

Abstract
Selenocysteine (Sec) is the 21st amino acid in the genetic code where this amino acid is primarily involved in redox reactions in enzymes due to its high reactivity towards oxygen and related reactive oxygen species. Sec has found wide utility in synthetic peptides, especially as a replacement for cysteine. One limitation of using Sec in synthetic peptides is that it can undergo β-syn elimination reactions after oxidation, rendering the peptide inactive due to loss of selenium. This limitation can be overcome by substituting Cα-H with a methyl group. The resulting Sec-derivative is α-methylselenocysteine ((αMe)Sec). Here we present a new strategy for the synthesis of (αMe)Sec by alkylation of an achiral methyl malonate through the use of a selenium-containing alkylating agent synthesized in the presence of dichloromethane. The seleno-malonate was then subjected to an enzymatic hydrolysis utilizing pig liver esterase followed by a Curtius rearrangement producing a protected derivative of (αMe)Sec that could be used in solid phase peptide synthesis. We then synthesized two peptides, one containing Sec, and the other containing (αMe)Sec, based on the sequence of glutathione peroxidase. This is the first reported incorporation of (αMe)Sec into a peptide as well as the first reported biochemical application of this unique amino acid. The (αMe)Sec-containing peptide had superior stability as it could not undergo β-syn elimination and it also avoided cleavage of the peptide backbone, which we surprisingly found to be the case for the Sec-containing peptide when it was incubated for 96 hours in oxygenated buffer at pH 8.0.
GRAPHICAL ABSTRACT
One limitation of using Sec in peptides is that it can undergo β-syn elimination reactions after oxidation, rendering the peptide inactive due to loss of selenium. This limitation can be overcome by substituting Cα-H with a methyl group. The resulting Sec-derivative is α-methylselenocysteine. Here we report a facile synthesis of α-methylselenocysteine and its use in a peptide as a glutathione peroxidase mimic. The mimic is resistant to loss of selenium when exposed to oxidant.
INTRODUCTION
Vertebrate glutathione peroxidase (GPX) is an enzyme that catalyzes the reduction of H2O2, organohydroperoxides, and lipid hydroperoxides. [1–4] Multiple forms of the enzyme contain the rare amino acid selenocysteine (Sec). [5, 6] The GPX catalytic cycle proceeds by nucleophilic attack of the selenolate on to the peroxyl oxygen of the substrate resulting in a selenenic acid intermediate, that is subsequently reduced back to the selenolate by two equivalents of glutathione (GSH) (Figure 1). [5] When concentrations of H2O2 are elevated, such as during reoxygenation after a period of ischemia, GPX can become overwhelmed and does not reduce H2O2 fast enough to prevent reperfusion injury. [7] Thus, it is desirable to design a small molecule GPX mimic that can serve a therapeutic function for these situations of high oxidative stress. [3]
Figure 1:

Catalytic cycle of GPX. The selenol of Sec first reduces oxidants resulting in a selenenic acid intermediate, which is then reduced by one equivalent of GSH forming the GPX-Sec-glutathione adduct. A second equivalent of GSH regenerates the enzyme by restoring the reduced selenol, forming oxidized glutathione (GSSG) as a byproduct. GSSG is then reduced by glutathione reductase (GR) in the presence of NADPH.
Ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one), first reported in 1924, [8] became a widely studied GPX mimic after its antioxidant properties were recognized in 1984 by Müller and coworkers. [9] Like GPX, ebselen relies on the redox properties of selenium to reduce harmful oxidants and proceeds through a similar catalytic mechanism (see Figure 1). Ebselen previously made it to clinical trials as a neuro- and cardioprotective agent, [10] and is currently in clinical trials for treatment of hearing loss that is related to oxidative stress. [11, 12] However, ebselen is not water soluble, making rapid intravenous administration impossible and has shown cellular toxicity in some studies. [13–15] Other selenium containing GPX mimics have been synthesized and some have potent GPX-like activity including cyclic seleninate and selenenate esters, [16–21] spirodioxyselenuranes, [22–24] pincer selenuranes, [22] and spirodiazaselenuranes. [25, 26] However, many of these compounds are expected to be toxic, are not water soluble, are unstable, or quickly lose catalytic activity in the presence of thiols. [3]
One possible reason that GPX cannot respond optimally to very high concentrations of H2O2, as occurs during reperfusion injury, is that it has been shown that GPX can be inactivated by conversion of the Sec residue to dehydroalanine (DHA). [27] DHA results when the Sec residue becomes overoxidized to the seleninic acid (Sec-SeO2–) state followed by rapid β-syn elimination.
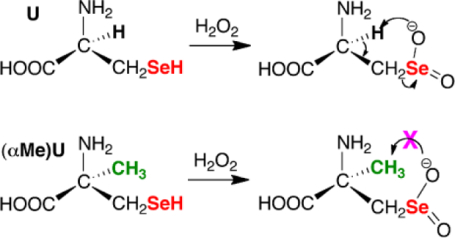
Ebselen is not as efficient of a catalyst as GPX, but it can be overoxidized without undergoing β-syn elimination because it contains an aromatic selenol. Sec-containing peptides however can undergo β-syn elimination, [28] so the design of any peptide based GPX mimic should avoid this problem. One useful derivative of Sec is α-methylselenocysteine ((αMe)Sec) in which the α-hydrogen has been replaced with a methyl group, removing the possibility of β-syn elimination (Figure 2). Thus, (αMe)Sec can resist inactivation by overoxidation while Sec cannot. These unique properties lend to many potential applications. Herein we report a new route for the synthesis of (αMe)Sec and we incorporate (αMe)Sec into a peptide that serves as a small molecule GPX mimic. Our peptide GPX mimic overcomes the problem of aqueous solubility, while avoiding β-syn elimination.
Figure 2: Avoidance of β-syn elimination in (αMe)Sec.

Sec and Sec-containing peptides can undergo deselenization due to oxidation of the selenol followed by rapid β-syn elimination. The (αMe)Sec analog avoids this complication as it lacks hydrogen on Cα.
MATERIALS AND METHODS
Materials.
Solvents for peptide synthesis were purchased from Fisher Scientific (Waltham, MA). Solvents for synthesis including N,N-dimethylformamide (DMF) and dichloromethane (DCM) were purchased from Fisher Scientific (Fair lawn, NJ) or Acros Organics (Morris Plains, New Jersey). N-Fmoc amino acids were purchased from RSsynthesis (Louisville, KY). 2-Chlorotrityl chloride resin SS (100–200 mesh) and 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) for solid-phase synthesis were purchased from Advanced ChemTech (Louisville, KY). Triisopropylsilane (98%) was purchased from ACROS Organics (Pittsburgh, PA). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and hydrogen peroxide for enzyme assays were purchased from ThermoFischer Scientific (Waltham, MA). NADPH was purchased from AppliChem (Darmstadt, Germany). Diphenylphosphoryl azide (DPPA) was purchased from TCI America (Portland, OR). 9-fluorenemethanol was purchased from Chempep Inc. (Miami, Fl). Titanium (IV) tert-butoxide was purchased from EMD Millipore. Silica gel (230–400 mesh) for purifications was purchased from Silicycle (Quebec City, QC, Canada). Deuterated solvents were purchased from Cambridge Isotope Laboratories (Andover, MA). All other chemicals were purchased from Sigma-Aldrich (Milwaukee, WI), Fisher Scientific (Waltham, MA), or ACROS Organics (Pittsburgh, PA). Mass spectral analysis for synthetic peptides was performed on an Applied Biosystems QTrap 4000 hybrid triple-quadrupole/linear ion trap liquid chromatograph-mass spectrometer (SciEx, Framingham, MA). High resolution mass spectral data on all synthesized compounds were obtained from positive electrospray ionization on a Bruker 12 Tesla APEX -Qe FTICR-MS with an Apollo II ion source (Norfolk, VA) and were either ran as solutions in tetrahydrofuran (THF) or THF/Methanol. NMR spectra were obtained from a 400 MHz Bruker spectrometer. 1H and 13C chemical shifts (δ) were reported as downfield from tetramethylsilane. Both 13C and 77Se NMR spectra were decoupled from 1H. IR spectra were obtained from a Nicolet Nexus 470 FT-IR spectrometer with a Smart Orbit Thermo™ diamond plate ATR attachment and samples were ran as a solution from CDCl3. Optical rotation data were obtained from a Rudolph Research Analytical Autopol IV Automatic Polarimeter at 589 nm with a 100 mm quartz cell. Enzyme kinetic assays were performed on a Cary50 UV-Vis spectrophotometer (Walnut Creek, CA) at room temperature.
Synthesis of tert-butyl(chloromethyl)selenide (2).
To a flask with a nitrogen atmosphere was added tert-butyldiselenide (1) (4.93 g, 18.1 mmol) and dry DCM (46.5 mL, 726 mmol). To this solution was added sodium bis(2-methoxyethoxy)aluminum hydride dropwise until the solution turned colorless and turbid and effervescence ceased. After stirring for an additional 30 min, the solution was diluted with diethyl ether (150 mL) and washed with saturated Rochelle salt solution (3 × 100 mL) followed by brine (100 mL). The organic phase was then dried over MgSO4, filtered, and the solvent removed in vacuo to afford 5.20 g (77%) of a colorless oil which was carried on directly to the following reaction. 1H NMR (400 MHz, CDCl3) δ 4.87 (s, 2H), 1.55 (s, 9H, Se satellites 2JSe-H 5.6 Hz); 77Se NMR (76 MHz, CDCl3) δ 518.8.
Synthesis of dimethyl 2-((tert-butylselanyl)methyl)-2-methylmalonate (3).
To a flask with a nitrogen atmosphere was added sodium hydride (60% dispersion in mineral oil) (1.12 g, 27.9 mmol), which was washed twice with pentane. To the flask was then added THF (40 mL) and dimethyl methylmalonate (3.00 mL, 22.5 mmol), which produced effervescence. After stirring for 45 min, 2 (4.23 g, 22.8 mmol) was added. After refluxing for 24 h, the reaction was cooled to room temperature and diluted with diethyl ether (100 mL), washed with water (4 × 100 mL), and brine (100 mL). The organic phase was then dried over MgSO4, filtered, and concentrated in vacuo. The crude oil was purified by gradient flash chromatography and eluted with hexanes/ethyl acetate (8:1 to 4:1; Rf = 0.50 in 4:1 hexanes/ethyl acetate). The product rich fractions were pooled and concentrated in vacuo to afford 3.79 g (57%) of a colorless oil. IR (CDCl3, cm−1) 2953, 2891, 2861, 1731; 1H NMR (400 MHz, CDCl3) δ 3.74 (s, 6H), 3.05 (s, 2H), 1.51 (s, 3H), 1.43 (s, 9H, Se satellites 2JSe-H 5.5 Hz); 13C (101 MHz, CDCl3) δ 171.7, 54.5 (Se satellites 2JSe-C 4.8 Hz), 52.7, 39.3 (Se satellites 1JSe-C 29.0 Hz), 32.2 (Se satellites 2JSe-C 6.6 Hz), 26.8 (Se satellites 1JSe-C 37.0 Hz), 21.0; 77Se NMR (76 MHz, CDCl3) δ 342.6; HRMS (ESI+) Calcd for C11H20O4Se [M+Na]+ 319.0419, found 319.0420.
Synthesis of (R)-2-((tert-butylselanyl)methyl)-3-methoxy-2-methyl-3-oxopropanoic acid (4).
To a beaker was added 3 (3.24 g, 11.0 mmol), pH 7.6 phosphate buffer (40 mL), and crude porcine liver esterase (52 mg, 18 units/mg). The reaction was stirred vigorously with the pH kept constant at 7.4 by titrating 1 M NaOH into the reaction. The reaction was complete after stirring for 4 h. The solution pH was raised to 9 with the addition of 1 M NaOH and was washed with diethyl ether (60 mL). The aqueous phase was reacidified to a pH of 3 with 4 M HCl and extracted with DCM (3 × 60 mL). The organic phase was filtered through a bed of celite, dried over MgSO4, and concentrated in vacuo to afford 3.03 g (98%, ee = 88%) of a slightly brown tinged oil. IR (CDCl3, cm−1) 2955, 2889, 1709, 1636; 1H NMR (400 MHz, CDCl3) δ 9.26 (br s, 1H), 3.77 (s, 3H), 3.09 (d, 2JH-H 11.6, 1H), 3.04 (d, 2JH-H 11.6, 1H), 1.54 (s, 3H), 1.43 (s, 9H, Se satellites 2JSe-H 5.5 Hz); 13C (101 MHz, CDCl3) δ 177.2, 171.5, 54.5 (Se satellites 2JSe-C 5.1 Hz), 52.9, 39.6 (Se satellites 1JSe-C 29.3 Hz), 32.2 (Se satellites 2JSe-C 6.6 Hz), 26.5 (Se satellites 1JSe-C 37.8 Hz), 21.1; 77Se NMR (76 MHz, CDCl3) δ 344.5; HRMS (ESI+) Calcd for C10H18O4Se [M+Na]+ 305.0263, found 305.0267; [α]D23 = - 2.1° (c = 1.07, CHCl3).
Synthesis of (R)-methyl 2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(tert-butylselanyl)-2-methylpropanoate (5).
To a flask with a nitrogen atmosphere was added 4 (578 mg, 2.06 mmol), dichloroethane (DCE) (5 mL), and dry triethylamine (0.35 mL, 2.5 mmol). After stirring for 5 min, diphenylphosphoryl azide (DPPA) (0.50 mL, 2.3 mmol) was added and the solution was stirred for 5 h at room temperature and then brought to reflux for 19 h. After cooling to room temperature the reaction was diluted with DCE (10 mL), washed with saturated NH4Cl (15 mL), dried over MgSO4, filtered, and concentrated in vacuo to afford a yellow tinged turbid oil. This oil was reconstituted in benzene (5 mL). To this solution was added 9-fluorenemethanol (416 mg, 2.12 mmol) and titanium (IV) tert-butoxide (0.10 mL, 260 μmol). After stirring at room temperature for 29 h the reaction was diluted with diethyl ether (15 mL) and washed with saturated NH4Cl (4 × 15 mL). The aqueous phases were extracted with DCM (25 mL). All organic phases were then pooled, dried over MgSO4, filtered, and concentrated in vacuo to afford a crude yellow oil. The crude product was purified by flash chromatography and eluted with 2:3 diethyl ether/hexanes (Rf = 0.36). The product rich fractions were pooled and concentrated in vacuo to afford 626 mg (64%) of a colorless oil. IR (CDCl3, cm−1) 3415, 3349, 2952, 2890, 1721, 1500, 1478, 1449; 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 7.5 Hz, 2H), 7.61 (d, J = 7.5 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.31 (td, J = 7.4, 1.1 Hz, 2H), 5.85 (br s, 1H), 4.29 (m, 3H), 3.78 (s, 3H), 3.48 (br d, 2JH-H = 11.2 Hz, 1H,), 3.09 (br d, 2JH-H = 11.1 Hz, 1H), 1.69 (s, 3H), 1.41 (s, 9H); 13C (101 MHz, CDCl3) δ 173.6, 154.6, 143.9, 141.3, 127.7, 127.1, 125.2, 120.0, 66.8, 60.1, 53.0, 47.2, 39.4, 32.3 (Se satellites 2JSe-C = 6.6 Hz), 28.7, 24.1; 77Se NMR (76 MHz, CDCl3) δ 334.9; HRMS (ESI+) Calcd for C24H29NO4Se [M+Na]+ 498.1154, found 498.1161; [α]D23 = + 8.9° (c = 1.01, CHCl3).
Synthesis of (R)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(tert-butylselanyl)-2-methylpropanoic acid (6).
To a flask was added 5 (558 mg, 1.18 mmol), 1,4-dioxane (7.5 mL) and 12.1 M HCl (7.5 mL). The solution was refluxed for 40 h and then cooled to room temperature. Solvents were removed in vacuo and the resulting viscous yellow residue was reconstituted with diethyl ether (20 mL). This solution was washed with water (4 × 20 mL) and brine (20 mL). The organic phase was dried over MgSO4, filtered, and concentrated in vacuo. Residual solvent was removed by azeotropic drying with pentane to afford 452 mg (84%) of a colorless fluffy solid. IR (CDCl3, cm−1) 3404, 2955, 1707, 1502, 1450; 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 7.8 Hz, 2H), 7.60 (d, J = 7.4 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.31 (td, J = 7.4, 0.8 Hz, 2H), 5.77 (br s, 1H), 4.28 (m, 3H), 3.39 (d, J = 11.1 Hz, 1H), 3.18 (d, J = 10.9 Hz, 1H), 1.71 (s, 3H), 1.41 (s, 9H); 13C (101 MHz, CDCl3) δ 177.7, 154.9, 143.8, 141.3, 127.7, 127.1, 125.2, 120.0, 67.0, 59.7, 47.1, 39.6, 32.3 (Se satellites 2JSe-C = 6.6 Hz), 28.7, 24.1; 77Se NMR (76 MHz, CDCl3) δ 331.0; HRMS (ESI+) Calcd for C23H27NO4Se [M+Na]+ 484.0998, found 484.1001; [α]D23 = + 2.6° (c = 1.01, CHCl3).
Synthesis of (S)-1-tert-butyl 3-methyl 2-((tert-butylselanyl)methyl)-2-methylmalonate (7).
To a flask was added DCM (13 mL), MgSO4 (1.77 g, 14.8 mmol), and concentrated sulfuric acid (0.20 mL, 3.7 mmol). Clumped magnesium sulfate was broken up by vigorously stirring. To this suspension was added a solution of 4 (1.03 g, 3.67 mmol) in DCM (7 mL). Tert-butanol (1.75 mL, 18.3 mmol) was then added dropwise and the flask was tightly sealed. After reacting for 47 h the reaction was filtered. The filtrate was washed with saturated NaHCO3 (20 mL) and brine (20 mL). The organic phase was dried over MgSO4, filtered, and concentrated in vacuo to afford 807 mg (65%) of an amber tinged liquid. IR (CDCl3, cm−1) 2975, 2952, 1728; 1H NMR (400 MHz, CDCl3) δ 3.73 (s, 3H), 3.03 (d, 2JH-H = 11.4 Hz, 1H), 3.00 (d, 2JH-H = 11.4 Hz, 1H), 1.46 (s, 3H), 1.45 (s, 9H), 1.43 (s, 9H, Se satellites 2JSe-H 5.3 Hz); 13C (101 MHz, CDCl3) δ 172.2, 170.3, 82.0, 55.0 (Se satellites 2JSe-C = 4.8 Hz), 52.4, 39.0 (Se satellites 1JSe-C = 29.3 Hz), 32.2 (Se satellites 2JSe-C = 6.6 Hz), 27.8, 26.8 (Se satellites 1JSe-C = 37.0 Hz), 20.8; 77Se NMR (76 MHz, CDCl3) δ 339.3; HRMS (ESI+) Calcd for C14H26O4Se [M+Na]+ 361.0889, found 361.0893; [α]D23 = - 9.6° (c = 1.00, CHCl3).
Synthesis of (S)-3-(tert-butoxy)-2-((tert-butylselanyl)methyl)-2-methyl-3-oxopropanoic acid (8).
To a flask containing 7 (591 mg, 1.75 mmol) was added THF (5.0 mL) and a solution of lithium hydroxide (130 mg, 3.1 mmol) in water (5.0 mL). After stirring at room temperature for 24 h the pH was reduced to 4 using 1 M HCl and extracted with diethyl ether (15 mL). The organic phase was then washed with water (3 × 15 mL) and brine (15 mL). The organic phase was then dried over MgSO4, filtered, and concentrated in vacuo to afford 496 mg (88%) of a yellow tinged liquid. IR (CDCl3, cm−1) 2977, 1707; 1H NMR (400 MHz, CDCl3) δ 10.69 (br s, 1H), 3.06 (d, 2JH-H = 11.4 Hz, 1H), 3.00 (d, 2JH-H = 11.4 Hz, 1H), 1.50 (s, 3H), 1.47 (s, 9H), 1.44 (s, 9H, Se satellites 2JSe-H 5.6 Hz); 13C (101 MHz, CDCl3) δ 177.3, 170.4, 82.7, 54.9 (Se satellites 2JSe-C = 4.8 Hz), 39.2 (Se satellites 1JSe-C = 29.0 Hz), 32.2 (Se satellites 2JSe-C = 6.6 Hz), 27.8, 26.7 (Se satellites 1JSe-C = 37.0 Hz), 21.1; 77Se NMR (76 MHz, CDCl3) δ 342.3; HRMS (ESI+) Calcd for C13H24O4Se [M+Na]+ 347.0732, found 347.0734; [α]D23 = - 2.4° (c = 1.00, CHCl3).
Synthesis of (S)-tert-butyl 2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(tert-butylselanyl)-2-methylpropanoate (9).
To a flask with a N2 atmosphere was added 8 (888 mg, 2.75 mmol), DCE (10 mL), and triethylamine (0.50 mL, 3.6 mmol). After stirring for 5 min, DPPA (650 μL, 3.0 mmol) was added. This solution was stirred for an additional hour and then refluxed for 17 h. After cooling the flask to room temperature, the solution was diluted with DCE (10 mL) and washed with saturated NH4Cl (20 mL). The organic phase was dried over MgSO4, filtered, and concentrated in vacuo to afford a turbid oil. The oil was reconstituted in a flask using benzene (10 mL). To this flask was added 9-fluorenemethanol (545 mg, 2.78 mmol) and titanium (IV) tert-butoxide (0.10 mL, 260 μmol). After stirring at room temperature for 47 h saturated NH4Cl (10 mL) was added and the reaction was stirred for 3.5 h. The reaction was diluted with diethyl ether (20 mL) and washed with saturated NH4Cl (2 × 20 mL) and water (20 mL). The emulsion was extracted with diethyl ether (20 mL). The organic phases were pooled, dried over MgSO4, filtered, and concentrated in vacuo to afford a crude oil. This oil was purified by gradient flash chromatography and eluted with hexanes/diethyl ether (8:1 to 4:1; Rf = 0.57 in 2:3 diethyl ether/hexanes). The product rich fractions were pooled and concentrated in vacuo to afford 786 mg (55%) of a colorless, gummy solid. IR (CDCl3, cm−1) 3413, 2973, 1715; 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 8.0 Hz, 2H), 7.62 (dd, J = 7.4, 2.9 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.31 (td, J = 7.4, 1.1 Hz, 2H), 5.96 (s, 1H), 4.28 (m, 3H), 3.51 (d, 2J = 11.2 Hz, 1H), 3.05 (d, 2J = 11.2 Hz, 1H), 1.69 (s, 3H), 1.50 (s, 9H), 1.41 (s, 9H); 13C (101 MHz, CDCl3) δ 172.2, 154.5, 144.1, 143.9, 141.3, 127.6, 127.0, 125.3, 119.9, 82.7, 66.7, 60.2 (Se satellites 2JSe-C = 4.0 Hz), 47.2, 39.0, 32.4 (Se satellites 2JSe-C = 6.6 Hz), 28.4, 27.9, 24.2; 77Se NMR (76 MHz, CDCl3) δ 336.8; HRMS (ESI+) Calcd for C27H35NO4Se [M+Na]+ 540.1624, found 540.1626; [α]D23 = + 7.7° (c = 1.00, CHCl3).
Synthesis of (S)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(tert-butylselanyl)-2-methylpropanoic acid (10).
To a flask was added 9 (546 mg, 1.06 mmol), DCM (10 mL), and trifluoroacetic acid (10.0 mL, 14.9 mmol). After stirring for 2 h at room temperature, the solvent was removed in vacuo. The residue was reconstituted in diethyl ether (10 mL) and washed with saturated NH4Cl (3 × 10 mL). The organic phase was dried over MgSO4, filtered, and concentrated in vacuo. Residual solvent was removed by azeotropic drying with pentane to afford 487 mg (quantitative) of a gummy, foamy solid. IR (CDCl3, cm−1) 3400, 3065, 2955, 1707, 1503, 1450; 1H NMR (400 MHz, CDCl3) δ 8.99 (br s, 1H), 7.76 (d, J = 7.5 Hz, 2H), 7.60 (d, J = 7.4 Hz, 2H), 7.39 (d, J = 7.4 Hz, 2H), 7.31 (td, J = 7.4, 0.8 Hz, 2H), 5.77 (br s, 1H), 4.29 (m, 3H), 3.36 (d, J = 10.5 Hz, 1H), 3.18 (d, J = 10.6 Hz, 1H), 1.70 (s, 3H), 1.41 (s, 9H); 13C (101 MHz, CDCl3) δ 178.0, 155.1, 143.7, 141.3, 127.7, 127.1, 125.1, 120.0, 67.2, 59.7, 39.8, 32.3 (Se satellites 2JSe-C = 6.2 Hz), 28.6, 24.0; 77Se NMR (76 MHz, CDCl3) δ 330.2; HRMS (ESI+) Calcd for C23H27NO4Se [M+Na]+ 484.0998, found 484.1000; [α]D23 = - 2.7° (c = 1.07, CHCl3).
Peptide Synthesis.
Either Fmoc-(αMe)Sec(But)-OH, or Fmoc-Sec(Mob)-OH were used to synthesize peptides of sequence H-(αMe)Sec-Gly-Thr-Thr-Val-Arg-Asp-Tyr-Thr-Gln-OH (H-(αMe)UGTTVRDYTQ-OH) (11), or H- Sec-Gly-Thr-Thr-Val-Arg-Asp-Tyr-Thr-Gln-OH (H-UGTTVRDYTQ-OH) (12). We note that we followed the recommendations of Jones for all of the abbreviations of amino acids and reagents for peptide synthesis in this paper. [29] All peptides were synthesized on a 0.1 mmol scale using a glass vessel that was shaken with a model 75 Burrell wrist action shaker. For each batch, 300 mg of 2-chlorotrityl chloride resin SS (100–200 mesh), was swelled in DCM for 30 min. The first amino acid was directly coupled to the resin using 2% N-methylmorpholine (NMM) in DCM, shaking for 1 h. The resin was then capped using 10 mL of 8:1:1 DCM:methanol:NMM. Subsequent amino acids were coupled using 0.2 mmol of Fmoc-protected amino acid, 0.2 mmol HATU, and 2% NMM in dimethylformamide (DMF), shaking for 1 h, rt. Fmoc-Sec(Mob)-OH or Fmoc-(αMe)Sec(But)-OH were coupled using a solution of 0.2 mmol amino acid, 1% N,N′-Diisopropylcarbodiimide (DIC), and 0.2 mmol 1-hydroxy-7-azabenzotriazole (HOAt) in DMF, shaking for 2 h, rt. Preactivation of any amino acid was not performed prior to coupling. Between amino acid couplings, the Fmoc protecting group was removed via two 10 min agitations with 10 mL of 20% piperidine in DMF. Success of Fmoc removal steps and amino acid couplings were monitored qualitatively using a ninhydrin test [30]. Removal of the final Fmoc protecting group completed the peptide synthesis. Peptides were cleaved from the resin via a 4 h reaction with a cleavage cocktail consisting of 0.4 mmol of 2,2′-dithiobis(5-nitropyridine) (DTNP), in 10 mL of 96:2:2 TFA/TIS/H2O. Following cleavage and side chain deprotection, the resin was washed with TFA and DCM and the volume of the cleavage solution was reduced by evaporation with nitrogen gas. The peptide solution was then transferred by pipette into cold, anhydrous diethyl ether, where the peptides were observed to precipitate. Centrifugation at 3000 rpm on a clinical centrifuge (International Equipment Co., Boston, MA) for 10 min pelleted the peptide. To wash the peptides, peptide pellets were dried, dissolved in minimal 96:2:2 TFA/TIS/H2O, then precipitated and pelleted as described previously. This wash process was repeated 5 times for each peptide. After washing, the final peptide pellet was dried, dissolved in a minimal amount of water, lyophilized, and used without further purification.
Measurement of peroxidase activity with a glutathione reductase coupled assay.
A coupled glutathione reductase (GR) assay was used to test the abilities of the peptides to function as GPX mimics. A 0.5 mL solution containing 100 mM potassium phosphate (KP) buffer, pH 8.0, 1 mM EDTA, 100 μM glutathione, 200 μM NADPH, 2 units of GR, and 10 μM of ebselen, H-UGTTVRDYTQ-OH, or H-(αMe)UGTTVRDYTQ-OH, were added to a quartz cuvette and mixed thoroughly. This was used to blank the spectrophotometer at 340 nm. Assays were initiated by addition of 200 μM H2O2. Activity was followed by the decrease of absorption at 340 nm using an extinction coefficient of 6220 M−1 cm−1, at 25 °C, and each initial rate was measured at least three times. Background NADPH consumption was corrected for by subtracting control experiment activities in which no peptide was present as well as an additional control in which no H2O2 was present.
Stability test of Sec-containing peptides.
To test the stability of peptides in the presence of oxygenated buffer, H-UGTTVRDYTQ-OH or H-(αMe)UGTTVRDYTQ-OH were incubated in 100 mM KP buffer, pH 8.0, at 25 °C. Aliquots were taken and centrifuged to pellet precipitated selenium after 24, 48, and 96 h. GPX-like activity of the aliquots was monitored as described above. To obtain mass spectra, aliquots were infused with a guard column at 10 μL/min into a 100 μL/min mobile phase flow consisting of 50/50 water/acetonitrile with 0.1% formic acid. The mobile phase flow was introduced into an Applied Biosystems QTrap 4000 hybrid triple-quadrupole/linear ion trap liquid chromatograph-mass spectrometer operating in positive ESI mode. Mass spectra were collected in linear ion trap mode, scanning from m/z 200–1800.
RESULTS AND DISCUSSION
Owing to the quaternary α-carbon, (αMe)Sec has proven challenging to synthesize. Previously, a racemic pathway by Reich [31] and a chiral pathway by Iwaoka [32, 33] have been executed for the synthesis of (αMe)Sec, however each have their limitations. Our synthetic method allows for quick synthesis, with moderate yields while also obtaining high enantiomeric excess. By utilizing this methodology, access to both enantiomers, as well as other relevant analogues, such as β-amino acids, are possible.
Our synthetic strategy starts with the alkylation of an achiral methyl malonate by a selenium-containing alkylating agent. Initially it was envisioned to create the alkylating agent based on our previous work synthesizing α-methylcysteine ((αMe)Cys), [34, 35] however due to the nucleophilicity of the selenolate anion, the alkylating agent could be directly synthesized in the presence of DCM (Scheme 1).
Scheme 1: Synthesis pathway of compound 4.

Reagents and conditions: (a) Red-Al, DCM, rt; (b) dimethyl methylmalonate, NaH, THF, reflux; (c) PLE, 7.4 pH phosphate buffer, rt.
Compound 1 was synthesized according to a modified version from Block and coworkers. [36] Several reducing agents were screened to reduce 1, with sodium bis(2-(methoxyethoxy)aluminum hydride (Red-Al) giving the best yields with minimal side products. Alkylating agent 2 was used to alkylate dimethyl methylmalonate to produce seleno-malonate 3 in 57% isolated yield. Malonate 3 was then subjected to an enzymatic hydrolysis utilizing pig liver esterase (PLE) producing 4, similar to our previous work. [34, 35, 37] Based on NMR after treatment with (S)-α-methylbenzylamine, PLE hydrolysis gave an 88% enantiomeric excess of the R-enantiomer by analysis of the now formed diastereomeric salts (see Figure S11 of the Supporting Information). The optical rotation and NMR data is consistent to the R-enantiomer when compared to the equivalent (R)-(αMe)Cys analogues. [35, 37, 38]
With the chirality of the molecule set, the amine was then installed. By using diphenylphosphoryl azide (DPPA) to convert 4 to an acyl azide, a Curtius rearrangement was used to prepare an isocyanate intermediate, which was used to generate carbamate 5 (Scheme 2).
Scheme 2: Synthesis of compound 6.

Reagents and conditions: (a) (i) DPPA, TEA, DCE, reflux; (ii) 9-fluorenemethanol, titanium (IV) tert-butoxide, benzene, rt; (b) HCl, 1,4-dioxane, reflux.
The transformation of 4 to a carbamate proved problematic due to the steric hindrance of the adjacent α-carbon as well as the bulkiness of the alcohols used. Copper (I) chloride, [39] titanium (IV) isopropoxide, and titanium (IV) tert-butoxide, [40] were tested as Lewis acids to assist in the conversion to the Fmoc protected amine, however titanium (IV) tert-butoxide was found to give the best yield. From 5, hydrolysis of the methyl ester yielded 6 which was used in synthesis of the target peptides.
Starting from the common synthetic intermediate 4, a divergent synthesis can be applied to access the S-enantiomer (Scheme 3) of the (αMe)Cys analogue. [37] To synthesize the S-enantiomer, the carboxylic acid of 4 was converted to a tert-butyl ester followed by the saponification of the methyl ester yielding 8. The carboxylic acid 8 is converted to an acyl azide, followed by a Curtius rearrangement to produce the isocyanate, which is then reacted with 9-fluorenemethanol to yield 9. The tert-butyl ester was then hydrolyzed with TFA to yield 10.
Scheme 3: Utilizing a divergent synthesis to form the opposite enantiomer of 6.

Reagents and conditions: (a) H2SO4, t-BuOH, DCM, rt; (b) LiOH, THF, H2O, rt; (c) (i) DPPA, TEA, DCE, reflux; (ii) 9-fluorenemethanol, titanium (IV) tert-butoxide, benzene, rt; (b) TFA, DCM, 0 °C.
With 6 in hand, we synthesized the peptide 11 of sequence: H-(αMe)Sec-Gly-Thr-Thr-Val-Arg-Asp-Tyr-Thr-Gln-OH, and the control peptide 12 of sequence: H-Sec-Gly-Thr-Thr-Val-Arg-Asp-Tyr-Thr-Gln-OH using standard solid phase Fmoc-based protocols. [41] Both peptides 11 and 12 correspond to amino acids 49–58 of the human GPX active site. For the control peptide, Fmoc-Sec(Mob)-OH was synthesized as reported previously. [42]
Sec and (αMe)Sec residues were coupled using HATU and DIC in DMF. [43] During cleavage of both peptides from the 2-cholortrityl resin, DTNP was included in the cleavage cocktail (TFA/TIS/H2O) to remove the Mob protecting groups, [44] and this deprotection afforded a mixture of three different forms of the peptide: (i) peptide diselenide, (ii) peptide selenol, and (iii) Sec(5-Npys) or (αMe)Sec(5-Npys) protected peptide. This crude peptide mixture containing these three redox states of the peptide were used without further purification for the enzyme assay. The HPLC chromatograms of each unpurified peptide is shown in Figure S36 of the Supporting Information.
A coupled glutathione reductase (GR) assay was used to test the abilities of the peptides to function as GPX mimics. [45] Both peptides 11 and 12 were able to reduce H2O2 significantly higher than the background rate. The rate for the (αMe)Sec-containing peptide (11) with 200 μM H2O2 was 23.50 ± 1.88 μM/min, while the rate for the Sec-containing peptide (12) was 17.96 ± 0.37 μM/min based on the slopes of the plot of A340 vs. time shown in Figure 3A. These values compare favorably to that of ebselen in our hands (38.84 ± 1.80 μM/min, data not shown). While the (αMe)Sec-containing peptide had only a slight kinetic advantage, a much larger difference in stabilities of the two peptides was observed as shown in Figure 3B, which shows the activity remaining after incubation in oxygenated KP buffer, pH 8.0, as a function of time.
Figure 3: GPX-like activity and stability of Sec- and (αMe)Sec-containing peptides.

(A) GPX-like activity of peptides 11 (blue) and 12 (red) assayed with H2O2. GSSG is formed as a by-product of the peptide catalytic cycle (Figure 1). GR reduces GSSG back to GSH using NADPH, and the consumption of NADPH can be measured via absorbance at 340 nm. (B) Percent activity remaining after incubation of 11 and 12 in 50 mM potassium phosphate buffer, pH 8.0, after 24 h and 96 h.
The loss in activity of the Sec-containing peptide in oxygenated buffer quantified in Figure 3B is most likely due to oxidation of the Sec residue followed by deselenization, which can occur by various mechanisms including β-syn elimination. Deselenization results in the appearance of a red precipitate due to the insolubility of elemental red selenium. Visual evidence for this phenomenon can be seen in Figure 4, which shows a comparison of deselenization of peptides 11 and 12. As can be seen, a red precipitate clearly forms after 96 hours in the tube with the Sec-containing peptide (12), but not in the (αMe)Sec-containing peptide (11). As is evident in Figure 4, no color change was observed in the reaction containing peptide 11 even after 96 hours of incubation. Fitting with these observations, the (αMe)Sec-containing peptide 11 maintained greater than 65% activity (Figure 3B).
Figure 4. β-syn elimination in a Sec-containing peptide exposed to oxygenated buffer, pH 8.0:

Peptides 11 and 12 (1 mM) were incubated in KP buffer, pH 8.0, for the indicated times above. After 96 hours β-syn elimination is clearly visible in peptide 12 (tube at right in each photo).
The (αMe)Sec-containing peptide cannot lose selenium through an oxidation/β-syn elimination pathway and thus this cannot be the reason for the modest loss in activity after 96 h in oxygenated buffer. Some possible explanations for the observed decrease in activity of peptide 11 are photo-degradation of the peptide due to the presence of selenium or oxidation of other amino acids.
To further investigate deselenization of peptides 11 and 12, we submitted them for MS analyses at the initial time point and after 96 hours of incubation in oxygenated buffer. The results of these analyses are shown in Figure 5. Initially both peptides exist as a mixture of three species: selenol (species A), diselenide (species B), and Sec(5-Npys) adduct (species C). The MS analysis of 12 provides clear evidence that β-syn elimination occurred upon 96 h of incubation in oxygenated buffer due to the presence of the dehydroalanine-containing peptide present in Figure 5D (species D). In contrast, the dehydroalanine species is absent from peptide 11 after incubation for the same time period (Figure 5B). In addition, the MS data in Figure 5D show that deselenization occurred by other mechanisms as evidenced by the presence of a truncated peptide (species E) and a peptide in which the Sec residue was converted into alanine via a radical mechanism (species F).[46] Species D-F are absent in the mass spectrogram of peptide 11 after incubation for the same time period as is evidenced by a much less complex mass spectrogram (compare Figures 5B and 5D).
Figure 5: Mass spectrograms of peptides 11 and 12 after 96 h of incubation in oxygenated buffer.

The arrow points to the observed m/z, given in the plot. The (αMe)Sec-containing-peptide (11) at time zero (A) and after 96 h incubation in KP buffer, pH 8.0 (B). For (αMe)Sec-peptides; species A is the peptide in selenol form (theoretical m/z = 1204.46), species A-NH3 is the ammonium adduct of A (theoretical m/z = 1220.46), species B is the peptide diselenide (theoretical m/z = 2406.91), and species C is the 5-Npys adduct (theoretical m/z = 1359.45). The Sec-containing-peptide (12) at time zero (C) and after 96 h incubation in KP buffer, pH 8.0 (D). For Sec-peptides; species A is the peptide in selenol form (theoretical m/z = 1190.45), species A-NH3 is the ammonium adduct of A (theoretical m/z = 1207.45), species B is the peptide diselenide (theoretical m/z = 2377.88), species C is the 5-Npys adduct (theoretical m/z = 1344.43), species D is dehydroalanine in place of Sec (theoretical m/z = 1108.51), species D-NH3 (theoretical m/z = 1125.52), species E is truncated peptide where Sec is missing (theoretical m/z = 1040.49), species E-potassium adduct (theoretical m/z = 1080.49), and species F is alanine in place of Sec (theoretical m/z = 1111.53).
While the mechanism of β-syn elimination of a seleninate and selenoxides are well understood, [2, 28, 47] we are unaware of any report where oxidation of a Sec-containing peptide results in cleavage of the peptide backbone. The cleavage product in which the Sec residue eliminated from peptide 12 completely (species E) is the most intense peak in the mass spectrogram visible in Figure 5D. This truncation is possible through a mechanism in which the seleninate oxygen attacks the carbonyl carbon of the peptide backbone, resulting in a 5-membered, cyclic seleninate ester. Deselenization of Sec to alanine (species F) has previously been reported and experimental evidence supported a radical deselenization mechanism. [46] Proposed mechanisms to explain the formation of species D-F from peptide 12 are shown in Figure 6.
Figure 6: Potential chemical mechanisms of deselenization of peptide 12.

The selenol in the sample can undergo oxidation to form the seleninate, which then undergoes rapid β-syn elimination to produce the dehydroalanine-containing peptide (species D). The truncated peptide (species E) is potentially explained by attack of the nucleophilic seleninate oxygen onto the carbonyl of the amide backbone, resulting in a cyclic seleninate ester and elimination of the Sec residue (lower pathway). Species F can be produced through a radical mechanism. Initiation of the radical could be photo-induced or alternatively, the selenolate can react with molecular oxygen to produce the selanyl radical and superoxide. The selanyl radical could recombine with molecular oxygen resulting in a seleninyl radical that can break down into selenium dioxide (SeO2) and an alanine-containing peptide.
As far as we are aware, the cleavage of the peptide backbone by a seleninate has not been reported in the literature and potentially represents a mechanism by which Sec-containing enzymes can be inactivated. The cleavage reaction is undoubtedly accelerated at basic pH as done here, and highlights the fact that Sec-containing enzymes may be better suited to work at acidic pH. [48] We do note however, that Adams and Macmillan report a related phenomenon whereby a side chain selenolate attacks the backbone carbonyl to form a selenoester, which can then be used for native chemical ligation. [49]
We believe a likely explanation for the absence of truncated product in peptide 11 is due to steric hindrance caused by the methyl group on the α-carbon. It is well known that the ϕ and ψ angles of α-methyl amino acids are restricted in comparison to amino acids lacking the methyl group at Cα. [50] The resulting rigidity introduced by the methyl group on Cα most likely prevents nucleophilic attack, explaining the absence of species E in peptide 11 upon prolonged incubation in oxygenated buffer.
An additional mechanism in which the Sec-containing peptide can be oxidized in buffer at basic pH is shown in Scheme 4. [51] Initially a significant fraction of both peptides are oxidized to the diselenide form. At basic pH, the diselenide can be oxidized to a selenoseleninate. The selenoseleninate can then undergo hydrolysis to yield the seleninate and a selenol. The selenol can reoxidize back to the diselenide to start the cycle over, or be converted to an alanine-containing peptide via a radical mechanism (species F). [46] The seleninate can breakdown via β-syn elimination or truncation to yield species D and E.
Scheme 4: Oxidation of the diselenide forms of peptides 11 and 12.

See the text for a description of the pathways.
In principle, the diselenide of the (αMe)Sec-containing peptide can undergo the same cycle of oxidation shown in Scheme 4 to yield the seleninate. However, the MS data in Figure 5B does not show evidence of the seleninate form being present. Either our analysis has not detected this intermediate form, or the diselenide of the (αMe)Sec-containing peptide is resistant to oxidation. Of course the seleninate form of the (αMe)Sec-containing peptide cannot undergo β-syn elimination or truncation due to the presence of the methyl group on Cα as discussed earlier. The selenol form of the (αMe)Sec-containing peptide could also be converted into α-methyl-alanine-containing peptide by a radical mechanism, but we also do not find evidence of this in our MS analysis.
CONCLUSIONS
In conclusion, we have synthesized (αM)Sec utilizing a common intermediate that can selectively access either enantiomer. The R-enantiomer of (αMe)Sec was incorporated into a 10-mer peptide corresponding to a fragment of GPX containing the catalytic Sec residue. This peptide demonstrated peroxidase activity that was 60% of that of ebselen, a known small molecule GPX mimic. Replacement of Sec with (αMe)Sec confers superior stability to the peptide as the oxidized form of (αMe)Sec cannot undergo β-syn elimination or cleavage of the backbone. This amino acid will undoubtedly find many chemical and biotechnological applications.
Supplementary Material
ACKNOWLEDGEMENT
We would like to thank Mr. Bruce O’Rourke of the Department of Chemistry at the University of Vermont for performing mass spectral analysis of the synthesized peptides reported here. These studies were supported by the National Science Foundation CAREER award MCB-0844478 to Doug Masterson, and the National Heart, Lung, and Blood Institute grant HL141146 to Robert J. Hondal. Robert J. Wehrle was supported by the National Science Foundation NRT grant 1449999. Emma J. Ste.Marie was supported by National Heart, Lung, and Blood Institute Grant 5T32HL007594–30 administered by Dr. Kenneth G. Mann and Dr. Robert J. Kelm.
These studies were supported by the National Science Foundation CAREER award MCB-0844478 to Douglas S. Masterson and and the National Heart, Lung, and Blood Institute grant HL141146 to Robert J. Hondal.
These authors made equal contributions to this work.
RJW was supported by the National Science Foundation NRT grant 1449999.
ESM was supported by National Heart, Lung, and Blood Institute Grant 5T32HL007594-30 administered by Dr. Kenneth G. Mann and Dr. Robert J. Kelm
ABBREVIATIONS
- A340
absorbance at 340 nm
- (αMe)Cys
alpha-methyl cysteine
- (αMe)Sec(5-Npys)
α-methyl Se-(2-thio(5-nitropyridyl))-L-selenocysteine
- (αMe)Sec
alpha-methyl selenocysteine
- Arg or R
arginine
- Asp or D
aspartic acid
- Cα
amino acid alpha carbon
- CDCl3
deuterated chloroform
- Cys
cysteine
- DCE
dichloroethane
- DCM
dichloromethane
- DHA
dehydroalanine
- DIC
N,N-diisopropylcarbodiimide
- DMF
N,N-dimethyl formamide
- DPPA
diphenylphosphoryl azide
- DTNP
2,2’-dithiobis(5-nitropyridine)
- ebselen
2-phenyl-1,2-benzisoselenazol-3(2H)-one
- EDTA
ethylenediaminetetraacetic acid
- ESI
electrospray ionization
- Fmoc
fluorenylmethyloxycarbonyl
- Fmoc-(αMe)Sec(But)-OH
N-Fmoc-S-(tert-butyl)-L-selenocysteine
- Fmoc-Sec(Mob)-OH
N-Fmoc-S-(4-methoxybenzyl)-L-selenocysteine
- FT-IR
Fourier-transform infrared spectroscopy
- Gln or Q
glutamine
- Gly or G
glycine
- GPX
vertebrate glutathione peroxidase
- GR
glutathione reductase
- GSH
glutathione
- GSSG
glutathione disulfide
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HOAt
1-hydroxy-7-azabenzotriazole
- HRMS
high resolution MS
- HCl
hydrochloric acid
- H2O2
hydrogen peroxide
- hν
light
- IR
infrared spectroscopy
- KP
potassium phosphate buffer
- LiOH
lithium hydroxide
- MgSO4
magnesium sulfate
- Mob
4-methoxybenzyl
- MS
mass spectrometry
- N2
nitrogen
- NADPH
β-nicotinamide adenine dinucleotide phosphate-reduced
- NaH
sodium hydride
- NaHCO3
sodium bicarbonate
- NaOH
sodium hydroxide
- H2SO4
sulfuric acid
- NH4Cl
ammonium chloride
- NMM
N-methyl morpholine
- NMR
nuclear magnetic resonance
- PLE
pig liver esterase
- Red-Al
bis(2-(methoxyethoxy)aluminum hydride
- rt
room temperature
- S
sulfur
- Se
selenium
- Sec or U
selenocysteine
- Sec(5-Npys)
Se-(2-thio(5-nitropyridyl))-L-selenocysteine
- Sec-SeO2–
seleninic acid
- SeO2
selenium dioxide
- SPPS
solid-phase peptide synthesis
- t-BuOH
tert-butyl alcohol
- TEA
triethylamine
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- Thr or T
threonine
- TIS
triisopropylsilane
- Tyr or Y
tyrosine
- UV-Vis
ultraviolet-visible
- Val or V
valine
Footnotes
SUPPORTING INFORMATION AVAILABLE
There are 36 figures in the Supporting Information consisting of: 1H-NMR, 13C-NMR, 77Se-NMR, and IR spectra for all of the synthesized compounds, HPLC chromatograms of the crude peptides, and a discussion of how the enantiomeric excess was determined.
DEDICATION
This paper is dedicated to Professor Ronald T. Raines on the occasion of this 60th birthday and his devotion to the field of peptide science.
REFERENCES
- [1].Flohe L, Günzler W, Schock H. Glutathione peroxidase: a selenoenzyme. FEBS lett. 1973; 32: 132–134. [DOI] [PubMed] [Google Scholar]
- [2].Rocher C, Lalanne JL, Chaudière J. Purification and properties of a recombinant sulfur analog of murine selenium‐glutathione peroxidase. Eur. J. Biochem 1992; 205: 955–960. [DOI] [PubMed] [Google Scholar]
- [3].Sands KN, Tuck TA, Back TG. Cyclic Seleninate Esters, Spirodioxyselenuranes and Related Compounds: New Classes of Biological Antioxidants That Emulate Glutathione Peroxidase. Chem. - Eur. J 2018; 24: 9714–9728. [DOI] [PubMed] [Google Scholar]
- [4].Tappel AL. Selenium–glutathione peroxidase: properties and synthesis. Current topics in cellular regulation: Elsevier; 1984. p. 87–97. [PubMed] [Google Scholar]
- [5].Toppo S, Flohe L, Ursini F, Vanin S, Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim. Biophys. Acta 2009; 1790: 1486–1500. [DOI] [PubMed] [Google Scholar]
- [6].Toppo S, Vanin S, Bosello V, Tosatto SC. Evolutionary and structural insights into the multifaceted glutathione peroxidase (Gpx) superfamily. Antioxid. Redox Signaling. 2008; 10: 1501–1514. [DOI] [PubMed] [Google Scholar]
- [7].Maulik N, Yoshida T, Das DK. Regulation of cardiomyocyte apoptosis in ischemic reperfused mouse heart by glutathione peroxidase. Stress Adaptation, Prophylaxis and Treatment: Springer; 1999. p. 13–21. [DOI] [PubMed] [Google Scholar]
- [8].Lesser R, Weiss R. Aromatic compounds containing selenium. VI. Ber. dtsch. Chem. Ges 1924; 57: 1077–1082. [Google Scholar]
- [9].Müller A, Cadenas E, Graf P, Sies H. A novel biologically active seleno-organic compound—1: Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (Ebselen). Biochem. Pharmacol 1984; 33: 3235–3239. [DOI] [PubMed] [Google Scholar]
- [10].Ogawa A, Yoshimoto T, Kikuchi H, Sano K, Saito I, Yamaguchi T, et al. Ebselen in acute middle cerebral artery occlusion: a placebo-controlled, double-blind clinical trial. Cerebrovasc. Dis 1999; 9: 112–118. [DOI] [PubMed] [Google Scholar]
- [11].Lynch E, Kil J. Development of ebselen, a glutathione peroxidase mimic, for the prevention and treatment of noise-induced hearing loss. Seminars in Hearing: © Thieme Medical Publishers; 2009. p. 047–55. [Google Scholar]
- [12].Kil J, Pierce C, Tran H, Gu R, Lynch ED. Ebselen treatment reduces noise induced hearing loss via the mimicry and induction of glutathione peroxidase. Hear. Res 2007; 226: 44–51. [DOI] [PubMed] [Google Scholar]
- [13].Yang CF, Shen HM, Ong CN. Intracellular thiol depletion causes mitochondrial permeability transition in ebselen-induced apoptosis. Arch. Biochem. Biophys 2000; 380: 319–330. [DOI] [PubMed] [Google Scholar]
- [14].Yang C-F, Shen H-M, Ong C-N. Ebselen induces apoptosis in HepG2 cells through rapid depletion of intracellular thiols. Arch. Biochem. Biophys 2000; 374: 142–152. [DOI] [PubMed] [Google Scholar]
- [15].Gogvadze V, Klein S, Shigenaga M, Ames B, Richter C. Effect of ebselen on Ca2+ transport in mitochondria. Redox Rep. 2000; 5: 359–363. [DOI] [PubMed] [Google Scholar]
- [16].Press DJ, Mercier EA, Kuzma Da, Back TG. Substituent effects upon the catalytic activity of aromatic cyclic seleninate esters and spirodioxyselenuranes that act as glutathione peroxidase mimetics. J. Org. Chem 2008; 73: 4252–4255. [DOI] [PubMed] [Google Scholar]
- [17].Tripathi SK, Sharma S, Singh HB, Butcher RJ. 2-Phenoxyethanol derived diselenide and related compounds; synthesis of a seven-membered seleninate ester. Org. Biomol. Chem 2011; 9: 581–587. [DOI] [PubMed] [Google Scholar]
- [18].Press DJ, McNeil NM, Hambrook M, Back TG. Effects of methoxy substituents on the glutathione peroxidase-like activity of cyclic seleninate esters. J. Org. Chem 2014; 79: 9394–9401. [DOI] [PubMed] [Google Scholar]
- [19].Selvakumar K, Singh HB, Butcher RJ. Aromatic ring strain in arylselenenyl bromides: Role in facile synthesis of selenenate esters via intramolecular cyclization. Chem. - Eur. J 2010; 16: 10576–10591. [DOI] [PubMed] [Google Scholar]
- [20].Selvakumar K, Singh HB, Goel N, Singh UP, Butcher RJ. Synthesis of Anionic Hypervalent Cyclic Selenenate Esters: Relevance to the Hypervalent Intermediates in Nucleophilic Substitution Reactions at the Selenium (II) Center. Chem. - Eur. J 2012; 18: 1444–1457. [DOI] [PubMed] [Google Scholar]
- [21].Singh VP, Singh HB, Butcher RJ. Synthesis of Cyclic Selenenate/Seleninate Esters Stabilized by ortho‐Nitro Coordination: Their Glutathione Peroxidase‐Like Activities. Chem. - Asian J 2011; 6: 1431–1442. [DOI] [PubMed] [Google Scholar]
- [22].McNeil NM, Press DJ, Mayder DM, Garnica P, Doyle LM, Back TG. Enhanced Glutathione Peroxidase Activity of Water-Soluble and Polyethylene Glycol-Supported Selenides, Related Spirodioxyselenuranes, and Pincer Selenuranes. J. Org. Chem 2016; 81: 7884–7897. [DOI] [PubMed] [Google Scholar]
- [23].Arai K, Kumakura F, Takahira M, Sekiyama N, Kuroda N, Suzuki T, et al. Effects of ring size and polar functional groups on the glutathione peroxidase-like antioxidant activity of water-soluble cyclic selenides. J. Org. Chem 2015; 80: 5633–5642. [DOI] [PubMed] [Google Scholar]
- [24].Back TG, Kuzma D, Parvez M. Aromatic derivatives and tellurium analogues of cyclic seleninate esters and spirodioxyselenuranes that act as glutathione peroxidase mimetics. J. Org. Chem 2005; 70: 9230–9236. [DOI] [PubMed] [Google Scholar]
- [25].Lamani DS, Bhowmick D, Mugesh G. Spirodiazaselenuranes: synthesis, structure and antioxidant activity. Org. Biomol. Chem 2012; 10: 7933–7943. [DOI] [PubMed] [Google Scholar]
- [26].Lamani DS, Bhowmick D, Mugesh G. Substituent effects on the stability and antioxidant activity of spirodiazaselenuranes. Molecules. 2015; 20: 12959–12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cho C-S, Lee S, Lee GT, Woo HA, Choi E-J, Rhee SG. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid. Redox Signaling. 2010; 12: 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ma S, Caprioli RM, Hill KE, Burk RF. Loss of selenium from selenoproteins: conversion of selenocysteine to dehydroalanine in vitro. J. Am. Soc. Mass Spectrom. 2003; 14: 593–600. [DOI] [PubMed] [Google Scholar]
- [29].Jones JH. Abbreviations and symbols in peptide science: a revised guide and commentary. J. Pept. Sci 2006; 12: 1–12. [DOI] [PubMed] [Google Scholar]
- [30].Kaiser E, Colescott R, Bossinger C, Cook P. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem 1970; 34: 595–598. [DOI] [PubMed] [Google Scholar]
- [31].Reich HJ, Jasperse CP, Renga JM. Organoselenium chemistry. Alkylation of acid, ester, amide, and ketone enolates with bromomethyl benzyl selenide and sulfide. Preparation of selenocysteine derivatives. J. Org. Chem 1986; 51: 2981–2988. [Google Scholar]
- [32].Iwaoka M, Ito S, Miyazaki I, Michibata M. Synthesis of l-Selenocysteine and α-Methyl-l-Selenocysteine Derivatives Using Woollins’ Reagent and Their Application as Chiral Selenium Catalysts. Proc. Natl. Acad. Sci., India, Sect. A 2016; 86: 499–509. [Google Scholar]
- [33].Makiyama A, Komatsu I, Iwaoka M, Yatagai M. One-Pot Conversion of Serine and α-Methylserine Derivatives to the Corresponding Cysteines and Selenocystines by Using Chalcogenophosphate Reagents. Phosphorus Sulfur Relat. Elem 2010; 186: 125–133. [Google Scholar]
- [34].Kotapati HK, Lawrence DR, Thames SO, Masterson DS. Enzyme mediated concise synthesis of NH-Fmoc-S-Trityl-Cα-Methyl Cysteine. Tetrahedron Lett. 2016; 57: 4389–4391. [Google Scholar]
- [35].Masterson D, Kedrowski B, Blair A. An Improved Method for the Preparation of Protected (R)-2-Methylcysteine: Solution-Phase Synthesis of a Glutathione Analogue. Synlett. 2010; 19: 2941–2943. [Google Scholar]
- [36].Block E, Birringer M, Jiang W, Nakahodo T, Thompson HJ, Toscano PJ, Uzar H, Zhang X, Zhu Z. Allium chemistry: synthesis, natural occurrence, biological activity, and chemistry of Se- alk(en)ylselenocysteines and their gamma-glutamyl derivatives and oxidation products. J. Agric. Food Chem 2001; 49: 458–470. [DOI] [PubMed] [Google Scholar]
- [37].Masterson DS, Roy K, Rosado DA, Fouche M. A divergent approach to the preparation of cysteine and serine analogs. J. Pept. Sci 2008; 14: 1151–1162. [DOI] [PubMed] [Google Scholar]
- [38].Kedrowski BL. Synthesis of orthogonally protected (R)- and (S)-2-methylcysteine via an enzymatic desymmeterization and Curtius rearrangement. J. Org. Chem 2003; 68: 5403–5406. [DOI] [PubMed] [Google Scholar]
- [39].Kapferer P, Vasella A. Electrophilic Bromination ofN-Acylated Cyclohex-3-en-1-amines: Synthesis of 7-Azanorbornanes. Helv. Chim. Acta 2004; 87: 2764–2789. [Google Scholar]
- [40].Spino C, Joly MA, Godbout C, Arbour M. Ti-catalyzed reactions of hindered isocyanates with alcohols. J. Org. Chem 2005; 70: 6118–6121. [DOI] [PubMed] [Google Scholar]
- [41].Ste.Marie EJ, Ruggles EL, Hondal RJ. Removal of the 5‐nitro‐2‐pyridine‐sulfenyl protecting group from selenocysteine and cysteine by ascorbolysis. J. Pept. Sci 2016; 22: 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hondal RJ, Raines RT. Semisynthesis of proteins containing selenocysteine. Methods Enzymol. 2002; 347:370. [DOI] [PubMed] [Google Scholar]
- [43].Han Y, Albericio F., Barany G. Occurance and minimization of cysteine racemization during stepwise solid-phase peptide synthesis. J. Org. Chem 1997; 62:65. [DOI] [PubMed] [Google Scholar]
- [44].Harris KM, Flemer S, Hondal RJ. Studies on deprotection of cysteine and selenocysteine side‐chain protecting groups. J. Pept. Sci 2007; 13: 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wilson SR, Zucker PA, Huang RRC, Spector A. Development of synthetic compounds with glutathione peroxidase activity. J. Am. Chem. Soc 1989; 111: 5936–5939. [Google Scholar]
- [46].Dery S, Reddy PS, Dery L, Mousa R, Dardashti RN, Metanis N. Insights into the deselenization of selenocysteine into alanine and serine. Chem. Sci 2015; 6: 6207–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Andreadou I, Menge WM, Commandeur JN, Worthington EA, Vermeulen NP. Synthesis of novel Se-substituted selenocysteine derivatives as potential kidney selective prodrugs of biologically active selenol compounds: evaluation of kinetics of β-syn elimination reactions in rat renal cytosol. J. Med. Chem 1996; 39: 2040–2046. [DOI] [PubMed] [Google Scholar]
- [48].Lacey BM, Flemer SJ, Eckenroth BE, Hondal RJ. Selenium in thioredoxin reductase: A mechanistic perspective. Biochemistry. 2008; 47: 12810–12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Adams AL, Macmillan D. Investigation of peptide thioester formation via N→Se acyl transfer. J. Pept. Sci 2013; 19: 65–73. [DOI] [PubMed] [Google Scholar]
- [50].Karle IL, Balaram P. Structural characteristics of. alpha.-helical peptide molecules containing Aib residues. Biochemistry. 1990; 29: 6747–6756. [DOI] [PubMed] [Google Scholar]
- [51].Caldwell K, Tappel A. Reactions of seleno-and sulfoamino acids with hydroperoxides, Biochemistry. 1964; 3: 1643–1647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.