

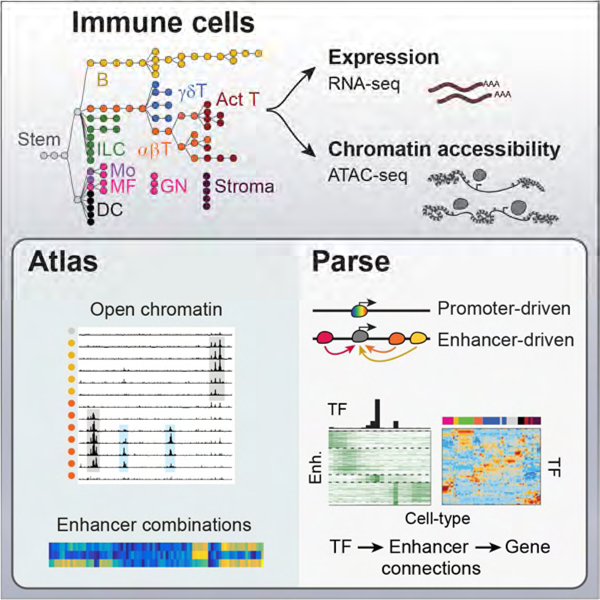
Summary
A complete chart of cis-regulatory elements and their dynamic activity is necessary to understand the transcriptional basis of differentiation and function of an organ system. We generated matched epigenome and transcriptome measurements in 86 primary cell-types that span the mouse immune system and its differentiation cascades. This breadth of data enables variance components analysis which suggests that genes fall into two distinct classes, controlled by either enhancer- or promoter-driven logic, and multiple regression that connects genes to the enhancers that regulate them. Relating transcription factor (TF) expression to the genome-wide accessibility of their binding motifs classifies them as predominantly openers or closers of local chromatin accessibility, pinpointing specific cis-regulatory elements where binding of given TFs is likely functionally relevant, validated by ChIP-seq. Overall, this cis-regulatory atlas provides a trove of information on transcriptional regulation through immune differentiation, and a foundational scaffold to define key regulatory events throughout the immunological genome.
Graphical Abstract
In Brief
A cis-regulatory map of the mouse immune system illuminates gene expression patterns and regulatory logic across 86 primary cell types and pairs immune transcription factors with cell type-specific regulatory elements.
INTRODUCTION
The establishment and maintenance of a cell’s transcriptional identity is largely driven by the specific activity of cis-regulatory elements: promoters at which initiation complexes are assembled around RNA Polymerase II (Pol-II), or distal enhancer elements that facilitate Pol-II loading and/or release from poised configuration. The time- and location-specific expression of a gene in differentiated states results from the combined activity of the several enhancers that control it, each of which may have a different regulatory logic, driven by the combinatorial activity of transcription factors (TFs) and chromatin remodelers. How enhancer activity is coordinated and integrated to define related, but functionally distinct, cell-types remains elusive, leaving two main questions: How do cis-regulatory landscapes vary between lineage-related cell-types to promote cellular identity? How do changes in the activity of cis-regulatory elements program the differentiation cascade of cell lineages? The mouse immune system represents an excellent setting to interrogate the interplay between epigenome and transcription: major cell states are well characterized, discrete cell populations can be readily purified, and the differentiation from common progenitors is well established, through pathways that can be parsed up to ten successive steps (Hardy and Hayakawa, 2001; Rothenberg, 2014), such that it is possible to address these questions and interrogate transitional stages at high granularity.
Previous large-scale efforts have profiled epigenomic differences across differentiation to reveal a highly diverse landscape of cis-regulatory element activity, point to master transcriptional regulators and key cis-regulatory elements (ENCODE Consortium, 2012; Roadmap Epigenomics, 2015). However, these have primarily involved cell lines in culture, or whole organs which mask regulatory heterogeneity. Few large scale programs have systematically paired epigenomic maps with gene expression measurements from primary cells, restricting the ability to infer the impact of epigenomic changes to functional consequences in gene expression. Other studies have focused on well-defined groups of cells ex vivo (Lavin et al., 2014; Yu et al., 2017), or run broader surveys of hematopoietic differentiation (Lara-Astiaso et al., 2014) but a cis-regulatory analysis that is both wide-ranging and fine-grained has not been carried out.
Here, we use low input epigenomic and transcriptomic profiling to generate matched measurements in 86 unique immune cell populations that span the entire immune system of the mouse, from granulocytes to terminally differentiated plasma cells. This atlas of open chromatin regions (OCR) is both comprehensive, defining the cis-regulatory space in the quasi-entirety of immunocytes, and highly granular by scanning closely related cell-types. This breadth and unique coherence of these data allows us to infer the activity of cis-regulatory elements and assign to many TFs specific locations in shaping the unique transcriptome of each cell-type, and reveal generally applicable insights on the relative roles of promoters and enhancers in differentiated gene expression.
RESULTS
We generated matched ATAC-seq and RNA-seq data for 86 immune cell populations, representing lymphoid and myeloid hematopoietic lineages, along with key stromal cell populations (Fig. 1A, Table S1). Lymphocytes included very granular differentiation cascades along the T and B lineages; myeloid cells included neutrophil (GN), macrophage (MF), monocyte (Mo) and dendritic cell (DC) populations stemming from either embryonic yolk sac or adult bone marrow precursors and sampled from different tissues (Guilliams et al., 2014). These cells were purified across 11 ImmGen participating laboratories, in biological duplicates (Table S1).
Figure 1. Overview for the chromatin and RNA profiling of broad immune cell populations.

(A) Cell-types in this study shown in a differentiation tree, color-coded by lineage. Stromal cells, and myeloid cell-types known to derive from embryonic precursors, are shown unconnected to the HSC-derived tree. (B) Representative pile-up traces of ATAC-seq signals, all to the same scale, for three genomic regions: Spi1 (encodes PU.1); Cd8, with previously determined enhancer elements shown (E8I to E8VI, top, red arrows denote novel OCRs in cDCs); the Hprt promoter as a housekeeping gene. mRNA levels are indicated by barplots at the right of each locus; *: no matching RNA-seq data. (C) A t-SNE representation of all OCRs identified in this study. Top panel: the Gini index characterizes OCRs that are broadly accessible (blue) or cell-type specific (red); middle: OCRs specifically open in progenitors or dendritic cells; bottom: OCRs at TSS or that contain CTCF motifs. See Fig. S1.
The sorted cells were jointly processed for expression profiling by low-input RNA-seq, and for chromatin accessibility analysis by fast-ATAC-seq (Corces et al., 2016), a simplified version of the original protocol (Buenrostro et al., 2013) which increases the proportion of reads within OCRs and allows lower cell inputs (10,000 cells), important here given the rarity of many immune cell-types (transitional stem/progenitor cell stages or innate-like lymphocytes (ILCs) could be analyzed). Rigorous quality control steps ensured data homogeneity (thresholds on mapped paired ends, on the enrichment of reads mapping to transcription start sites, and on depth-adjusted inter-replicate correlation). We obtained high quality ATAC-seq profiles for 86 cell-types (Table S1; only mast cells failed, likely from interference by heparin). To determine the full atlas of open chromatin across the immune system, we first called OCRs in individual datasets with usual thresholds (MACS FDR 0.01), supplemented by additional OCRs identified by merging reads from related cell-types. We thus identified 512,595 OCRs (FDR 0.01), whose activity index was normalized across cell-types by quantile normalization (Table S2). We then parsed 14,292 OCRs connected to transcriptional startsites (TSS, RefSeq) vs 498,303 mapping to more distal locations (hereafter “distal enhancers” (DE), acknowledging that not all are necessarily true enhancers in the functional sense).
The results, a virtually complete perspective on accessible chromatin across immune lineages, present a fascinating portrait of enhancer and promoter activity (Fig. 1, ImmGen Chromatin databrowser). Several match known immunogenomics, but others were novel and unexpected. For example, many of the OCRs detected in the Cd8 locus correspond to (and help position) the enhancer elements mapped in classic studies of T cell differentiation (Issuree et al., 2017): some OCRs are active prior to transcription (E-8II), others only in mature CD8+ T cells (E8-VI) (Fig. 1B). We also identified previously unknown elements: Cd8 expression in DCs coincides with a novel OCR specific to CD8+ classic DCs (cDCs) and another solely active in plasmacytoid DCs (pDCs) (Fig. 1B). The difference in regulatory strategies among DCs was also visible in many other loci, e.g. the defining Itgax locus (encodes CD11c; Fig. S2). Another example was the activity of the Spi1 enhancer (encodes PU.1), which extinguished as expected at the DN2a-DN2b transition, coincident with commitment to T cell fate (Yui and Rothenberg, 2014)
To visualize the genome-wide diversity of OCR activity across immune cell-types, we used a tSNE algorithm to project every OCR into a 2-dimensional space, revealing distinct substructures in the data and its variability (Gini index, Fig. 1C). OCRs particularly active in a lineage naturally tended to cluster, as shown for progenitors and DCs. Interestingly, many of the OCRs mapping to TSS regions clustered together in an eccentric region of low variability, suggesting a degree of conformity within promoters as a group (many others did scatter throughout, though, denoting some cell-type specificity). As detailed below, we mapped the TF binding motifs associated with each OCR. OCRs associated with the structural factor CTCF, an essential anchor of chromatin loops, mapped homogeneously to a central region of limited variability. This pattern is consistent with the notion that topological domains and loops are conserved between cell lineages, even if their transcriptional activity differs (Dixon et al., 2016; Hnisz et al., 2016).
Expression variance explained by chromatin accessibility
Our extensive data provide an opportunity to determine, on a large scale, the relationship between chromatin accessibility and gene expression. In keeping with previous reports (Corces et al., 2016), cell/cell correlation matrices computed from chromatin accessibility at DE OCRs yielded sharper distinctions between differentiated cell-types than those drawn from expression profiles (Fig. 2A). Furthermore, DE OCRs showed more discrimination between cell-types than TSS OCRs, consistent with the isolation and limited variance of TSS OCRs on the tSNE plots (Fig. 1C).
Figure 2. Integrated ATAC-RNA variance decomposition: parsing enhancer influence.

(A) Matrices of Pearson correlation between cell-types, based on ATAC signal intensity at all TSS-OCR, all DE OCRs, or mRNA levels in RNA-seq. Color-coding of cell-types at right per Fig. 1A. (B) Variance component decomposition of the mRNA expression for every gene (as column), in a variance component model that discretizes the explanatory power of DE- or TSS-OCRs (blue and green, respectively), the proportion of unexplained variance being shown in red. (C) Enrichment in TF-binding sequence motifs (signed -log10 p, Fisher test) in the promoter-proximal region (−1000 to +1 bp) of genes with DE-logic and TSS-logic determinism (from 2B). See Fig. S2.
Packaging and accessibility of DNA in chromatin are the first level of control on gene expression in differentiated cells, setting patterns that are secondarily modified by splicing or differential mRNA stability. We sought to determine how much differences in chromatin accessibility can explain differences in expression of individual genes. Variance component models, such as those used in genome-wide association and eQTL studies (Chen et al., 2016; Ye et al., 2014), can identify quantitative variables associated with relatedness between observations by explicitly modeling sample covariance as random effects. Here, we applied an analogous concept using variance component models to quantify the proportion of variation in gene expression that could be attributed to covariance in chromatin accessibility. For each of the 15,600 expressed genes, we fit a set of variance components models, including both TSS OCR covariance and DE OCR covariance in the model, to attribute the expression levels variance of each gene to either promoter or enhancer covariance patterns. For clarification, these relationships are not between a gene and the accessibility of its own promoter or enhancers, but to the overall status of all enhancers or promoters. For most the genes, more than 90% of the expression variation could be explained, confirming that gene expression generally follows chromatin accessibility (Fig. S3A, Table S3A). These results were robust with respect to the number of OCRs used in computing covariance matrices, as assessed by iterative downsampling of the OCR sets (Fig. S3B). Strikingly, this analysis revealed two distinct groups of genes (Fig 2B): one for whom >99.0% of the expression variance could be best explained by TSS OCR covariance (943 genes, including Cdca3, Hprt); and a larger group (4,409 genes, including known lineage specifying genes like Pax5 or Foxp3) best explained by DE OCR covariance (DE OCRs mapping to gene bodies or to extragenic regions behaved identically). This observation suggests a dichotomy between sets of active genes, whose expression follows an “enhancer logic” or a “promoter logic”. These sets differed significantly in their range and variability of expression (higher and less variable for the TSS-logic set) but not in GC content (Fig. S3C), with a surfeit of housekeeping and cell cycle-related genes in the TSS-logic group (Table S3B–D).
This dichotomy suggested fundamentally different modes of transcriptional regulation, and we hypothesized that these groups may coopt different sets of TFs. We compared the enrichment of TF-binding motifs in the −1kb>TSS region of the two gene sets (a span which would encompass the promoter and some proximal enhancers). Members of the ETS/ELK family were more associated with TSS-logic genes, while members of the KLF family were over-represented in the promoters of genes of the DE-logic group (Fig. 2C, Table S3E). Altogether, these results suggest that gene expression in differentiated mouse immunocytes is cued by global patterns of chromatin covariation, but follow two different modes.
Cis regulation of gene expression: linking enhancers to genes
One of the recurring difficulties in mapping enhancer elements is in establishing the link between a regulatory element and the gene(s) it regulates. Although enhancers often map inside or within a few kb of genes they regulate, and “closest gene” is often taken as a rough proxy to hypothesize an enhancer’s target, there are documented instances of enhancers mapping megabases away from their target gene (Bahr et al., 2018). We hypothesized that correlation across cell-types between the accessibility of an enhancer and the expression of a given gene denotes a functional connection, a determination facilitated by the unique breadth and granularity of the present data. For illustration, such a correlation could be detected between the expression of Samd3 and accessibility of an OCR located 1,320 bp upstream of its TSS (Fig. 3A). This correlation extended genome-wide (Fig. 3B, Table S3F). By globally assessing accessibility/expression correlation we identified at least one significantly associated cis OCR within a 1Mb window from the TSS for 7,444 of the 15,601 expressed genes (Bonferroni p<0.05). Predictably, the remaining genes that were not associated with a cis OCR were enriched in the set of TSS logic genes identified above (p<10−20). These correlated OCRs preferentially mapped in the close vicinity of the correlated gene (50% of best correlated OCRs are within 13Kb of the gene’s TSS), in essence vindicating the usual approach, with an exponentially decaying relationship between distance and correlation (Fig. 3C). Given this observation, to reduce the number of spurious associations, we restricted our further analysis to 334,879 OCRs that fall within 100 Kb of some TSS. In many instances, each gene was associated with several significantly correlated cis OCRs (Fig. 3D), including some highly complex regions with multiple associations such as the Il7r locus, correlated to 21 nearby OCRs (Fig. 3E). For these genes with multiple correlated OCRs, the OCRs tended to be correlated to each other (64% of the OCRs correlated with one gene were themselves correlated (at Bonferroni p<0.05)). These multiple OCRs likely represent repeated regions that operate with the same regulatory controls.
Figure 3. Landscape of cis-regulation: associating genes with specific enhancers.

(A) Example of correlated mRNA expression (x-axis) and OCR accessibility (y-axis) at the Samd3 locus. (B) As in A, but correlation between expression and activity of a strongly associated DE-OCR for 1000 genes. (C) Distance distribution of DE-OCRs that are strongly correlated (Bonferroni p<0.05) to a given gene, relative to the gene’s TSS. (D) Number of significantly associated DE-OCRs for each gene. (E) Chromatin accessible landscape of the Il7r locus for all cell-types (histogram of expression at right). Red arcs correspond to 21 OCRs that share significant correlation with Il7r expression; non Il7r associations are shown in black (height reflects association p-value). (F) ATAC-seq signal in the promoter and enhancer regions of Rag1 and Rag2 loci in B and T precursors (right: mRNA expression). Previously reported DNAse-I hypersensitivity sites are indicated below. *: newly identified OCRs. (G, H) Identification by multiple regression of OCRs that complementarily explain the expression patterns of Tyrobp and Cd28; heatmaps denote accessibility at these OCRs; the bar histogram mRNA expression. See Fig. S3.
In other instances, genes were surrounded by OCRs with clearly different patterns of activity. An anecdotal but striking example was noted upstream of Rag1-Rag2, where two distinct OCRs are activated in tight connection with the appearance of Rag transcripts in either B or T cells (Fig. 3F), suggesting that T and B lineages have different solutions to tightly control B or T expression. We thus sought to broadly identify sets of independent OCRs for each target gene, using the ability of stepwise regression to identify independent explanatory variables. For a substantial number of genes (n=493), two or more independent signals were identified in the 100 Kb regions (Fig. S2D, Table S3G). These “secondary” OCRs may contain cell-specific regulatory elements to fine tune expression in different lineages. For example in the Tyrobp locus (encodes DAP12), the regression identified a second OCR associated with expression in the B cell lineage, and a third active in NK cells (Fig. 3G). Around Cd28, an OCR whose range of activity includes plasma cells [likely related to CD28 function there (Delogu et al., 2006)] is complemented by another OCR uniquely active in T cells (Fig. 3H).
Timing of OCR activation during lymphocyte differentiation
We then investigated more closely the changes in OCR activity that accompany T and B lymphocyte differentiation, attempting to track changes in regulatory elements that underlie these multistep cascades. At the two main cell-specific loci in the T lineage, Cd4 and Cd8, classic analyses have mapped a number of functionally important enhancer elements (Issuree et al., 2017). As noted above, several of the OCRs at the Cd8 locus showed activity in the differentiation series prior to the appearance of Cd8a transcripts (i.e. E8-I and E8-II in DN3 and DN4). OCRs were also found at several known enhancer elements of Cd4, with the expected timing of activation [e.g. E4T, E4p and E4D (Issuree et al., 2017); Fig. S4A]. The S4 silencer was accessible in mature CD8+ T cells, indicating that silencing here is likely an ongoing process. Several hitherto unrecognized elements were also observed (red labels in Fig. S4A), whose function begs to be elucidated: an OCR very close to the S4 silencer and specifically active in CD4+ cells, several OCRs active in cDC or pDCs.
To consider more generally how OCR opening relates to changing gene expression, we selected a broad set of transcripts that are stably induced or extinguished during T differentiation, most at the point of T cell fate commitment (DN2a/DN2b, (Yui and Rothenberg, 2014), or at CD4+CD8+ “double-positives” (DPs) (Fig. 4A). Aggregating DE OCR accessibility in a 10 kb window around the TSS (but excluding TSS OCRs, and constitutively accessible DE OCRs) showed that this aggregate accessibility largely tracked with gene expression (Fig. 4B); in contrast, there was little relation with accessibility at these genes’ TSS (not shown). Thus, the T cell differentiation cascade also seems to follow an enhancer-driven logic, rather than a TSS-driven one. Bolstering the significance of these correlated OCRs was that they showed significant enrichment in binding motifs for TFs known to be involved in controlling T differentiation (Fig. S4B), such as Tcf12(HEB), Lef1, Tcf7(TCF-1), Tcf3(E2A), and Zbtb7b (ThPOK).
Figure 4. Timing of OCR activation.

(A) Heatmap representing expression of 496 genes that vary most through T cell differentiation (ordered by k-means, color-coded relative to the 95th expression quantile). (B) Integrated accessibility of variable DE-OCR in the −10 kb>−250 bp region of these genes (order as in A). C: Timing of enhancer activation: for genes which are induced during T differentiation, the dots denote the cell-stage at which mRNA expression first reaches 50% of max (x-axis) vs the stage at which the best correlated OCR (from Fig. 3) first reaches 50% of max accessibility. See Fig. S4.
We then investigated the dynamics of OCR activation, asking whether the early enhancer activation relative to transcription observed with several Cd8 enhancers is a general rule. We compared the differentiation stage at which a given gene’s mRNA level, or the accessibility at its most correlated OCR, reach 50% of their maxima (and conversely drop to 50% or their initial max for repressed loci). Strongly skewed patterns were observed, wherein OCRs mainly became open before the onset of transcription (Fig. 4C). Genes whose expression increased sharply at the DP stage already had active enhancers after the DN2a>DN2b transition, while activation of expression at the time of positive selection to T4 single-positives was foretold by activation of their enhancers in late DN stages. Thus, consistent with prior observations in early B and myeloid differentiation (Mercer et al., 2011), turning on enhancers precedes the actual activation of the loci, at several steps in T cell differentiation.
Trans regulation: TF effects on chromatin accessibility
Paired epigenomic and transcriptomic data across a large set of cell-types provides a powerful opportunity to relate epigenomic variation to the activity of specific TFs, by correlating the activity of an OCR to the TF binding sites (TFBS) it contains. To identify cell- and lineage-specific TFs that may influence chromatin accessibility, we first mapped TFBS present in each of the 334,879 robust OCRs (curated TFBS list from CisBP, per Schep et al., 2017; Table S4 for TF motifs associated to each OCR at p<0.1). We compared the aggregate accessibility in each cell-type of all OCRs containing a given motif, relative to a background set of OCRs matched in %GC and average accessibility, (Schep et al., 2017). This deviation analysis, which yields a “TFBS accessibility score” for each TF motif in each cell, identified 76 TF motifs significantly associated with chromatin accessibility differences (Fig. 5A).
Figure 5. Regulators of chromatin accessibility.

(A) Transcription factor motif accessibility scores, TFs in rows (z-normalized), cell-types in columns (hierarchically clustered). (B–E) Relationship between the expression of a TF and its motif accessibility score for representative factors; each point represents a cell-type, color-coded per Fig. 1A. (F) Accessibility score of Bcl11a motifs (top; cells arranged per Fig. 1A) in relation to the expression of Bcl11a or Bcl11b (bottom). (G) Pearson coefficient and significance of correlations between TF motif score and TF expression (generalized from B–E); known regulators of immune cell development and function are highlighted in red. (H) Expression patterns of the TFs determined to significantly correlate with changes in chromatin accessibility (positive correlation: top block; negative: bottom). Side bars: motif variability and correlation coefficient. See Fig. S5.
TFs of the same family tend to bind the same or similar motifs (e.g. Gata family members all bind the canonical Gata motif). To disambiguate the relationship between the accessibility of a motif and the actual TFs that binds it, we compared the TFBS accessibility score to the expression of the corresponding TFs. For some like Pax5, the key regulator of B cell differentiation, there was a simple correlation between expression of the TF and the accessibility of its motif (Fig. 5B). For Tbx21 (encodes T-Bet, Fig. 5C) the relationship was less linear, the motif only becoming accessible at the highest expression levels, possibly denoting dose-dependence, cofactor requirement, or competitive displacement effects. This analysis also identified several repressive relationships: Pbx1, a negative regulator of stem cell differentiation (Ficara et al., 2008), and Zbtb7b, the classic repressor of the CD8-lineage transcriptome in CD4+ T cells (Wang et al., 2008) both showed decreased accessibility of their motif at higher TF expression (Fig. 5D, E). In other cases, by calculating the similarity of the TF motifs used, we defined pairs of TFs that cooperate to modulate the accessibility of target enhancers. For instance, the hematopoietic regulator Bcl11a correlated positively with accessibility of the Bcl11a motif, but Bcl11b, which shuts down the B or myeloid differentiation potential in early thymic progenitors, negatively correlated with accessibility (Fig. 5F).
Such correlations between accessibility and expression were generalized to all TFs with associated TFBS accessibility scores (Fig. 5G), identifying 61 activators and 18 repressors (Table S5) confirmed by .permutation analysis (Fig. S5A). The expression of these chromatin accessibility regulators (Fig. 5H) paints a uniquely rich portrait of immune cell differentiation, with regulators which appear to act individually (EOMES, Pax5) and are uniquely correlated to activity of OCRs that contain their motifs, and regulators that operate interchangeably to regulate a common motif such as RUNX1/2/3, which all promote accessibility of the Runx motif. Some TFs are expected to be refractory to such an analysis and showed no such correlation: FoxP3 because of its binding to previously open elements (Samstein et al., 2012), Nfatc1 because its regulation is based in intracellular localization rather than abundance, Stat1 because the present dataset may poorly capture its rapid induction. With this reservation, this integrated approach enabled us to define some of the key TFs which positively or negatively shape chromatin accessibility in immune cells.
Transcriptional regulation in myeloid cells
Having broadly characterized the trans regulatory relationships between TFs and OCRs, we looked more specifically at OCR activity patterns in the myeloid compartment. Previous studies profiled the epigenomic state of tissue resident macrophages and other myeloid cells (Bornstein et al., 2014; Lavin et al., 2014), but the breadth of the present data provided additional perspective. Clustering of myeloid cells based on OCR activity grouped them largely according to lineage and tissue residency (Fig. 6A, Table S6). There was differential OCR activity between resident macrophages of different tissues, consistent with prior reports (Lavin et al., 2014). GN and pDCs were more distant from other myeloid lineages, somewhat unexpectedly for pDCs, as they are closely related with cDCs (Reizis et al., 2011). Indeed, pDCs had a high number of distinct OCRs from cDCs and other myeloid cells, and pDCs were more similar to T cells for Cd8 OCRs (Fig. 1B), supporting the proposition that pDCs arise from a spectrum of progenitors with myeloid and lymphoid potential (Reizis et al., 2011).
Figure 6. Regulatory factors in myeloid cells inferred from chromatin accessibility.

(A) Differential OCR signals across all steady-state myeloid cells (right: numbers of distinguishing OCRs; top: correlation tree). (B) TF motif enrichment scores (chromVAR z-test) in myeloid group-specific OCRs from A, filtered for TF expression levels and statistical significance, with signed -log10 p values capped at 100 for display; bars shaded by TF mRNA expression. (C) Comparison of TF enrichment scores for OCRs accessible CD4+ and CD8+ cDCs, points are shaded according to the TF’s mRNA expression fold change between the two DC subsets. See Fig. S6.
A major question in myeloid biology is what factors drive the programs of closely related but functionally distinct cells. We used the TFBS resource described above to identify motifs enriched in OCRs uniquely active in certain cell types (Fig. 6B). Several enriched motifs corresponded to TFs with established roles. For example, the binding motif for Tcf4 (encodes E2–2) was amongst the most enriched in pDCs, and E2–2 has an essential role in pDC biology (Cisse et al., 2008). Enrichment of critical tissue macrophage regulators was apparent: Gata6 in peritoneal cavity (PC) macrophages (Rosas et al., 2014), Mef2c in microglia (Deczkowska et al., 2017; Lavin et al., 2014). These served as useful validation, we also noted a number of novel associations that may warrant further investigation: Bach1 in PC macrophages, Egr2 in alveolar macrophages, and Zeb1 in Neutrophils. The motif for Ehf, a gene with expression restricted mostly to cDCs and epithelial cells (Fig S6A), was enriched in cDC specific OCRs. This gene has roles in the regulation of inflammation and antigen transport in epithelial cells (Asai and Morrison, 2013; Fossum et al., 2017), and may have a similar function in cDCs.
CD4+ and CD8+ cDCs are developmentally and functionally related, yet have subtle but important differences in antigen presentation and in their transcriptomes (Miller et al., 2012). We identified several thousand differential OCRs between them, with dissimilar enrichments in some TF motifs (Fig. 6C). The differentiation of CD8+ cDCs is dependent on Irf8 and Batf3 (Hildner et al., 2008; Tamura et al., 2005) and those motifs were correspondingly enriched in CD8+ DC OCRs. In contrast, CEBP family motifs were enriched in OCRs specific to CD4+ cDCs. Motifs enriched in CD103+CD11b− intestinal DCs, which are also Irf8-dependent (Ginhoux et al., 2009), were similar to those of CD8+ DCs (Fig S6B), including strong enrichments of transducers of type I IFN signaling Stat2 and Irf9. The differential accessibility at steady-state of OCRs predicted to bind Stat2 and Irf9, suggests that Irf8-dependent cDCs may be specifically poised to respond to IFN-I. This type of signaling is specifically required by CD8+ and CD103+ DCs to promote cross presentation and an increased anti-viral state (Diamond et al., 2011; Helft et al., 2012).
TFs that control OCR dynamics during lymphocyte differentiation
We applied the same powerful logic as for Fig. 5 of correlating the presence of a TF motif, the activity of the OCR, and the expression of the corresponding TF, to discover relevant sites of action for particular TFs in T and B lymphocyte differentiation. In Fig. 7A, we plot the activity of OCRs with the best score for the RORγ-binding. Clustering these OCRs based on ATAC-seq intensity identified 6 patterns of OCR accessibility. Among those, one cluster (cl3) exhibited a clear relationship to the expression of Rorc gene in thymic DPs (Fig. 7A, Table S7A; p<10–4), and these OCRs coincided precisely with demonstrable RORγ binding from ChIP-seq data (Guo et al., 2016) (Fig. 7A, right). Another cluster (cl4) seemed to respond to RORγ in colonic T regulatory (Treg) cells; some of the specific OCRs fall in close proximity with genes that are differentially expressed in RORγ+ Tregs, such as Il23r (Table S7A). That different RORγ-binding OCRs are active in DP thymocytes and colonic RORγ+ Tregs is consistent with the notion that RORγ controls different transcriptional targets in a context-dependent manner (Sefik et al., 2015). The OCR clusters whose activity does not correlate with RORγ expression may correspond to false-positives from motif prediction, or to TFs that share the same binding motif. To validate the prediction that cl3 represents OCRs whose accessibility depends on RORγ, we performed ATAC-seq in DPs from Rorc-deficient mice (Rorcgfp/gfp). Strikingly, almost all OCRs from the DP-specific cl3 disappeared in RORγ–deficient DPs (Fig. 7B), while those of the uncorrelated cl6 were unaffected. Thus, RORγ seems to operate as a pioneer factor.
Figure 7. Dynamics of chromatin accessibilities on TF-motif containing OCRs along differentiation.

(A–D) Cell-type-dependent accessibility for OCRs that contain RORγ, Zbtb7b or Pax5 motifs (top 1000 predicted OCRs, clustered by k-means; top: mRNA levels; right: TF motif scores and ChIP-seq signals averaged per cluster). (B) Normalized ATAC-seq intensity for OCRs that contain an RORγ-binding motif of cluster3 or cluster6 (from A), in immature DP thymocytes of Rorc-deficient mice or -positive littermates. (E) Chromatin accessibility for 1080 DE-OCRs known to bind FoxP3 in ChIP-seq experiments. Distal OCRs are classified as constitutive or dynamic (2-fold higher signal in Tregs than in precursor cell-types). ChIP-seq signal in these OCRs for Mediator, Cohesin, or histone marks in Tregs are shown below. (E) TF motif enrichment score in constitutive and dynamic FoxP3-binding OCRs. See Fig. S7.
ThPOK (Zbtb7b) and Runx3 are key TFs for the branched differentiation of CD4+ and CD8+ T cells (Ellmeier and Taniuchi, 2014; Xiong and Bosselut, 2012). For Runx3, which is under dominant translation control, our approach would not be informative, but for ThPOK the correlative approach proved highly suggestive: accessibility of many of the OCRs that contain its cognate motif was curtailed in T cells in which ThPOK was present (Fig. 7C and Table S7B), especially those in cl2. This negative correlation (see also Fig. 5G) is consistent with the dominant suppressive function of ThPOK, suggesting that it shuts down the CD8+ T lineage program not only by inhibiting Runx3 expression but also by directly inhibiting a swath of enhancer elements.
Together with EBF1, Pax5 is the major TF defining B lymphocyte identity, essential for both early development and to maintain the function of mature B cells (Horcher et al., 2001; Medvedovic et al., 2011). It is expressed throughout B cell differentiation before being silenced in plasma cells (Shi et al., 2015). Pax5’s aggregated OCR scores directly correlate with its expression (Fig. 5B, validated by prior ChIP-seq data (Revilla-I-Domingo et al., 2012)), but a clustered analysis of OCRs that contain its binding motif revealed a striking dynamic variation: OCRs in cl2 and cl3 became active at the proB.FrBC stage, only transiently for cl2, more stably for cl3. OCRs of cl5 only became active later, in germinal center stages after immune activation, before being extinguished in plasma cells concomitant with the loss of expression (Fig. 7D, Table S7C). These different behaviors of Pax5-binding OCRs are consistent with its context-specific activity (Revilla-I-Domingo et al., 2012).
FoxP3 is the key controller of Treg development and function (Ramsdell and Ziegler, 2014). It is not considered as a pioneer factor, but binds and modifies active enhancer elements (Samstein et al., 2012). With the unique landscape available here, we revisited the status of FoxP3 binding sites across immunocyte differentiation. Among 2,000 high-confidence FoxP3-binding sites (Kitagawa et al., 2017; Kwon et al., 2017; Samstein et al., 2012) we identified 1,080 DE OCRs that were accessible in Tregs (Table S7D,E). Consistent with prior conclusions (Samstein et al., 2012), many of these DE OCRs (80%) were constitutively accessible, from stem cells onwards, and even in B or myeloid cells (Fig. 7E); all FoxP3-binding OCRs that mapped to TSSs behaved similarly (Fig. S7A). Another set of FoxP3-binding OCRs behaved more dynamically, only becoming active after the DP stage, suggesting control by thymic positive selection events. A small but distinct minority of these FoxP3-binding OCRs were Treg-specific (Fig. 7E, right), suggesting that FoxP3 opens these regions, alone or with other Treg determining cofactors. Interestingly, the constitutive and dynamic OCR sets were markedly distinguished by their associated histone marks [Fig. 7E, bottom; data from (Kitagawa et al., 2017)]. All were H3K27Ac-positive enhancer elements, but the active enhancer mark H3K4Me1 was much more prevalent among dynamic than constitutive OCRs (Fig 7E, bottom, Fig S7b). In addition, Nfkb(1/2)-binding motifs were specifically enriched in dynamic FoxP3 OCRs (Fig 7F), consistent with the role of NF-kB family members in Treg differentiation (Oh et al., 2017). Ets and Lef1 binding sites were enriched in both classes on OCRs, while Forkhead or Runx motifs were preferentially present in constitutive FoxP3-binding OCRs. Thus, these analyses reveal the existence of two classes of FoxP3-binding enhancer elements. One is constitutively open in many immunocytes, while the other is activated with final Treg differentiation, and seems to electively involve the NF-kB family of TFs. These examples highlight the power of these data to map OCRs that truly respond to a given TF and identify relevant binding sites (see immgen.org for a larger set of 300 TFs).
DISCUSSION
We profiled chromatin accessibility and gene expression in 90 cell types to generate a cis-regulatory atlas that encompasses the entire span of lineages that compose the mouse immune system. The paired chromatin/transcriptome approach, the focus on immunocytes, and the unprecedented granularity of the data enabled us to move beyond an epigenomic roadmap, providing a platform to infer causal regulatory interactions. Besides providing a deep resource of great value to understand immunological differentiation and function, the data bring insights of broad relevance on the role and positions of enhancer elements, reveal a deep dichotomy within mammalian gene regulation, and illuminate the relation between transcription factor activity and chromatin configuration.
Establishing this “complete” landscape of 512,595 cis-regulatory elements was enabled by sampling a large repertoire of closely related cell types, borderline significance of an ATAC peak in one cell-type being bolstered by related cells. This fine mapping of cell state transitions enabled the analysis of regulatory interactions, which would not be possible with epigenomic data obtained from whole tissues, or from a partial sampling of specific cell-types. We anticipate that future efforts with even finer parsing of some lineages, as well as single-cell approaches, may lead to an even more precise atlas. In the discussion and interpretation of the present data we have assumed that OCRs distant from known TSS are likely to be enhancer elements. While some OCRs may correspond to other structures (e.g. TSS of unrecognized transcriptional units, or non-enhancer structural elements), the “rediscovery” of known enhancers in the vicinity of the Cd4 and Cd8 loci support the validity of this assumption.
We connected a number of OCRs to the expression of a nearby gene, based on the plausible assumption that such a correlation between accessibility of a cis-regulatory element and expression of a gene signifies a functional relationship. It has long been a conundrum to formally associate a cis-regulatory element with the gene(s) it might regulate. The “closest gene” is usually the default call, even though it is known that some enhancers can be effective from very long distances. The results of Fig. 3C give some support to this general notion, by showing that genes are mostly associated with enhancers within 20kb or less of their TSS. Indeed, the “closest gene” assignment is likely correct 90.2 % of the time (from Table S3F). Widespread redundancy was another aspect of enhancer activity revealed by this analysis, as most genes with an enhancer correlated to their expression actually had several correlated enhancers (Fig. 3E), themselves inter-connected. That enhancers are often repeated has been recognized from their first description (Banerji et al., 1981; Benoist and Chambon, 1981), and a recent study showed that 64% of D. melanogaster loci have redundant “shadow” enhancers (Cannavò et al., 2016). Redundancy may provide functional buffering and evolutionary flexibility and robustness (Hong et al., 2008; Osterwalder et al., 2018), or allow fine-tuning of a gene’s transcription in slightly different stages or states, or be mechanistically more efficient, synergistic binding of the same TFs at closely spaced sites helping to stabilize an enhanceosome complex. Finally, we observed very few cases of silencer elements (defined as accessibility negatively correlated with expression of the target gene), suggesting that positive enhancement is the predominant mode of gene regulation in mammalian transcription.
We observed a striking partition between one set of genes whose activity seemed cued by the overall pattern of activity of all distal enhancers and another that was aligned to activity in promoter regions, with enrichment for different TFBS in the promoters of each class. There are precedents for such divergence. For instance, the housekeeping Hprt locus contains no discernible enhancer (Gasperini et al., 2017), and enhancer catalogs have generally shown them to be tissue-specific (Shen et al., 2012). This dichotomy may be related to the demonstration in Drosophila of enhancer/promoter specificities that distinguish housekeeping and differentiation-linked regulatory programs (Zabidi et al., 2015). More generally, it relates to the long-lasting debate on the differences between promoters and enhancers. Promoters are classically defined as sites that focus transcription initiation by recruiting Pol-II and basal transcription factors, while enhancers supercharge the promoters they target to increase the rate of transcription. But whether they truly represent different entities has been nuanced or challenged (Kim and Shiekhattar, 2015). The present results suggest that they do play fundamentally different roles in orchestrating transcription. .
Beyond establishing the rich landscape of cis-regulatory regions, the three-way correlation between activity of an OCR, the TF motifs it contains, and the expression of the TFs allows for accurate and pointwise predictions of how TFs orchestrate immunocyte differentiation and function. We note that this approach is blind to some classes of TFs: opportunistic TFs that exploit already accessible chromatin regions (e.g. FoxP3), those whose regulation is post-transcriptional, those controlled by modification or localization (STATs, NF-AT). Our analyses accurately predicted TFs whose activity is associated with specific lineages and stages (Fig. 5), and which specific OCRs (and genes) are actually activated by these TFs (Fig. 7, S7), this on an unprecedented scale. These include well-known TFs for which existing ChIP-seq data provide valuable validation, but also TFs for which no such data were available (Gata2, Nfe2 or Eomes). Several of these profiles reveal a strong context dependence for TF action, e.g. for RORγ and Pax5, consistent with RORγ’s different footprint in Th17 vs colonic Tregs (Sefik et al., 2015), and with Pax5’s variable involvement along the B cell lineage (Revilla-I-Domingo et al., 2012), perhaps depend on cell-type specific post-translational modifications, co-factors or ligands.
The analysis also reveals that TFs can have either positive or negative consequence on accessibility of an OCR. The former is readily conceptualized (docking of the factor displacing nucleosomes and/or recruiting additional chromatin modifiers), the latter less so, since closing of the element would potentially hinder the TF from binding. Some “hit-and-run” mechanism that instructs stable repressive histone marks or DNA methylation may be at play. It is generally thought that individual TFs can both activate or repress transcription, depending on local context. Our analyses (Fig. 5G) suggest that many TFs dominantly play either activating or repressive roles (since dual function would result in no correlation overall). TFs with negative correlation between expression and motif accessibility includes a Who’s Who of known repressors (Pbx1, Bcl11b, Zbtb7b). A majority of TFs, however, had positive effects on accessibility, prompting the speculation that opening of chromatin is the dominant mode of control for the unfolding of gene expression through immune cell differentiation. This coonclusion is consistent with observations that target activation by Pax5 and PU.1 correlated positively with DNA binding (Champhekar et al., 2015; Revilla-I-Domingo et al., 2012). Mechanistically, PU.1 has recently been shown to indirectly repress genes in T cell progenitors via TF theft; recruiting partner TFs to its own directly activated genes and thus depleting them from their own targets (Hosokawa et al., 2018).
In conclusion, this resource provides an atlas of cis-regulatory elements that will be leveraged by the community to guide focused experiments to understand the regulation of a particular locus through immune function or disease. This cis-regulatory atlas may serve as an initial scaffold on which to systematically build, through complementary “multi-omics” strategies, additional knowledge towards a complete understanding of genomic regulation in immune cells.
STAR METHODS
Contact for reagent and resource sharing:
Further information and requests for resources and reagents should be directed to the Lead Contact, Christophe Benoist (cbdm@hms.harvard.edu).
Experimental model and subject details:
Mice
C57BL/6 mice were obtained from the Jackson Laboratory, as were B6.Rorctm2Litt (Jax7572), housed under SPF conditions (HMS IACUC protocol 02954). Young adult males or females (as listed in Table S1) were used at 5–6 weeks of age. Rorc-deficient mice were generated in (+/− x +/−) crosses, and +/+ or +/− littermates were used as controls.
Method details:
ATAC-seq Data Generation
To ensure consistency in the data, the different immune cell populations were sorted and frozen in each participating laboratory, but all processing, library construction and sequencing were performed jointly in the ImmGen core lab. As a pilot for this multi-site program, all participating labs generated samples of total splenic CD19+ B cells, a readily sorted cell population (which led to some refinement of the procedure and provided a baseline of inter-replicate variance).
Mice were sacrificed and immunocytes were isolated to high purity by flow cytometry according to ImmGen SOP using the antibodies and gates indicated in Table S1, Fig S1A. two rounds of sorting were performed to collect 10,000 cells (exceptions for Cd34-LTHSC, Cd34+LTHSC and STHSC populations for which 677, 2483 and 3660 cells were sorted, respectively) in 1.5mL DNA lo-bind tubes (#022431021, Eppendorf) containing 100uL of BAMBANKER (serum-free cell freezing medium, No.302–14681, Wako). Cells were kept on ice at most 30 minutes and immediately stored at −80°C following a slow-freeze procedure; (cell freezing container with isopropyl alcohol at a rate of −1°C/minute with temperatures decreasing from 4°C to −80°C).
ATAC-seq libraries were prepared as previously reported (Corces et al., 2016) with the following modifications. Frozen cells were thawed, washed with 1mL of PBS containing protease inhibitors (Complete EDTA-free protease inhibitor cocktail, Roche Diagnostics, Basel, Switzerland) and cell pellets were resuspended in 10uL of Tn5 transposase mixture: 1x Tagment DNA Buffer, 0.5uL Tagment DNA Enzyme (Nextera DNA Library Preparation Kit, Illumina) and 0.2mg/ml digitonin (#G9441, Promega) on ice. Cells were incubated at 37°C for 30 minutes with agitation followed by DNA isolation using the MinElute Reaction Cleanup Kit (Qiagen, Hilden, Germany). Construction of ATAC-seq libraries included an initial round of PCR in a total volume of 50uL using the NEBNext High-Fidelity 2X PCR Master Mix (New England Biolabs, MA, USA) with primers (0.5uM each) from (Buenrostro et al., 2015) with the following thermal cycles: 5 minutes at 72°C, 30 seconds at 98 °C, followed by 7 cycles [98°C for 10 seconds, 63°C for 30 seconds and 72°C for 60 second s] and a final extension at 72°C for 5 minutes. PCR products were purified and size-selected using Agencourt AMPure XP beads (Beckman Coulter) (0.65x and 1.8x volume to remove long and short fragments respectively) and eluted in 18uL of EB (Qiagen). To avoid over amplification of libraries which result in GC bias, 2uL of the eluted DNA were subjected to qPCR (StepOnePlus Real-Time PCR System, Life Technologies) in a volume of 20uL using SYBR GreenI dye (final 0.6x SYBR GreenI, Life Technologies) and with the respective primers (1.25uM each), as performed in the first round of PCR. Following qPCR [30 seconds at 98°C, followed b y 30 cycles (98°C for 10 seconds, 63°C for 30 seconds and 72°C for 60 seconds)], amplifica tion curves were analyzed and the optimal number of PCR cycles for each sample were estimated with cycle thresholds reaching ¼ of the maximum. Upon selecting the cycle threshold, 12.5uL of the eluted DNA were subjected to a second round of PCR in a volume of 50uL with NEBNext High-fidelity 2x PCR master mix, respective primers (1.25uM each) and the following thermal cycles: 30 seconds hot-start at 98°C, followed by 7~13 cycles [98°C for 10 seconds, 63°C for 30 seconds and 72°C for 60 seconds] and a final extension at 72°C for 5 minute s. The libraries were purified by Agencourt AMPure XP beads (x1.8 vol.), quantified by qPCR using Power SYBR Green PCR Master mix (ThermoFisher) and universal sequencing primers (P5_FW:5’AATGATACGGCGACCACCGA and P7_RV:5’CAAGCAGAAGACGGCATACGA, 0.2uM each) and pooled, which were sequenced as paired-end (38+37bp) on an Illumina NextSeq 500 instrument in high-output mode.
ATAC-seq Quantification and Normalization
After trimming adapter sequences and low quality reads using sickle1.2 (https://github.com/najoshi/sickle), short reads were mapped to mm10 reference genome using bowtie2 with the following options; -X 1000 –fr, while non-unique, ChrM mapping (0.1~20%, median=4.1%) and duplicated reads (7~69%, median=22%) were filtered out using samtools view -q 30 [samtools 0.1.19] and Picard Tools (Picard MarkDuplicates, http://broadinstitute.github.io/picard). The summary of ATAC-seq read statistics can be found in Table S1. Paired-end reads spanning less than 120 bp were used for determining the peak summits in all populations using MACS2 functions (--call-summits) (https://github.com/taoliu/MACS). Open chromatin regions (OCR) of a 250 bp width were centered on all summits selecting the peak summit with the most significant q-value, when compared with ATAC-seq signals in 332,233 regions. Formally, 2 to 181 samples were grouped according to a hierarchical clustering with various cut-offs in order to achieve sequencing depth and estimate the peak summits for all populations. A window of 250 bp was used onset based off centered summits similarly in the first step, which resulted in 518,845 ATAC-seq OCRs. As some OCRs can arise as sequencing-based artifacts (ENCODE Project Consortium, 2012) and may also share sequence homology with the mitochondrial genome, we removed possible artifact OCRs by filtering blacklisted genomic regions and chrM homologous regions (a blacklist was downloaded from: https://sites.google.com/site/anshulkundaje/projects/blacklists).
ChrM homologous regions were identified by mapping short mitochondrial DNA sequences to the mouse nuclear chromosomes, consisting of 7,889 genomic regions in total. We report 512,595 cumulative OCRs (ImmGenATAC1219.peak_1 ~ 512595) across our cis-regulatory atlas. For the analysis of TSS (transcriptional start sites) and DE (distal enhancers) connected OCRs, we designated these OCRs as TSS connected (i.e., OCRs of which summit is within 125bp upstream or downstream of TSS, as all OCRs are 250bp width centered on the summit) and all others as DE connected OCRs. 27,921 TSS positions were defined from UCSC annotation data on mm10 (http://hgdownload.cse.ucsc.edu/goldenPath/mm10/database/refFlat.txt.gz, downloaded Jan. 2017). We employed the same reference data to assign close-by genes for each OCR as reported in Table S2.
To compute signal intensity in each OCR, reads mapped to the plus strand were shifted by +4 bp and reads mapped to the minus strand by −5 bp. Secondly, edges of fragments corresponding to paired reads were tested for OCR overlapping using BEDTools2.25.0 [bedtools intersect (Quinlan and Hall, 2010)]. A fragment edge in an OCR was counted unless the other edge of the fragment mapped to the same OCR in order to avoid counting non-independent Tn5 insertion events. A pseudo count of 0.1 was added to edge counts in peaks, log2-transformed and normalized by quantile normalization. For calculating the cell population mean, the quantile-normalized counts were converted back to linear scale and means of replicates were calculated (Table.S2A). Backgrounds were estimated based on the ATAC-seq signals of regions through random sampling with p-values for each OCR computed using a negative binominal distribution of the background in each sample. Data were also adjusted for multiple hypothesis testing using the Benjamini and Hochberg (BH) method. All population p-values are supplied in Table S2.
Replicates for each population were merged and paired reads spanning less than 120 bp regions were used to compute ATAC-seq pile-up traces for each population using MACS2, which were further normalized by quantile normalization across 25bp bins. To visualize the data, we used IGV (http://software.broadinstitute.org/software/igv/). The assembled data can be analyzed interactively on the USCS platform via the ImmGen Chromatin browser (http://rstats.immgen.org/Chromatin/chromatin.html).
ATAC-seq QC
Data quality control analyses were performed for each sample and across the projects by: (1) counting the number of properly mapped paired ends, setting a threshold of 2,470,102 as acceptable across this project (range 2,470,102 to 16,029,540, median 6,841,995). (2) computing signal enrichments around the TSS relative to genomewide average, a metric which identifies datasets with high signal to noise ratios (Corces et al., 2017) (Table S1); a value > 3.9% was considered acceptable across this project (range 3.9% to 31.8%, median 12.5%). (3) Concordance between the two biological replicates. We selected, for each cell-type, a subset of OCRs in which the raw edge counts were >= 10 in at least one replicate, which was used to compute a Pearson correlation between the two replicates. The Pearson coefficient is sensitive to the total number of reads (as evidenced by the B cell pilot, Fig. S1B left). The samples retained for the analysis exhibited comparable inter replicates correlations to the trend estimated from B cell pilots (green line, Fig. S1B right).
RNA-seq
RNA-seq was performed with the standard ImmGen low-input protocol. A total of 1,000 cells were sorted directly into 5ul of lysis buffer (TCL Buffer (Qiagen) with 1% 2-Mercaptoethanol). Smart-seq2 libraries were prepared as previously described (Picelli et al., 2014) with slight modifications. Briefly, total RNA was captured and purified on RNAClean XP beads (Beckman Coulter). Polyadenylated mRNA was then selected using an anchored oligo(dT) primer (5′–AAGCAGTGGTATCAACGCAGAGTACT30VN-3′) and converted to cDNA via reverse transcription. First strand cDNA was subjected to limited PCR amplification followed by Tn5 transposon-based fragmentation using the Nextera XT DNA Library Preparation Kit (Illumina). Samples were then PCR amplified for 18 cycles using barcoded primers such that each sample carries a specific combination of eight base Illumina P5 and P7 barcodes for subsequent pooling and sequencing. Paired-end sequencing was performed on an Illumina NextSeq 500 using 2 × 25bp reads.
Low quality reads were trimmed using sickle1.2 and the adapter sequence with TrimGalore (version0.4.0,http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Short reads were then mapped to mm10 genome using hisat2 [version2.0.4 (https://ccb.jhu.edu/software/hisat2/manual.shtml)] with --transcriptome-mapping-only --no-discordant options. Unmapped and low quality scoring (MAPQ<5) reads were removed using samtools. Moreover, duplicated reads were removed using the Picard MarkDuplicates function. Properly paired reads were selected by samtools view -f 0×02 and counted for each gene using htseq-count (version0.6.1) with -s no option and a GTF file from UCSC mm10 refGene downloaded from UCSC table browser (https://genome.ucsc.edu/cgi-bin/hgTables). Genes with a minimum read count of 5 in all replicates of a population (17,535 genes) were retained. A pseudo count of 1 was added and log2-transformed prior to quantile normalization. Quantile-normalized counts were converted back to a linear scale and means of replicates were calculated for each population (Table S2C). The number of reads for each processing step can be found in Table S1B.
Quantification and statistical analysis:
Dimensionality Reduction and Visualization with t-SNE
To visualize the 334,879 OCRs sampled from our collection of populations, we performed a t-Stochastic Neighbor Embedding of the OCR x cell-type count matrix. Specifically, we derived the top 25 principal components from the 334,870 × 86 matrix using the implicitly restarted Lanczos bidiagonalization algorithm (irlba). Next, we embedded this high-dimensional chromatin accessibility landscape into a two-dimensional coordinate system using the Barnes-Hut implementation of t-SNE through the Rtsne package with default parameters (perplexity = 30). Individuals peaks were assigned binary TF motif matches based on predicted binding affinities of the mm10 sequence and correspondingly colored (see below for motif matching analysis). For sample populations-based coloring, each peak was assigned a population with the maximum chromatin accessibility observed from the normalized counts matrix. Finally, for each peak in our data set, we computed the Gini Index over the populations, yielding a per-peak measure of “chromatin inequality” about the populations. The Gini Index for peak i, denoted Gi, was computed as:
where xi,j represents an element in the the log2 normalized counts matrix for peak i and population j.
OCR Variance Component Analysis
We applied variance component models to characterize how patterns of chromatin covariance (Fig. 2a) explained observed gene expression variance within our sorted populations. As a variance components model assumes normally distributed noise, we utilized a variance stabilizing transformation proposed by Anscombe (Anscombe, 1948) to model the empirical a negative binomial distribution of RNA-seq count data. Specifically, for each gene (indexed by i), the vector of normalized gene expression counts per cell-type, Yi, was transformed into a new vector from centering and scaling using Anscombe’s transformation:
where ϕ is the dispersion chosen so as to minimize the ratio of the dispersion of the residual standard deviation as implemented in the Varistran package (Harrison, P.F. The Journal of Open Source Software 2). With our transformed gene expression vector j, we then fit the following variance component model:
where D and T are the sample-sample correlation matrices computed from the distal enhancers and transcription start site OCRs respectively (see Fig. 2a) and I is the identify matrix. Average information restricted likelihood estimation (AIREML) was used to estimate the values of the parameters of the variance component models, σd, σt, and σe. To then determine the proportion of the variance explained by each variance component, we generated a vector Vi, which by definition sums to 1:
Here, the proportion of the variance in expression for gene i explained by the DE logic would be represented by the first element in the Vi vector.
Associating OCRs with Target Genes
Data normalization and aggregation: We defined “expressed” genes as those with at least 10 reads in at least one cell population. Using this filter, we removed lowly expressed genes, and retained expression data for 15,601 genes. This filtered gene expression data was then log transformed, quantile normalized, and averaged across replicates. Similarly, ATAC-seq data was filtered to exclude OCRs with low intensity (BH adjusted MACS2 p-value > 0.05). The intensities across the cell populations were log transformed, quantile normalized, and averaged across replicates. We excluded Stromal cells from this analysis: because of the large biological differences, data from this population has very different distributional properties compare to the others.
Association analysis: In simple association analysis, for each expressed gene, we identified all OCRs that are within 1Mb of the gene’s TSS. Then, for each gene and “cis” OCR pair, we computed the Pearson correlation coefficient and the associated p-value to quantify the association between activity (intensity) of the OCR and expression level of the gene across all 81 cell populations (samples). We used Bonferroni correction, to adjust the resulting p-values. In addition, we used stepwise regression to identify independently associated OCRs nearby each gene. For each gene, we performed stepwise regression analysis with the gene expression level of a particular gene as outcome and the intensity of OCRs within 100Kb of the corresponding gene as the predictors. Similarly, we accounted for multiple testing using Bonferroni correction.
Computing Aggregated OCR Scores
As demonstrated in Fig. 3C, we observed a “distance biased” relationship between OCR intensity and gene expression levels, whereby those cis OCR that are closer to TSSs are more strongly associated with gene expression levels. On the basis of this observation, we constructed an aggregated OCR score for each gene as the inverse weighted sum of OCR intensities within 100Kb of its TSS. More specifically, for a given gene, each OCR within 100Kb of the gene’s TSS was given a weight 1/d where d is the absolute distance (in bp) between the gene’s TSS and the center of the OCR.
Annotating OCRs with motifs
To annotate OCRs with putative transcription factor binding motifs, we used the motifmatchr package as part of the chromVAR suite of tools (Schep et al., 2017). Motifs were defined from a set of curated mouse position weight matrices (PWMs) from the cisBP database (http://cisbp.ccbr.utoronto.ca/) publicly available at (https://github.com/buenrostrolab/chromVARmotifs). For each OCR and motif pair, we determined a binary annotation for compatibility of the motif PWM in the mm10 reference sequence from the OCR. Specifically, our background nucleotide frequency was the total nucleotide content over all OCRs, and a motif match was called for sequences with a p-value < 5×10^−6. Note that these choices are identical the defaults provided in the motifmatchr package.
Associating Aggregated Motif Scores with Transcription Factor Expression
Deviation scores, referred to as “TFBS accessibility scores” throughout the text, were calculated using chromVAR with the default parameters (Schep et al., 2017) and the chromVAR motif database “mouse_pwms_v2”. To compute the correlation between scores and TF expression we excluded epithelial cells whose patterns were too divergent, then filtered TFs for motif-TF expression pairs, wherein the maximum TF expression in a measured cell type was greater than 4 (log2 scale), resulting in 430 TFs (see Table S5G for the full list of TF motif and expression pairs). To calculate correlation between deviation scores and TF expression, log2 transformed gene expression counts were correlated (Pearson) to raw deviation scores. To calculate the statistical significance of the correlation two permutation tests were performed: we either permuted the sample labels or the TF labels (100 permutations with replacement), P-values were calculated using a z-test comparing the observed TF motif-expression correlation coefficient to the permuted correlation coefficient. Reported values represent the max (least significant) of the two permutation approaches, TFs with P-values less than 0.1 are called as significant. Notably, we found the two permutation approaches provided correlated P-values however, permuting TFs labels generally provided less significant P-values.
To compute correlation for myeloid and lymphoid TFs, the same approach was repeated for samples identified as lymphoid (LTHSC.34-.BM; LTHSC.34+.BM; STHSC.150-.BM; MMP4.135+.BM; preT.DN1.Th; preT.DN2a.Th; preT.DN2b.Th; preT.DN3.Th; DN4.Th; T.ISP.Th; T.DP.Th; T.4.Th; T.4.Nve.Sp; Treg.4.25hi.Sp; Treg.4.FP3+.Nrplo.Co; T.8.Th; T.8.Nve.Sp; T8.TN.P14.Sp; T8.TE.LCMV.d7.Sp; T8.MP.LCMV.d7.Sp; T8.Tcm.LCMV.d180.Sp; T8.Tem.LCMV.d180.Sp) or myeloid (Mo.6C+II-.Bl;Mo.6C-II-.Bl; MF.PC;MF.RP.Sp; MF.Alv.Lu;DC.103+11b-.SI; DC.103+11b+.SI; DC.4+.Sp;DC.8+.Sp; MF.microglia.CNS; GN.BM;GN.Sp;DC.pDC.Sp; MF.226+II+480lo.PC; MF.ICAM+480hi.PC) cell types.
Motif Enrichment in TSS
To determine motifs associated with DE-logic and TSS-logic genes identified from the variance components analysis (Fig. 2), we performed two Fisher Exact tests per-motif. For each of the 15,600 expressed genes, we determined all motif matches from the motif collection within 1kb upstream of the annotated TSS. We then determined which motifs were enriched in the set of 943 TSS-logic genes from a first set of Fisher Tests (y-axis Fig. 2C), or enriched in the set of 4,409 DE-logic genes in a second set of Fisher Tests (x-axis Fig. 2C).
To assess of motif enrichment in selected sets of OCRs in the myeloid and T cell lineages (Fig.6, Fig.S4) we employed a parametric test using motif frequency distributions calculated from GC-content matched background sets of OCRs, otherwise referred to as “chromVAR z-test for motif enrichment”. First, after identifying OCRs to be tested (i.e. GN specific OCRs), 200 sets of GC-content matched OCRs were selected, using the ‘getBackgroundPeaks’ function from chromVAR, out of the robust set of 334,879 OCRs in the study. Background frequency distributions for each motif were then calculated from the background OCR sets using the OCR to motif pairing described above. Signed P values were then determined by the probability of obtaining the test set motif frequency in the background distribution and multiplying by the sign of the direction of effect, assuming a normal probability distribution for the background.
Analysis of Myeloid Lineage
Myeloid clustering and peak selection: OCRs were filtered for only those detected in at least one myeloid cell sample (BH adjusted MACS2 p-value < .05; 215,583 peaks). The filtered peak signals (log2 + 1) were used for hierarchical clustering with 1 – Pearson correlation distance values and average linkage between clusters. Cell groups were formed by performing a tree cut on the dendrogram at a distance height of .21. Sets of peaks for each cluster were then identified by looking either for peaks specifically detected in a given cell group and no other myeloid cell types or for peaks with a minimum log2 peak signal fold change greater than 2 of that group compared to all other myeloid cells.
Myeloid motif enrichment: chromVAR z-test motif enrichment was performed on the selected sets of peaks for each group. Only the top 15 or fewer motifs having an unadjusted log10 signed p-value greater than 5, and linear normalized gene counts greater than 100 in the population showing the enrichment were displayed in Fig. 6B. SI macrophages not displayed due to lack of sequencing data.
cDC comparisons: Peaks for CD4 and CD8 DCs were selected by looking for peaks that were detected in one subset and not the other (BH adjusted MACS2 p-value < .05), a minimum peak signal of 4, and a log2 peak signal fold change greater than 2 between the two cell types. chromVAR z-test motif enrichment was then run on these sets independently. The same analysis was run between the CD103+CD11b+ and CD103+CD11b− DC populations in the SI.
Analysis of T Lineage
We identified 836 “T cell differentiation genes” whose expression varied the most during the T cell differentiation by combining the following groups: 1) 543 differential expressed genes by computing the mean and coefficient of variation from 12 cell populations along the T lineage ranging from MPP4.135+.BM to T.4.Nve.Sp and T.8.Nve.Sp and fitting generalized linear model on mean and squared CV (top 5% variable of the expressed genes); 2) 345 CD4 T cell related genes whose expression are significantly different between (MPP4 and STHSC) and (T.4.Th and T.4.Nve.Sp), (FC >=5 or <=0.2, P.value <=0.05); and 3) 358 CD8 T cell related genes whose expression are significantly different between (MPP4 and STHSC) and (T.8.Th and T.8.Nve.Sp), (FC and P.value same as 2). Then, to examine the associated OCRs with these T cell genes, we targeted the most varied 1,232 OCRs within 10Kb from the TSS of these genes (TSS-OCRs were excluded and of 4,105 significant OCRs with P.value <=0.05 in at least one population. For the analysis of Fig. 5B, we excluded the constitutively open OCRs, and selected the 30% OCRs with highest variability through the T cell dataset (by fitting generalized linear model on mean vs squared CV of ATAC-seq signals).
For the analysis of OCR activation, we focused on the DE-OCRs with the highest positive correlation to each gene by computing Pearson’s correlation between ATAC-seq signal and the corresponding gene expression within 12 T cell populations spanning differentiation from MPP4.135+.BM to T.4.Nve.Sp populations (because some genes had no correlated OCR within 10 Kb, 429 genes were retained). We then determined the population in which ATAC-seq signal and gene expression exceeded a 50% maximum and fell below 50% of the maximum along T cell differentiation. Genes were counted for the respective timing of OCR and gene activation/inactivation and represented as bubble plots in Fig4B. Genes in which the expression was already maximum in MPP4.135+.BM progenitors were not considered in the analysis.
To relate TF expression and motif accessibility (Fig. 7A–C), OCRs containing a TF motif were selected from the table of significant OCRs (P.value <=0.05 in at least one population) and 1,000 OCRs with the highest motif score were clustered using k-means. For TF motifs where ChIP-seq data are available at the NCBI GEO database, raw data were downloaded (https://www.ncbi.nlm.nih.gov/geo/, SRR4431502 and SRR4431506 for RORγ and SRR499696 ~ SRR499708 for Pax5) with a corresponding control data and analyzed by 1) mapping to mm10 reference using bowtie2, 2) discarding reads of non-unique mapping (samtools view -q 30), 3) removing duplicated reads by Picard.MarkDuplicates, 4) counting number of reads overlapping the OCRs, 5) normalizing reads by RPM (reads per million mapped reads) and 6) computing ChIP-seq signal as fold changes (ChIP-seq samples /control) after adding a pseudo count of 0.1.
FoxP3 Analysis
ChIP-seq datasets (Kitagawa et al., 2017), Database accession DRA003955) for H3K27Me3, H3K27Ac, H3K4Me1, H3K27Me3, Mediator and Smc1a (Cohesin) in Tregs were mapped to mm10 genome using bowtie. ChIP-seq peaks were called using HOMER (http://homer.ucsd.edu/homer/) with corresponding biological replicates and respective input controls. Additionally, H3K4Me1 ChIP-seq data was analyzed in the same manner for Tconv cells [(Placek et al., 2017), GSE69162]. A robust set of FoxP3 ChIP-seq binding sites were previously defined in Treg cells (Kwon et al., 2017). Briefly, fastq files (GSE40684, DRA003955) were mapped to the mm10 reference genome using bowtie. FoxP3 peaks from both studies were called using HOMER findPeaks function with an FDR of 1% using the parameter (-style factor) and respective input background peaks. Intersection of FoxP3 peaks were derived from both data sets Intersection of FoxP3 peaks were derived from both data sets using the BEDtools intersect function with a 50% reciprocal overlap requirement, yielding 5,047 robust FoxP3 peaks. Our analysis maps the cis-regulatory landscape during T cell differentiation for the top 2,000 FoxP3 ChIP-seq binding sites. FoxP3 peaks were parsed into promoter-proximal (920) and distal (1080) OCRs. Treg ChIP-seq histone mark and TF data were used to annotate all 2,000 FoxP3 peaks by binarizing each chromatin feature as being absent or present in each respective FoxP3 peak. Distal FoxP3 peaks were ordered based on accessibility differences Treg/LTHSC and Treg/DP. Distal FoxP3 OCRs were then parsed into constitutive (no differences in accessibility during T cell differentiation; 860 peaks) or dynamic (> 2 fold ATAC-seq signal in at least one cell type upstream of Tregs; 220 peaks). TF motif enrichment was performed on FoxP3 constitutive and distal OCRs using chromVAR functions.
Data availability:
The GEO accession number for the RNAseq and ATACseq data reported in this paper is GSE100738. Processed ATAC-seq data and called peaks can be found at: https://sharehost.hms.harvard.edu/immgen/ImmGenATAC18_AllOCRsInfo.csv
Additional resources:
The data can be visualized in the UCSC genome browser, the link to these data can be found here: http://rstats.immgen.org/Chromatin/chromatin.html.
Supplementary Material
Supplementary Figure. 1 (Related to Figure 1).
(A) Representative FACS page demonstrating cell isolations for profiling. The entire collection is assembled in a very large PDF which can be consulted at http://www.immgen.org/DOI366.S1A (B) Relationship between data depth and inter-replicate correlation. Left: Inter-replicate correlations for ATAC-seq signals in OCRs for splenic B cells (21 replicates of B.Sp, generated in different labs). Blue dots: optimal relationship between sequencing depth and inter-replicate correlation, computed by iterative subsampling of the entire merged B.Sp data. Subsampled data were applied to simulate the correlations in shallower depth and the trend line by a logistic regression model depicted in green. Blue dots indicate the correlations after different subsampling from all merged data with the trend line in blue. Right: Inter-replicate correlations for cell types employed in the analysis. The green line and the blue dotted line in the left panel are superimposed.
Supplementary Figure. 5 (Related to Figure 5).
(A,B) Correlation of expression and TF motif accessibility scores and permuted correlation, permuted either by their (A) sample labels or (B) TF labels. (C) –log10 p-values for TF expression and motif accessibility scores after permutation of the sample or TF labels. (D) Hierarchical clustering for significantly correlated TF motif accessibility scores, motif name is labeled, motif family is denoted by parentheses. (E,F) Correlation by –log10 p-value of TF expression and motif accessibility scores filtering for (E) myeloid or (F) lymphoid samples.
Supplementary Figure. 6 (Related to Figure 6).
(A) ChromVAR z-test motif enrichment for distinct peaks between CD103+CD11b+ and CD103+ CD11b- DCs. (B) Mean Ehf mRNA counts from RNA-seq data in all samples profiled.
Supplementary Figure. 7 (Related to Figure 7).
(A) Constitutive chromatin accessibility (log2 ATAC-seq signal) for FoxP3 bound TSS OCRs (920). ChIP-seq data in Tregs for H3K27Ac, H3K4Me1, H3K4Me3, H3K27Me3, Mediator and Cohesin are marked as being present or absent for each respective TSS OCR.
(B) Chromatin accessibility (log2 Treg/Tconv ATAC-seq signal) and H3K4Me1 ChIP-seq (log2 Treg/Tconv ChIP-seq signal) from Treg and Tconv cells, reflects a shift in activity for dynamic FoxP3 OCRs.
TableS1 (Related to Fig. 1): Summary of immune cell populations profiled by ATAC-seq and their QC matrices.
First tab: Individual row indicates the properties of each biological replicate. Columns are as below, respectively:
1.SampleName – an abbreviation for the cell type and a replicate number,
2.CellType – an abbreviation for the cell type,
3.ImmGenLab – a lab contributed to cell isolation,
4.Lineage – lineage group adopted in phylogram (Fig1A),
5.CellFamily – description of the cell population,
6.Organ – origin of isolated cells,
7.SortingMarkers – criteria for cell isolation by FACS,
8.InputCellNumber – number of cells sorted to be profiled by ATAC-seq,
9.PF.reads - sum of read1 and read2 which passed Illumina filter,
10.%chrM.mapped – proportion of reads aligned to chrM among all mapped reads (%),
11.Paired.read.after.removing.PCR.duplication – total number of paired reads after removing PCR duplication, which were employed for downstream analysis,
12.%fragment.1Kb_TSS - proportion of reads aligned within 1Kb windows centered on each TSS of RefSeq gene among Paired.read.after.removing.PCR.duplication (%), 13.Replicate.cor – Pearson correlation between biological replicates.
Table S2 (Related to Fig. 1) All OCRs, genomic location and activity in different cells https://sharehost.hms.harvard.edu/immgen/ImmGenATAC18_AllOCRsInfo.csv
Table S3 (related to Figs. 2 & 3): (A): Gene expression explained by genome-wide DE or TSS OCRs. (B–D): Gene ontology enrichment for genes with DE-logic, TSS-logic or unexplained regulation (E): Motif enrichment in promoter region of TSS or DE-logic genes. (F): Significant associations between gene expression and OCRs activity (regression). (G) Significant association between gene expression and combinations of OCRs (multiple regression results)
Table S4 (related to Fig. 5): Transcription factor motifs found in all OCRs. Chromvar TF motifs for all OCRs (keyed to same ImmGenATAC1219.peakID of Table S2) https://sharehost.hms.harvard.edu/immgen/ImmGenATAC18_AllTFmotifsInOCRs.txt
Table S5 (related to Fig. 5): List of TF whose expression correlates, positively or negatively, with the accessibility of OCRs that include them (underlies Fig. 5G)
TableS6 (related to Fig. 6): OCRs specifically active in myeloid cell-types and TF enrichment.
Table S7 (related to Fig. 7) (A–C): Tables of RORg, ThPOK and Pax5-binding OCRs, with scores and correlation to TF expression (underlying Fig7 A, C, D). (D–E) FoxP3-binding OCRs with FoxP3 signal and ATAC values in different T cells (underlies Fig. 7E). The presence of Cohesin, Mediator or particular histone marks, inferred from published ChIPseq data (see Key Resources for refs), is shown at right.
Supplementary Figure. 2 (Related to Figure 1).
Pile-up traces of ATAC-seq signals in Itgax locus. Blue bars in the first row indicate the positions of identified peaks (Pval <=0.05) and the graph in the 2nd row conservation score among vertebrates. RNA expression for Itgax (Cd11c) gene are indicated by barplots with * where RNA-seq data was not acquired.
Supplementary Figure. 3 (Related to Figure 2). (A) Summary of variance components results for each expressed gene using a single variance component of cell type relatedness from open chromatin data. (B) Results of variance components model for two permuted matrices per gene model, showing minimal gene expression explained. Compare to Fig. 2b. (C) Attributes of genes where 99% of expression variance could be explained by DE (blue; n = 4,409) or TSS (green; n = 955) covariance. Genes in red (n = 223) had > 99% of variance unexplained from the variance components model. Shown are boxplots for promoter GC content, transcript inequality measured by the Gini Index, and mean log2 gene expression. (C) Histogram of number of independent associated OCRs per gene. To determine independent associations, stepwise regression was used and the associated p-values were adjusted for multiple testing using the Bonferroni approach.
Supplementary Figure. 4 (Related to Figure 4).
(A) Pile-up traces of ATAC-seq signals around Cd4 locus. Blue bars in the first row indicate the positions of identified OCRs and the graph in the 2nd row conservation score among vertebrates. RNA expression for Cd4 gene are indicated by barplots. LCR/E4T: locus control region/thymocyte enhancer, E4M: maturity enhancer, S4: silencer, E4P: proximal enhancer, E4D: distal enhancer (B) TF motif enrichment (Left) normalized gene expressions for differentially expressed genes displayed after k-means clustering, same as Fig.4A (Right) Enriched TF binding motif in most variable OCRs within 10Kb from TSS for genes in each cluster (same set of OCRs as in Fig.4B) ChromVAR z-test motif enrichment was performed and motifs having an p-value greater than or equal to 0.05 are displayed after k-means clustering.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD4 APC | Thermo Fisher Scientific | clone: RM4–5; cat#: 17–0042-81; lab: Brown |
| MHC II APC-eFluor780 | Thermo Fisher Scientific | clone: M5/114.15.2; cat#: 47–5321-82; lab: Brown |
| CD45 APC-eFluor780 | Thermo Fisher Scientific | clone: 30-F11m; cat#: 47–0451-82; lab: Brown |
| CD11c PE-Cy7 | Thermo Fisher Scientific | clone: N418; cat#: 25–0114-82; lab: Brown |
| MHC II eFluor450 | Thermo Fisher Scientific | clone: M5/114.15.2; cat#: 48–5321-82; lab: Brown |
| CD103 PE | Thermo Fisher Scientific | clone: 2E7; cat#: 12–1031-82; lab: Brown |
| CD64 APC | Thermo Fisher Scientific | clone: X54–5/7.1; cat#: 17–0641-82; lab: Brown |
| CD11b FITC | Thermo Fisher Scientific | clone: M1/70; cat#: 11–0112-82; lab: Brown |
| Siglec F PE | Thermo Fisher Scientific | clone: 1RNM44N; cat#: 12–1702-82; lab: Brown |
| CD11c PE-Cy7 | Thermo Fisher Scientific | clone: N418; cat#: 25–0114-82; lab: Brown |
| CD45 BV510 | BD Biosciences | clone: 30-F11; cat#: 563891; lab: Brown |
| CD8 PE | Thermo Fisher Scientific | clone: 53–6.7; cat#: 12–0081-82; lab: Brown |
| CD3 FITC | Thermo Fisher Scientific | clone: eBio500A2; cat#: 11–0033-82; lab: Brown |
| CD19 FITC | Thermo Fisher Scientific | clone: eBio1D3; cat#: 11–0193-82; lab: Brown |
| CD11c PE | Thermo Fisher Scientific | clone: N418; cat#: MA5–16878; lab: Brown |
| B220 eFluor450 | Thermo Fisher Scientific | clone: RA3–6B2; cat#: 48–0452-82; lab: Brown |
| PDCA1 APC | Thermo Fisher Scientific | clone: eBio927; cat#: 17–3172-82; lab: Brown |
| Siglec H PE-Cy7 | Thermo Fisher Scientific | clone: eBio440c; cat#: 25–0333-82; lab: Brown |
| cd45 PE-Cy7 | Biolegend | clone: 30F11; cat#: 103114; lab: Turley |
| Epcam PE-Cy7 | Biolegend | clone: G8.8; cat#: 118216; lab: Turley |
| Ter11 PE-Cy7 | Biolegend | clone: TER119; cat#: 116222; lab: Turley |
| CD21/35 FITC | Biolegend | clone: 7E9; cat#: 123407; lab: Turley |
| Madcam 488 | Biolegend | clone: MECA-367; cat#: 120708; lab: Turley |
| PDPN APC | Biolegend | clone: 8.1.1; cat#: 127410; lab: Turley |
| CD140a PE | BD Pharmingen | clone: APA5; cat#: 562776; lab: Turley |
| CD31 PE-Dazzle | Biolegend | clone: 390; cat#: 102430; lab: Turley |
| Calcein Blue | Molecular Probes | clone: NA; cat#: C1429; lab: Turley |
| Live/Dead Near-IR633 | Molecular Probes | clone: NA; cat#: L10119; lab: Turley |
| CD45.2 FITC | Invitrogen | clone: 104; cat#: 11–0454-85; lab: Goldrath |
| CD127 PE-Cy7 | Invitrogen | clone: A7R34; cat#: 25–1271-82; lab: Goldrath |
| CD8a APC-eFlour780 | Invitrogen | clone: GK1.5; cat#: 47–0041-82; lab: Goldrath |
| CD45.1 APC | Invitrogen | clone: A20; cat#: 17–0453-82; lab: Goldrath |
| KLRG1 E450 | Invitrogen | clone: 2F1; cat#: 48–5893-82; lab: Goldrath |
| CD44 PE-Cy7 | Invitrogen | clone: IM7; cat#: 25–0441-82; lab: Goldrath |
| CD62L APC | BioLegend | clone: MEL-14; cat#: 104412; lab: Goldrath |
| Rat anti-mouse CD4 PE-Cy7 | BD Biosciences | clone: RM4–5; cat#: 552775; lab: Kang |
| Rat anti-mouse CD8a PE-Cy7 | BD Biosciences | clone: 53–6.7; cat#: 552877; lab: Kang |
| Hamster anti-mouse CD3e PerCP-Cy5.5 | BD Biosciences | clone: 145–2C11; cat#: 551163; lab: Kang |
| Hamster anti-mouse TCRd BV 421 | Biolegend | clone: GL3; cat#: 118120; lab: Kang |
| Hamster anti-mouse Vg2-FITC | Biolegend | clone: UC3–10A6; cat#: 137703; lab: Kang |
| Hamster anti-mouse Vg1.1-APC | Biolegend | clone: 2.11; cat#: 141108; lab: Kang |
| Rat anti-mouse CD24 APC-eFluor780 | eBioscience | clone: M1/69; cat#: 48–0242-82; lab: Kang |
| Rat anti-mouse Scart2 | J. Kisielow | clone: NA; cat#: NA; lab: Kang |
| Hamster anti-mouse CD27 PE | eBioscience | clone: LG.7F9; cat#: 12–0271-81; lab: Kang |
| CD19 | eBioscience | clone: MB19–1; cat#: NA; lab: Edy Kim |
| Ter119 | eBioscience | clone: TER119; cat#: NA; lab: Edy Kim |
| Ly6G/Gr1 | eBioscience | clone: A18; cat#: NA; lab: Edy Kim |
| CD8α | eBioscience | clone: 53/6.7; cat#: NA; lab: Edy Kim |
| TCRβ | eBioscience | clone: H58–597; cat#: NA; lab: Edy Kim |
| mCD1d tetramer PBS-57 APC | NIH Tetramer Core Facility | clone: NA; cat#: NA; lab: Edy Kim |
| CD3 FITC | eBioscience | clone: eBio500A2; cat#: 11–0033-82; lab: Merad |
| CD19 FITC | eBioscience | clone: eBio1D3; cat#: 11–0193-82; lab: Merad |
| CD8 PE | BioLegend | clone: 53–6.7; cat#: 100707; lab: Merad |
| CD4 APC | BioLegend | clone: GK1.5; cat#: 100411; lab: Merad |
| CD11c PE/Cy7 | BioLegend | clone: N418; cat#: 117317; lab: Merad |
| CD45 BV510 | BioLegend | clone: 30-F11; cat#: 103137; lab: Merad |
| MHCII APC/Cy7 | BioLegend | clone: M5/114.15.2; cat#: 107627; lab: Merad |
| PDCA1 APC | eBioscience | clone: ebio927; cat#: 17–3172-82; lab: Merad |
| B220 eF450 | BioLegend | clone: RA3–6B2; cat#: 103239; lab: Merad |
| Siglec H Pe/Cy7 | eBioscience | clone: ebio440c; cat#: 25–0333-82; lab: Merad |
| CD64 APC | BioLegend | clone: X54–5/7.1; cat#: 139305; lab: Merad |
| CD103 PE | BioLegend | clone: 2E7; cat#: 121405; lab: Merad |
| CD11b FITC | BioLegend | clone: M1/70; cat#: 101205; lab: Merad |
| Siglec F PE | BD Biosciences | clone: E50–2440; cat#: 552126; lab: Merad |
| CD4 | UCSF Ab core | clone: GK1.5; cat#: AM012; lab: Nabekura |
| CD5 | UCSF Ab core | clone: 53–7.3; cat#: AM018; lab: Nabekura |
| CD8a | UCSF Ab core | clone: 2.43; cat#: AM023; lab: Nabekura |
| CD19 | UCSF Ab core | clone: 1D3; cat#: AM005; lab: Nabekura |
| Gr-1 | UCSF Ab core | clone: RB6–8C5; cat#: AM051; lab: Nabekura |
| Ter110 | UCSF Ab core | clone: Ter119; cat#: AM030; lab: Nabekura |
| BioMag goat anti-rat IgG beads | Qiagen | clone: NA; cat#: 310107; lab: Nabekura |
| CD49b FITC | BioLegend | clone: DX5; cat#: 108906; lab: Nabekura |
| NK1.1 PerCP-Cy5.5 | BioLegend | clone: PK136; cat#: 108728; lab: Nabekura |
| CD3e PE-CY7 | BioLegend | clone: 145–2C11; cat#: 100320; lab: Nabekura |
| CD19 PE-CY7 | BD Biosciences | clone: 1D3; cat#: 552854; lab: Nabekura |
| CD11b PE | BD Biosciences | clone: M1/70; cat#: 553311; lab: Nabekura |
| CD27 APC | BioLegend | clone: LG.3A10; cat#: 124212; lab: Nabekura |
| CD49a Biotin | Miltenyi Biotec | clone: REA493; cat#: 130–107-587; lab: Nabekura |
| CD127 Biotin | BioLegend | clone: A7R34; cat#: 135006; lab: Nabekura |
| CD51 Biotin | BD Biosciences | clone: RMV-7; cat#: 551380; lab: Nabekura |
| Streptavidin-BV421 | BioLegend | clone: NA; cat#: 405226; lab: Nabekura |
| Propidium Iodide | Sigma-Aldrich | clone: NA; cat#: P4170; lab: Nabekura |
| CD138 PECy7 | BioLegend | clone: 281–2; cat#: 142514; lab: Nutt |
| CD138 PE | BDBiosciences | clone: 281–2; cat#: 553714; lab: Nutt |
| CD38 Alexa fluor 680 | in house | clone: 90; cat#: NA; lab: Nutt |
| NK1.1 APC | BD Biosciences | clone: PK136; cat#: 550627; lab: Nutt |
| CD11b (MAC-1) Alexa fluor 647 | in house | clone: M1/70; cat#: NA; lab: Nutt |
| TCRb APC | eBioscience | clone: H57–597; cat#: 17–5961-83; lab: Nutt |
| TCRb PE | BD Biosciences | clone: H57–597; cat#: 553172; lab: Nutt |
| MHC-II APC-eFluor780 | eBioscience | clone: M5/114.15.2; cat#: 47–5321-82; lab: Nutt |
| Gr-1 (Ly-6G) PE | in house | clone: RB6–8C5; cat#: NA; lab: Nutt |
| B220 (CD45R) FITC | in house | clone: RA3–6B2; cat#: NA; lab: Nutt |
| CD95 (Fas) PECy7 | BD Biosciences | clone: Jo2; cat#: 557653; lab: Nutt |
| CXCR4 BV421 | BD Biosciences | clone: 2B11; cat#: 585522; lab: Nutt |
| CD86 BV605 | BD Biosciences | clone: GL1; cat#: 563055; lab: Nutt |
| IgM APC-eFluor780 | eBioscience | clone: II/41; cat#: 47–5790-82; lab: Nutt |
| IgD APC-eFluor780 | eBioscience | clone: 11–26C; cat#: 47–5993-80; lab: Nutt |
| IgG BV421 | BioLegend | clone: Poly4053; cat#: 405317; lab: Nutt |
| CD117 PE-Cy7 | BioLegend | clone: 2B8; cat#: 105814; lab: Benoist |
| CD11b PerCPcy5.5 | BioLegend | clone: M1/70; cat#: 101228; lab: Benoist |
| CD11b PE | BioLegend | clone: M1/70; cat#: 101208; lab: Benoist |
| CD11C A700 | BioLegend | clone: N418; cat#: 117320; lab: Benoist |
| CD11c APC-Cy7 | BioLegend | clone: N418; cat#: 117324; lab: Benoist |
| CD19 APC-ef780 | eBiosciences | clone: 1D3; cat#: 47–0193-82; lab: Benoist |
| CD19 PE-TR | eBiosciences | clone: 1D3; cat#: 61–0193-82; lab: Benoist |
| CD19 PE-Cy7 | BioLegend | clone: 1D3; cat#: 115520; lab: Benoist |
| CD19 APC-Cy7 | BioLegend | clone: 1D3; cat#: 115530; lab: Benoist |
| CD24 Fitc | BioLegend | clone: M1/69; cat#: 101806; lab: Benoist |
| CD25 APC | BioLegend | clone: PC61; cat#: 101910; lab: Benoist |
| CD25 PE | BioLegend | clone: PC61; cat#: 101904; lab: Benoist |
| CD28 Bio | BioLegend | clone: E18; cat#: 102104; lab: Benoist |
| CD4 APC | eBiosciences | clone: RM4–5; cat#: 17–0042-82; lab: Benoist |
| CD4 PE | eBiosciences | clone: RM4–5; cat#: 12–0042-82; lab: Benoist |
| CD44 Fitc | BioLegend | clone: IM7; cat#: 103022; lab: Benoist |
| CD45 PE CF594 | BioLegend | clone: 30 F11; cat#: 562420; lab: Benoist |
| CD45 APC-Cy7 | BioLegend | clone: 30 F11; cat#: 103116; lab: Benoist |
| CD45R P.B. | BioLegend | clone: RA3 6B2; cat#: 193227; lab: Benoist |
| CD62L PE-Cy7 | BioLegend | clone: MEL14; cat#: 104418; lab: Benoist |
| CD69 A700 | BioLegend | clone: H1.2F3; cat#: 104539; lab: Benoist |
| CD8 A700 | BioLegend | clone: 53 6.7; cat#: 100730; lab: Benoist |
| CD8 PE-Cy7 | BioLegend | clone: 53 6.7; cat#: 100722; lab: Benoist |
| CD8 APC | BioLegend | clone: 53 6.7; cat#: 100712; lab: Benoist |
| EpCAM APC | BioLegend | clone: G8.8; cat#: 118214; lab: Benoist |
| F4/80 APC-Cy7 | BioLegend | clone: BM8; cat#: 123118; lab: Benoist |
| F4/80 PE-Cy7 | BioLegend | clone: BM8; cat#: 123114; lab: Benoist |
| F4/80 PE | BioLegend | clone: BM8; cat#: 123110; lab: Benoist |
| Ly6G/Gr1 APC-ef780 | eBiosciences | clone: RB6 8C5; cat#: 47–5931-82; lab: Benoist |
| Ly6G/Gr1 APC-Cy7 | BioLegend | clone: RB6 8C5; cat#: 108424; lab: Benoist |
| Ly6G/Gr1 APC | BioLegend | clone: RB6 8C5; cat#: 108412; lab: Benoist |
| ICAM2/CD102 Fitc | eBiosciences | clone: mlC2/4; cat#: 11–1029-42; lab: Benoist |
| IgM Fitc | eBiosciences | clone: eB121 15F9; cat#: 11–5890-82; lab: Benoist |
| Ly51 PE | BioLegend | clone: 6C3; cat#: 108308; lab: Benoist |
| MHCII Fitc | BioLegend | clone: M5/114; cat#: 107606; lab: Benoist |
| NK1.1 APC-Cy7 | BioLegend | clone: PK136; cat#: 108710; lab: Benoist |
| NK1.1 APC | BioLegend | clone: PK136; cat#: 108724; lab: Benoist |
| Nrp1 APC | BioLegend | clone: 3E12; cat#: 145206; lab: Benoist |
| TCRb ef450 | eBiosciences | clone: H57 597; cat#: 47–5961-82; lab: Benoist |
| TCRb PE-Cy7 | BioLegend | clone: H57 597; cat#: 109222; lab: Benoist |
| TCRb P.B. | BioLegend | clone: H57 597; cat#: 109226; lab: Benoist |
| TCRb PerCPcy5.5 | BioLegend | clone: H57 597; cat#: 109228; lab: Benoist |
| TCRgd PerCPcy5.5 | BioLegend | clone: GL3; cat#: 118118; lab: Benoist |
| Ter119 APC-ef780 | eBiosciences | clone: Ter119; cat#: 47–5921-82; lab: Benoist |
| Ter119 APC-Cy7 | BioLegend | clone: Ter119; cat#: 116223; lab: Benoist |
| CD45-APC-cy7 | Biolegend | clone: NA; cat#: NA; lab: Colonna |
| CD3-FITC | Biolegend | clone: NA; cat#: NA; lab: Colonna |
| CD19-FITC | eBioscience | clone: NA; cat#: NA; lab: Colonna |
| THy1.2-APC | eBioscience | clone: NA; cat#: NA; lab: Colonna |
| TCRb-PE | Pharmingen/BD | clone: NA; cat#: NA; lab: Colonna |
| KLRG1-PE | Biolegend | clone: NA; cat#: NA; lab: Colonna |
| CD5-PE | Biolegend | clone: NA; cat#: NA; lab: Colonna |
| CCR6-BV421 | Pharmingen/BD | clone: NA; cat#: NA; lab: Colonna |
| NKp46-biotin | Colonna Lab | clone: NA; cat#: NA; lab: Colonna |
| CD3-PercP-Cy5.5 | eBioscience | clone: NA; cat#: NA; lab: Colonna |
| CD19-PercP-Cy5.5 | ebioscience | clone: NA; cat#: NA; lab: Colonna |
| NK1.1-PercP-Cy5.5 | Biolegend | clone: NA; cat#: NA; lab: Colonna |
| SCA1-PacBlue | Biolegend | clone: NA; cat#: NA; lab: Colonna |
| CD127-FITC | eBioscience | clone: NA; cat#: NA; lab: Colonna |
| KLRG1-APC | eBioscience | clone: NA; cat#: NA; lab: Colonna |
| ST2-biotin | eBioscience | clone: NA; cat#: NA; lab: Colonna |
| SAV-PE-Cy7 | eBioscience | clone: NA; cat#: NA; lab: Colonna |
| CD3e PE | eBioscience | clone: 145–2C11; cat#: 12–0031-82; lab: Randolph |
| CD3e PECy7 | eBioscience | clone: 145–2C11; cat#: 25–0031-82; lab: Randolph |
| CD11b APCCy7 | Biolegend | clone: M1/70; cat#: 101226; lab: Randolph |
| CD19 PE | Biolegend | clone: 6D5; cat#: 115508; lab: Randolph |
| CD45 PB | Biolegend | clone: 30-F11; cat#: 103126; lab: Randolph |
| CD64 APC | BD Biosciences | clone: X54–5/7.1; cat#: 558539; lab: Randolph |
| CD102 (ICAM2) Alexa 488 | eBioscience | clone: 3C4; cat#: 53–1021-82; lab: Randolph |
| CD115 PE | eBioscience | clone: AFS98; cat#: 12–1152-82; lab: Randolph |
| CD115 APC | eBioscience | clone: AFS98; cat#: 17–1152-82; lab: Randolph |
| CD206 FITC | Biolegend | clone: C068C2; cat#: 141704; lab: Randolph |
| CD226 PE | Biolegend | clone: 1000000; cat#: 128806; lab: Randolph |
| B220 PECy7 | eBioscience | clone: RA3–6B2; cat#: 25–0452-82; lab: Randolph |
| Ly6C BV421 | Biolegend | clone: HK1.4; cat#: 128032; lab: Randolph |
| Ly6G PE | BD Biosciences | clone: 1A8; cat#: 553128; lab: Randolph |
| Ly6G PE-Cy7 | BD Biosciences | clone: 1A8; cat#: 560601; lab: Randolph |
| F4/80 FITC | Biolegend | clone: BM8; cat#: 123108; lab: Randolph |
| F4/80 PE-Cy7 | Biolegend | clone: BM8; cat#: 123114; lab: Randolph |
| MHCII (I-A/I-E) PB | Biolegend | clone: M5/114.15.2; cat#: 107620; lab: Randolph |
| MerTK PECy7 | eBioscience | clone: DSSMMER; cat#: 25–5751-82; lab: Randolph |
| CD3 Biotin | Biolegend | clone: 145–2C11; cat#: 100304; lab: Wagers |
| CD4 Biotin | Biolegend | clone: GK1.5; cat#: 100404; lab: Wagers |
| CD5 Biotin | ebioscience | clone: 53–7.3; cat#: 12–0051-85; lab: Wagers |
| CD8 Biotin | Biolegend | clone: 53–6.7; cat#: 100704; lab: Wagers |
| CD19 Biotin | Biolegend | clone: 6D5; cat#: 115504; lab: Wagers |
| B220 Biotin | Biolegend | clone: RA3–6B2; cat#: 103204; lab: Wagers |
| GR-1 Biotin | ebioscience | clone: RB6–8C5; cat#: 13–5931-82; lab: Wagers |
| Mac-1 Biotin | ebioscience | clone: M1/70; cat#: 13–0112-85; lab: Wagers |
| Ter-119 Biotin | Biolegend | clone: TER-119; cat#: 116204; lab: Wagers |
| Streptavidin Pacific Orange | Thermo Fisher | clone: NA; cat#: S32365; lab: Wagers |
| Sca-1 PECy7 | Biolegend | clone: D7; cat#: 108113; lab: Wagers |
| c-Kit APC | ebioscience | clone: 2B8; cat#: 17–1171-83; lab: Wagers |
| CD48 APCCy7 | Biolegend | clone: HM48–1; cat#: 103431; lab: Wagers |
| CD150 PE | Biolegend | clone: TC15–12F12.2; cat#: 115904; lab: Wagers |
| CD34 FITC | ebioscience | clone: RAM34; cat#: 11–0341-85; lab: Wagers |
| Flk2 PEcf594 | BD biosciences | clone: A2F10.1; cat#: 562537; lab: Wagers |
| Bacterial and Virus Strains | ||
| NA | ||
| Biological Samples | ||
| Sorted cell populations | This paper | Table S1 and http://www.immgen.org/ImmGenATAC1219Sorts.S1A.pdf |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NA | ||
| Critical Commercial Assays | ||
| Nextera DNA Library Preparation Kit | Illumina | FC-121–1030 |
| Deposited Data | ||
| Raw sequencing data | This paper | GEO: GSE100738 |
| Experimental Models: Cell Lines | ||
| NA | ||
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice | Jackson Laboratory | Jax0664 |
| B6.Rorctm2Litt | Jackson Laboratory | Jax7572 |
| Foxp3-ires-gfp reporter mice | Bettelli E et.al, 2006 | PubMed: 16648838 |
| Oligonucleotides | ||
| NA | ||
| Recombinant DNA | ||
| NA | ||
| Software and Algorithms | ||
| Genome mm10 | http://hgdownload.cse.ucsc.edu/goldenpath/mm10/bigZips/mm10.2bit | mm10 |
| Transcription start sites | http://hgdownload.cse.ucsc.edu/goldenPath/mm10/database/refFlat.txt.gz | downloaded Jan. 2017 |
| Blacklisted regions | http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/mm10-mouse/mm10.blacklist.bed.gz | NA |
| RNAseq GTF file | https://genome.ucsc.edu/cgi-bin/hgTables | NA |
| Genome conservation scores | http://hgdownload.cse.ucsc.edu/goldenpath/mm10/phastCons60way/mm10.60way.phastCons.bw | NA |
| sickle1.2 (Version 1.33) | Joshi NA, Fass JN. (2011) | https://github.com/najoshi/sickle |
| TrimGalore version 0.4.0 | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ | Krueger, 2015 |
| Bowtie2 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | Langmead and Salzberg, 2012 |
| Samtools 0.1.19 | http://samtools.sourceforge.net | Li et al., 2009 |
| Picard Tools | http://broadinstitute.github.io/picard/ | NA |
| hisat2 version 2.0.4 | https://ccb.jhu.edu/software/hisat2/manual.shtml | Kim et al., 2015 |
| Htseq version 0.6.1 | https://htseq.readthedocs.io | Anders et al., 2015 |
| Gaston version 1.5.3 | https://cran.r-project.org/web/packages/gaston/index.html | Dandine-Ro and Perdry, 2015 |
| IGV | http://software.broadinstitute.org/software/igv/ | Robinson et al. 2011 |
| MACS2 | https://github.com/taoliu/MACS/wiki | Zhang et al., 2008 |
| BEDTools | https://bedtools.readthedocs.io/en/latest/ | Quinlan and Hall, 2010 |
| HOMER | http://homer.ucsd.edu/homer/ | Heinz et al., 2010 |
| chromVAR | https://bioconductor.org/packages/release/bioc/html/chromVAR.html | Schep et al., 2017 |
| chromVARmotifs version 0.2.0 | https://github.com/buenrostrolab/chromVARmotifs | Schep et al., 2017 |
| Other | ||
| H3K4Me1 ChIP-seq | Placek et al., 2017 | GSE69162 |
| FoxP3 ChIP-seq | Samstein et al., 2012 | GSE40684 |
| FoxP3, H3K27Ac, H3K4Me1/3, Mediator, Cohesin ChIP-seq | Kitagawa et al., 2017 | DRA003955 |
| PAX5 ChIPseq | Revilla-I-Domingo R et.al, 2012 | GSE38046 |
| RORγ ChIPseq | Yanxia Guo et.al, 2016 | GSE88916 |
Highlights.
Atlas of 512,595 cis-regulatory elements active in 86 immunologic cell-types
Two classes of loci, controlled by either promoter- or enhancer-driven logic
Inference of enhancer elements that activate each gene across differentiation
Context-specificity of enhancer activation by transcription factors
ACKNOWLEDGEMENTS
We thank M. Aryee, H. Finucane, R. Bosselut, A. Stark for valuable discussions. Immgen is supported by NIH R24-AI072073, BDB and SAR by R01AI113221 and T32AI007605.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Authors declare no competing interests.
REFERENCES
- Anscombe FJ (1948). The Transformation of Poisson, Binomial and Negative-Binomial Data. Biometrika 35, 246–254. [Google Scholar]
- Asai T, and Morrison SL (2013). The SRC family tyrosine kinase HCK and the ETS family transcription factors SPIB and EHF regulate transcytosis across a human follicle-associated epithelium model. J. Biol. Chem 288, 10395–10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, Murison A, Langenfeld K, Petretich M, Scognamiglio R, et al. (2018). A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 553, 515–520. [DOI] [PubMed] [Google Scholar]
- Banerji J, Rusconi S, and Schaffner W (1981). Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 299–308. [DOI] [PubMed] [Google Scholar]
- Benoist C, and Chambon P (1981). In vivo sequence requirements of the SV40 early promotor region. Nature 290, 304–310. [DOI] [PubMed] [Google Scholar]
- Bornstein C, Winter D, Barnett-Itzhaki Z, David E, Kadri S, Garber M, and Amit I (2014). A negative feedback loop of transcription factors specifies alternative dendritic cell chromatin States. Mol. Cell 56, 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavò E, Khoueiry P, Garfield DA, Geeleher P, Zichner T, Gustafson EH, Ciglar L, Korbel JO, and Furlong EEM (2016). Shadow Enhancers Are Pervasive Features of Developmental Regulatory Networks. Curr. Biol 26, 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champhekar A, Damle SS, Freedman G, Carotta S, Nutt SL, and Rothenberg EV (2015). Regulation of early T-lineage gene expression and developmental progression by the progenitor cell transcription factor PU.1. Genes Dev 29, 832–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Ge B, Casale FP, Vasquez L, Kwan T, Garrido-Martín D, Watt S, Yan Y, Kundu K, Ecker S, et al. (2016). Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell 167, 1398–1414.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, den Hollander NS, Kant SG, et al. (2008). Transcription factor E2–2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 135, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. (2016). Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet 48, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, Spinrad A, Singer O, David E, Winter DR, Smith LK, et al. (2017). Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat. Commun 8, 717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, and Busslinger M (2006). Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity 24, 269–281. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. (2011). Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med 208, 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Gorkin DU, and Ren B (2016). Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 62, 668–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellmeier W, and Taniuchi I (2014). The role of BTB-zinc finger transcription factors during T cell development and in the regulation of T cell-mediated immunity. Curr. Top. Microbiol. Immunol 381, 21–49. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficara F, Murphy MJ, Lin M, and Cleary ML (2008). Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell 2, 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossum SL, Mutolo MJ, Tugores A, Ghosh S, Randell SH, Jones LC, Leir S-H, and Harris A (2017). Ets homologous factor (EHF) has critical roles in epithelial dysfunction in airway disease. J. Biol. Chem 292, 10938–10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini M, Findlay GM, McKenna A, Milbank JH, Lee C, Zhang MD, Cusanovich DA, and Shendure J (2017). CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions. Am. J. Hum. Genet 101, 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, Price J, Yin N, Bromberg J, Lira SA, et al. (2009). The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med 206, 3115–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, and Yona S (2014). Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat. Rev. Immunol 14, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, MacIsaac KD, Chen Y, Miller RJ, Jain R, Joyce-Shaikh B, Ferguson H, Wang I-M, Cristescu R, Mudgett J, et al. (2016). Inhibition of RORγT Skews TCRα Gene Rearrangement and Limits T Cell Repertoire Diversity. Cell Rep 17, 3206–3218. [DOI] [PubMed] [Google Scholar]
- Hardy RR, and Hayakawa K (2001). B cell development pathways. Annu. Rev. Immunol 19, 595–621. [DOI] [PubMed] [Google Scholar]
- Helft J, Manicassamy B, Guermonprez P, Hashimoto D, Silvin A, Agudo J, Brown BD, Schmolke M, Miller JC, Leboeuf M, et al. (2012). Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J. Clin. Invest 122, 4037–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Day DS, and Young RA (2016). Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 167, 1188–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J-W, Hendrix DA, and Levine MS (2008). Shadow enhancers as a source of evolutionary novelty. Science 321, 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horcher M, Souabni A, and Busslinger M (2001). Pax5/BSAP maintains the identity of B cells in late B lymphopoiesis. Immunity 14, 779–790. [DOI] [PubMed] [Google Scholar]
- Hosokawa H, Ungerbäck J, Wang X, Matsumoto M, Nakayama KI, Cohen SM, Tanaka T, and Rothenberg EV (2018). Transcription Factor PU.1 Represses and Activates Gene Expression in Early T Cells by Redirecting Partner Transcription Factor Binding. Immunity 48, 1119–1134.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issuree PDA, Ng CP, and Littman DR (2017). Heritable Gene Regulation in the CD4:CD8 T Cell Lineage Choice. Front. Immunol 8, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-K, and Shiekhattar R (2015). Architectural and Functional Commonalities between Enhancers and Promoters. Cell 162, 948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, Yasuda K, Motooka D, Nakamura S, Kondo M, et al. (2017). Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat. Immunol 18, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H-K, Chen H-M, Mathis D, and Benoist C (2017). Different molecular complexes that mediate transcriptional induction and repression by FoxP3. Nat. Immunol 18, 1238–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. (2014). Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, and Amit I (2014). Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedovic J, Ebert A, Tagoh H, and Busslinger M (2011). Pax5: a master regulator of B cell development and leukemogenesis. Adv. Immunol 111, 179–206. [DOI] [PubMed] [Google Scholar]
- Mercer EM, Lin YC, Benner C, Jhunjhunwala S, Dutkowski J, Flores M, Sigvardsson M, Ideker T, Glass CK, and Murre C (2011). Multilineage priming of enhancer repertoires precedes commitment to the B and myeloid cell lineages in hematopoietic progenitors. Immunity 35, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, et al. (2012). Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol 13, 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, Wang J, Bhatt DM, Heise N, Schmid RM, et al. (2017). An NF-κB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 47, 450–465.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder M, Barozzi I, Tissières V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, Lee EA, Zhu Y, Plajzer-Frick I, Pickle CS, et al. (2018). Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Placek K, Hu G, Cui K, Zhang D, Ding Y, Lee J-E, Jang Y, Wang C, Konkel JE, Song J, et al. (2017). MLL4 prepares the enhancer landscape for Foxp3 induction via chromatin looping. Nat. Immunol 18, 1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsdell F, and Ziegler SF (2014). FOXP3 and scurfy: how it all began. Nat. Rev. Immunol 14, 343–349. [DOI] [PubMed] [Google Scholar]
- Reizis B, Bunin A, Ghosh HS, Lewis KL, and Sisirak V (2011). Plasmacytoid dendritic cells: recent progress and open questions. Annu. Rev. Immunol 29, 163–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revilla-I-Domingo R, Bilic I, Vilagos B, Tagoh H, Ebert A, Tamir IM, Smeenk L, Trupke J, Sommer A, Jaritz M, et al. (2012). The B-cell identity factor Pax5 regulates distinct transcriptional programmes in early and late B lymphopoiesis. EMBO J 31, 3130–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas M, Davies LC, Giles PJ, Liao C-T, Kharfan B, Stone TC, O’Donnell VB, Fraser DJ, Jones SA, and Taylor PR (2014). The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science 344, 645–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg EV (2014). Transcriptional control of early T and B cell developmental choices. Annu. Rev. Immunol 32, 283–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. (2012). Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J, et al. (2015). MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 349, 993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. (2012). A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, Smyth GK, Hodgkin PD, Nutt SL, and Corcoran LM (2015). Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat. Immunol 16, 663–673. [DOI] [PubMed] [Google Scholar]
- Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O’Shea JJ, Singh H, and Ozato K (2005). IFN regulatory factor-4 and −8 govern dendritic cell subset development and their functional diversity. J. Immunol 174, 2573–2581. [DOI] [PubMed] [Google Scholar]
- Wang L, Wildt KF, Castro E, Xiong Y, Feigenbaum L, Tessarollo L, and Bosselut R (2008). The zinc finger transcription factor Zbtb7b represses CD8-lineage gene expression in peripheral CD4+ T cells. Immunity 29, 876–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, and Bosselut R (2012). CD4-CD8 differentiation in the thymus: connecting circuits and building memories. Curr. Opin. Immunol 24, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye CJ, Feng T, Kwon H-K, Raj T, Wilson MT, Asinovski N, McCabe C, Lee MH, Frohlich I, Paik H-I, et al. (2014). Intersection of population variation and autoimmunity genetics in human T cell activation. Science 345, 1254665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Zhang K, Milner JJ, Toma C, Chen R, Scott-Browne JP, Pereira RM, Crotty S, Chang JT, Pipkin ME, et al. (2017). Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation. Nat. Immunol 18, 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui MA, and Rothenberg EV (2014). Developmental gene networks: a triathlon on the course to T cell identity. Nat. Rev. Immunol 14, 529–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabidi MA, Arnold CD, Schernhuber K, Pagani M, Rath M, Frank O, and Stark A (2015). Enhancer-core-promoter specificity separates developmental and housekeeping gene regulation. Nature 518, 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure. 1 (Related to Figure 1).
(A) Representative FACS page demonstrating cell isolations for profiling. The entire collection is assembled in a very large PDF which can be consulted at http://www.immgen.org/DOI366.S1A (B) Relationship between data depth and inter-replicate correlation. Left: Inter-replicate correlations for ATAC-seq signals in OCRs for splenic B cells (21 replicates of B.Sp, generated in different labs). Blue dots: optimal relationship between sequencing depth and inter-replicate correlation, computed by iterative subsampling of the entire merged B.Sp data. Subsampled data were applied to simulate the correlations in shallower depth and the trend line by a logistic regression model depicted in green. Blue dots indicate the correlations after different subsampling from all merged data with the trend line in blue. Right: Inter-replicate correlations for cell types employed in the analysis. The green line and the blue dotted line in the left panel are superimposed.
Supplementary Figure. 5 (Related to Figure 5).
(A,B) Correlation of expression and TF motif accessibility scores and permuted correlation, permuted either by their (A) sample labels or (B) TF labels. (C) –log10 p-values for TF expression and motif accessibility scores after permutation of the sample or TF labels. (D) Hierarchical clustering for significantly correlated TF motif accessibility scores, motif name is labeled, motif family is denoted by parentheses. (E,F) Correlation by –log10 p-value of TF expression and motif accessibility scores filtering for (E) myeloid or (F) lymphoid samples.
Supplementary Figure. 6 (Related to Figure 6).
(A) ChromVAR z-test motif enrichment for distinct peaks between CD103+CD11b+ and CD103+ CD11b- DCs. (B) Mean Ehf mRNA counts from RNA-seq data in all samples profiled.
Supplementary Figure. 7 (Related to Figure 7).
(A) Constitutive chromatin accessibility (log2 ATAC-seq signal) for FoxP3 bound TSS OCRs (920). ChIP-seq data in Tregs for H3K27Ac, H3K4Me1, H3K4Me3, H3K27Me3, Mediator and Cohesin are marked as being present or absent for each respective TSS OCR.
(B) Chromatin accessibility (log2 Treg/Tconv ATAC-seq signal) and H3K4Me1 ChIP-seq (log2 Treg/Tconv ChIP-seq signal) from Treg and Tconv cells, reflects a shift in activity for dynamic FoxP3 OCRs.
TableS1 (Related to Fig. 1): Summary of immune cell populations profiled by ATAC-seq and their QC matrices.
First tab: Individual row indicates the properties of each biological replicate. Columns are as below, respectively:
1.SampleName – an abbreviation for the cell type and a replicate number,
2.CellType – an abbreviation for the cell type,
3.ImmGenLab – a lab contributed to cell isolation,
4.Lineage – lineage group adopted in phylogram (Fig1A),
5.CellFamily – description of the cell population,
6.Organ – origin of isolated cells,
7.SortingMarkers – criteria for cell isolation by FACS,
8.InputCellNumber – number of cells sorted to be profiled by ATAC-seq,
9.PF.reads - sum of read1 and read2 which passed Illumina filter,
10.%chrM.mapped – proportion of reads aligned to chrM among all mapped reads (%),
11.Paired.read.after.removing.PCR.duplication – total number of paired reads after removing PCR duplication, which were employed for downstream analysis,
12.%fragment.1Kb_TSS - proportion of reads aligned within 1Kb windows centered on each TSS of RefSeq gene among Paired.read.after.removing.PCR.duplication (%), 13.Replicate.cor – Pearson correlation between biological replicates.
Table S2 (Related to Fig. 1) All OCRs, genomic location and activity in different cells https://sharehost.hms.harvard.edu/immgen/ImmGenATAC18_AllOCRsInfo.csv
Table S3 (related to Figs. 2 & 3): (A): Gene expression explained by genome-wide DE or TSS OCRs. (B–D): Gene ontology enrichment for genes with DE-logic, TSS-logic or unexplained regulation (E): Motif enrichment in promoter region of TSS or DE-logic genes. (F): Significant associations between gene expression and OCRs activity (regression). (G) Significant association between gene expression and combinations of OCRs (multiple regression results)
Table S4 (related to Fig. 5): Transcription factor motifs found in all OCRs. Chromvar TF motifs for all OCRs (keyed to same ImmGenATAC1219.peakID of Table S2) https://sharehost.hms.harvard.edu/immgen/ImmGenATAC18_AllTFmotifsInOCRs.txt
Table S5 (related to Fig. 5): List of TF whose expression correlates, positively or negatively, with the accessibility of OCRs that include them (underlies Fig. 5G)
TableS6 (related to Fig. 6): OCRs specifically active in myeloid cell-types and TF enrichment.
Table S7 (related to Fig. 7) (A–C): Tables of RORg, ThPOK and Pax5-binding OCRs, with scores and correlation to TF expression (underlying Fig7 A, C, D). (D–E) FoxP3-binding OCRs with FoxP3 signal and ATAC values in different T cells (underlies Fig. 7E). The presence of Cohesin, Mediator or particular histone marks, inferred from published ChIPseq data (see Key Resources for refs), is shown at right.
Supplementary Figure. 2 (Related to Figure 1).
Pile-up traces of ATAC-seq signals in Itgax locus. Blue bars in the first row indicate the positions of identified peaks (Pval <=0.05) and the graph in the 2nd row conservation score among vertebrates. RNA expression for Itgax (Cd11c) gene are indicated by barplots with * where RNA-seq data was not acquired.
Supplementary Figure. 3 (Related to Figure 2). (A) Summary of variance components results for each expressed gene using a single variance component of cell type relatedness from open chromatin data. (B) Results of variance components model for two permuted matrices per gene model, showing minimal gene expression explained. Compare to Fig. 2b. (C) Attributes of genes where 99% of expression variance could be explained by DE (blue; n = 4,409) or TSS (green; n = 955) covariance. Genes in red (n = 223) had > 99% of variance unexplained from the variance components model. Shown are boxplots for promoter GC content, transcript inequality measured by the Gini Index, and mean log2 gene expression. (C) Histogram of number of independent associated OCRs per gene. To determine independent associations, stepwise regression was used and the associated p-values were adjusted for multiple testing using the Bonferroni approach.
Supplementary Figure. 4 (Related to Figure 4).
(A) Pile-up traces of ATAC-seq signals around Cd4 locus. Blue bars in the first row indicate the positions of identified OCRs and the graph in the 2nd row conservation score among vertebrates. RNA expression for Cd4 gene are indicated by barplots. LCR/E4T: locus control region/thymocyte enhancer, E4M: maturity enhancer, S4: silencer, E4P: proximal enhancer, E4D: distal enhancer (B) TF motif enrichment (Left) normalized gene expressions for differentially expressed genes displayed after k-means clustering, same as Fig.4A (Right) Enriched TF binding motif in most variable OCRs within 10Kb from TSS for genes in each cluster (same set of OCRs as in Fig.4B) ChromVAR z-test motif enrichment was performed and motifs having an p-value greater than or equal to 0.05 are displayed after k-means clustering.
Data Availability Statement
The GEO accession number for the RNAseq and ATACseq data reported in this paper is GSE100738. Processed ATAC-seq data and called peaks can be found at: https://sharehost.hms.harvard.edu/immgen/ImmGenATAC18_AllOCRsInfo.csv