

Abstract
Nociceptive sensory neurons (SNs) in Aplysia provide useful models to study both memory and adaptive responses to nerve injury. Induction of long-term memory in many species, includingAplysia, is thought to depend on activation of cAMP-dependent protein kinase (PKA). Because Aplysia SNs display similar alterations in models of memory and after nerve injury, a plausible hypothesis is that axotomy triggers memory-like modifications by activating PKA in damaged axons. The present study disproves this hypothesis. SN axotomy was produced by (1) dissociation of somata from the ganglion [which is shown to induce long-term hyperexcitability (LTH)], (2) transection of neurites of dissociated SNs growing in vitro, or (3) peripheral nerve crush. Application of the competitive PKA inhibitor Rp-8-CPT-cAMPS at the time of axotomy failed to alter the induction of LTH by each form of axotomy, although the inhibitor antagonized hyperexcitability produced by 5-HT application. Strong activation of PKA in the nerve by coapplication of a membrane-permeant analog of cAMP and a phosphodiesterase inhibitor was not sufficient to induce LTH of either the SN somata or axons. Furthermore, nerve crush failed to activate axonal PKA or stimulate its retrograde transport. Therefore, PKA activation plays little if any role in the induction of LTH by axotomy. However, the expression of LTH was reduced by intracellular injection of the highly specific PKA inhibitor PKI several days after nerve crush. This suggests that long-lasting activation of PKA in or near the soma contributes to the maintenance of long-term modifications produced by nerve injury.
Keywords: sensitization, nerve injury, long-term memory, cAMP, protein kinase inhibitor (PKI), cell dissociation
After nerve injury, some somatosensory neurons display electrophysiological alterations, growth, and transcriptional responses that resemble alterations associated with memory (Titmus and Faber, 1990; Walters, 1994; Zimmermann and Herdegen, 1996). An interesting question is whether common signal transduction pathways are critical for inducing both injury-related and memory-related responses. The most extensively analyzed signaling pathway for the induction of long-term memory involves the synthesis of cAMP, activation of protein kinase A (PKA), translocation of PKA into the nucleus, and activation of a Ca2+/cAMP response element binding protein (CREB) (for review, see Abel and Kandel, 1998; Dubnau and Tully, 1998; Silva et al., 1998). Some have suggested that cAMP regulation of gene expression is a universal requirement for induction of long-term memory (Goda, 1995).
Nociceptive sensory neurons (SNs) in the pleural ganglia ofAplysia provide an opportunity to compare learning-related and injury-related plasticity within the same neurons. In co-cultures with dissociated motor neurons, SNs have revealed mechanisms that may underlie long-term memory. They display presynaptic facilitation and synaptic growth 24 hr after prolonged application of serotonin (5-HT) (Montarolo et al., 1986; Dale et al., 1988; Bailey et al., 1992) or cAMP analogs (Schacher et al., 1988, 1993). SNs also display long-term hyperexcitability (LTH) of the soma after 5-HT or cAMP application (Dale et al., 1987; Scholz and Byrne, 1988; Lewin and Walters, 1996,1999). Procedures that injure axons of Aplysia SNs elicit long-term responses indistinguishable from those elicited by 5-HT or cAMP treatment: synaptic facilitation, growth, and LTH (Walters, 1991;Clatworthy and Walters, 1994; Steffensen et al., 1995; Bedi et al., 1998). Because axotomy procedures usually release neuromodulators that may activate adenylyl cyclase, and because Ca2+influx into injured axons (Ziv and Spira, 1993) may also activate adenylyl cyclase (Weisskopf et al., 1994), a plausible hypothesis is that long-term plasticity after axotomy is induced by the cAMP–PKA–CREB pathway (Walters and Ambron, 1995; Bedi et al., 1998;Dash et al., 1998). Indeed, Bedi et al. (1998) reported that a PKA inhibitor blocked LTH induced by transecting neurites of SNs in culture, although this study did not distinguish between effects of the inhibitor on maintenance and induction of LTH.
We have investigated contributions of PKA to LTH of AplysiaSNs induced by three forms of axotomy (Fig.1). First is the disconnection of SN somata from their axons during dissociation. Second is in vitro transection of neurites of previously dissociated SNs. Third is crush of peripheral nerves containing SN axons, in vivoor in vitro. We find that PKA inhibitors do not affect the induction of LTH by axotomy. However, they reduce the expression of LTH when applied days after its induction. In addition, PKA is not activated at a site of nerve crush, and its activation in the nerve is neither sufficient nor necessary for the induction of LTH.
Fig. 1.

Three forms of sensory neuron (SN) axotomy. A, Dissociation of SN somata, which severed the major axon(s) of each SN close to the soma. Neurites began to grow within hours after plating.B, Transection close to the soma of outgrowing neurites from isolated SNs 2 d after dissociation. C, Axotomy produced by crushing pedal nerves containing SN axons, eitherin vivo or in an in vitro preparation (in which all nerves were left as long as possible). In both cases nerves innervating the midbody region and tail (notably p7, p8, and p9) were crushed ∼1 cm from the pedal ganglion. In some studies PKA activity was measured in segments of the nerve or axoplasm extruded from nerve segments (Fig. 7) after crushing pedal nerves ∼3 cm from the ganglion. Some of the nerves were ligated (data not shown) midway between the crush site and the ganglion to accumulate material transported retrogradely from the crush site (see measurement of PKA activity in Materials and Methods).
Some of these results have been published previously in abstract form (Liao et al., 1997).
MATERIALS AND METHODS
General. Aplysia californica (70–200 gm) were supplied by the National Institutes of Health-AplysiaResource Facility (Miami, FL) and Alacrity Marine Biological Services (Redondo Beach, CA). Animals were housed in aquaria containing artificial seawater (ASW) (Instant Ocean, Burlington, NC) at 15–18°C for 1–7 d before use. Constant body weight was maintained on a diet ofGracilaria seaweed. Animals were dissected after injection of isotonic MgCl2 (equivalent to ∼50% of body volume), and the ganglia were excised and desheathed in a 1:1 solution of isotonic MgCl2 and ASW.
Axotomy by cell dissociation. Cultures of pleural SNs (Walters et al., 1983) were prepared using methods modified from those of Schacher and Proshansky (1983), Montarolo et al. (1986), and Rayport and Schacher (1986). Pleural VC clusters were excised (Fig.1A) and placed in polystyrene dishes coated with poly-l-lysine containing Liebowitz-15 (L15) (Sigma, St. Louis, MO) supplemented with appropriate salts. Cells were dissociated by gently vibrating two micropipettes within each cluster. No protease was used. The dissociated SNs were then cultured at 16–18°C in a 1:1 mixture of L15 and filtered hemolymph until they were tested for excitability 1–5 d later.
Axotomy by neurite transection. Cultures of dissociated pleural SNs were prepared using the methods of Ambron et al. (1996). Briefly, polystyrene dishes coated with poly-l-lysine were exposed to hemolymph for 3 hr at room temperature. The dishes were washed thoroughly to remove soluble proteins, and isotonic L15 lacking hemolymph was used as the culture medium. Pleural SNs were dissociated using protease, and individual neurons were added to the dish and maintained at 16°C for 3 d. On day 2, most of the major neurites of each SN were transected (Fig. 1B) with a sharp micropipette. On day 3 (24 hr later), each cell was tested for excitability.
Axotomy by nerve crush. For in vitro nerve injury, the longest pedal nerves (p7, p8, p9) on one side were crushed (Fig. 1C) ∼1 cm from the ipsilateral pedal ganglion in 1:1 ASW/isotonic MgCl2 solution, whereas all other nerves were cut as far as possible from the ganglion (Gunstream et al., 1995). Forin vivo nerve injury, a small incision was made in the intact, anesthetized animal (injected into the head with ∼30% of its volume of isotonic MgCl2), and all major pedal nerves were crushed ∼1 cm from the ganglion on one side of the animal (Walters et al., 1991). The incision was sutured, and the animal was returned to its home tank for 4–5 d.
Electrophysiological tests. Intracellular recordings from SN somata were made with glass microelectrodes filled with 3 mpotassium acetate (electrode resistance 8–20 MΩ). Recordings were made at 19–21°C while the preparation was bathed in buffered ASW, L15 medium, or a 1:1 mixture of ASW and L15, pH 7.6. These different test solutions had no apparent effects on excitability. Soma spike threshold was measured with a standard series of 20 msec depolarizing pulses. Repetitive firing (spike accommodation) was quantified by counting the number of spikes evoked by a 1 sec intracellular depolarizing pulse using 2.5× the threshold current determined with the 20 msec pulse. In some experiments, repetitive firing was examined by counting the number of spikes evoked by a series of 1 sec depolarizing pulses at 1.25, 2.5, and 5× the threshold current, or by 1, 2, 3, and 5 nA. Input resistance (Rin) was determined from the voltage change produced during injection of a 1 sec hyperpolarizing pulse (0.5 nA). Axon excitability was tested by passing current between two compartments through a narrow, Vaseline-sealed opening containing nerves p7, p8, and p9. Threshold was determined with a rapid series of 2 msec pulses, and then repetitive firing was tested by applying two 1 sec pulses at 0.4 and 0.8× the 2 msec threshold current.
Pharmacological treatments. The membrane-permeant PKA inhibitors Rp-8-CPT-cAMPS and Rp-cAMPS (Biolog) were applied in the bath to a final concentration of 100–1500 μm 0.5–1 hr before axotomy or testing, and left in the bath for the times indicated. Protein kinase inhibitor peptide PKI(6–22)amide (Life Technologies, Gaithersburg, MD) was pressure-injected into SN somata at a concentration of 1.5 mm after being dissolved in a solution of 350 mm KCl containing 0.05% fast green (FG) dye with pH adjusted to 7.3. Injection electrodes were beveled from 10 to 7 MΩ and backfilled with PKI by suction. Pulses (10 msec at 2–10 psi) were rapidly repeated until the soma became visibly green. All injections were performed in isolated ganglia preparations. 5-HT (Sigma) was applied in the bath or by pressure ejection (with 0.05% FG dye for visibility) onto the SN soma.
Measurement of PKA activity. In protocol 1, the CNS and major peripheral nerves were exposed, and pedal nerves p8 and p9 on one side were crushed 3 cm from the ganglion. Five minutes later the nerves were rapidly frozen, and the 1 cm segment immediately proximal to the crush site, the segment from the same nerves adjacent to the ganglion, and a segment from an uninjured nerve on the contralateral side were removed. The appropriate segments from three animals were pooled, homogenized in 50 mm Tris-HCl, 5 mm EDTA, pH 7.6, and centrifuged at 16,000 × g to obtain the soluble and membrane fractions. In protocol 2 (Ambron et al., 1995), the pedal nerves on one side were crushed 3 cm from the ganglion and were then ligated 1 cm proximal to the crush site. Control nerves were ligated only. Twenty hours later, axoplasm was extruded from the 0.5 cm segment located proximal to the crush site (Cr), distal to the ligation on the crushed nerve (Cr/Lig), and distal to the ligation on the control nerve (Lig). Axoplasm from each site from two or more animals was pooled for analysis. PKA activity was determined with a kit from Life Technologies, using methods that have been used effectively withAplysia tissue (Hooper et al., 1994a,b). The reaction mixture contained 5 μg of nerve fractions or axoplasm, 50 μm of a peptide containing a consensus PKA phosphorylation site, 50 μm γ–32P-ATP, 10 mm MgCl2, 250 μg BSA, in 10 mm Tris-HCl, pH 7.5, in a total volume of 50 μl. The reaction was performed at 30°C: incorporation is linear for more than 20 min. At 15 min a sample was removed and distributed on a phosphocellulose disk. The disk was washed with acid, and radioactivity was measured by liquid scintillation. The total amount of holoenzyme was determined using a duplicate reaction mixture containing 10 μm cAMP to dissociate the catalytic and regulatory subunits, and specificity was determined using another sample containing 1 μm of a PKA pseudosubstrate. PKA activity was calculated after subtracting the radioactivity incorporated into the pseudosubstrate and was expressed as picomoles of phosphate transferred from ATP to the substrate per minute per microgram of protein.
Statistical analysis. Pretests and post tests of the same group were compared with paired t tests. Comparisons of test responses of two separate groups were made with unpaired ttests, whereas comparisons of more than two groups were made with one-way ANOVA followed by Newman–Keuls or Dunnett’s multiple comparisons tests.
RESULTS
PKA inhibitors reduce 5-HT-induced hyperexcitability of SN somata
Before examining effects of PKA inhibitors on axotomy-induced hyperexcitability, we wanted to confirm that these inhibitors affect properties of Aplysia SNs previously linked to PKA activation. One such property is hyperexcitability of the SN soma that appears in the presence of 5-HT (Hochner et al., 1986; Baxter and Byrne, 1990; Ghirardi et al., 1992; Goldsmith and Abrams, 1992). We examined the effect of bath-applied Rp-cAMPS on hyperexcitability of SNs in excised ganglia produced by (1) applying 5-HT in the bath and (2) puffing 5-HT directly onto the soma. We were surprised to find that bath application of Rp-cAMPS at 0.5 mm caused little or no depression of hyperexcitability of SNs produced by either form of delivery of 5-HT, and only partially attenuated hyperexcitability at 2 mm. In addition, when we tested dissociated SNs, depression of 5-HT-induced hyperexcitability by 0.5 mm Rp-cAMPS was not reliable. However, the more permeant analog, Rp-8-CPT-cAMPS (1 mm), clearly reduced 5-HT-induced hyperexcitability in excised ganglia (Fig.2A). Excitability was quantified by counting the spikes (repetitive firing) evoked by a 1 sec depolarizing test pulse. The repetitive firing evoked in the presence of both 5-HT (5–10 μm) and this PKA inhibitor was less than the firing evoked in the same cells in the presence of 5-HT alone (mean ± SEM number of spikes, 7.3 ± 0.9 and 12.9 ± 0.9, respectively; p < 0.001; n = 10 and 9 cells). We also observed that bath application of Rp-8-CPT-cAMPS (0.5–1 mm) consistently reduced 5-HT-induced hyperexcitability in dissociated SNs in culture (data not shown), extending observations of Ghirardi et al. (1992), who used the less permeant PKA inhibitor, Rp-cAMPS. We then confirmed, as shown previously by Goldsmith and Abrams (1992), that injection of the highly specific PKA inhibitor PKI (1.5 mm in micropipette) into SNs in the isolated ganglion preparation reduces hyperexcitability produced by puffing 5-HT (50 μm) onto the soma (Fig.2B). This reduction by injected PKI was observed in every cell examined (n = 5). Together, these results indicate that either bath application of Rp-8-CPT-cAMPS or injection of PKI into SNs reduces soma hyperexcitability that is thought to depend on PKA activation.
Fig. 2.
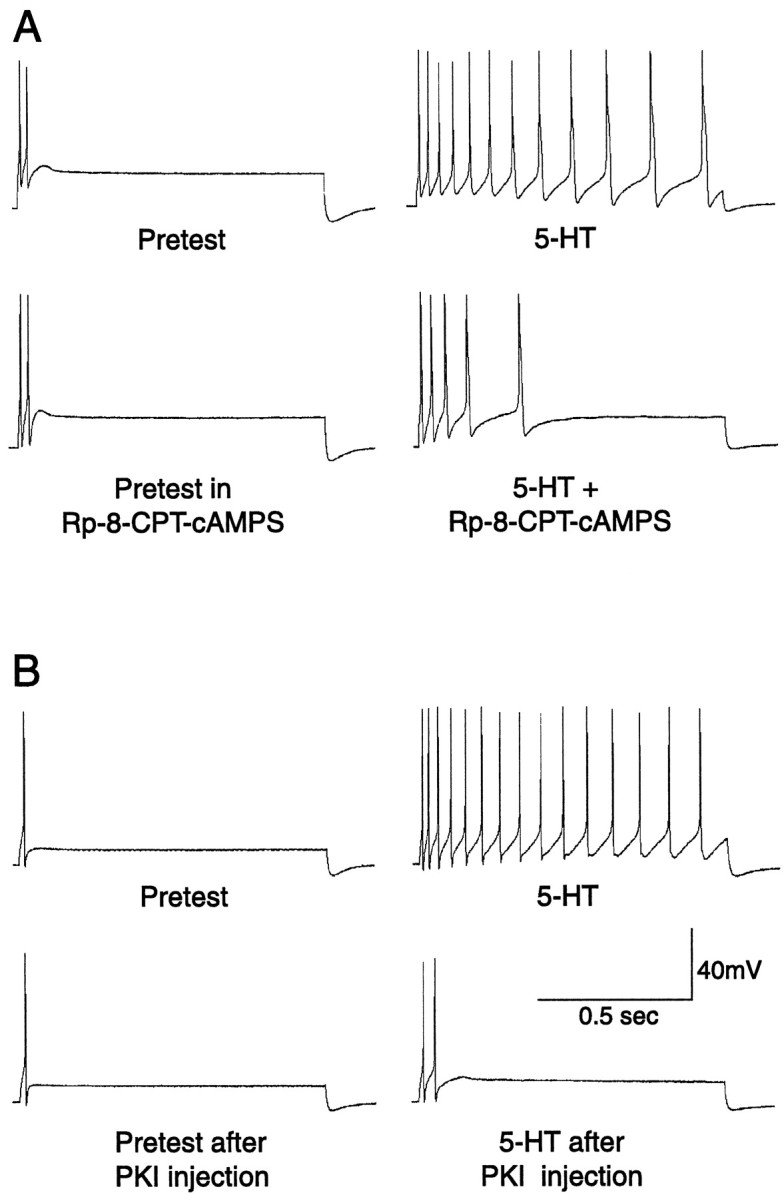
Examples of the reduction of immediate 5-HT-induced hyperexcitability by PKA antagonists. A, Bath application of 1 mm Rp-8-CPT-cAMPS reduced the increase in repetitive firing (compared with the pretest) caused by applying 5 μm 5-HT to the bath. B, Injection of PKI (1.5 mm, with 0.05% FG dye) into the SN reduced the increase in repetitive firing produced by puffing 50 μm 5-HT onto the cell soma.
Dissociation of SNs induces LTH with a latency of several days
LTH of Aplysia SNs is produced by several forms of axon injury, including excision of the cluster of SN somata from the ganglion (Gunstream et al., 1995). This suggested that axotomy caused by dissociating individual SNs might also induce LTH. We dissociated pleural SNs using standard methods (in particular, [Mg2+] was elevated during dissection and desheathing to prevent neuromodulator release, but not during dissociation). Figure 3A shows the effects on repetitive firing evoked by a 1 sec test pulse normalized to spike threshold. This normalization factored out alterations attributable to changes in spike threshold (see below), leaving alterations caused primarily by changes in spike accommodation (Walters et al., 1991). No significant differences were seen in repetitive firing between dissociated SNs and SNs in control ganglia for 3 d after dissociation. However, on days 4–7, dissociated SNs displayed significant enhancement of repetitive firing compared with SNs in control ganglia (F8,60 = 18.2;p < 0.0001; with p < 0.01 for each day between days 4 and 7 by Dunnett’s test). Dissociation also enhanced excitability by lowering spike threshold (Fig. 3B); lower thresholds were observed in dissociated SNs compared with thresholds in control ganglia 2–7 d after dissociation (F8,60 = 5.35; p < 0.0001; withp < 0.01 for each of days 2–7). The decrease in threshold was caused, at least in part, by an increase in input resistance, Rin. Because of large variability inRin after dissociation, we pooled measurements on days 0–3 and measurements on days 4–7. Rinon days 4–7 (117 ± 9 MΩ, n = 20) was significantly greater than Rin on days 0–3 (90 ± 9 MΩ, n = 35), and both groups of dissociated SNs showed significantly greater Rinthan SNs tested in control ganglia attached to long pedal nerves during the same period (46 ± 5 MΩ, n = 21;F2,75=13.6; p < 0.0001, ANOVA;p < 0.001 for each dissociated group vs SNs in ganglia, and p < 0.05 for days 4–7 vs days 0–3; Newman–Keuls tests). For purposes of comparison (see next section), this figure also shows the previously reported LTH on day 3 produced by transecting neurites of dissociated SNs on day 2 (data from Ambron et al., 1996). These experiments were run during the same period in the same populations of animals as reported by Ambron et al. (1996).
Fig. 3.

Long-term hyperexcitability of SNs produced by dissociation. Repetitive firing (A) and spike threshold (B) were tested in SNs sampled at the indicated times after dissociation. G, Control cells tested in excised ganglia preparations during the same period. Data in this and other figures are shown as means ± SEM. *Significant differences (p < 0.05) compared with control SNs in ganglia. In addition to producing changes in repetitive firing and spike threshold, dissociation increasedRin (see Results). Also shown is the previously reported enhancement of excitability 24 hr later produced by transecting SN neurites 2 d after dissociation (Ambron et al., 1996).
Rp-8-CPT-cAMPS blocks expression but not induction of dissociation-induced LTH
Four days after dissociation, repetitive firing was reduced and spike threshold was increased when SNs were tested in the presence of Rp-8-CPT-cAMPS (0.5–1.2 mm) (Fig.4A,B). Depression of firing was observed at each of the three test currents used (p < 0.001 in each case; n = 47, 14, and 10 for the 1, 2, and 3 nA groups tested in ASW, andn = 32, 23, and 21 for the same groups tested in Rp-8-CPT-cAMPS). These effects were rapidly reversed by washout of the inhibitor (data not shown). In contrast to its immediate depressive effect on SNs made hyperexcitable by previous dissociation, the Rp-8-CPT-cAMPS did not affect the induction of LTH by dissociation. When Rp-8-CPT-cAMPS (1 mm) was present from 30 min before to 24 hr after dissociation, LTH 4 d later was unaffected (Fig. 4C) (n = 37 each for the 1, 2, and 3 nA groups axotomized in L15 alone; n = 32 each for the same groups axotomized in Rp-8-CPT-cAMPS). These data indicate that PKA activity is not necessary for the induction of LTH by the axotomy that occurs during dissociation, but they suggest that it contributes to the later expression of LTH.
Fig. 4.

Rp-8-CPT-cAMPS applied during dissociation does not prevent the induction of LTH, although it reduces the expression of LTH when applied later. A, Examples of repetitive firing tested at three test current intensities in the presence and absence of Rp-8-CPT-cAMPS 4–5 d after dissociation. B, Significant depression of repetitive firing and elevation of spike threshold when SNs (dissociated 4–5 d previously) were tested in the presence of Rp-8-CPT-cAMPS (Rp). Thresh, Mean threshold in nanoamperes for eliciting a single spike in each group (horizontal error bars that would indicate variability of threshold current are too small to be seen). C, Lack of effect of Rp-8-CPT-cAMPS (present during dissociation) on the induction of LTH measured 4–5 d later.
Rp-8-CPT-cAMPS blocks expression but not induction of LTH induced by neurite transection
Soma hyperexcitability also appears 24 hr after transecting the neurites of previously dissociated SNs growing in vitro(Ambron et al., 1996; Bedi et al., 1998). We asked whether this form of LTH requires PKA activation, as suggested by Bedi et al. (1998). Two days after plating, the SN neurites were severed (Fig. 1), and the cells were then tested for excitability 24 hr later. Cells tested in the presence of Rp-8-CPT-cAMPS (0.5–1 mm) were significantly less excitable than cells tested in the absence of inhibitor (Fig. 5). To examine effects on induction of LTH, we superfused Rp-8-CPT-cAMPS (0.5–1 mm) onto dissociated SNs 0.5 hr before transection of SN neurites and washed it out either 5.5 hr or 22.5 hr later. SN excitability was tested 24 hr after transection. When transection occurred in the presence of the PKA inhibitor, but testing was performed in its absence, no significant reduction of LTH was found, even when the inhibitor was left in until 90 min before testing. For the data shown in Figure 5, only the untransected cells and the cells tested in the presence of Rp-8-CPT-cAMPS were significantly less excitable than the transected cells (F4,34 = 24.8;p < 0.0001; with p < 0.01 for these two groups compared with the group transected in the absence of inhibitor; Dunnett’s test). These data suggest that PKA activity is not necessary for the induction of LTH by axotomy of SNs growing in dissociated cell culture but might contribute to the later expression of LTH.
Fig. 5.

Rp-8-CPT-cAMPS (Rp) applied during neurite transection does not prevent the induction of LTH, although it significantly reduces the expression of LTH if present during the test 24 hr after transection. Times indicate duration of Rp-8-CPT-cAMPS treatment after transection.
Axonal PKA activation is neither sufficient nor necessary for induction of delayed LTH by nerve crush
Crushing of pedal nerves containing the axons of SNs causes LTH of both the SN soma and axon, and in each case the LTH depends on protein synthesis within the ganglia (Walters et al., 1991; Gunstream et al., 1997). LTH occurs even if spike activity and synaptic transmission in the ganglia are blocked while the nerve is crushed. Under these conditions, rapid activation of PKA in the soma is unlikely, and the crush-induced LTH depends on retrograde transport of macromolecular signals from the axonal injury site to the soma (Ambron et al., 1995;Gunstream et al., 1995). An interesting possibility is that the generation of such retrogradely transported injury signals depends on activation of PKA in the axons (Schmied et al., 1993). We tested this possibility by conducting the study summarized in Figure6. Both sets of pleural–pedal ganglia were dissected from eight animals, leaving all pedal nerves as long as possible so that retrogradely transported injury signals from the site of surgical transection would not reach the soma during the period of the experiment (Gunstream et al., 1995). One of four different treatments was then given to each of the 16 pleural–pedal ganglion preparations (with attached nerves) (Fig. 1), as described below. After 24 hr, the excitability of both the soma and axon of sampled SNs was tested while the nerve was bathed with ASW and the ganglion was bathed with a solution containing altered concentrations of Ca2+ (0.01% normal) and Mg2+(200% normal). This prevented release of neuromodulators in the ganglia during testing, as well as generation of afterdischarge in or near the SN soma that could confound measurements of axonal excitability.
Fig. 6.

PKA activation in the nerve is neither sufficient nor necessary for the induction of LTH. A, Activation of PKA by bathing the nerve in 8-CPT-cAMP + IBMX (cAMP) for 2 hr failed to increase repetitive firing of the soma or axons when tested 24 hr later. In addition, bathing the nerves in Rp-8-CPT-cAMPS (Rp) for 30 min before and 2 hr after they were crushed failed to prevent LTH 24 hr later. B, Bathing the nerve in 8-CPT-cAMP + IBMX also failed to reduce spike threshold in the soma 24 hr later, and bathing the nerves in Rp-8-CPT-cAMPS during and after nerve crush failed to prevent the reduction in soma threshold 24 hr later.
We first tested the sufficiency of PKA activation in the nerve to induce LTH in the absence of proximal nerve injury. To strongly stimulate axonal PKA in each experimental preparation, pedal nerves 7, 8, and 9 were bathed 1–2 cm from the pedal ganglion with a solution containing both the membrane-permeant cAMP analog 8-CPT-cAMP (100 μm) and a phosphodiesterase inhibitor, IBMX (500 μm), for 2 hr (Schacher et al., 1988). In the control (contralateral) preparation, the nerves were treated with ASW. Although 8-CPT-cAMP and IBMX produced an immediate increase in SN axon excitability that persisted for as long as the drugs were present (X. Liao, C. Brou, and E. Walters, unpublished observations), this treatment did not alter the repetitive firing (Fig.6A) or spike threshold (Fig. 6B) of either the soma or the axons 24 hr after treatment (n = 4 preparations in each group, with six SNs sampled per preparation).
We then tested the necessity of PKA activation in the nerve for axon and soma LTH induced by nerve injury. Pedal nerves 7, 8, and 9 were crushed 1 cm from the pedal ganglion in eight pleural–pedal ganglion preparations. In half the preparations, the crush was administered to a region of nerve that was bathed in Rp-8-CPT-cAMPS (1 mm); in the contralateral preparations the crush was administered in ASW. The Rp-8-CPT-cAMPS had no effect on the induction of axonal LTH, because there was no significant difference between the two groups (Fig. 6A), and both nerve-crushed groups displayed significant long-term enhancement of evoked repetitive firing compared with evoked firing in the uncrushed preparations (p < 0.01; n = 8 preparations in each group, with six SNs sampled per preparation). To assess soma LTH in these sensitized preparations, we measured spike threshold but did not test repetitive firing. We did this to avoid the intense repetitive firing that is often evoked by prolonged stimulation of the soma in such preparations, which could alter responses to subsequent test stimuli applied to the axon. Although nerve crush significantly decreased SN soma spike threshold compared with threshold in uncrushed preparations (Fig. 6B) (p < 0.01; n = 8 preparations in each group, with six SNs sampled per pleural ganglion), soma spike thresholds in SNs whose axons were crushed in Rp-8-CPT-cAMPS were not significantly different from those of SNs whose axons were crushed in ASW (Fig.6B) (n = 4 pleural ganglia in each group, with six SNs sampled per ganglion).
Nerve crush does not activate axonal PKA or stimulate its retrograde transport
The lack of a contribution of PKA activation to the induction of LTH by nerve crush received further support from measurements of PKA activity in the region of the crush. Pedal nerves were crushed 3 cm from the ganglion. Five minutes later crushed and uncrushed nerves were rapidly frozen and 1 cm segments were taken from three regions: (1) immediately proximal to the crush site, 2–3 cm from the ganglion, (2) proximal to the crush site, adjacent to the ganglion, and (3) 2–3 cm from the ganglion in uncrushed nerves. These nerve segments from three animals were pooled, homogenized, and centrifuged to obtain soluble and membrane fractions. No differences in constitutive activity were found among the different regions in either fraction. This constitutive activity was similar to levels of PKA activity reported inAplysia muscle in the absence of stimulation (Hooper et al., 1994a,b). All nerve segments also contained large amounts of inactive PKA, which could be activated by adding exogenous cAMP to the extracts. Despite the abundance of the holoenzyme, there was no significant difference in total activity among the nerve segments (Fig.7A). The distribution of the catalytic subunit between the supernatant and pellet was also essentially the same in the different segments. Because activation of a small amount of axonal PKA might be masked by a lack of activation of a potentially larger pool of PKA in surrounding glia and connective tissue, we also examined PKA activity in axoplasm extruded from nerve segments. Roughly one-third of the total kinase in the segment was found in the extrudate, but again there was no difference in activity of the axoplasm between the crushed segment and the two control segments (data not shown).
Fig. 7.

Nerve injury does not activate PKA or stimulate its transport toward the soma. A, Lack of injury-activated PKA at the crush site 5 min after crushing pedal nerves. No increase in the constitutive activity of the catalytic subunit in the crushed region was found in either the supernatant (Sup) or pellet (Pellet) compared with activity in a region of the same nerves near the ganglion (Proximal control) or in the region in uncrushed contralateral nerves corresponding to the crushed region (Contralateral control). Each segment contained a considerable amount of the PKA holoenzyme, which was revealed by adding exogenous cAMP to the extracts (Total activity). No transfer of active catalytic subunit from the soluble fraction (Sup) to the membrane fraction (Pellet) occurred in the crushed region, as indicated by similar ratios ofSup/Pellet activity in each region. B, C, Lack of injury-induced transport of PKA. Activity of the PKA catalytic subunit was assessed 20 hr after pedal nerve crush in segments of whole nerve (B) and in extruded axoplasm (C). Segments (supernatant) or extruded axoplasm were assayed on the proximal side adjacent to the crush site (Cr) and immediately distal to a ligation that was midway between the ganglion and the crush site (Cr/Lig). Any PKA that was activated and transported retrogradely would accumulate at the Cr/Lig site. As controls, segments and axoplasm immediately distal to a ligation on an uncrushed nerve (Lig), or from a corresponding section of a control nerve that was neither crushed nor ligated (Co), were examined (Ambron et al., 1995).
To determine whether PKA had been transported toward the soma 20 hr after nerve crush, we collected axoplasm that had accumulated at a ligation near the pedal ganglia in two animals (Ambron et al., 1995). The amount of PKA activity found in whole nerve segments (Fig.7B) or in extruded axoplasm (Fig. 7C) from the ligated site on crushed nerves was no higher than the corresponding activity from the ligated site in uncrushed nerves. This shows that axotomy does not trigger the transport of PKA toward the soma.
PKI injection reduces expression of central LTH induced by in vivo nerve crush
Our studies found no evidence that PKA activation in the nerve contributes to the induction of LTH after axotomy. The experiments shown in Figures 4 and 5, however, suggest that persistent PKA activity in the soma does contribute to the maintenance orexpression of LTH. To test this possibility further, pedal nerves were crushed unilaterally in vivo. Both pleural–pedal ganglia were excised and exposed to Rp-8-CPT-cAMPS (0.5–1 mm in the bath) 4–5 d later. We found a significant depression of repetitive firing in the presence of the inhibitor (data not shown). Unexpected, however, was the observation that this inhibitor (and Rp-cAMPS) also reduced repetitive firing in SNs whose axons had not been crushed. Among other possibilities (see Discussion), this suggested that these monophosphorothioate PKA inhibitors might have immediate effects on excitability that are independent of PKA activity. Therefore, we then tested the effects of injecting PKI, an unrelated and more specific PKA inhibitor. Pedal nerves were crushed unilaterally in vivo under anesthesia. Pleural ganglia were excised 4–5 d later from each animal under heavy anesthesia, with the nerves left as long as possible, to prevent effects on excitability from either rapid or slow injury signals generated during the dissection. Individual SNs were then tested before (baseline) and after being injected with either FG dye or PKI + FG. Figure 8A shows the effects of these injections on repetitive firing measured 1–2 min after injection. SNs whose axons had not been crushed 4–5 d earlier showed no significant changes in repetitive firing responses after PKI injection, and responses of these cells (n = 17) were not significantly different from those of SNs in the same ganglion that were injected with FG alone (n = 14). In contrast, previously axotomized SNs injected with PKI showed significant depression of repetitive firing compared with both their pre-injection responses (p < 0.001; n = 20) and responses of axotomized cells in the same ganglia injected with FG alone (p < 0.005; n = 22). PKI injection also altered spike threshold (Fig. 8B); the increase in threshold was significantly greater in previously axotomized SNs than in SNs whose axons had not been crushed (p < 0.005; n = 20 and 17) and was larger than the increase in threshold in both groups injected with FG alone (p < 0.05 in each case;n = 20, 14, and 22). There was also a small but significant increase of threshold of previously crushed nerves after injection of FG alone, indicating that FG, the injection per se, or the pre-injection test can increase threshold even if no PKA inhibitor is injected. However, the larger increase in threshold by PKI injection in previously axotomized SNs than in uninjured SNs (Fig.8B) and the greater reduction of repetitive firing in previously axotomized SNs (compared with uninjured SNs) (Fig.8A) suggest that nerve crush under anesthesia results in a maintained activation of PKA in or near the SN soma 4–5 d later.
Fig. 8.

Evidence that LTH induced by nerve crush 4–5 d earlier is partially maintained by PKA activity. A,Left, Neither injection of Fast Green (FG) dye nor FG + protein kinase inhibitor (FG + PKI) affected repetitive firing responses tested in somata of SNs whose axons had not been crushed in vivo.Right, SNs whose axons had been crushed displayed greater firing responses than SNs with uncrushed axons, but this injury-induced hyperexcitability of the soma was significantly reduced by injection of PKI. B, Left, Injection of PKI significantly increased spike threshold in SNs whose axons had not been crushed. Right, Injection of both FG and PKI + FG significantly increased threshold in SNs with previously crushed axons. However, the percentage increase (black bars) after PKI + FG injection into previously axotomized SNs (Crushed nerves, right side) was significantly greater than the increase after FG injection, or the increases in previously unaxotomized SNs (Uncrushed nerves, left side).
DISCUSSION
Dissociation induces delayed long-term hyperexcitability of SNs
Dissociated SNs from Aplysia are commonly used to study memory-related alterations, including changes in excitability (Dale et al., 1987). Although dissociation severs SN axons and other forms of axotomy trigger LTH (Walters et al., 1991; Gunstream et al., 1995), it was not known whether dissociation also triggers LTH. Dissociation-induced LTH is not expressed for 3–4 d and peaks after an additional 2–3 d. Thus, LTH is probably still developing 4–5 d after dissociation, when synaptic growth has plateaued and studies of dissociated SNs are typically conducted. The latency for dissociation-induced LTH is 1–2 d longer than that for LTH induced by either nerve crush close to the ganglion or excision of the whole SN cluster (Gunstream et al., 1995), suggesting that the long latency is not simply a consequence of the time required to transport injury signals to the soma. The latency for the decrease in spike threshold after dissociation was shorter than the latency for the increase in repetitive firing (Fig. 3), perhaps because of the early increase inRin after axotomy. Conductance changes that persist long after axotomy may be complex and have yet to be characterized. It should be noted that SNs were dissociated under conditions that allow Ca2+ influx. Therefore, PKA might have been activated during dissociation not only by indirect effects of Ca2+ flux into axotomized SNs but also by release of neuromodulators from terminals near SN somata (Zhang et al., 1991).
PKA activation is not important for induction of long-term hyperexcitability by axotomy
An extensively documented pathway for induction of transcription-dependent plasticity involves synthesis of cAMP, activation of PKA, translocation of PKA into the nucleus, and activation of CREB transcription factors (Abel and Kandel, 1998; Dubnau and Tully, 1998; Silva et al., 1998). Because much evidence for this pathway in memory formation was obtained from Aplysia SNs, and 5-HT (which stimulates cAMP–PKA–CREB in these cells) produces long-term effects similar to those of axotomy (Walters and Ambron, 1995; Ambron and Walters, 1996), it seemed likely that PKA activation would contribute to induction of LTH by axotomy. Strengthening this assumption was a report that C/EBP, an immediate-early gene induced inAplysia CNS by in vivo treatment with 5-HT or cAMP analogs, is also induced by tissue dissection (Alberini et al., 1994). This suggested that some SN genes can be induced by both PKA and injury.
Although research on long-term plasticity has focused on synaptic alterations, a parsimonious assumption has been that long-term changes in soma conductances and synapses are induced by the same signal pathways (Dale et al., 1987; Scholz and Byrne, 1987; Walters, 1987). Indeed, Scholz and Byrne (1988) showed that injection of cAMP into individual SNs depressed outward currents in the soma 24 hr later, andLewin and Walters (1996, 1999) found that cAMP injection caused soma LTH as revealed by the tests used in the present study. Furthermore,Bedi et al. (1998) observed that application of Rp-cAMPS during neurite transection reduced 24 hr LTH in dissociated cell culture. These findings, however, did not prove that PKA contributes to the induction of LTH by axotomy. The concentrations of cAMP used to induce LTH were quite high (200–400 mm in the micropipette) (Scholz and Byrne, 1988; Lewin and Walters, 1996, 1999) and thus might have had nonspecific effects. Moreover, the Rp-cAMPS applied during neurite transection by Bedi et al. (1998) was not washed out until shortly before testing. Because Rp-cAMPS is not highly membrane permeant and is resistant to phosphodiesterases, intracellular Rp-cAMPS levels would be unlikely to change rapidly after washout of extracellular Rp-cAMPS. Consequently, the reduction in LTH observed by Bedi et al. (1998) might have represented an effect of the Rp-cAMPS on expression rather than induction of LTH.
Our results show that PKA makes little or no contribution to the induction of LTH by axon injury. Prolonged application of Rp-8-CPT-cAMPS or Rp-cAMPS during axotomy, at concentrations effective for blocking 5-HT-induced hyperexcitability, had no effect on LTH 1–5 d later. These PKA inhibitors failed to reduce LTH of the soma (or, in some cases, the axon) when applied during (1) dissociation of SNs, (2) transection of neurites growing from dissociated SNs, or (3) nerve crush. Activating PKA by exposing uninjured nerve segments to solutions containing 8-CPT-cAMP and IBMX failed to induce LTH of the soma or axon. Furthermore, despite substantial inactive PKA in pedal nerves, nerve crush failed to activate PKA in the nerve or induce its retrograde transport. Interestingly, LTH induced by brief noxious stimulation of the body is also independent of PKA activation (Lewin and Walters, 1998, 1999).
These findings suggest that protein kinases other than PKA induce long-term changes after axotomy. Among candidate protein kinases are several that have been implicated in long-term plasticity of the SNs. For example, a mitogen-activated protein kinase (MAPK) is necessary for 5-HT-induced long-term synaptic facilitation (Martin et al., 1997). Injection of MAPK into the soma can induce LTH, and a possible MAPK homolog in Aplysia nerve is activated by nerve crush and transported to the soma (Sung et al., 1998). In addition, PKC can activate MAPK cascades (Berra et al., 1995), and an activator of PKC can induce LTH in the SNs (Manseau et al., 1998). An important role for PKC is further suggested by observations that PKC inhibitors, but not a MAPK inhibitor, significantly reduce induction of LTH by dissociation (Liao et al., 1998). Another potential contributor to the induction of LTH by injury is PKG, because LTH induced by body pinch depends on PKG activation, and injection of cGMP into the SN soma induces LTH (Lewin and Walters, 1999). Given the high metabolic cost of injury responses (especially regenerative growth), it may be adaptive for LTH and other long-term reactions to axotomy to be contingent on multiple signals.
An unanswered question is why 5-HT-induced long-term synaptic facilitation in dissociated cell culture depends on PKA activation, whereas LTH induced by axotomy or by noxious cutaneous stimulation does not. Answers to this question may have to consider not only differences in types of plasticity and induction conditions, but also the possibility that such plasticity involves intermediate consolidation phases that connect early induction to late expression phases. Delayed PKA activity might contribute to some consolidation phases after axotomy [also see Lewin and Walters (1999)].
PKA activity contributes to the late expression of axotomy-induced hyperexcitability
Injection of the PKA inhibitor PKI into SNs 4–5 d after axotomy decreased repetitive firing and increased spike threshold, whereas injection of PKI into SNs that had not been axotomized did not. This indicates that nerve injury causes an activation of PKA in the soma that lasts for days and contributes to the maintenance of LTH. A similar role for prolonged PKA activation in the maintenance of long-term plasticity is indicated by enhanced PKA activity in the SNs 1 d after 5-HT treatment (Mueller et al., 1998). PKA activation produces immediate hyperexcitability of Aplysia SNs (Baxter and Byrne, 1990; Goldsmith and Abrams, 1992; Hochner and Kandel, 1992), in part by closing S-type K+ channels (Siegelbaum et al., 1982). Thus, persistent activation of PKA after nerve crush should continuously maintain hyperexcitability. Maintained PKA activation could be produced by persistent activation of interneurons that continuously release neuromodulators (e.g., 5-HT) that activate PKA in SNs. This possibility is supported by observations of a reduction in the expression of nerve crush-induced LTH by treatments that block ongoing spike activity (Gasull et al., 1997), or by a 5-HT antagonist (Liao, Brou, and Walters, unpublished observations). A second possibility is that noxious stimulation causes loss of regulatory subunits of PKA, thereby maintaining activity of the catalytic subunits (Greenberg et al., 1987; Hegde et al., 1993). Although this effect lasts <24 hr after 5-HT treatment (Hegde et al., 1997), it might last longer after nerve crush or dissociation.
Application of Rp-8-CPT-cAMPS several days after axotomy reduced soma excitability, providing additional evidence that continuing PKA activity maintains LTH. However, Rp-8-CPT-cAMPS also reduced excitability of SNs that had not been axotomized previously. The action on uninjured SNs could be explained by either (1) inhibition of basal PKA activity causing a reduction in background excitability or (2) PKA-independent effects on membrane excitability. In separate studies (Liao and Walters, unpublished observations) we found that both Rp-8-CPT-cAMPS and Rp-cAMPS have transient effects on SN membrane properties that differ somewhat from those of PKI. Because PKI is a highly specific inhibitor of PKA (Kemp et al., 1988) and because all of the electrophysiological effects of PKI, unlike some of those of the monophosphorothioate analogs of cAMP, oppose known actions of cAMP inAplysia SNs, we have relied on PKI to assess contributions of PKA to the expression of LTH. On the other hand, using Rp-8-CPT-cAMPS to test the role of PKA activity in theinduction of LTH (see above) is appropriate because the monophosphorothioate analogs of cAMP effectively inhibit PKA (Van Haastert et al., 1984). Any transient, nonspecific effects on excitability should not affect measurements of LTH made several days after washout of the inhibitor.
Footnotes
This work was supported by National Institutes of Health Grants NS35979 and NS35882 to E.T.W. and NS22150 to R.T.A. Animals were supplied by the National Center for Research Resources, National Resource forAplysia, at the University of Miami under National Institutes of Health Grant RR10294.
Correspondence should be addressed to Dr. Edgar T. Walters, Department of Integrative Biology, Pharmacology and Physiology, University of Texas–Houston Medical School, Houston, TX 77030.
REFERENCES
- 1.Abel T, Kandel E. Positive and negative regulatory mechanisms that mediate long-term memory storage. Brain Res Brain Res Rev. 1998;26:360–378. doi: 10.1016/s0165-0173(97)00050-7. [DOI] [PubMed] [Google Scholar]
- 2.Alberini CM, Ghirardi M, Metz R, Kandel ER. C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Cell. 1994;76:1099–1114. doi: 10.1016/0092-8674(94)90386-7. [DOI] [PubMed] [Google Scholar]
- 3.Ambron RT, Walters ET. Priming events and retrograde injury signals: a new perspective on the cellular and molecular biology of nerve regeneration. Mol Neurobiol. 1996;13:61–79. doi: 10.1007/BF02740752. [DOI] [PubMed] [Google Scholar]
- 4.Ambron RT, Dulin MF, Zhang X-P, Schmied R, Walters ET. Axoplasm enriched in a protein mobilized by nerve injury elicits memory-like alterations in Aplysia neurons. J Neurosci. 1995;15:3440–3446. doi: 10.1523/JNEUROSCI.15-05-03440.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ambron RT, Zhang X-P, Gunstream JD, Povelones M, Walters ET. Intrinsic injury signals enhance growth, survival, and excitability of Aplysia neurons. J Neurosci. 1996;16:7469–7477. doi: 10.1523/JNEUROSCI.16-23-07469.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey CH, Montarolo P, Chen M, Kandel ER, Schacher S. Inhibitors of protein and RNA synthesis block structural changes that accompany long-term heterosynaptic plasticity in Aplysia. Neuron. 1992;9:749–758. doi: 10.1016/0896-6273(92)90037-e. [DOI] [PubMed] [Google Scholar]
- 7.Baxter DA, Byrne JH. Differential effects of cAMP and serotonin on membrane current, action-potential duration, and excitability in somata of pleural sensory neurons of Aplysia. J Neurophysiol. 1990;64:978–990. doi: 10.1152/jn.1990.64.3.978. [DOI] [PubMed] [Google Scholar]
- 8.Bedi SS, Salim A, Chen S, Glanzman DL. Long-term effects of axotomy on excitability and growth of isolated Aplysia sensory neurons in cell culture: potential role of cAMP. J Neurophysiol. 1998;79:1371–1383. doi: 10.1152/jn.1998.79.3.1371. [DOI] [PubMed] [Google Scholar]
- 9.Berra E, Diaz-Meco MT, Lozano J, Frutos S, Municio MM, Sanchez P, Sanz L, Moscat J. Evidence for a role of MEK and MAPK during signal transduction by protein kinase C zeta. EMBO J. 1995;14:6157–6163. doi: 10.1002/j.1460-2075.1995.tb00306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clatworthy AL, Walters ET. Comparative analysis of hyperexcitability and synaptic facilitation induced by nerve injury in two populations of mechanosensory neurones of Aplysia californica. J Exp Biol. 1994;190:217–238. doi: 10.1242/jeb.190.1.217. [DOI] [PubMed] [Google Scholar]
- 11.Dale N, Kandel ER, Schacher S. Serotonin produces long-term changes in the excitability of Aplysia sensory neurons in culture that depend on new protein synthesis. J Neurosci. 1987;7:2232–2238. doi: 10.1523/JNEUROSCI.07-07-02232.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dale N, Schacher S, Kandel ER. Long-term facilitation in Aplysia involves increase in transmitter release. Science. 1988;239:282–285. doi: 10.1126/science.2892269. [DOI] [PubMed] [Google Scholar]
- 13.Dash PK, Tian LM, Moore AN. Sequestration of cAMP response element-binding proteins by transcription factor decoys causes collateral elaboration of regenerating Aplysia motor neuron axons. Proc Natl Acad Sci USA. 1998;95:8339–8344. doi: 10.1073/pnas.95.14.8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubnau J, Tully T. Gene discovery in Drosophila: new insights for learning and memory. Annu Rev Neurosci. 1998;21:407–444. doi: 10.1146/annurev.neuro.21.1.407. [DOI] [PubMed] [Google Scholar]
- 15.Gasull X, Gunstream JD, Lewin MR, Walters ET. Long-term hyperexcitability of Aplysia sensory neurons after nerve injury involves activity-dependent induction signals. Soc Neurosci Abstr. 1997;23:1958. [Google Scholar]
- 16.Ghirardi M, Braha O, Hochner B, Montarolo PG, Kandel ER, Dale N. Roles of PKA and PKC in facilitation of evoked and spontaneous transmitter release at depressed and nondepressed synapses in Aplysia sensory neurons. Neuron. 1992;9:479–489. doi: 10.1016/0896-6273(92)90185-g. [DOI] [PubMed] [Google Scholar]
- 17.Goda Y. Memory mechanisms. A common cascade for long-term memory. Curr Biol. 1995;5:136–138. doi: 10.1016/s0960-9822(95)00034-0. [DOI] [PubMed] [Google Scholar]
- 18.Goldsmith BA, Abrams TW. cAMP modulates multiple K+ currents, increasing spike duration and excitability in Aplysia sensory neurons. Proc Natl Acad Sci USA. 1992;89:11481–11485. doi: 10.1073/pnas.89.23.11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenberg SM, Castellucci VF, Bayley H, Schwartz JH. A molecular mechanism for long-term sensitization in Aplysia. Nature. 1987;329:62–65. doi: 10.1038/329062a0. [DOI] [PubMed] [Google Scholar]
- 20.Gunstream JD, Castro GA, Walters ET. Retrograde transport of plasticity signals in Aplysia sensory neurons following axonal injury. J Neurosci. 1995;15:439–448. doi: 10.1523/JNEUROSCI.15-01-00439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunstream JD, Castro GA, Walters ET. Region-specific, protein synthesis-dependent long-term hyperexcitability of Aplysia sensory neurons suggests local tagging of axons by 5HT. Soc Neurosci Abstr. 1997;23:1958. [Google Scholar]
- 22.Hegde AN, Goldberg AL, Schwartz JH. Regulatory subunits of cAMP-dependent protein kinases are degraded after conjugation to ubiquitin: a molecular mechanism underlying long-term synaptic plasticity. Proc Natl Acad Sci USA. 1993;90:7436–7440. doi: 10.1073/pnas.90.16.7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH. Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell. 1997;89:115–126. doi: 10.1016/s0092-8674(00)80188-9. [DOI] [PubMed] [Google Scholar]
- 24.Hochner U, Kandel ER. Modulation of a transient K+ current in the pleural sensory neurons of Aplysia by serotonin and cAMP: implications for spike broadening. Proc Natl Acad Sci USA. 1992;89:11476–11480. doi: 10.1073/pnas.89.23.11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hochner B, Klein M, Schacher S, Kandel ER. Action-potential duration and the modulation of transmitter release from the sensory neurons of Aplysia in presynaptic facilitation and behavioral sensitization. Proc Natl Acad Sci USA. 1986;83:8410–8414. doi: 10.1073/pnas.83.21.8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hooper SL, Probst WC, Cropper EC, Kupfermann I, Weiss KR. Myomodulin application increases cAMP and activates cAMP-dependent protein kinase in the accessory radula closer muscle of Aplysia. Neurosci Lett. 1994a;179:167–170. doi: 10.1016/0304-3940(94)90960-1. [DOI] [PubMed] [Google Scholar]
- 27.Hooper SL, Probst WC, Cropper EC, Kupfermann I, Weiss KR. SCP application or B15 stimulation activates cAPK in the ARC muscle of Aplysia. Brain Res. 1994b;657:337–341. doi: 10.1016/0006-8993(94)90988-1. [DOI] [PubMed] [Google Scholar]
- 28.Kemp BE, Cheng HC, Walsh DA. Peptide inhibitors of cAMP-dependent protein kinase. Methods Enzymol. 1988;159:173–183. doi: 10.1016/0076-6879(88)59018-3. [DOI] [PubMed] [Google Scholar]
- 29.Lewin MR, Walters ET. Long-term hyperexcitability of Aplysia sensory neurons following cAMP injection: involvement of Ca2+ and other signals. Soc Neurosci Abstr. 1996;22:1445. [Google Scholar]
- 30.Lewin MR, Walters ET. Cyclic GMP pathway is critical for inducing long-term sensitization of nociceptive sensory neurons. Nature Neurosci. 1999;2:18–23. doi: 10.1038/4520. [DOI] [PubMed] [Google Scholar]
- 31.Liao X, Gunstream JD, Lewin MR, Ambron RT, Walters ET. Injury-induced long-term hyperexcitability of Aplysia sensory neurons is not due to persistent activation of PKA: differences between Rp-cAMPS and PKI treatments. Soc Neurosci Abstr. 1997;23:1332. [Google Scholar]
- 32.Liao X, Grose E, Walters ET. Activity of PKC but not PKA is important for induction of dissociation-induced long-term hyperexcitability of Aplysia sensory neurons. Soc Neurosci Abstr. 1998;24:701. [Google Scholar]
- 33.Manseau F, Sossin WS, Castellucci VF. Long-term changes in excitability induced by protein kinase C activation in Aplysia sensory neurons. J Neurophysiol. 1998;79:1210–1218. doi: 10.1152/jn.1998.79.3.1210. [DOI] [PubMed] [Google Scholar]
- 34.Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 35.Montarolo PG, Goelet P, Castellucci VF, Morgan J, Kandel ER, Schacher S. A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia. Science. 1986;234:1249–1254. doi: 10.1126/science.3775383. [DOI] [PubMed] [Google Scholar]
- 36.Müeller U, Carew TJ. Serotonin induces temporally and mechanistically distinct phases of persistent PKA activaty in Aplysia sensory neurons. Neuron. 1998;21:1423–1434. doi: 10.1016/s0896-6273(00)80660-1. [DOI] [PubMed] [Google Scholar]
- 37.Rayport SG, Schacher S. Synaptic plasticity in vitro: cell culture of identified Aplysia neurons mediating short-term habituation and sensitization. J Neurosci. 1986;6:759–763. doi: 10.1523/JNEUROSCI.06-03-00759.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schacher S, Proshansky E. Neurite regeneration by Aplysia neurons in dissociated cell culture: modulation by Aplysia hemolymph and the presence of the initial axonal segment. J Neurosci. 1983;12:2403–2413. doi: 10.1523/JNEUROSCI.03-12-02403.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schacher S, Castellucci VF, Kandel ER. cAMP evokes long-term facilitation in Aplysia sensory neurons that requires new protein synthesis. Science. 1988;240:1667–1669. doi: 10.1126/science.2454509. [DOI] [PubMed] [Google Scholar]
- 40.Schacher S, Kandel ER, Montarolo P. cAMP and arachidonic acid simulate long-term structural and functional changes produced by neurotransmitters in Aplysia sensory neurons. Neuron. 1993;10:1079–1088. doi: 10.1016/0896-6273(93)90056-w. [DOI] [PubMed] [Google Scholar]
- 41.Schmied R, Huang C-C, Zhang X-P, Ambron DA, Ambron RT. Endogenous axoplasmic proteins and proteins containing nuclear localization signal sequences use the retrograde axonal transport/nuclear import pathway in Aplysia neurons. J Neurosci. 1993;13:4064–4071. doi: 10.1523/JNEUROSCI.13-09-04064.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scholz KP, Byrne JH. Long-term sensitization in Aplysia: biophysical correlates in tail sensory neurons. Science. 1987;235:685–687. doi: 10.1126/science.2433766. [DOI] [PubMed] [Google Scholar]
- 43.Scholz KP, Byrne JH. Intracellular injection of cAMP induces a long-term reduction of neuronal K+ currents. Science. 1988;240:1664–1666. doi: 10.1126/science.2837826. [DOI] [PubMed] [Google Scholar]
- 44.Siegelbaum SA, Camardo JS, Kandel ER. Serotonin and cyclic AMP close single K+ channels in Aplysia sensory neurones. Nature. 1982;299:413–417. doi: 10.1038/299413a0. [DOI] [PubMed] [Google Scholar]
- 45.Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- 46.Steffensen I, Dulin MF, Walters ET, Morris CE. Peripheral regeneration and central sprouting of sensory neurone axons in Aplysia following nerve injury. J Exp Biol. 1995;198:2067–2078. doi: 10.1242/jeb.198.10.2067. [DOI] [PubMed] [Google Scholar]
- 47.Sung YJ, Povelones M, Zhang X-P, Zhu D-F, Ambron RT. Injury-activated kinase (IAK-1): a MAPK homologue that is retro- gradely transported after injury and which recognizes the transcription factor C/EBP. Soc Neurosci Abstr. 1998;24:65. [Google Scholar]
- 48.Titmus MJ, Faber DS. Axotomy-induced alterations in the electrophysiological characteristics of neurons. Prog Neurobiol. 1990;35:1–51. doi: 10.1016/0301-0082(90)90039-j. [DOI] [PubMed] [Google Scholar]
- 49.Van Haastert PJ, Van Driel R, Jastorff B, Baraniak J, Stec WJ, De Wit RJ. Competitive cAMP antagonists for cAMP-receptor proteins. J Biol Chem. 1984;259:10020–10024. [PubMed] [Google Scholar]
- 50.Walters ET. Multiple sensory neuronal correlates of site-specific sensitization in Aplysia. J Neurosci. 1987;7:408–417. doi: 10.1523/JNEUROSCI.07-02-00408.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walters ET. A functional, cellular, and evolutionary model of nociceptive plasticity in Aplysia. Biol Bull. 1991;180:241–251. doi: 10.2307/1542394. [DOI] [PubMed] [Google Scholar]
- 52.Walters ET. Injury-related behavior and neuronal plasticity: an evolutionary perspective on sensitization, hyperalgesia and analgesia. Int Rev Neurobiol. 1994;36:325–427. doi: 10.1016/s0074-7742(08)60307-4. [DOI] [PubMed] [Google Scholar]
- 53.Walters ET, Ambron RT. Long-term alterations induced by injury and by 5-HT in Aplysia sensory neurons: convergent pathways and common signals? Trends Neurosci. 1995;18:137–142. doi: 10.1016/0166-2236(95)93891-z. [DOI] [PubMed] [Google Scholar]
- 54.Walters ET, Byrne JH, Carew TJ, Kandel ER. Mechanoafferent neurons innervating tail of Aplysia. I. Response properties and synaptic connections. J Neurophysiol. 1983;50:1522–1542. doi: 10.1152/jn.1983.50.6.1522. [DOI] [PubMed] [Google Scholar]
- 55.Walters ET, Alizadeh H, Castro GA. Similar neuronal alterations induced by axonal injury and learning in Aplysia. Science. 1991;253:797–799. doi: 10.1126/science.1652154. [DOI] [PubMed] [Google Scholar]
- 56.Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- 57.Zimmermann M, Herdegen T. Plasticity of the nervous system at the systemic, cellular and molecular levels: a mechanism of chronic pain and hyperalgesia. Prog Brain Res. 1996;110:233–259. doi: 10.1016/s0079-6123(08)62578-9. [DOI] [PubMed] [Google Scholar]
- 58.Zhang ZS, Fang B, Marshak DW, Byrne JH, Cleary LJ. Serotoninergic varicosities make synaptic contacts with pleural sensory neurons of Aplysia. J Comp Neurol. 1991;311:259–270. doi: 10.1002/cne.903110207. [DOI] [PubMed] [Google Scholar]
- 59.Ziv NE, Spira ME. Spatiotemporal distribution of Ca2+ following axotomy and throughout the recovery process of cultured Aplysia neurons. Eur J Neurosci. 1993;5:657–668. doi: 10.1111/j.1460-9568.1993.tb00531.x. [DOI] [PubMed] [Google Scholar]