

Abstract
Background
Huntington disease (HD) is a devastating neurodegenerative autosomal dominant genetic condition. Predictive testing (PT) is available through a defined protocol for at‐risk individuals. We analyzed the over‐24‐years evolution of practices regarding PT for HD in a single center.
Methods
We gathered data from the files of all individuals seeking PT for HD in Lyon, France, from 1994 to 2017.
Results
448 out of 567 participants had exploitable data. Age at consultation dichotomized over 24 years toward an eightfold increase in individuals aged >55 (2/94 vs. 30/183; 2% to 16%; p < .0001) and twice as many individuals aged 18–20 (3/94 vs. 12/183; 3%–7%; p < .05). Motives for testing remained stable. The rate of withdrawal doubled over 24 years (9/94 vs. 38/183; 9%–21%; p < .02). Independently of the time period, less withdrawal was observed for married, accompanied, at 50% risk, and symptomatic individuals, and in those able to explicit the motives for testing or taking the test to inform their children. We also assessed the consistency between the presence of subtle symptoms compatible with HD found before the test by the team's neurologist, and the positivity of the molecular test. The concordance was 100% (17/17) for associated motor and cognitive signs, 87% (27/31) for isolated motor signs, and 70% (7/10) for isolated cognitive signs. Furthermore, 91% (20/22) of individuals who requested testing because they thought they had symptoms, were indeed found carriers.
Conclusion
This over‐24 years study underlines an increasing withdrawal from protocol and a dichotomization of participants’ age. We also show a strong concordance between symptoms perceived by the neurologist or by the patient, and the subsequent positivity of the predictive molecular test.
Keywords: genetics, Huntington disease, neurogenetics, predictive testing, presymptomatic testing
1. INTRODUCTION
Huntington disease (HD) is a devastating neurodegenerative disorder, typically presenting as a recognizable association of choreiform movements, behavioral manifestations, and cognitive impairment (Bates et al., 2015). After an insidious prodromal phase of usually more than 10 years (Liu et al., 2015; Ross et al., 2014), the manifest phase begins at a variable age, typically around 45 years (Bates et al., 2015). Patients then suffer from a progressive and ultimately fatal loss of motor and cognitive skills, with an average survival time of 18 years after onset (Bates et al., 2015). While there is neither curative nor preventive treatment, symptomatic pharmacological therapy along with supportive care, has proven helpful (Bates et al., 2015).
HD is a genetic disease, corresponding to an unstable expansion of polyglutamine (CAG) triplets in exon 1 of the Huntingtin (HTT,MIM:613004) gene (Macdonald, 1993). The length of CAG repeats determines the occurrence of the disease: ≥40 repeats is a fully penetrant HD‐causing allele, while having <26 repeats is non‐pathogenic (i.e., normal). Alleles between these two values are infrequent (Duyao et al., 1993; Lee et al., 2012; Alicia Semaka et al., 2013). Alleles with 36–39 repeats are pathogenic with a reduced penetrance (RP) (Kay et al., 2016; Quarrell et al., 2006). Alleles with 26–35 repeats are called intermediate alleles: while not disease‐causing, they may expand during the gametogenesis (Wheeler et al., 2007), especially during spermatogenesis, and lead to the transmission of a pathogenic allele to the offspring (Semaka & Hayden, 2014; Semaka et al., 2013). The repeats length is by far the leading predictor of the variance of age at onset, with longer CAG repeats responsible for earlier onset (Aziz, van der Burg, Tabrizi, & Landwehrmeyer, 2018; Duyao et al., 1993; Keum et al., 2016; Lee et al., 2012; Sun, Zhang, & Wu, 2017). The remaining variance is thought to be roughly equally shared by other genetic factors and by environment (Gem‐HD, 2015; Hensman Moss et al., 2017; Pinto et al., 2013).
The transmission of HD is autosomal dominant: mono‐allelic (heterozygous) carriers of a fully penetrant pathogenic allele have a 50% theoretical risk of having an affected child. The normal‐size allele in trans does not seem to participate to the disease trajectory (Keum et al., 2016). Bi‐allelic carriers of a pathogenic allele show a more rapid disease progression, but not an earlier onset (Lee et al., 2012; Squitieri et al., 2003).
Predictive testing (PT) of HD has been available for asymptomatic at‐risk‐individuals since 1986 by family linkage analysis, and since 1993 by direct study of the HTT gene (Nance, 2017). It follows a multi‐step protocol based on international recommendations from both the medical community and patient's associations, updated in 2013 (MacLeod et al., 2013; Nance, 2017). This protocol aims at protecting the participant against an unfavorable result, by giving information about the disease and the social consequences of the test and helping him prepare for the future. This protocol puts emphasis on the liberty of choice for the patient and the possibility to opt‐out of the protocol at any step, and to re‐enter the protocol.
Multiple centers offer PT for HD in France, evenly geographically distributed. Our center has been offering PT for neurogenetic diseases since 1993, and recruits individuals from an approximately 100 km radius. The multidisciplinary team composition has been relatively stable over the years, with currently a geneticist, a clinical psychologist, a psychiatrist, a nurse, a neurologist, and a molecular biologist. Our protocol consists in a minimum of four consultations over at least three months before the test, and three recommended follow‐up consultations.
In this study, we review the main data from 16 previously published cohorts, to summarize the average expected global outcome of PT for HD nowadays, then take advantage of our unique 24‐years’ experience to describe the evolution of practices in a stable setup. We assess multiple aspects of PT, notably the modifications of the age of participants, the reasons for and evolution of withdrawal, the age‐motive patterns, the effects of social environment, and the relationship between symptoms perceived before the test and actual genetic status.
2. METHODS
2.1. Editorial Policies and Ethical Considerations
This observational study was ethically approved by a committee of healthcare practitioners from our University Hospital.
2.2. Inclusion and exclusion criteria
Inclusion criteria were as follows: every person seeking PT for Huntington disease at our local center of Lyon, France, between 1994 and 2017 included. Exclusion criteria: persons seeking only information about the disease, minors, individuals not at risk.
2.3. Parameters studied
Data were collected anonymously and retrospectively from each patient's files by a single author and transcribed into a computer file for analysis (Excel, Microsoft). The following parameters were queried for each individual: gender, domiciliation (postal code), occupational category according to the French national statistics institute standard (INSEE), age at first consultation, duration of the protocol defined by the delay between first consultation and the consultation for results, relationship, number of children, gender of the affected or carrier parent, number of known affected relatives (alive or deceased), motive for testing defined as the single main motive for seeking PT reported by the individual based on data from the medical and psychological files, presence of motor and cognitive symptoms compatible with HD as evaluated by an expert neurologist, nature of the accompanying person during the protocol, theoretical risk of being carrier according to family history, result of the molecular test (lengths of both allele), and reasons for not completing the protocol (assigned to “loss to follow‐up” after 3 years of absence of contact). Frequencies were compared according to χ 2 test, with a significance level set at .05. For clarity purpose, most percentages are rounded to the closest integer.
2.4. Molecular analysis
Molecular analysis was performed in the same laboratory at Lyon University Hospital for all patients using two specific Polymerase Chain Reactions according to protocols described by Warner et al. and McDonald et al.(Macdonald, 1993; Warner, Barron, & Brock, 1993). An expansion was defined as an increase of CAG repeats by ≥2 from parent to child, and a contraction as a decrease of ≥2 repeats.
3. RESULTS
3.1. Global results and demographics
Out of the 567 individuals who had asked PT for HD between 1994 and 2017, 119 were excluded, and 448 were analyzed (see Methods). The median age of the participants was 35 years‐old (Table 1). Individuals were predominantly females (282; 63%), involved in a stable relationship (336; 75%), already having children (242; 54%), and when known, having a maternal rather than paternal ancestor history (216 vs. 158; ratio 1.4). 399 (89%) were at 50% risk of being carrier, while the remaining were mostly at 25% risk. The gender bias toward females did not significantly increase over 24 years (53/94, 57% in 1994–2001; vs. 123/183, 67% in 2010–2017; p = .09, Supplementary Figure S1). There was an overrepresentation of individuals living close to our center (37% of cohort, vs. an expected 15% from demographic data, i.e. 2.4‐fold more than expected; p < 10e‐5, Supplementary Figure S2), an overrepresentation of upper occupational classes (x1.9; p < .001) and of middle classes (x1.2–1.7; p < .01) compared other occupational categories, and an under‐representation (x0.25; p < .0001) of retired individuals (not shown). The observed duration of the protocol (6 month mean), remained unchanged over the years.
Table 1.
Main characteristics of published cohorts of presymptomatic testing for Huntington disease
| Cohorts of presymtomatic testing participants | Cohorts of presymptomatic testees | Average of all cohortsh | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| First author | Present study | Holman | Mandich | Scuffham | Krukenberg | Wedderburn | Sizer | Dufrasne | Bernhart | Alonso | Trembath | Goizet | Baig | Panas | Peterlin | Tassicker | Creighton | – |
| Publication year | 2019 | 2018 | 2016 | 2014 | 2013 | 2013 | 2012 | 2011 | 2009 | 2009 | 2006 | 2002 | 2016 | 2011 | 2009 | 2006 | 2003 | – |
| Country | France | USA | Italy | Australia | USA | Australia | South Africa | Canada | Germany | Mexico | Australia | France | UK | Greece | Slovenia | Australia | Canada | – |
| Center | Lyon | Multicentric | Genoa | Queensland | Indianapolis | Western | Johannesburg | Montreal | Bochum | Mexico City | Victoria | Multicentric | Whole country | Athens | Ljubljana | Queensland | Multicentric | – |
| Studied period | 1994–2017 | 1996–2014 | 1993–2014 | 2006–2010 | 1990–2000 | 1993–2012 | 1998–2006 | 1994–2008 | 1993–2004 | 1995–2007 | 1989–2004 | 1994–2000 | 1993–2014 | 1995–2008 | 1997–2007 | 1994–2003 | 1987–2000 | – |
| Study duration | 24 year | 4 yearsa | 22 years | 5 years | 11 years | 20 years | 9 years | 15 years | 11 years | 13 years | 15 years | 7 years | 21 years | 14 years | 11 years | 10 years | 14 years | – |
| Applicants (N) | 448 | 135 | 299 | 152 | 141b | 466 | 57 | 181 | 478 | 75 | 756 | 712 | 9,407 | 256 | 68 | 2036 | 1,061 | – |
| Average age | 35 | 34 | 35 | 39 | 34 | 49 | 30 | 36 | 35 | 34 | 40 | 34 | 37 | 34 | 33 | – | 39 | 36 |
| Gender (Women %) | 63% | 55% | 55% | 54% | 65% | 58% | 67% | 57% | 57% | 63% | 58% | 63% | 56% | 55% | 54% | – | 60% | 59% |
| Stable relationship (%) | 75% | 70% | 66% | 70% | 61% | – | – | 70% | 74% | 43% | – | 69% | – | – | – | – | – | 66% |
| Have children | 54% | 47% | 41% | 59% | 54% | – | 44% | 57% | 44% | 48% | 67% | 53% | – | – | – | – | – | 52% |
| At 50% risk (%) | 89% | 90% | 85% | 82% | – | – | – | 99% | 92% | 100% | 89% | 100%c | 90% | – | – | 94% | 89% | 91% |
| marternal/paternal history (ratio) | 1.4 | – | 1.2 | 1.3 | 1.2 | – | – | – | – | – | 1.5 | 1.3 | – | 2.2 | – | – | – | 1.4 |
| Protocol completed (%) | 81% | – | 60% | 63% | – | 61%d | 67% | 75% | 52% | 88% | 86% | 57% | 100%f | 100f | 100%f | 100%f | 100%f | i70% |
| Pathogenic test result (% at CAG ≥ 36) | 40% | 42% | 38% | 55% | – | 37% | ≥ 37%e | 42% | – | 38% | 38% | 41% | 46% | 48% | 43% | 41% | 45% | 42% |
| Reduced penetrance alleles (%) | 0.8% | – | – | 3.0% | – | – | – | 2.2% | – | 2.6% | 3.2% | – | g4.5% | – | 7% | 2.9% | – | 3.60% |
| Median time of protocol (months) | 6 | – | 1.5 | 4.5 | – | – | – | 3 | >1 | – | – | – | – | – | – | – | – | 3.8 |
| Symptomatic applicants | 16% | – | 11% | – | – | – | – | – | – | – | – | 10% | – | – | – | – | – | 12% |
Demographics and principle results of present cohort, compared to previous cohorts of >50 individuals published since 2001, are presented. The center of the table is divided in two large result panels: cohorts of individuals seeking presymptomatic diagnosis for Huntington disease (left center panel), and cohorts of individuals who had a presymptomatic molecular testing for Huntington disease (right center panel). The rightmost panel indicates the average results calculated from all cohorts reported in this table, not ponderated according to the number of individuals in each cohort. For clarity, percentages have been approximated to the closest percent, except for the proportion of reduced penetrance allele.
Four time‐points of one year each.
Answering to survey.
Only at‐50% risk individuals were included.
Of PT for HD and (marginally) for other neurodegenerative diseases.
Apparently restricted to ≥40 CAG values.
Defined by inclusion criteria (only tested individuals).
From 2010 to 2014 data only.
Not ponderated by N.
Exluding data from testees‐only cohorts.
3.2. Withdrawal from predictive testing: evolution and risk factors
364 out of 448 (81%) individuals completed the protocol, and 85 (19%) did not. Of those who did not complete the protocol, 69 (15.5% of cohort) withdrew, 11 (2.5%) were dissuaded from being tested for medical or psychosocial reasons, and 4 (1%) were oriented to symptomatic testing because they needed immediate care (Figure 1a). Over 24 years analysis revealed a progressive and significant ≥2‐fold increase in the global rate of withdrawal, from 9% in 1994–2001 to 21% in 2010–2017 (corresponding to 9/94 and 38/183 respectively; p < .02, Figure 1b), while the number of participants and the number of tests increased over the same period (Supplementary Figure S3). Reasons for withdrawal were varied (Figure 1c), with a predominance of loss to follow‐up (40% of the 85 individuals that did not complete the protocol). 30 individuals reentered the protocol after a withdrawal, and all of them except one completed the protocol (not shown).
Figure 1.
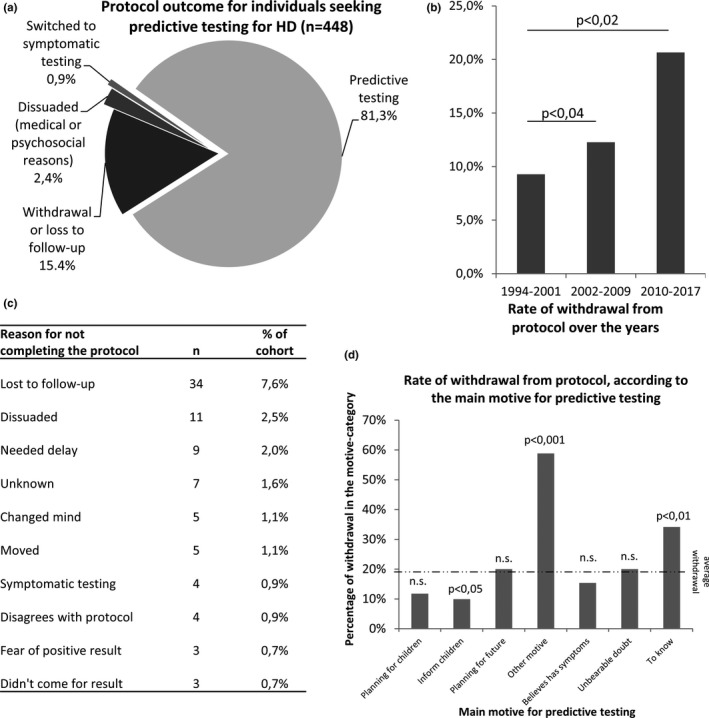
Global outcomes of protocol and predictive testing in our cohort. (a) Protocol outcome for individuals seeking predictive testing for Huntington disease (n = 448). (b) Global rate of withdrawal from protocol, in three time‐periods of 8 years each. (c) Disclaimed or default (loss to follow‐up) reasons for not completing the protocol, listed from the most to the least frequent. (d) Rate of withdrawal from the predictive testing protocol according to the main motive for predictive testing. Dotted lines indicate the overall withdrawal rate in the cohort. Statistical significance is indicated above histograms as a p‐value, in (a) and (c). n.s. = not statistically significant
The rate of withdrawal varied depending on the motives for PT (Figure 1d). Compared to the average 15.5% in our cohort, the withdrawal rate was increased by threefold for participants that were unable to give a reason for testing (59%, 10/17; p < .001) and by twofold for those who had no other motive than the desire to know their status (34%, 14/41; p < .01). On the contrary, individuals seeking PT to inform their children of their risk, were those with the lowest rate of withdrawal (10%, 10/101; p < .05).
Withdrawal rate was also influenced by social environment (Supplementary Figure S4). Married individuals, but surprisingly not individuals involved in other stable relationships, were about twice less prone to quit the protocol compared to other relationship status (11% vs. 23%, 22/196 vs. 54/238; p < .0001; Supplementary Figure S4a). Individuals following the protocol without the help of an accompanying person, opted‐out twice as frequently as accompanied individuals (30% vs. 16%, p < .01; Supplementary Figure S4b). Interestingly, those who were accompanied by a friend (18 participants), never withdrew from the protocol, but this did not reach statistical significance (p < .055) (Supplementary Figure S4b).
Withdrawal rate was also significantly modified by the theoretical risk of being carrier: withdrawal was twice as frequent in individuals at 25%‐risk compared to those at 50%‐risk (28% vs. 14%, p < .02, data not shown).
Furthermore, withdrawal was decreased by about 3‐fold in individuals showing both motor and cognitive signs compatible with HD after neurological evaluation, compared to asymptomatic individuals (5% vs. 15%; p < .05, data not shown), but unmodified in individuals seeking PT because they believed they were symptomatic (Figure 1d).
Finally, the withdrawal rate was not significantly modified by the gender of the participant (male 17%, female 14%, p > .05), occupational category, kindred (18% for childless individuals, 13% for individuals with children), sex of the affected ancestor, and age of the participant (16% for 18–20 years‐old; 18% for 20–35; 12% for 35–50; and 14% for ≥ 51 years‐old, p > .05) (data not shown).
3.3. Molecular test outcome
Of the 364 individuals who had a PT for HD, 198 (54%) had a strictly normal/negative result (<27 CAG repeats), 146 (40%) had a full‐penetrance HD causing allele (>39 CAG), 5% had an intermediate allele (26–35 CAG), and only 3 (0.8%) had a reduced‐penetrance allele (36–39 CAG) (Supplementary Figure S5a). As expected from the unstable status of intermediate alleles, the distribution of CAG repeats length on the longest allele was uneven, with few individuals carrying intermediate or reduced‐penetrance alleles (Supplementary Figure S5b, dark dots). The clear majority (142 out of 146, ≥97%) of carriers had ≤50 repeats, with only 4 individuals above this threshold. The shortest allele (Supplementary Figure S5b) had an ordinary distribution, with only 6 individuals being carriers of a >26 repeats on this allele: 5 with intermediate alleles, and 1 with a RP allele. No carrier of two fully‐penetrant disease‐causing alleles was found in our cohort. As expected from autosomal dominant transmission, individuals at 25%‐risk had more negative results (73%) than those at 50%‐risk (53%) (Not shown).
3.4. Anticipation phenomenon
We had access to the affected ancestor's genotype for only 58 out of 149 (39%) individuals found carriers, allowing a limited analysis of the anticipation phenomenon in our cohort. 38 of these 58 individuals (65%) had inherited from an allele of the same size of their parent's, 16 (28%) had a small expansion ranging from +2 to +4 CAG units, and 4 (7%) had a small contraction ranging from −2 to −3 units (not shown). No larger amplification or contraction was found. Expansions were twice as frequent when the affected parent was a male (18% vs. 10%; p = .05).
3.5. Evolution of the age of participants
The over 24 years median age of participants has been stable (Figure 2a, dotted line) around 34 years old. However, we found a progressive and significant dichotomization of the age of participants (Figure 2b–e): the proportion of very young participants (18–20 years old) increased by twofold in the latest years compared to earliest (3% in 1997–2001 (3/94) and 7% in 2010–2017 (12/183); p < .05), while the proportion of older participants, aged >55, increased by eightfold (2% in 1994–2001 (2/94) and 16% in 2010–2017 (30/183); p < .001).
Figure 2.
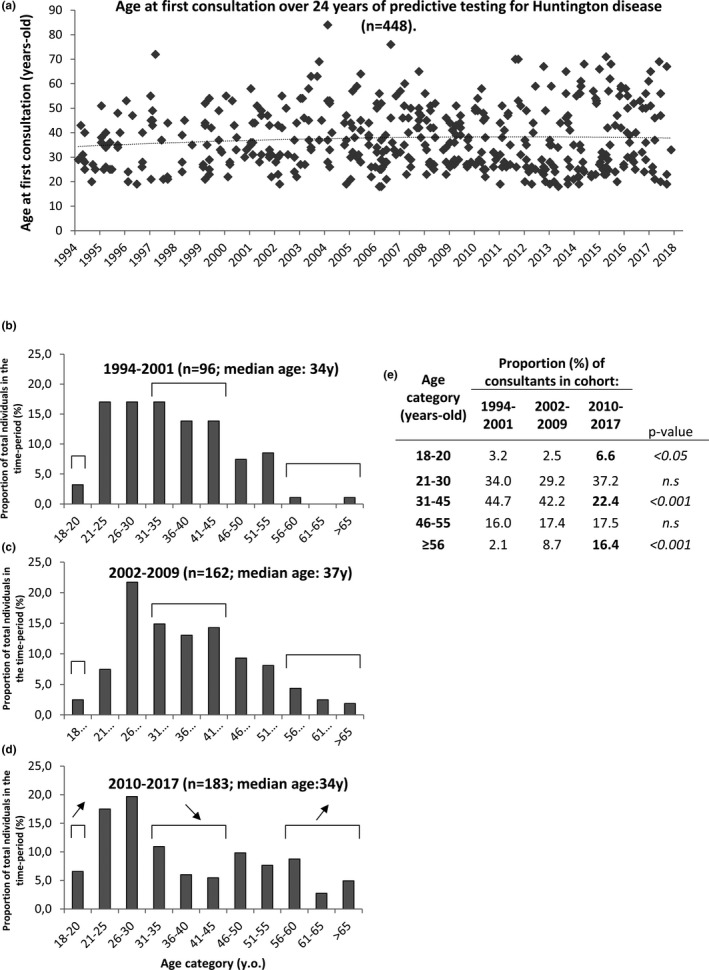
Evolution of the age of participants for predictive testing of Huntington disease, over 24 years (1994–2017). (a) Age of participants over 24 years. Each point represents a single individual. Dashed line corresponds to mean age of participants. (b–d): repartition of participants by age categories, at three time‐periods of 8 years each: 1994–2001 (b), 2002–2009 (c), 2010–2017 (d). Total number of individuals (n) and median age are indicated in title for each time period. Significant changes between time‐periods are indicated by brackets and up‐arrows (increase in last time‐period) or down‐arrow (decrease in last time period). (e): same data as in (b–d), shown in a table. Indicated p‐values account for the comparison between the last time period (2010–2017) versus the 2 other periods. n.s = not statistically significant. PT = predictive testing
3.6. Motives for testing
Participants usually had several motives for seeking PT for HD. To get a clearer picture, we decided to select the single main motive of each individual (Figure 3a). Three major main motives emerged: 27% planning for children (i.e., individuals planning for a pregnancy with potentially a prenatal diagnosis if found carrier), 25% planning for future (i.e., individuals that would organize their lifestyle according to their genetic status), and 23% wanting to inform their children of their risk (i.e., individuals taking the test not mainly for themselves, but rather for the life decisions of their children). These motives remained very stable over 24 years (Figure 3b).
Figure 3.
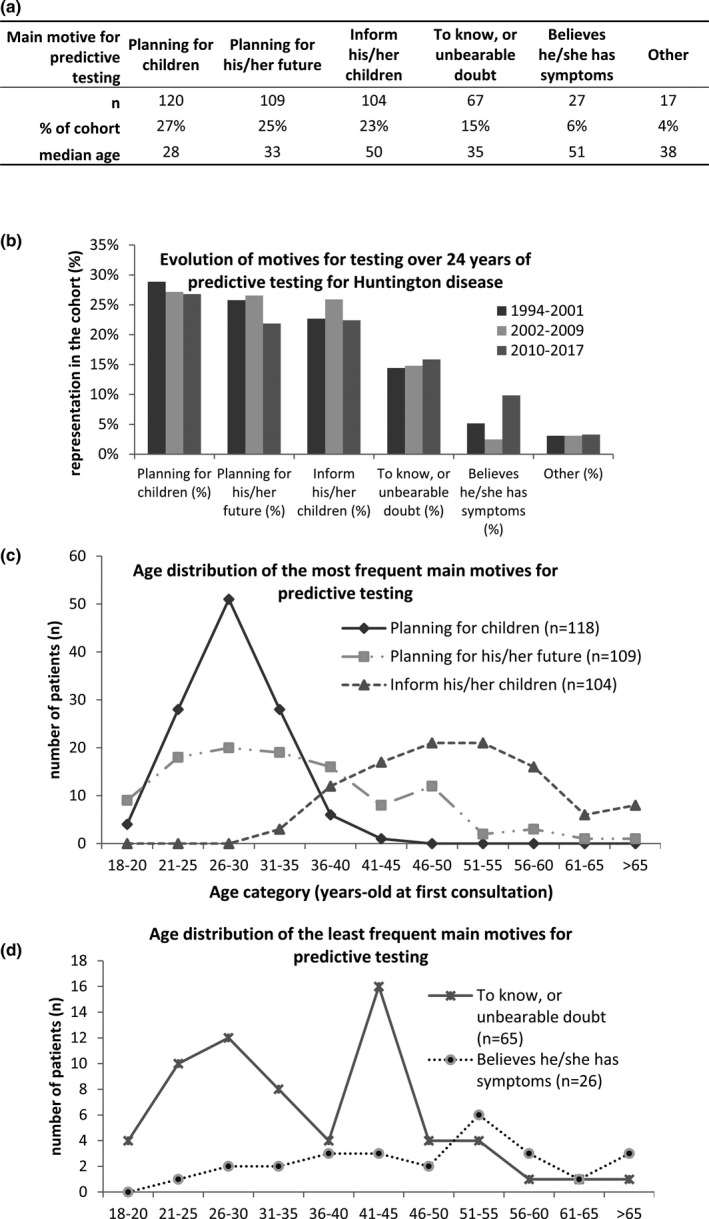
Age‐dependent motives for predictive testing, and evolution of motives over 24 years of predictive testing for Huntington disease. (a) Overall repartition of main motives for predictive testing in the cohort. (b) Over‐24‐years evolution of main motives for predictive testing. (c–d). Age‐dependent distribution of the three most frequents (c) and of the two least frequent (d) main motives for testing
Strikingly, each main motive for PT had a precise pattern of age‐distribution (Figure 3c,d). There was a single‐peak distribution around 28 years old for individuals planning for children, and around 50 years old for those who wanted to inform their children of their risk. There was a bifid distribution of those who wanted to organize their life, with two peaks at 26–30 and 46–50 years old. Interestingly, those who only “wanted to know” or had an unbearable doubt about their status, had a frequency peak at 26 years old and another at 41–45 years old, the latter corresponding to the average age‐at‐onset (Figure 2c). Those believing they had symptoms were distributed among all ages, with an increase in older individuals (Figure 2d). The motives were not significantly gender‐dependent, except for planning for children (females 31%, males 20%, p < .03, not shown).
3.7. Rate of pathogenic results according to perceived symptoms before test
We lastly focused on the relationship between the symptoms of HD perceived either by the participant or by the team before the test, and the actual genetic status found after molecular testing. Of the 448 persons seeking PT for HD, 72 (16%) were found symptomatic after examination by our neurologist (Figure 4a), of which 4 were switched to diagnostic testing and 68 kept in the predictive protocol. The signs were categorized either in motor or cognitive (including psychiatric) categories, regardless of their intensity. Every (17/17, 100%) tested individual showing both motor and cognitive symptoms, was found carrier of a fully penetrant HD causing allele, while 27/31 (87%) of those exhibiting isolated motors symptoms and 7/10 (70%) of those with isolated cognitive symptoms (Figure 4a, bottom panel) were found positive.
Figure 4.
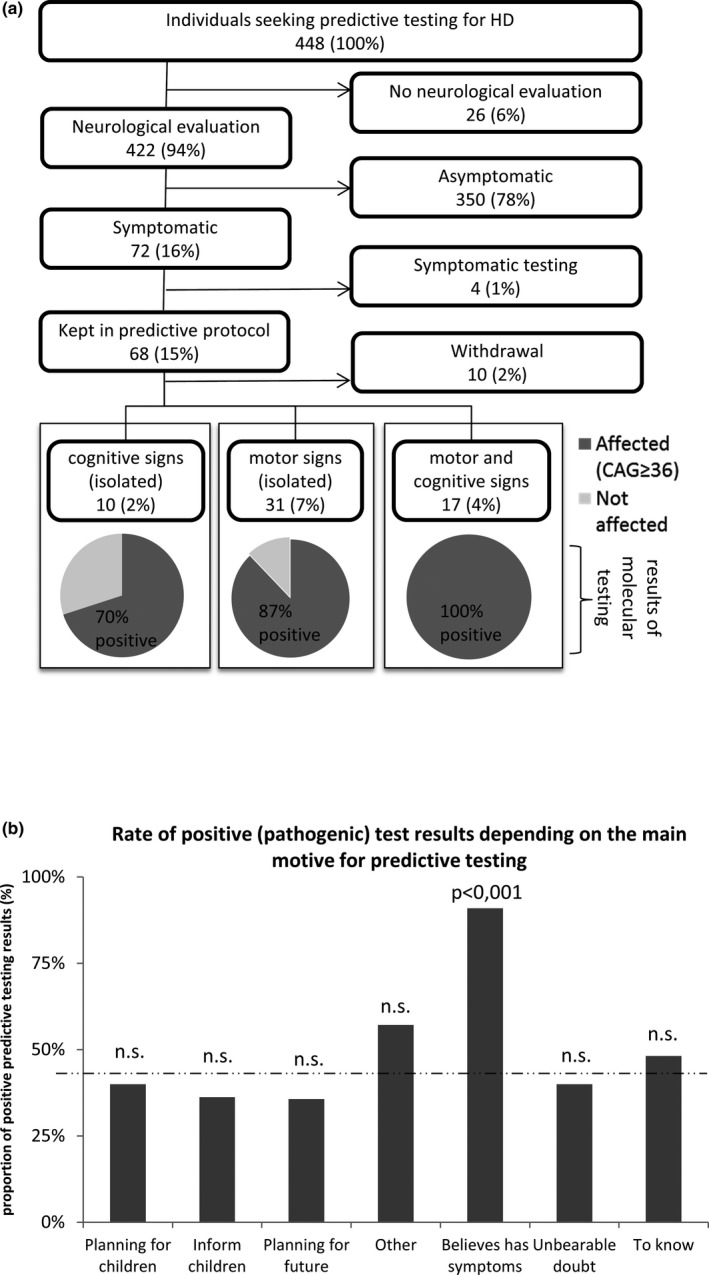
Outcomes of predictive molecular testing for Huntington disease (HD), in suspected symptomatic patients. (a) Analytic flow‐chart of patients found symptomatic after examination by a trained neurologist (motor and cognitive signs compatible with the diagnosis of manifest HD). Number of individuals in each category is indicated in boxes, along with the absolute percentage of these individuals in the cohort. The bottom panel shows pie charts of the molecular results obtained after predictive testing, with dark gray and light gray areas corresponding to affected (pathogenic allele, ≥36 CAG repeats) and unaffected individuals, respectively. Percentages indicated on pie charts refer to the proportion of positive and negative results in each category of symptoms: isolated cognitive signs, isolated motor signs, and combined motor and cognitive signs. (B) Proportion of positive (i.e., pathogenic) results from predictive testing, depending on the main motive for testing. Statistical significance is indicated above histograms as a p‐value. n.s. = not statistically significant. Dotted lines indicate the overall mean value of y‐axis parameter
On the “patient side,” out of the 22 individuals who entered the protocol because they felt they had symptoms of HD and then had a molecular testing, 20 (91%) were indeed found carrier of pathogenic allele, and 2 (9%) were found negative (Figure 4b).
4. DISCUSSION
In this study, reporting 448 individuals requesting PT for HD, we establish a detailed profile of the participants and follow its evolution over 24 years. The protocol for PT remained stable, setting‐up satisfactory conditions for studying the evolution of the nature of the participants and their decisions. We analyze multiple parameters, and more specifically the causes for withdrawal, the motives for testing, and the relationship between the signs perceived before the test and the actual status after the molecular test.
4.1. Review of previous cohorts
Table 1 shows that most of the characteristics of previously published cohorts are remarkably conserved between countries and time periods, including the age of participants, the gender bias towards females, overrepresentation of maternal history, and the lower‐than‐50% rate of pathogenic results. This is probably due in part to the strict application of international recommendations for this PT, and to the natural history of the disease in families. However, we observe notable differences regarding the rates of protocol completion and the median time of protocol. Interestingly, the shortest protocol durations seems to correlate with the highest rate of withdrawal. This suggests that the duration of the protocol could influence the decision of a participant to pursue or cancel its undertaking. Some participant might opt‐out because they do not have enough time to prepare for the result. Further studies would be required, since the liberty of choice is of utmost importance in this context. Our cohort is comparable to previously reported cohorts in most aspects (Bernhardt, Schwan, Kraus, Epplen, & Kunstmann, 2009; Creighton et al., 2003; Dufrasne, Roy, Galvez, & Rosenblatt, 2011; Goizet, Lesca, Durr, & French Group for Presymptomic Testing in Neurogenetic Disorders, 2002; Panas et al., 2011; Peterlin, Kobal, Teran, Flisar, & Lovrečić, 2009; Trembath et al., 2006), expect for a moderately high completion rate, a long duration of protocol, and a lower frequency of RP alleles, discussed below.
4.2. Withdrawal from protocol: causes and evolution
In our study, the rate of withdrawal was lower when the individual was: married; accompanied during the protocol, irrespective of the type of accompanying person; had explicit motives for PT; and had a prior risk of 50% at birth. These findings confirm those form previous observations (Bernhardt et al., 2009; Krukenberg, Koller, Weaver, Dickerson, & Quaid, 2013; Mandich et al., 2017; Scuffham & MacMillan, 2014), and underline the importance of social environment and motivations of a participant when faced to such a difficult process. Interestingly, we did not observe a single withdrawal from participants being accompanied by a friend (0%, 0/18), but this did not reach statistical significance probably because of small numbers of individuals in this category. If confirmed, this finding could imply that friends accompanying the participants are either very supportive, or might compromise the freedom of choice of the participant.
One of the most salient results from our study is the gradually increasing rate of withdrawal, which was multiplied by two in 24 years. This phenomenon in not unheard of, since Bernhardt et al. (2009) showed a progressive decrease in test uptake from 1993 to 2004 in a single center in Germany, and hypothesized that this could be partly related to the influence of team members on the decision making of participants. In our cohort, this influence cannot be ruled out, but is unlikely: the gradual increase in withdrawal does not fit the timing of the limited changes that took place in the team. This might rather be explained by some changes in the society, for example, regarding the changes in the prevalence of factors known to affect the withdrawal rate. Indeed, there was significantly less marriage in 2010–2017 compared to 1994–2001 in our cohort (35% vs. 51% respectively, p < .04), and we've shown that being married is associated with a notably higher completion rate of the protocol. There were also more unaccompanied individuals in the later years, although not reaching statistical significance (26% in 2010–2017% vs. 17% in 1994–2000, p = .09), and we've shown that unaccompanied individuals during protocol, opt‐out more frequently.
We also show that symptomatic individuals showing isolated motor or associated motor and cognitive signs, were notably less prone to quit the protocol (3‐fold, p < .05), despite their unawareness of their symptoms. To our knowledge, this was not reported before. It might be explained by a higher pressure applied to the participant by his relatives, when they feel that he is symptomatic. It might also reflect an involuntary bias from the team members to suggest the participant completes the protocol, or even originate from the participant's subconscious mind.
Overall, the withdrawal rate in our cohort is moderately low compared to previous cohorts (Table 1). This difference could be related to the long duration of our protocol, as discussed above, with a very stable 6 months median duration compared to 1–4.5 months in other cohorts. However, only limited date is available on this subject, and more specific investigations would be required to conclude. This relatively low withdrawal rate could also result from the intrinsic characteristics of the population, however this is not probable since in the only other French cohort published so far (Goizet et al., 2002), the withdrawal was notably more prevalent than in ours. The long duration of our study could by itself have slightly increased the observed completion rate, since 30 individuals out of 448 re‐entered the protocol after several years.
While there were various reasons for not completing the protocol (Figure 1c), the loss‐to‐follow up was the most prevalent (34/85 uncompleted protocols). It would be interesting to understand the actual reasons behind this loss‐to follow‐up, but by definition is it not accessible to direct assessment. Our analysis of this subgroup was limited by the low number of individuals. No specific risk factor was identified, including geographical distance and gender. A much larger cohort is probably required to conclude on this aspect.
4.3. Evolution of age at consultation
We show a significant dichotomization of the age of individuals seeking PT for HD, with a twofold increase in very young participants (aged 18–20) and an eightfold increase in participants aged >55. Similarly, the UK consortium showed an excess in younger individuals having had a PT for HD in the later years (2010–2014) compared to earlier years (1994–1998) (Baig et al., 2016). A small over‐time decrease in the age of participants was also reported between 1996 and 2014 from 7 testing centers in USA (Holman et al., 2018). There are now multiple lines of arguments pointing toward a moderate and gradual increase in younger adults requesting PT for HD, and this data should be integrated in our practice in PT centers. This might reflect the evolution of global cultural and social practices, with the younger requesting more immediate information.
Regarding the proportion of older participants, more contrasted findings have be reported, with a gradually lower proportion of older individuals taking the test in the United Kingdom (Baig et al., 2016). This might be explained by cultural differences between UK and France since the authors also observe a notable decrease in the number of tests over the years, while we observe the opposite. Also, the UK cohort included testees only, and not the participants who withdrew from the testing protocol. While there is no indication that this methodological difference could affect the age of individuals, this cannot be ruled out. In our cohort, the increasing proportion of older participants could be related to the increasing knowledge about the disease, including the existence of late‐onset HD that was probably less obvious 20 years ago. The varying age of participant cannot be explained by an evolution their motives for testing, since they are remarkably stable over time. Our approach based on age‐distribution rather than median‐age allowed a more detailed characterization, hopefully providing more significant data.
4.4. Reduced penetrance alleles
The RP alleles are very infrequent in our cohort (0.8%), compared to 3.6% in average in other cohorts (Table 1). We do not know if this is a peculiarity of the French population because the only other cohort from the same country did not report the frequency of RP alleles (Goizet et al., 2002). The majority of previously reported frequencies of RP alleles in PT cohorts originate from Anglo‐Saxon populations (Baig et al., 2016; Dufrasne et al., 2011; Scuffham & MacMillan, 2014; Trembath et al., 2006), and one from Mexico (Alonso et al., 2009). Thus, the ancestry might explain a difference between these allele frequencies. The unusually low frequency of RP alleles reported in our study is closer to the frequency reported in the general population of British‐Columbia (Semaka et al., 2013) (0.4%), but higher than in the general population of Portugal (Sequeiros et al., 2010) (0.1%). It is unlikely that this low frequency of RP alleles is related to randomness, since we report a large cohort. We describe the length of both the largest and the shortest allele, allowing us to avoid underestimation of the frequency of RP and IA alleles. We've found only one RP allele on the shortest allele, and two on the longest.
4.5. Symptomatic patients
During the presymptomatic testing process, participants can display signs compatible with the condition, ranging from obvious HD, to subtle and non‐specific motor or cognitive signs that may or may not be related to HD. The presence of these signs was not disclosed to the participant unless specifically requested. Like in other centers (Alonso et al., 2009; Trembath et al., 2006), our team thought that in many cases the benefit from the protocol would be greater than an unaccompanied symptomatic test, for participants not in need of immediate medical care. Individuals requiring a switch toward symptomatic testing were rare in our cohort (4 individuals), indicating that the clear majority of participants were faultlessly referred. Importantly, for the analysis all the symptomatic individuals were purposely kept in the cohort, because their presence reflects the actual practice of presymptomatic testing that we assess in this article. Excluding them would have introduced a notable bias.
In our cohort as in others, a significant proportion of symptomatic HD patients are taking a presymptomatic rather than symptomatic test, unaware of their symptoms. This is not a surprise since HD usually begins insidiously, and since anosognosia is a recognized hallmark of the disease (McCusker & Loy, 2014). The rather high proportion of symptomatic individuals found in our cohort (72/448, 16%, Table 1) could reflect the difference in criteria chosen between studies: in our article, every participant showing a sign compatible with HD, detected by our trained team, and unexplained by a differential diagnosis, has been labeled as symptomatic. Further analysis of these symptomatic participants showed a very strong correlation with a subsequent positive (pathogenic) molecular testing (Figure 4a,b), confirming that a trained physician is able to accurately establish a diagnosis accurately based on the combination of motor and cognitive signs in this context of high risk. However, the incomplete correlation between the presence of isolated motor or cognitive signs and the actual carrier status, reminds us that some signs can be very unspecific, and that we should invariably refrain ourselves from drawing conclusions based on clinical data in the context of PT for HD.
Maybe more surprisingly, when the participant believed he had symptoms, it was also strongly indicative of an actual carrier status, since 91% (20/22) of those participants were indeed found carrier of a pathogenic allele. Because of the natural history of the disease, the individuals that request PT for HD are frequently well aware of the symptoms, and we show that their clinical impressions should indeed not be taken lightly. It also indicates that anosognosia does not affect every HD patient.
4.6. Motives for testing
Using a “principle motive”‐approach, rather than a “all‐motives”‐approach, when questioning the reasons for seeking PT, we managed to obtain a simple picture of age‐dependent motives for testing (Figure 3b,c). The results correspond to what could be expected: young adults are mostly planning for pregnancies and for their future life, middle‐aged tend to organize their future life and worry about their status when approaching the average age of onset, and a majority of older adults want to inform their children of their risk for family planning. Previous publications pointed out the importance of lifestage regarding individuals’ motivations for PT (“Holding your breath”, n.d.; Holman et al., 2018; Taylor, 2005). To our surprise, the motives for testing were remarkably stable over 24 years, resisting the changes of the society. Individuals who were unable to explain the reasons for testing and those who had no precise motive, where those who had by far the highest withdrawal rate (59%, p < .001, and 34%, p < .01, respectively, Figure 1d). This probably illustrates that when an individual has no tangible impact on his life decisions from knowing his status, he can be less determined to pursue this psychologically demanding protocol.
5. CONCLUSION
In this study conducted on 448 individuals seeking PT for HD over 24 years between 1993 and 2017 in a single center, we report a progressive twofold increase in the withdrawal rate from the protocol, possibly related to changes in the society, and more specifically partly due to a reduction in the proportion of married individuals. This finding is in line with other observations (Bernhardt et al., 2009), and should be known by the professionals involved in HD PT. The main motives for testing were stable over 24 years, and we describe specific patterns of age‐dependent motives for testing, giving a clear picture of the already suspected profiles of participants. The age of individuals evolved through this 24‐years period, with gradually more (twofold) very young adults and more (eightfold) older individuals seeking predictive diagnosis for HD. This can prove challenging especially when very young adults seek PT with no precise goal besides knowing the genetic status, and therefore do not have “actionable” decisions to help to cope with a pathogenic result. We also report a strong but not complete correlation between symptoms of HD perceived or detected before the test, and the actual carrier status after the test, reminding us both of the importance of anosognosia in HD, and of the expertise developed by patients exposed to the disease through family history.
CONFLICT OF INTEREST
All authors (FR, IS, LL, HC, FB, MB, and EO) declare an absence of conflicts of interest related to the research covered in the article.
Supporting information
ACKNOWLEDGMENTS
We wish to thank all the participants and their relatives, the Association pour le Développement de la Neurogénétique, the French associations for Huntington disease including Huntington France and Huntington Avenir, the staff of our hospital department for their support, and Dr Renaud Touraine for critical reading of the manuscript.
Ramond F, Quadrio I, Le Vavasseur L, et al. Predictive testing for Huntington disease over 24 years: Evolution of the profile of the participants and analysis of symptoms. Mol Genet Genomic Med. 2019;7:e881 10.1002/mgg3.881
Funding information
The charge for article publication was fully supported by the non‐profit association Association pour le Développement de la Neurogénétique.
REFERENCES
- Alonso, M. E. , Ochoa, A. , Sosa, A. L. , Rodríguez, Y. , Chávez, M. , Boll, C. , … Rasmussen, A. (2009). Presymptomatic diagnosis in Huntington’s disease: The Mexican experience. Genetic Testing and Molecular Biomarkers, 13(6), 717–720. 10.1089/gtmb.2009.0032 [DOI] [PubMed] [Google Scholar]
- Aziz, N. A. , van der Burg, J. M. M. , Tabrizi, S. J. , & Landwehrmeyer, G. B. (2018). Overlap between age‐at‐onset and disease‐progression determinants in Huntington disease. Neurology, 90(24), e2099–e2106. 10.1212/WNL.0000000000005690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig, S. S. , Strong, M. , Rosser, E. , Taverner, N. V. , Glew, R. , Miedzybrodzka, Z. , … Quarrell, O. W. (2016). 22 Years of predictive testing for Huntington’s disease: The experience of the UK Huntington’s Prediction Consortium. European Journal of Human Genetics, 24(10), 1396–1402. 10.1038/ejhg.2016.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates, G. P. , Dorsey, R. , Gusella, J. F. , Hayden, M. R. , Kay, C. , Leavitt, B. R. , … Tabrizi, S. J. (2015). Huntington disease. Nature Reviews Disease Primers, 1, 15005 10.1038/nrdp.2015.5 [DOI] [PubMed] [Google Scholar]
- Bernhardt, C. , Schwan, A.‐M. , Kraus, P. , Epplen, J. T. , & Kunstmann, E. (2009). Decreasing uptake of predictive testing for Huntington’s disease in a German centre: 12 years’ experience (1993–2004). European Journal of Human Genetics, 17(3), 295–300. 10.1038/ejhg.2008.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton, S. , Almqvist, E. W. , MacGregor, D. , Fernandez, B. , Hogg, H. , Beis, J. , … Hayden, M. R. (2003). Predictive, pre‐natal and diagnostic genetic testing for Huntington’s disease: The experience in Canada from 1987 to 2000. Clinical Genetics, 63(6), 462–475. 10.1034/j.1399-0004.2003.00093.x [DOI] [PubMed] [Google Scholar]
- Dufrasne, S. , Roy, M. , Galvez, M. , & Rosenblatt, D. S. (2011). Experience over fifteen years with a protocol for predictive testing for Huntington disease. Molecular Genetics and Metabolism, 102(4), 494–504. 10.1016/j.ymgme.2010.12.001 [DOI] [PubMed] [Google Scholar]
- Duyao, M. , Ambrose, C. , Myers, R. , Novelletto, A. , Persichetti, F. , Frontali, M. , … MacDonald, M. (1993). Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nature Genetics, 4(4), 387–392. 10.1038/ng0893-387 [DOI] [PubMed] [Google Scholar]
- Gem‐HD Consortium (2015). Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell, 162(3), 516–526. 10.1016/j.cell.2015.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goizet, C. , Lesca, G. , & Durr, A. , & French Group for Presymptomatic Testing in Neurogenetic Disorders (2002). Presymptomatic testing in Huntington’s disease and autosomal dominant cerebellar ataxias. Neurology, 59(9), 1330–1336. 10.1212/01.WNL.0000032255.75650.C2 [DOI] [PubMed] [Google Scholar]
- “Holding your breath”: Interviews with young people who have undergone predictive genetic testing for Huntington disease—Duncan—2007. (n.d.). American Journal of Medical Genetics Part A ‐ Wiley Online Library. Retrieved from https://onlinelibrary.wiley.com/doi/abs/10.1002/ajmg.a.31720 [DOI] [PubMed] [Google Scholar]
- Holman, M. A. , Quillin, J. , York, T. P. , Testa, C. M. , Rosen, A. R. , & Norris, V. W. (2018). The changing age of individuals seeking presymptomatic genetic testing for Huntington disease. Journal of Genetic Counseling, 27, 1157–1166. 10.1007/s10897-018-0233-9 [DOI] [PubMed] [Google Scholar]
- Kay, C. , Collins, J. A. , Miedzybrodzka, Z. , Madore, S. J. , Gordon, E. S. , Gerry, N. , … Hayden, M. R. (2016). Huntington disease reduced penetrance alleles occur at high frequency in the general population. Neurology, 87(3), 282–288. 10.1212/WNL.0000000000002858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keum, J. W. , Shin, A. , Gillis, T. , Mysore, J. S. , Abu Elneel, K. , Lucente, D. , … Lee, J.‐M. (2016). The HTT CAG‐expansion mutation determines age at death but not disease duration in Huntington disease. American Journal of Human Genetics, 98(2), 287–298. 10.1016/j.ajhg.2015.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krukenberg, R. C. , Koller, D. L. , Weaver, D. D. , Dickerson, J. N. , & Quaid, K. A. (2013). Two decades of Huntington disease testing: Patient’s demographics and reproductive choices. Journal of Genetic Counseling, 22(5), 643–653. 10.1007/s10897-013-9596-0 [DOI] [PubMed] [Google Scholar]
- Lee, J.‐M. , Ramos, E. M. , Lee, J.‐H. , Gillis, T. , Mysore, J. S. , Hayden, M. R. , … Gusella, J. F. (2012). CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology, 78(10), 690–695. 10.1212/WNL.0b013e318249f683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D. , Long, J. D. , Zhang, Y. , Raymond, L. A. , Marder, K. , Rosser, A. , … Paulsen, J. S. (2015). Motor onset and diagnosis in Huntington disease using the diagnostic confidence level. Journal of Neurology, 262(12), 2691–2698. 10.1007/s00415-015-7900-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald, M. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell, 72(6), 971–983. 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- MacLeod, R. , Tibben, A. , Frontali, M. , Evers‐Kiebooms, G. , Jones, A. , Martinez‐Descales, A. , … Editorial Committee and Working Group ‘Genetic Testing Counselling’ of the European Huntington Disease Network (2013). Recommendations for the predictive genetic test in Huntington’s disease. Clinical Genetics, 83(3), 221–231. 10.1111/j.1399-0004.2012.01900.x [DOI] [PubMed] [Google Scholar]
- Mandich, P. , Lamp, M. , Gotta, F. , Gulli, R. , Iacometti, A. , Marchese, R. , … Ferrandes, G. (2017). 1993–2014: Two decades of predictive testing for Huntington’s disease at the Medical Genetics Unit of the University of Genoa. Molecular Genetics & Genomic Medicine, 5(5), 473–480. 10.1002/mgg3.238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker, E. , & Loy, C. T. (2014). The many facets of unawareness in huntington disease. Tremor and Other Hyperkinetic Movements (New York, N.Y.), 4, 257 7916/D8FJ2FD3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss, D. J. H. , Pardiñas, A. F. , Langbehn, D. , Lo, K. , Leavitt, B. R. , Roos, R. , … Tan, L. (2017). Identification of genetic variants associated with Huntington’s disease progression: A genome‐wide association study. The Lancet. Neurology, 16(9), 701–711. 10.1016/S1474-4422(17)30161-8 [DOI] [PubMed] [Google Scholar]
- Nance, M. A. (2017). Genetic counseling and testing for Huntington’s disease: A historical review. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics: the Official Publication of the International Society of Psychiatric Genetics, 174(1), 75–92. 10.1002/ajmg.b.32453 [DOI] [PubMed] [Google Scholar]
- Panas, M. , Karadima, G. , Vassos, E. , Kalfakis, N. , Kladi, A. , Christodoulou, K. , & Vassilopoulos, D. (2011). Huntington’s disease in Greece: The experience of 14 years. Clinical Genetics, 80(6), 586–590. 10.1111/j.1399-0004.2010.01603.x [DOI] [PubMed] [Google Scholar]
- Peterlin, B. , Kobal, J. , Teran, N. , Flisar, D. , & Lovrečić, L. (2009). Epidemiology of Huntington’s disease in Slovenia. Acta Neurologica Scandinavica, 119(6), 371–375. 10.1111/j.1600-0404.2008.01110.x [DOI] [PubMed] [Google Scholar]
- Pinto, R. M. , Dragileva, E. , Kirby, A. , Lloret, A. , Lopez, E. , St. Claire, J. , … Wheeler, V. C. (2013). Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: Genome‐wide and candidate approaches. PLoS Genetics, 9(10), 10.1371/journal.pgen.1003930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarrell, O. W. J. , Rigby, A. S. , Barron, L. , Crow, Y. , Dalton, A. , Dennis, N. , … Warner, J. (2006). Reduced penetrance alleles for Huntington’s disease: A multi‐centre direct observational study. Journal of Medical Genetics, 44(3), e68–e68. 10.1136/jmg.2006.045120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, C. A. , Aylward, E. H. , Wild, E. J. , Langbehn, D. R. , Long, J. D. , Warner, J. H. , … Tabrizi, S. J. (2014). Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nature Reviews Neurology, 10(4), 204–216. 10.1038/nrneurol.2014.24 [DOI] [PubMed] [Google Scholar]
- Scuffham, T. M. , & MacMillan, J. C. (2014). Huntington disease: Who seeks presymptomatic genetic testing, why and what are the outcomes? Journal of Genetic Counseling, 23(5), 754–761. 10.1007/s10897-013-9678-z [DOI] [PubMed] [Google Scholar]
- Semaka, A. , & Hayden, M. R. (2014). Evidence‐based genetic counselling implications for Huntington disease intermediate allele predictive test results: Genetic counselling implications for IAs. Clinical Genetics, 85(4), 303–311. 10.1111/cge.12324 [DOI] [PubMed] [Google Scholar]
- Semaka, A. , Kay, C. , Doty, C. , Collins, J. A. , Bijlsma, E. K. , Richards, F. , … Hayden, M. R. (2013). CAG size‐specific risk estimates for intermediate allele repeat instability in Huntington disease. Journal of Medical Genetics, 50(10), 696–703. 10.1136/jmedgenet-2013-101796 [DOI] [PubMed] [Google Scholar]
- Sequeiros, J. , Ramos, E. M. , Cerqueira, J. , Costa, M. C. , Sousa, A. , Pinto‐Basto, J. , & Alonso, I. (2010). Large normal and reduced penetrance alleles in Huntington disease: Instability in families and frequency at the laboratory, at the clinic and in the population. Clinical Genetics, 78(4), 381–387. 10.1111/j.1399-0004.2010.01388.x [DOI] [PubMed] [Google Scholar]
- Squitieri, F. , Gellera, C. , Cannella, M. , Mariotti, C. , Cislaghi, G. , Rubinsztein, D. C. , … Donato, S. D. (2003). Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain, 126(4), 946–955. 10.1093/brain/awg077 [DOI] [PubMed] [Google Scholar]
- Sun, Y.‐M. , Zhang, Y.‐B. , & Wu, Z.‐Y. (2017). Huntington’s disease: Relationship between phenotype and genotype. Molecular Neurobiology, 54(1), 342–348. 10.1007/s12035-015-9662-8 [DOI] [PubMed] [Google Scholar]
- Taylor, S. D. (2005). Predictive genetic test decisions for Huntington’s disease: Elucidating the test/no‐test dichotomy. Journal of Health Psychology, 10(4), 597–612. 10.1177/1359105305053442 [DOI] [PubMed] [Google Scholar]
- Trembath, M. K. , Tassicker, R. J. , Collins, V. R. , Mansie, S. , Sheffield, L. J. , & Delatycki, M. B. (2006). Fifteen years of experience in predictive testing for Huntington disease at a single testing center in Victoria, Australia. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 8(11), 673–680. 10.1097/01.gim.0000245633.97952.f1 [DOI] [PubMed] [Google Scholar]
- Warner, J. P. , Barron, L. H. , & Brock, D. J. (1993). A new polymerase chain reaction (PCR) assay for the trinucleotide repeat that is unstable and expanded on Huntington’s disease chromosomes. Molecular and Cellular Probes, 7(3), 235–239. 10.1006/mcpr.1993.1034 [DOI] [PubMed] [Google Scholar]
- Wheeler, V. C. , Persichetti, F. , McNeil, S. M. , Mysore, J. S. , Mysore, S. S. , MacDonald, M. E. , … US‐Venezuela Collaborative Research Group (2007). Factors associated with HD CAG repeat instability in Huntington disease. Journal of Medical Genetics, 44(11), 695–701. 10.1136/jmg.2007.050930 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials