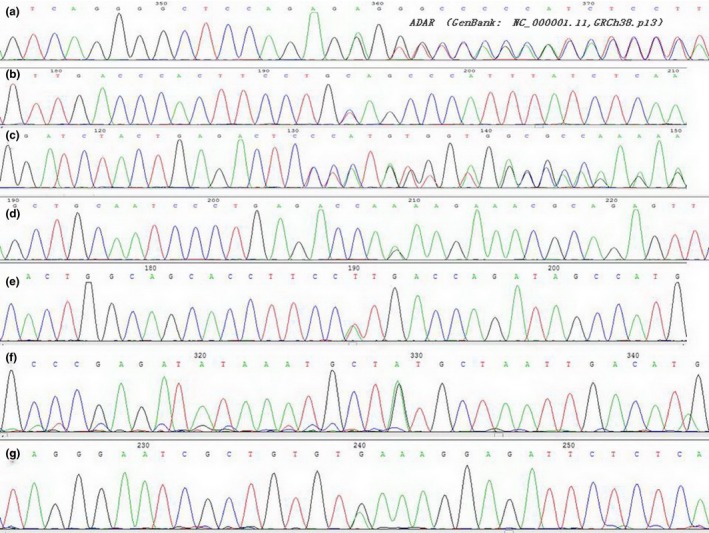
Mutations of the ADAR1 gene in multi‐ethnic pedigrees with DSH in China were seven novel mutations including four missense mutations (p.K326E, p.H841L, p.V332M and p.R892H), frameshift mutations (p.R105fs and p.S1181fs) and one nonsense mutation (p.G1058*) in seven pedigrees from different ethnicities
3.1. Bioinformatics analysis results
The seven novel mutations in this study is mostly damage by Polyphen‐2, SIFT and Provean software, located in conservative regions of ADAR (Table 2). ADAR sequence comparison among multispecies using UniProtKB showed mutation regions located in conservative regions (Figure 3).
Figure 3.

ADAR1 sequence comparison among multispecies using UniProtKB showed mutation regions located in conservative regions
3.2. Phenotype
Patients in this study had a typical mixture of hyperpigmented and hypopigmented macules on the dorsal aspect of hands and feet (Figure 4). Ages of onset were a few months and childhood. Patients in Family 3 had characteristic clinical features on the hands and feet and hyperpigmented and hypopigmented macules on the ankles. The patients in the same family had different degrees of skin lesions and the same patients’ degree of skin lesions was highest in summer and lowest in winter. No reports were found of any relationship between phenotypes and genotypes. In our study, the degree of skin lesions varied. Our data showed mostly lesions that were more severe after exposure to UV light. These results indicated that the environment may affect the phenotype.
Figure 4.
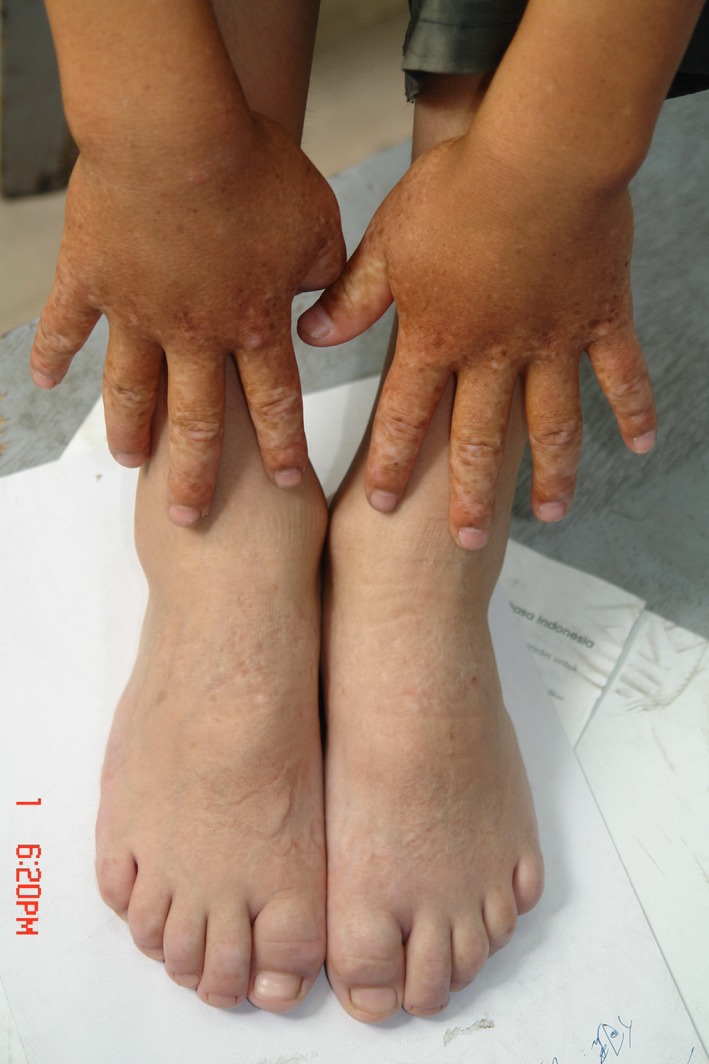
Patients in this study had a typical mixture of hyperpigmented and hypopigmented macules on the dorsal aspect of hands and feet
In the literature, 203 different mutations of the ADAR gene for DSH are reported (Figure 5), mostly concentrated in the Asian region of Japan, China and Taiwan. The mutations include 89 (43.84%) missense mutations, 20 (9.85%) splicing mutations, 36 (17.73%) nonsense mutations, 56 (27.59%) frameshift mutations, 1 (0.49%) synonymous mutations and 1 (0.49%) nonstop mutation. Among the 203 unique mutations, 2 (0.99%) are located in Zα, 7 (3.45%) in the Zβ domain, 14 (6.90%) in the dsRBDI domain, 6 (2.96%) in the dsRBDII domain, 11 (5.42%) in the dsRBDIII domain, 122 (60.10%) in deaminase domain of the ADAR protein, and 41 (20.20%) on the other domain.
Figure 5.
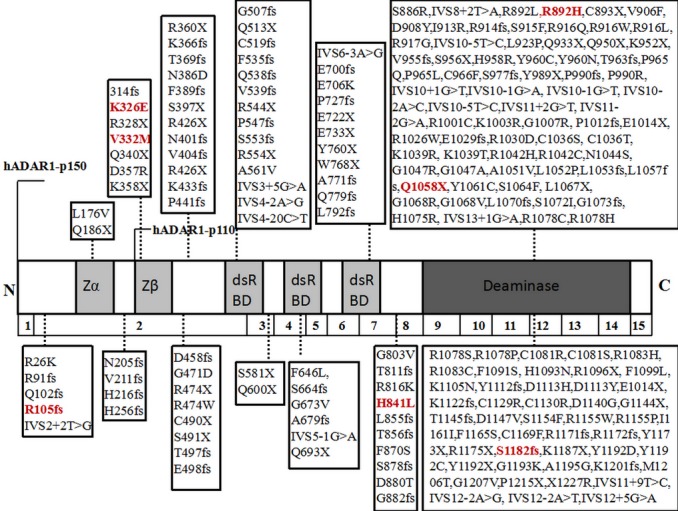
Two‐hundred and three different mutations of the ADAR1 gene for DSH
4. DISCUSSION
ADAR is also named double‐stranded RNA specific adenosine deaminase. It is important for RNA editing, mostly adenosine (A)‐to‐ inosine (I) RNA editing of post‐transcriptional modifications, modifying to A to I in pre‐mRNA. ADAR reduces double‐strandedness and amino acid substitutions, which may affect sequence information and RNA structure. ADAR contains six functional domains: two adenosine deaminase Z‐alpha domains (exon 2), three double‐stranded (ds) RNA‐binding domains (exons 2‐7) and a dsRNA adenosine deaminase domain (exons 9‐15). ADAR protein contains 1,226 amino acid residues, translating to 139 kDa molecular mass.
ADAR protein has a structure of two Z‐DNA‐binding motifs and three DRBMs affecting the efficiency of RNA editing. Translation of mRNA efficiency is influenced by DRBMs and the C‐terminal portion of the catalytic domain. Theoretically, mutations in specific regions such as DRBMs and the catalytic domain may have more severe clinical phenotypes than mutations in the two Z‐DNA‐binding domains.
ADAR is expressed as two isoforms. ADARp150 is an IFN‐inducible full‐length isoform 150‐kDa protein containing a methionine initiation codon for a 1,226 amino acid and an N‐terminal Za domain with a nuclear localization signal in the DRBMs. This protein is mainly distributed in the cytoplasm. ADARp110 is an N‐terminally truncated 110‐kDa protein. The AUG at codon 296 initiates the 931 amino acid protein containing one Za domain and three DRBMs and localizes in the nucleus. The Za domain is reported to be important in antiviral progress, but the clear mechanism is still unknown. The two isoforms have different promoters. The p150 and p110 proteins are expressed in different locations in cells: p150 is expressed in the nucleus and cytoplasm and p110 is expressed only in the nucleus.
In our study, we found seven novel mutations in different populations in China: c.1330A>G(p.Lys326Glu), c.2702A>T (p.His841Leu), c.1176G>A(p.Val332Met) and c.2861G>A(p.Arg892His) are missense mutations that may induce amino acid changes. Functional prediction software analysis showed damaging mutations in ADAR that may affect ADAR protein function by altering the activity of ADAR or interfering with the formation of ADAR homodimers. Four novel missense mutations were predicted to be damaging by both Polyphen 2.0 and SFIT. c.497delA(p.Arg105fs) and c.3722delT(p.Ser1181fs) may cause ADAR truncations lacking the important ADEAMc domain, while c.3352C>T(p.Gln1058*) may change the reading frame, causing ADAR truncations with partial ADEAMc domains and producing inactive ADAR enzymes. We presumed that mutant ADAR proteins causing disease may not all be based on nonsense‐mediated mRNA decay, which is surveilled by cells recognizing and degrading premature translation termination codons in mRNAs (Song et al., 2016). The clinical features and mutation analyses showed no clear relationship between them.
Mutation c.497delA(p.Arg105fs) in our study was upstream in the ADAR frameshift mutations. It created PTCs upstream of codon 296, which may affect normal p150 protein function, which is also located in the 3‐UTR region in the P110 protein and may affect P110 protein expression. Reports state that p.R91fs, p.Q102fs, p.N205fs, p.V211fs and p.H216fsX261 are in those regions and p.R91fs is a p150 protein transcript due to NMD (Consigli et al., 2010; Tang et al., 2018). Mutation c.497delA(p.Arg105fs) may have a loss‐of‐function effect, inducing haploinsufficiency as the mechanism of DSH dominant inheritance( Hayashi & Suzuki, 2013). Mutations c.1330A>G(p.Lys326Glu) and c.1176G>A(p.Val332Met), located on exon 2, are missense mutations, expressing the Zβ domain. P150 and p110 have this domain, which affected the function of these two isoforms.
To date, more than 200 mutations in ADAR have been reported among DSH patients, most of whom were in East Asia; Japan and China. Analysis of these mutations shows that most are located in the ADEAMc, suggesting the domain may be a hot spot of ADAR mutations among patients with DSH. Although Zα, DSRM, and ADEAMc are essential for the A‐to‐I modification activity, Zβ and interval areas are also important regions for the ADAR protein. Of mutations, R892H, Q1058X, and S1182fs on the ADEAM; K326E and V332M were in the Zβ domain; and R105fs and H841L were in the interval area of ADAR. This result may suggest that mutations in multi‐ethnic families may be consistent with previous reports. Missense mutations (43.84%) and frameshift mutations (27.59%) were more frequent than other mutation types, but had one synonymous mutation I1161I (Liu et al., 2006) and one nonstop mutation (X1227R) (Li et al., 2005; Luo et al., 2012). We predict that the p.X1227R mutation may lead to an open reading frame and a mutant‐type ADAR protein containing 1,247 amino acid residues. The region between Zα and Zβ has all frameshift mutations, with the translated area of p150 and regulation of p110.
The clinical features of DSH vary in different families. A few families have small freckle‐like pigmented and hypopigmented spots on the back of the feet and hands and on the face and knees. Some skin lesions have severe chilblains, blisters, and erosion. Although no exact relationship is known between genotype and phenotype, most patients show hypopigmented macules that are aggravated during the summer, suggesting that genotype is not the only factor to explain phenotype (Zhang et al., 2008). Viral infection during in utero or in childhood and exposure to UV light may also contribute to phenotype (Hayashi & Suzuki, 2013). Electron microscopy of pigmented lesions showed that glut melanin pigment deposited on the basal layer, with smaller and immature melanosomes in the cytoplasm. Electron microscopy of hypopigmented areas showed a decreased number of melanocytes and a large number of degraded cytoplasmic vacuoles that indicated apoptosis participated in melanocyte degeneration. ADAR gene mutation expression affects melanocyte function leading to clinical appearance (Tojo et al., 2006). Viral infection induced melanocytes to express ADAR mutations, so viral infection may be a factor in DSH. Zhang et al. used a minigene strategy and dual‐luciferase of ADAR c.271_272delAG to investigate the functionality of p150 and p110. Findings showed mutated p150 transcripts led to nonsense‐mediated mRNA decay, but p110 protein had no significant influence (Zhang et al., 2013). Expression of the p150 isoform induced by IFN accelerated translation. The level of p150 increased 2‐ to 3‐fold over basal expression when treated with IFN‐alpha, IFN‐beta or IFN‐gamma. UV light‐induced IFN‐gamma promoted melanocyte survival or immune evasion in mice (Zaidi et al., 2011). After exposure to UV light, macrophages infiltrated pups' skin and produced IFN‐gamma, by which melanocytes accelerated proliferation and migrated to the epidermis (Natarajan et al., 2014). The hypothesis is that the mutation type of p150 may participate in melanogenesis pathways affected by exposure to UV light and IFN‐gamma on the epidermis.
Various complications accompany DSH. Kondo and Tojo reported that two patients developed neurological symptoms, dystonia and brain calcification. The identified mutation was ADAR c.3019G>A(p.G1007R) (Kondo, Suzuki, Ito, et al., 2008; Tojo et al., 2006). No neurological or mental disease was found in another patient with ADAR c.3019G>A(p.G1007R). A Japanese girl with DSH with ADAR c.3444‐1G>A(p.Arg534X) developed brain calcification at 4 months of age (Suzuki et al., 2005). Developmental regression and torsion dystoration developed in patients with DSH, but genetic analysis was not performed. Reviewing the literature, we could not find a clear relationship in ADAR mutations with neurological disease. Other complications are psoriasis, limb hypertrophy, and depression, however, no significant correlations were reported. Our study did not find neurological symptoms or complications in patients.
In conclusion, seven novel mutations of the ADAR gene in multi‐ethnic pedigrees with DSH in China were analyzed for potential functions using software. Our research further enriched the ADAR gene database for DSH, contributing to our understanding of the mechanisms of DSH.
CONFLICT OF INTEREST
The authors have no conflict of interests to declare.
ACKNOWLEDGEMENT
All participants agreed on peripheral blood collection, approved by the Research Ethics Committee of China Medical University. All participants gave written informed consent. This study was supported by the National Natural Science Foundation of China (Grant No.81360235).
Wang P, Yu S, Liu J, Zhang D, Kang X. Seven novel mutations of ADAR in multi‐ethnic pedigrees with dyschromatosis symmetrica hereditaria in China. Mol Genet Genomic Med. 2019;7:e905 10.1002/mgg3.905
REFERENCES
- Consigli, J. , Zanni, M. S. , Ragazzini, L. , & Danielo, C. (2010). Dyschromatosis symmetrica hereditaria: Report of a sporadic case. International Journal of Dermatology, 49(8), 918–920. 10.1111/j.1365-4632.2010.04472.x [DOI] [PubMed] [Google Scholar]
- Hayashi, M. , & Suzuki, T. (2013). Dyschromatosis symmetrica hereditaria. The Journal of Dermatology, 40(5), 336–343. 10.1111/j.1346-8138.2012.01661.x [DOI] [PubMed] [Google Scholar]
- Kondo, T. , Suzuki, T. , Ito, S. , Kono, M. , Negoro, T. , & Tomita, Y. (2008). Dyschromatosis symmetrica hereditaria associated with neurological disorders. Journal of Dermatology, 35(10), 662–666. 10.1111/j.1346-8138.2008.00540.x [DOI] [PubMed] [Google Scholar]
- Kondo, T. , Suzuki, T. , Mitsuhashi, Y. , Ito, S. , Kono, M. , Komine, M. , … Tomita, Y. (2008). Six novel mutations of theADAR1 gene in patients with dyschromatosis symmetrica hereditaria: Histological observation and comparison of genotypes and clinicalphenotypes. Journal of Dermatology, 35(7), 395–406. 10.1111/j.1346-8138.2008.00493.x [DOI] [PubMed] [Google Scholar]
- Li, C. R. , Li, M. , Ma, H. J. , Luo, D. , Yang, L. J. , Wang, D. G. , … Zhu, W. Y. (2005). A new arginine substitution mutation of DSRAD gene in a Chinese family with dyschromatosis symmetrica hereditaria. Journal of Dermatological Science, 37(2), 95–99. 10.1016/j.jdermsci.2004.11.004 [DOI] [PubMed] [Google Scholar]
- Liu, Q. , Jiang, L. , Liu, W. L. , Kang, X. J. , Ao, Y. , Sun, M. , … Zhang, X. (2006). Two novel mutations and evidence for haploinsufficiency of the ADAR gene in dyschromatosis symmetrica hereditaria. British Journal of Dermatology, 154(4), 636–642. 10.1111/j.1365-2133.2006.07133.x [DOI] [PubMed] [Google Scholar]
- Luo, S. , Zheng, Y. , Ni, H. , Liu, Y. , Liu, Y. , Li, X. , & Liu, Q. (2012). Novel clinical and molecular findings in Chinese families with dyschromatosis symmetrica hereditaria. Journal of Dermatology, 39(6), 556–558. 10.1111/j.1346-8138.2011.01431.x [DOI] [PubMed] [Google Scholar]
- Miyamura, Y. , Suzuki, T. , Kono, M. , Inagaki, K. , Ito, S. , Suzuki, N. , & Tomita, Y. (2003). Mutations of the RNA‐specificadenosine deaminase gene (DSRAD) are involved indyschromatosis symmetrica hereditaria. American Journal of Human Genetics, 73(3), 693–699. 10.1086/378209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan, V. T. , Ganju, P. , Singh, A. , Vijayan, V. , Kirty, K. , Yadav, S. , … Gokhale, R. S. (2014). IFN‐γ signaling maintains skin pigmentation homeostasis through regulation of melanosome maturation. Proceedings of the National Academy of Sciences of the United States of America, 111(6), 2301–2306. 10.1073/pnas.1304988111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura, R. , Fukai, K. , Sugawara, K. , Tsuruta, D. , Okamura, K. , Hozumi, Y. , & Suzuki, T. (2017). Ultrastructural study of dyschromatosis symmetricahereditaria with widespread pigmentary eruption. Journal of Dermatology., 44(7), e150–e151. 10.1111/1346-8138.13849 [DOI] [PubMed] [Google Scholar]
- Patterson, J. B. , & Samuel, C. E. (1995). Expression and regulation byinterferon of a double‐stranded‐RNA‐specific adenosine deaminasefrom human cells: Evidence for two forms of the deaminase. Molecular and Cellular Biology, 15(10), 5376–5388. 10.1128/mcb.15.10.5376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, C. , Sakurai, M. , Shiromoto, Y. , & Nishikura, K. (2016). Functions of theRNA editing enzyme ADAR1 and their relevance to humandiseases. Genes, 7(12), 129– 10.3390/genes7120129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, N. , Suzuki, T. , Inagaki, K. , Ito, S. , Kono, M. , Fukai, K. , … Tomita, Y. (2005). Mutation analysis of the ADAR1 gene in dyschromatosis symmetrica hereditaria and genetic differentiation from both dyschromatosis universalis hereditaria and acropigmentatio reticularis. The Journal of Investigative Dermatology, 124(6), 1186–1192. 10.1111/j.0022-202X.2005.23732.x [DOI] [PubMed] [Google Scholar]
- Tang, Z.‐L. , Wang, S. , Chen, T. U. , Wang, T. , Ma, C.‐W. , Liu, Y. , … Xiao‐peng, W. (2018). Eight novel mutations of the ADAR1 gene in Chinese patients with dyschromatosis symmetrica hereditaria. Genetic Testing and Molecular Biomarkers, 22(2), 104–108. 10.1089/gtmb.2017.0207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojo, Kana , Sekijima, Yoshiki , Suzuki, Tamio , Suzuki, Noriyuki , Tomita, Yasushi , Yoshida, Kunihiro , Hashimoto, Takao , & Ikeda, Shu‐ichi (2006). Dystonia, mental deterioration, and dyschromatosis symmetrica hereditaria in a family withADAR1 mutation. Movement Disorders, 21(9), 1510–1513. 10.1002/mds.21011 [DOI] [PubMed] [Google Scholar]
- Tomita, Y. , & Suzuki, T. (2004). Genetics of pigmentary disorders. American Journal of Medical Genetics, 131C(1), 75–81. 10.1002/ajmg.c.30036 [DOI] [PubMed] [Google Scholar]
- Zaidi, M. R. , Davis, S. , Noonan, F. P. , Graff‐Cherry, C. , Hawley, T. S. , Walker, R. L. , … Merlino G. (2011). Interferon‐γ links ultraviolet radiation to melanomagenesis in mice. Nature, 469(7331), 548–553. 10.1038/nature09666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F. , Liu, H. , Jiang, D. , Tian, H. , Wang, C. , & Yu, L. (2008). Six novel mutations of the ADAR1 gene in Chinese patients with dyschromatosis symmetrica hereditaria. Journal of Dermatological Science, 50(2), 109–114. 10.1016/j.jdermsci.2007.11.011 [DOI] [PubMed] [Google Scholar]
- Zhang, G. , Shao, M. , Li, Z. , Gu, Y. , Du, X. , Wang, X. , & Li, M. (2016). Genetic spectrum of dyschromatosis symmetrica hereditaria in Chinese patients including a novel nonstop mutation in ADAR1 gene. BMC Medical Genetics, 17(1), 10.1186/s12881-015-0255-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. Y. , Chen, X. D. , Zhang, Z. , Wang, H. L. , Guo, L. , Liu, Y. , … Shao, F. M. (2013). The adenosine deaminase acting on RNA 1 p150 isoform is involved in the pathogenesis of dyschromatosis symmetrica hereditaria. British Journal of Dermatology, 169(3), 637–644. 10.1111/bjd.12401 [DOI] [PubMed] [Google Scholar]
- Zhang, X. J. , Gao, M. , Li, M. , Li, M. , Li, C. R. , Cui, Y. , … Huang, W. (2003). Identification of a locus for dyschromatosissymmetrica hereditaria at chromosome 1q11‐1q21. The Journal of Investigative Dermatology, 120(5), 776–780. 10.1046/j.1523-1747.2003.12130.x [DOI] [PubMed] [Google Scholar]