

Abstract
Adenovirus-mediated gene transfer into the brain is associated with significant inflammation and activation of anti-vector and anti-transgene immune responses that curtail the gene delivery of adenoviruses and therapeutic efficacy. Elucidating the molecular mediators of inflammatory and immune responses to adenoviruses injected into the brain should allow us to inhibit their inflammatory actions, thereby reducing vector clearance and enhance adenoviral-mediated gene transfer into the CNS. Cytokines are primary mediators of the immune response and are released during inflammation. Here we report for the first time that injection of replication-deficient adenovirus vectors into the cerebral ventricles of rats causes a rapid increase in body temperature. This fever response precedes any vector-encoded transgene expression and occurs with vectors encoding no transgene, as well as with vectors encoding a therapeutic transgene i.e., HSV1-thymidine kinase. No fever is detected after infection of the striatum, an important brain target in studies on neurodegeneration. After infection of the brain ventricles, CSF levels of immunoreactive tumor necrosis factor (TNF)-α and interleukin (IL)-1β increase significantly (up to 300-fold). In the hypothalamus, the locus of thermoregulation in the brain, only IL-1β and IL-6 are significantly elevated. A neutralizing TNF-α antibody has no effect on adenovirus-induced fever. However, pretreatment with either the IL-1 receptor antagonist or the cyclooxygenase inhibitor flurbiprofen completely abolishes adenovirus-induced fever, suggesting that IL-1 and prostaglandins are direct mediators of this response. These results are the first to demonstrate that IL-1, but not TNF-α, is the main mediator of a very early inflammatory response to adenovirus in the brain.
Keywords: adenovirus, inflammation, cytokines, brain, rat, fever
Despite their many advantages (Chen et al., 1994; Eck et al., 1996; Lowenstein et al., 1996; Choi-Lundberg et al., 1997; Geddes et al., 1997), adenoviral vectors induce innate inflammatory and adaptive immune responses on delivery either to peripheral organs such as the liver (Elkon et al., 1997; Lieber et al., 1997; Worgall et al., 1997), respiratory tract (Devergne et al., 1991;Ginsberg et al., 1991; McCoy et al., 1995), salivary glands (Adesanya et al., 1996), or the CNS (Wood et al., 1996). After the transduction of the liver or lungs, inflammatory and immune responses lead to the complete elimination of first generation vector particles and transduced cells within 2–3 weeks (Elkon et al., 1997). In the brain, however, low level persistence of biologically relevant amounts of transgene expression can be sustained (Wood et al., 1996) despite inflammatory, immune responses, and delayed-type hypersensitivity phenomena that can be triggered through the peripheral readministration of recombinant vectors (Byrnes et al., 1996a). Nevertheless, the mechanisms by which inflammatory and immune responses affect adenovirus-encoded transgene expression and clear viral vectors from the brain remain to be determined. This is reflected in contradictory data regarding long-term transgene expression. Thus, although one study reported a 4 months stable reversion of the diabetes insipidus phenotype of Brattelboro rats infected with adenoviral vectors expressing vasopressin (Geddes et al., 1997), a separate study failed to detect any adenovirally mediated β-galactosidase expression at 6 months after infection (Blomer et al., 1997).
Immune response priming occurs efficiently after infection of the CSF, but not the brain parenchyma, with replicating viruses (Stevenson et al., 1997a). This indicates that immune presentation is deficient in the brain parenchyma, but not in the CSF, suggesting that the elicitation of inflammatory or immune responses to viruses in the brain may be brain region-specific (Stevenson et al., 1997b). Thus, whether inflammatory and immune responses to viral vectors are elicited in the brain could depend on the anatomical area infected, the microbiological purity of viral vectors used, or the surgical technique used during virus delivery.
So far, the early molecular responses underlying the activation of inflammatory and immune responses after the administration of adenovirus vectors to target tissues are not well understood. In the liver and lung, despite rapid macrophage-mediated clearing of a large percentage of adenoviral genomes, a vast majority of hepatocytes become transduced (Elkon et al., 1997; Worgall et al., 1997). Longer term transgene expression, however, is further restricted by the cellular arm of the immune system; thus, in immunodeficient mice lacking T and B lymphocytes, transgene expression is prolonged (Zsengeller et al., 1995). The same does not apply to adenoviral infection of the CNS, where transgene expression is only moderately enhanced either in nude rats, in the absence of T and B cells, or after treatment with dexamethasone (Byrnes et al., 1996b; Hermens and Verhaagen, 1997). However, no information exists on the molecular basis responsible for triggering adenovirus-induced inflammation and immune clearance in the brain.
In this study, we examined early inflammatory responses after the injection of first generation (E1/E3 deleted) adenovirus vectors into the CSF or brain parenchyma (striatum) and detected extremely rapid increases in core body temperature and in the concentrations of proinflammatory cytokines, tumor necrosis factor (TNF)-α, interleukin (IL)-β, and IL-6 in the CNS. We obtained identical responses with first generation adenoviral vectors encoding: (1) the marker enzyme β-galactosidase, (2) the therapeutic transgene thymidine kinase of herpes simplex virus type 1 (HSV1-TK) under the control of the short major immediate early human cytomegalovirus (sMIEhCMV) promoter (RAd128), or (3) no transgene (RAd0).
MATERIALS AND METHODS
All animal procedures conformed with the requirements of the British Home Office Animal Licensing Inspectorate.
Animals and injections. All experiments were performed on adult male Sprague Dawley rats (Charles River Laboratories) of 250–300 gm body weight. The animals were housed individually in a temperature-controlled room (21 ± 2°C) artificially lit from 8:00 A.M. to 8:00 P.M. and were provided with food (pelleted rat chow; Beekay International) and water ad libitum.
Core body temperature was monitored in free-moving animals by remote radiotelemetry via small battery-operated temperature-sensitive radiotransmitters (Data Sciences International). Rats were anesthetized with halothane (3% in oxygen) and transmitters implanted into the abdominal cavity. The animals were allowed to recover for 7 d before experimentation. The output frequency (in Hertz) was monitored by an antenna, mounted in a receiver board situated beneath the individually caged animals, and converted to degrees centigrade (°C). Central injections were administered via an indwelling guide cannula, stereotaxically implanted into either the right lateral cerebral ventricle (intracerebroventricular; bregma, −0.8 mm; lateral, 1.5 mm; ventral, 3.5 mm) or the striatum (bregma, +0.3 mm; lateral, 3.6 mm; ventral, 5.5 mm) (Paxinos and Watson, 1986), during the same procedure as transmitter implantation. Intracerebroventricular and striatal injections were administered at 10:00 A.M. in a volume of 2 μl to conscious, free-moving animals (n = 5–6 per treatment).
Adenovirus construction and purification. Construction and characterization of RAd35 and the sMIEhCMV promoter, was described earlier (Wilkinson and Akrigg, 1992), and viruses were grown up and purified as previously described (Shering et al., 1997; Morelli et al., 1999). Briefly, the transfer vector pAL119/lacZ was constructed from pXCX2 with the addition of a linker containing theHindIII cloning site at the XbaI cleavage site.lacZ was cloned under sMIEhCMV promoter control and upstream of a polyadenylation signal on a HindIII expression cassette cotransfected with pJM17 (Microbix Biosystems Inc., Toronto, Canada) into HEK-293 cells by calcium phosphate precipitation. Homologous recombination resulted in the recombinant adenovirus, RAd35. The virus was propagated on HEK-293 cells, purified on previously prepared CsCl gradients (using a modified protocol with densities 1.45 and 1.33), dialyzed twice against a buffer of 10 mm Tris, 1 mm MgCl2, 135 mm NaCl, pH 7.5 and once against the same buffer plus 10% glycerol. The virus was titrated by plaque assay on 293 cells, and the viral titer was determined to be 6.55 × 1011 pfu/ml. Levels of endotoxin were measured using the E-toxate assay (Sigma, St. Louis, MO) and, in all virus preparations used, were below 0.3 endotoxin U/ml. The characterization of RAd128 is described in Dewey et al. (1998) and RAd0 in David et al. (1997).
Drugs. RAd35, RAd128, or RAd0 were diluted in sterile PBS and administered at a dose of 1.31 × 108pfu/rat (intracerebroventricular or striatum). Control animals were injected with vehicle (sterile PBS) (intracerebroventricular or striatum). The concentration of CsCl, as used in the viral purification gradient, was dialyzed against the purification buffers and injected intracerebroventricularly to control for the possible pyrogenic effect of any remaining CsCl in the viral preparation. Recombinant human IL-1ra (200 μg/rat; Peprotech, Rocky Hill, NJ) was administered intracerebroventricularly in saline vehicle at 0 and 1 hr. The cyclooxygenase inhibitor flurbiprofen (kindly provided by Dr. M. Dascombe, Manchester, UK), was dissolved in 1% sodium bicarbonate and 0.9% sterile saline, and administered intraperitoneally (1 mg/kg) 0.5 hr before intracerebroventricular treatment of adenovirus or vehicle. Rabbit murine TNF-α antiserum (kindly provided by Dr Steve Kunkel, Ann Arbor, MI) was administered intracerebroventricularly 24 hr before treatment with adenovirus or vehicle.
ELISA. At specific time points after injection of adenovirus or vehicle (intracerebroventricular or striatum; n = 5 per treatment per time point) CSF was collected from the cisterna magna of rats that had been anesthetized by halothane (3% in oxygen). Thereafter, the animals were killed by cervical dislocation, and the hypothalami and striati were removed. CSF samples containing any trace of blood after centrifugation (10,500 rpm, 10 min, 4°C) were discarded from all subsequent analyses. Brain samples were placed in sterile PBS containing a protease inhibitor cocktail [0.2 mm 4-[2-aminoethyl]benzenesulfonylfluoride, HCl (AEBSF), 1 μg/ml aprotinin, 1 mm benzamidine, 1 mmEDTA, 10 μg/ml leupeptin, and 10 μg/ml of pepstatin], homogenized, centrifuged (10,500 rpm, 15 min, 4°C), and the supernatant removed and stored at −70°C. All samples were assayed for immunoreactive TNF-α, IL-1β, and IL-6 using validated rat specific sandwich ELISAs (Safieh-Garabedian et al., 1995; Rees et al., 1998). The assay sensitivity for TNF-α, IL-1β, and IL-6 in CSF was 3.8 pg/ml and in brain tissue was 10 pg/ml.
Data analysis. All results are reported as mean values ± SEM. Body temperature data were analyzed for statistical significance according to the integrated hyperthermic response, calculated as the deviation from baseline over the 8 hr period after administration of adenovirus (8 hr fever index in degrees centigrade per hour). Differences between more than two groups were determined by ANOVA followed by Tukey–Kramer multiple comparisons post hoc test, and differences between two groups were identified by an unpaired Student’s t test. Changes in cytokine levels were analyzed using unpaired Student’st test. A two-tailed probability < 0.05 was considered statistically significant.
RESULTS
Intracerebroventricular, but not intrastriatal, injections of adenovirus vectors cause fever
Injection of 1.3 × 108 pfu of RAd35 into the CSF resulted in a rapid increase in body temperature starting 1–2 hr after vector injection (Fig.1A). At this time, no transgene can be detected by either X-gal histochemistry or immunocytochemistry (data not shown). Expression of β-galactosidase can be detected from 6 to 8 hr after infection (data not shown). The rise in temperature after the adenoviral intracerebroventricular injection was sustained, reaching a peak 7 hr after infection (vehicle 37.2 ± 0.2 vs RAd35 39.0 ± 0.1°C; t test;p < 0.001; n = 6). When the same amount of virus was injected into the striatum, no fever response was detected (Fig. 1B). Body temperature changes were monitored for 48 hr after injection. The fever after intracerebroventricular injection lasted for ∼10 hr. By 24 hr after infection, body temperature was back to normal and remained so for another 24 hr (data not shown).
Fig. 1.
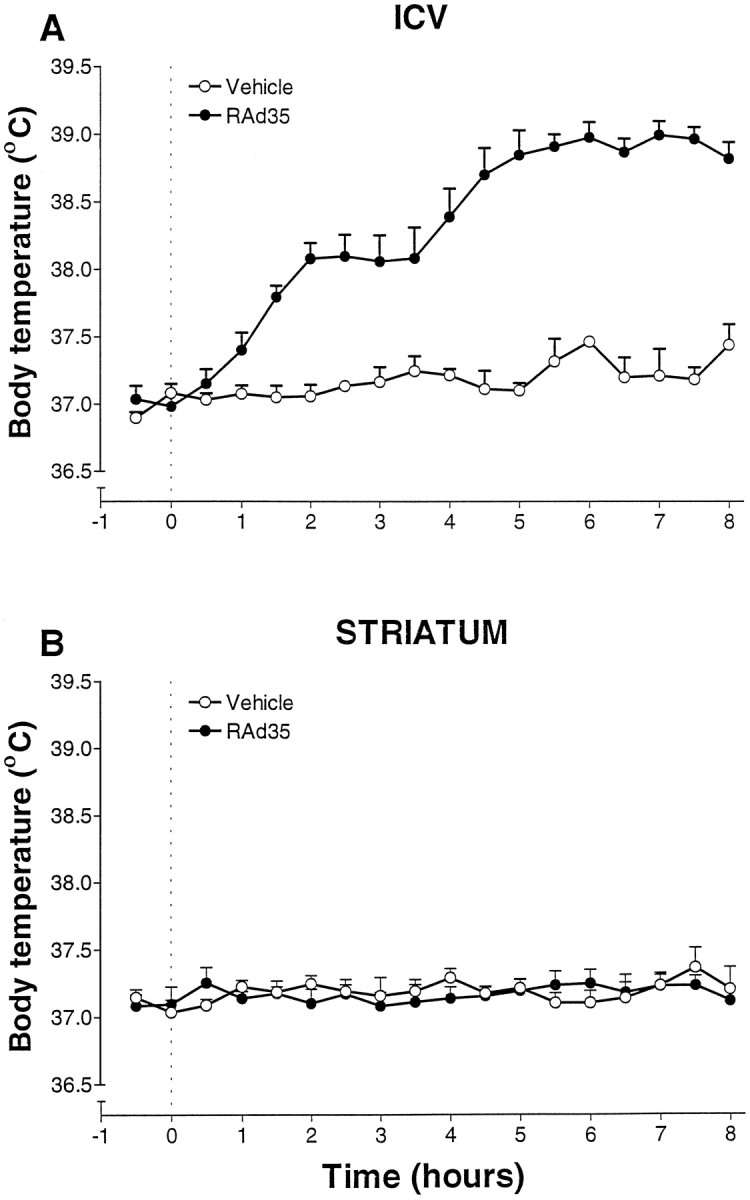
A, A replication-deficient recombinant adenovirus expressing β-galactosidase (RAd35) injected intracerebroventricularly (1.3 × 108 pfu in 2 μl) caused a significant increase in core body temperature that was maximal 7 hr after injection (***p < 0.001 vs vehicle; n = 6). Dotted lineindicates time of injection (0 hr, 10:00 A.M.). B, Striatal injection of the same recombinant adenovirus vector resulted in no significant change in core body temperature (vs vehicle;n = 5) for the duration of the experiment (48 hr).Dotted line indicates time of injection (0 hr).
Importantly, identical results were obtained with other adenoviral recombinants: (1) RAd128, a replication-deficient adenovirus expressing the herpes simplex virus type 1 thymidine kinase (Dewey et al., 1998), and (2) RAd0, a replication-deficient adenovirus encoding no transgene (David et al., 1997). All vectors induced fever of similar magnitude (Fig. 2). All viral preparations used in this study were endotoxin-free, as defined by Cotten et al. (1994)(<6 × 10−4 endotoxin units per dose of adenovirus administered into the brain). Thus, because vectors were purified on a double CsCl gradient-purified vector, and intracerebroventricular injection of CsCl alone did not induce fever, it is highly unlikely that the febrile response is caused by endotoxin or any other contaminant. Furthermore, heat treatment of Rad0 (30 min at 90°C) completely abolished adenovirus-induced fever (Fig. 2). This procedure abolishes adenovirus infectivity but does not inactivate endotoxin.
Fig. 2.

Temperature responses to intracerebroventricular (2 μl) injection of 1.3 × 108 pfu/rat of RAd35 (encoding the marker transgene β-galactosidase), RAd0 (containing no transgene), or RAd128 (encoding HSV1-TK) were significantly different from that of vehicle (ANOVA, ***p < 0.001). The concentration of CsCl, as used in the viral purification gradient, was dialyzed against the purification buffers and injected intracerebroventricularly to control for the possible pyrogenic effect of any remaining CsCl in the viral preparation. Neither the vehicle nor CsCl induced fever. Furthermore, heat treatment of RAd0 (30 min at 90°C) completely eliminated the fever response.
Adenovirus vectors induce increases in TNF-α, IL-1β, and IL-6 concentration in the brain
The proinflammatory cytokines, TNF-α, IL-1β and IL-6, are strong candidates for inducing fever in response to adenovirus infection of the brain. TNF-α can display pyrogenic effects (Stefferl et al., 1996) and has been reported recently to mediate the clearing of adenovirus from the liver after intravenous virus administration (Elkon et al., 1997). Both IL-1 and IL-6 are potent pyrogens when administered into the brain in many different experimental models both in rats and mice (Kluger, 1991). We, therefore, measured the levels of these cytokines in the CSF and hypothalamus after injection of adenovirus into the lateral ventricle (Fig. 3).
Fig. 3.
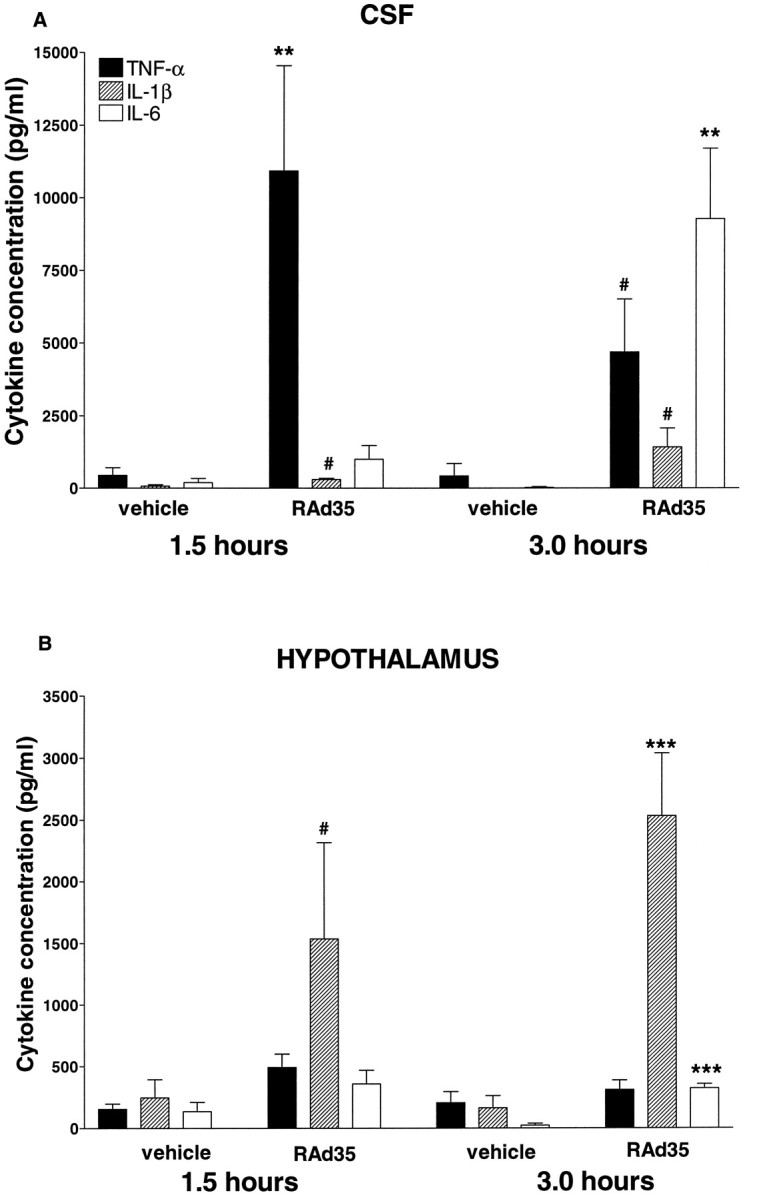
A, Intracerebroventricular injection of adenovirus elicited a marked increase in the levels of TNF-α (**p < 0.01) and IL-1β (#p < 0.05) in the CSF 1.5 hr after injection, followed at the 3 hr time point by a reduction in the TNF-α levels (by 61%) and a continued increase in IL-1β levels (#p < 0.05) when compared with vehicle-injected controls. IL-6 increased significantly (**p < 0.01 vs vehicle) 3 hr after adenovirus injection. B, Hypothalamic IL-1β increased significantly at the 1.5 hr time point (#p < 0.05 vs vehicle). Both IL-1β (***p < 0.001) and IL-6 (***p< 0.001) levels were significantly elevated 3 hr after adenovirus injection, compared with vehicle-injected controls.
In the CSF (Fig. 3A) at 1.5 hr after infection, we detected a 25-fold increase of TNF-α (vehicle 443.5 ± 261.5 vs RAd35 10,923.6 ± 3623.1, p < 0.01) and a smaller, fivefold increase of IL-1β (vehicle 74.8 ± 46.2 vs RAd35 296.2 ± 43.7; p < 0.05), whereas IL-6 levels remained unchanged. At 3 hr after infection, TNF-α levels had decreased by 61% but were still 11 times higher than uninfected controls; both IL-1β (vehicle 4.2 ± 4.2 vs RAd35 1412.0 ± 657.3; p < 0.05) and IL-6 (vehicle 24.4 ± 24.4 vs RAd35 9278.1 ± 2425.1; p < 0.01) levels had increased by >300-fold compared with uninfected controls.
In the hypothalamus (Fig. 3B), however, IL-1β was the only cytokine that was significantly elevated at the 1.5 hr time point (vehicle 390.6 ± 76.6 vs RAd35 1537.0 ± 779.4;p < 0.05). By 3 hr after adenovirus-induced infection, both IL-1β (vehicle 163.5 ± 98.1 vs RAd35 2533.6 ± 509.8;p < 0.001) and IL-6 (vehicle 22.6 ± 16.3 vs RAd35 322.3 ± 34.4; p < 0.001) levels were significantly elevated in comparison to control animals. Importantly, injection of RAd35 into the striatum induced a local (striatal) increase in IL-1β (vehicle 416.60 ± 268.6 vs RAd35 1706.40 ± 509.0 pg/ml; p < 0.05) but did not induce increased IL-1β in the hypothalamus (vehicle 113.6 ± 23.3 vs RAd35 89.5 ± 22.3 pg/ml). Similarly, intracerebroventricular injection did not increase striatal IL-1β levels (data not shown).
IL-1 but not TNF-α, is a necessary mediator of adenovirus-induced fever
Having established that TNF-α, IL-1β, and IL-6 are rapidly released into the CSF in response to adenovirus administration, we examined if these cytokines were necessary and/or sufficient to mediate the adenovirus-induced fever. We first used a neutralizing antibody to TNF-α injected intracerebroventricularly 24 hr earlier. This procedure significantly attenuated the temperature response to intramuscular turpentine [the schedule of injection of TNF-α antiserum is described in detail in Luheshi et al. (1997)] (data not shown). Intramuscular turpentine-induced fever has been previously shown to be mediated by brain TNF-α (Luheshi et al., 1997). However, this TNF-α antiserum had no effect on adenovirus-induced fever (Fig.4A).
Fig. 4.
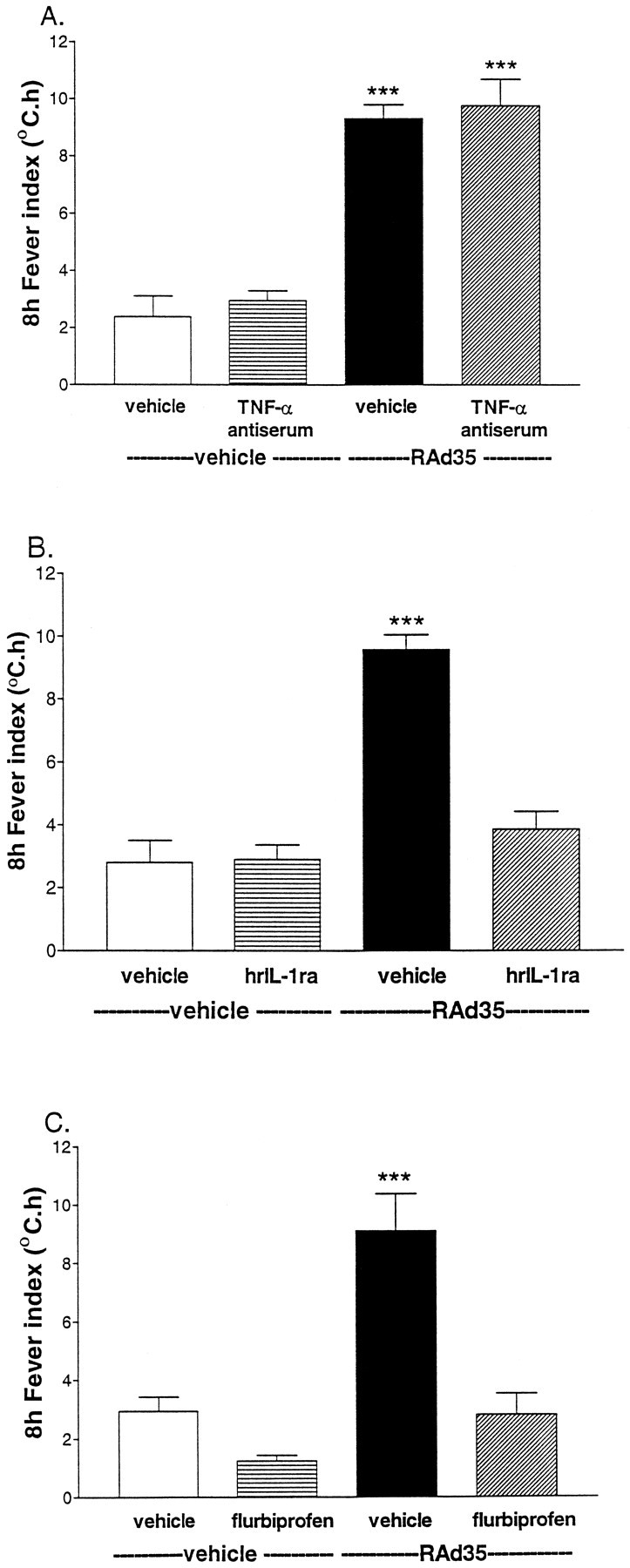
A, The temperature response to intracerebroventricular injection of adenovirus (0 hr) after pretreatment with vehicle or TNF-α antiserum was only significantly different (ANOVA; ***p < 0.001) from that of vehicle or TNF-α antiserum injected 24 hr earlier plus vehicle injected at 0 hr. B, The increase in core temperature elicited by adenovirus (i.c.v., 0 hr) was abolished by injection of IL-1ra (200 μg/rat, i.c.v.; 0 and 1 hr). The temperature response to adenovirus was significantly different from that of vehicle, IL-1ra, or adenovirus plus IL-1ra (ANOVA; ***p < 0.001).C, Intracerebroventricular injection of adenovirus (0 hr) elicited a marked and sustained increase in core temperature that was totally abolished by intraperitoneal injection of flurbiprofen (1 mg/kg, −0.5 hr). Data for vehicle, flurbiprofen, or adenovirus plus flurbiprofen were all significantly different from the temperature response to adenovirus (ANOVA; ***p < 0.001).
The involvement of IL-1 was then examined using the highly selective endogenous IL-1 receptor antagonist (IL-1ra) (Eisenberg et al., 1990). IL-1ra injected into the lateral ventricle at a dose previously shown to inhibit IL-1-induced fever (data not shown) was coadministered with RAd35 (Fig. 4B), and this completely abolished the adenovirus-induced fever. It has been previously established that IL-1-induced fever is prostaglandin-dependent (Elmquist et al., 1997). Thus, we investigated the role of the cyclooxygenase pathway in the fever response to adenovirus (Fig. 4C). Intraperitoneal injection of flurbiprofen (1 mg/kg), a cyclooxygenase inhibitor, 30 min before adenovirus injection completely inhibited the adenovirus-induced fever. This regimen of flurbiprofen administration was previously shown to inhibit intracerebroventricular IL-1-induced fever (data not shown). These data demonstrate that both IL-1 and prostaglandins are necessary mediators of the febrile response to RAd35. It is possible that IL-6, which is known to be increased in response to IL-1 (Klir et al., 1994), contributes to the fever response detected (Kluger, 1991). This possibility was not explored further in the present study.
DISCUSSION
The inflammatory responses detected occurred very early after vector administration. They were independent of the particular vector used and whether the virus encoded or expressed a therapeutic marker or no transgene. Furthermore, heat treatment of adenoviral vectors at a temperature that abolishes viral infectivity without affecting endotoxin integrity completely abolished adenovirus-induced fever. Importantly, given that transgene expression can only be detected by histochemical or immunocytochemical methods at 6–12 hr after infection, the responses observed do not depend on the encoded transgene but on the virion particle itself. Similar observations on the proinflammatory potential of nonreplicating and inactivated adenovirus vectors have been previously shown to occur after the administration of such viruses to the respiratory tract (Ginsberg et al., 1991; McCoy et al., 1995).
The initial site of action through which the adenovirus acts to trigger the fever response must lie within the ventricles themselves. The lack of histochemically detectable β-galactosidase activity in the hypothalamus after infection of the CSF demonstrates that RAd35 does not reach this brain area after intracerebroventricular injection. These data confirm that the ependymal cell layer prevents the virus itself from entering the brain from the CSF, a finding also reported by other authors (Bajoccki et al., 1993). Thus, initially, adenovirus must interact with one of several potential target cells accessible within the CSF (e.g., the ependymal cell layer, the choroid plexus, or immune cells present within the CSF) to induce the CSF increase of TNF-α, IL-1β, and IL-6. The concomitant increase of hypothalamic IL-1β and IL-6 could be caused by either a diffusion of these cytokines from the ventricle or secondary to an intraventricular proinflammatory signal that is transferred to the hypothalamus and eventually leads to the intrahypothalamic increase of interleukins.
It is most likely that in our experiments IL-1 is acting within the hypothalamus, its recognized main site of action as a pyrogen (Kluger, 1991), to induce fever in response to the intraventricular adenovirus injection. Although intracerebroventricular IL-1ra was very effective in blocking adenovirus-induced fever, we believe IL-1ra inhibits the intrahypothalamic pyrogenic action of IL-1 by diffusing from the CSF to the hypothalamus. Whether IL-1 and IL-6 diffuse from the ventricle to the hypothalamus or are produced within the hypothalamus itself in response to a proinflammatory signal originating within the ventricles remains to be determined. Prostaglandins are also involved in adenovirus-induced fever. If produced within the ventricle, they could diffuse to the hypothalamus to convey the proinflammatory signal.
The striatum has not been shown to have a direct role in thermoregulation. Anatomically, the striatum is not directly interconnected with the thermoregulatory centers in the hypothalamus, and thus, the intrastriatal increase in IL-1β is unlikely to reach the hypothalamus in high enough concentrations to induce fever. Thus, our results predict different inflammatory responses to adenovirus injection into different brain areas.
The adenovirus-induced increase in brain IL-1 causes fever. TNF-α is not involved in this response, and the increase in IL-6 is most likely induced by IL-1 (Klir et al., 1994). The increase in prostaglandins is downstream of IL-1, and prostaglandins have not been shown to increase IL-1 secretion or production on their own (Elmquist et al., 1997). The increase in CSF and hypothalamic IL-1β at the 1.5 hr time point was comparable, whereas at the 3 hr time point the increase was substantially larger in the CSF compared with the hypothalamus. Furthermore, IL-1β synthesis within the hypothalamus itself has been demonstrated previously (Tringali et al., 1996), and its receptors are also present therein (Takao et al., 1990; Yabuuchi et al., 1994). Importantly, another virus, certain strains of vaccinia, express a soluble IL-1 receptor that binds IL-1β but not IL-1α. Deletion of this gene induces fever, showing that vaccinia-induced fever is mediated by IL-1β (Alcami and Smith, 1996). This suggests that several viruses can induce fever via IL-1. Furthermore, even if the direct intrahypothalamic injection of IL-6 can induce fever on its own (Klir et al., 1993), we believe that IL-6 is not the final mediator of adenovirus-induced fever, because at the 1.5 hr time point, only hypothalamic IL-1β is significantly elevated. Nevertheless, the putative role of IL-6 in adenovirus-induced fever remains to be examined.
We favor the hypothesis that hypothalamic IL-1β and IL-6 are produced endogenously and are not diffusing from the ventricle and that hypothalamic IL-1 is responsible for the adenovirus-mediated fever response. If cytokines could diffuse freely from the CSF to the hypothalamus, we would have expected TNF-α levels to increase within the hypothalamus. However, despite the large increase of intraventricular TNF-α, we did not detect any intrahypothalamic increase of this cytokine. An in situ hybridization study of IL-1β mRNA in the hypothalamus in response to intracerebroventricular adenovirus injection could clarify whether the hypothalamic increase in IL-1β is effectively caused by locally enhanced IL-1 synthesis.
The role of TNF-α in response to adenovirus injection into the brain remains to be assessed. We have demonstrated that despite an increase in the CSF levels of TNF-α, there is no direct evidence to suggest that this cytokine is involved in mediating the febrile response to adenovirus, by, for example acting as a trigger for IL-1 release, as has been suggested in other models of inflammation (Stefferl et al., 1996). Importantly, Klir et al. (1993) report that the direct injection of TNF-α into the anterior hypothalamus did not increase body temperature. In addition, recent studies in TNF-α knock-out mice suggest TNF-α has an anti-inflammatory role in the brain (Liu et al., 1998).
Previous studies have shown that the production of another proinflammatory cytokine, IL-8, is rapidly increased in HeLa cells after adenovirus infection (Bruder and Kovesdi, 1997). Intracerebroventricular injection of IL-8 can induce fever in rats, however, unlike IL-1 and adenovirus-induced fever, IL-8-induced fever in the rat is prostaglandin-independent (Zampronio et al., 1994). Given that flurbiprofen efficiently blocked adenovirus-induced fever, this response is unlikely to be mediated by induction of CSF IL-8.
This is the first study to report the extremely rapid increase in brain concentrations of proinflammatory cytokines in response to injection of replication-deficient adenovirus vectors. Previous studies have investigated the immune responses to adenovirus vectors at later stages after infection, thus leaving the early inflammatory changes poorly characterized. These early inflammatory changes are likely to be extremely important in the brain, where most of the initial response eliminating viral particles is likely to be performed by cells of the innate immune system. The rapid cytokine and fever response to adenovirus, independently of whether or not these express a transgene, suggests that either the viral particle by itself, or through direct interactions with the plasma membrane of target cells (Bruder and Kovesdi, 1997), rapidly stimulates inflammatory cytokine secretion. If the adenoviral particle per se triggers a fever response, we would predict that new generation “gutless” adenoviral vectors would do so also. Our work demonstrates that administration of replication-deficient adenoviruses into the CSF, but not the striatum, induces an extremely rapid cytokine release and a febrile response that is mediated by IL-1 but is independent of TNF-α. The role of IL-6 remains to be determined.
Footnotes
This work was supported by The Wellcome Trust, Medical Research Council, Biotechnology and Biological Sciences Research Council, The Parkinson’s Disease Society, the European Community Concerted Action Programme (BIOMED 1, Cytokines in the Brain PL96, and BIOMED 2, Suicide Gene Therapy PL95), and the University of Manchester. We thank Mrs. Anthea Hughes for technical assistance and Ms. Tricia Maleniak for preparation of viral stocks and endotoxin assays. T.C. is a Wellcome Trust International Postdoctoral Travel Fellow, T.S. a predoctoral Action Research Fellow, and P.R.L. a Research Fellow of The Lister Institute for Preventive Medicine.
Correspondence should be addressed to Professor P. R. Lowenstein, Room 1.302 Stopford Building, Molecular Medicine Unit, Department of Medicine, University of Manchester, Oxford Road, Manchester M13 9PT, UK.
REFERENCES
- 1.Adesanya MR, Redman RS, Baum BJ, O’Connell BC. Immediate inflammatory responses to adenovirus-mediated gene transfer in rat salivary glands. Hum Gene Ther. 1996;7:1085–1093. doi: 10.1089/hum.1996.7.9-1085. [DOI] [PubMed] [Google Scholar]
- 2.Alcami A, Smith GL. A mechanism for the inhibition of fever by a virus. Proc Natl Acad Sci USA. 1996;93:11029–11034. doi: 10.1073/pnas.93.20.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bajocchi G, Feldman SH, Crystal RG, Mastrangeli A. Direct in vivo gene transfer to ependymal cells in the central nervous system using recombinant adenovirus vectors. Nat Genet. 1993;3:229–234. doi: 10.1038/ng0393-229. [DOI] [PubMed] [Google Scholar]
- 4.Blomer U, Naldini L, Kafri T, Trono D, Verma IM, Gage FH. Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J Virol. 1997;71:6641–6649. doi: 10.1128/jvi.71.9.6641-6649.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruder JT, Kovesdi I. Adenovirus infection stimulates the Raf/MAPK signaling pathway and induces interleukin-8 expression. J Virol. 1997;71:398–404. doi: 10.1128/jvi.71.1.398-404.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrnes AP, MacLaren RE, Charlton HM. Immunological instability of persistent adenovirus vectors in the brain: peripheral exposure to vector leads to renewed inflammation, reduced gene expression, and demyelination. J Neurosci. 1996a;16:3045–3055. doi: 10.1523/JNEUROSCI.16-09-03045.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrnes AP, Wood MJ, Charlton HM. Role of T cells in inflammation caused by adenovirus vectors in the brain. Gene Ther. 1996b;3:644–651. [PubMed] [Google Scholar]
- 8.Chen SH, Shine HD, Goodman JC, Grossman JC, Grossman RG, Woo SLC. Gene therapy for brain tumors. Regression of experimental gliomas by adenovirus-mediated gene transfer in vivo. Proc Natl Acad Sci USA. 1994;91:3054–3057. doi: 10.1073/pnas.91.8.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi-Lundberg DL, Lin Q, Chang YN, Chiang YL, Hay CM, Mohajeri H, Davidson BL, Bohn MC. Dopaminergic neurons protected from degradation by GDNF gene therapy. Science. 1997;275:838–841. doi: 10.1126/science.275.5301.838. [DOI] [PubMed] [Google Scholar]
- 10.Cotten M, Baker A, Saltik M, Wagner E, Buschle M. Lipopolysaccharide is a frequent contaminant of plasmid DNA preparations and can be toxic to primary human cells in the presence of adenovirus. Gene Ther. 1994;1:239–246. [PubMed] [Google Scholar]
- 11.David A, Chetritt J, Coupel H, Tesson L, Cassard A, Blancho G, Charreau B, Charreau JB, Sigalla J, Buzelin F, Le Mauff B, Soulillou JP, Anegon I. Adenovirus-mediated gene transfer in rat liver of interleukin 4 but not interleukin 10 produces severe acute hepatitis. Cytokine. 1997;9:818–829. doi: 10.1006/cyto.1997.0234. [DOI] [PubMed] [Google Scholar]
- 12.Devergne O, Peuchmaur M, Humbert M, Navratil E, Leger-Ravet MB, Crevon MC, Petit MA, Galanaud P, Emilie D. In vivo expression of IL-1 beta and IL-6 genes during viral infections in human. Eur Cytokine Netw. 1991;2:183–194. [PubMed] [Google Scholar]
- 13.Dewey RA, Southgate T, Morelli A, Klatzmann D, Castro MG, Lowenstein PR. Adenoviral-mediated suicide gene therapy using the CNS-1 rat glioma model. Soc Neurosci Abstr. 1998;24:859.9. [Google Scholar]
- 14.Eck SL, Alavi JB, Alavi A, Davis A, Hackney D, Judy K, Mollman J, Phillips PC, Wheeldon EB, Wilson JM. Treatment of advanced CNS malignancies with the recombinant adenovirus H5.010RSVTK: a phase I trial. Hum Gene Ther. 1996;7:1465–1482. doi: 10.1089/hum.1996.7.12-1465. [DOI] [PubMed] [Google Scholar]
- 15.Eisenberg SP, Evans RJ, Arend WP, Verderber E, Brewer MT, Hannum CH, Thompson RC. Primary structure and functional expression from complementary DNA of a human interleukin-1 receptor antagonist. Nature. 1990;343:341–346. doi: 10.1038/343341a0. [DOI] [PubMed] [Google Scholar]
- 16.Elkon KB, Liu CC, Gall JG, Trevejo J, Marino MW, Abrahamsen KA, Song X, Zhou JL, Old LJ, Crystral RG, Falck-Pedersen E. Tumor necrosis factor alpha plays a central role in immune-mediated clearance of adenoviral vectors. Proc Natl Acad Sci USA. 1997;94:9814–9819. doi: 10.1073/pnas.94.18.9814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elmquist JK, Scammell TE, Saper CB. Mechanisms of CNS response to systemic immune challenge: the febrile response. Trends Neurosci. 1997;20:565–570. doi: 10.1016/s0166-2236(97)01138-7. [DOI] [PubMed] [Google Scholar]
- 18.Geddes BJ, Harding TC, Lightman SL, Uney JB. Long-term gene therapy in the CNS: reversal of hypothalamic diabetes insipidus in Brattleboro rat by using an adenovirus expressing arginine vasopressin. Nat Med. 1997;3:1402–1404. doi: 10.1038/nm1297-1402. [DOI] [PubMed] [Google Scholar]
- 19.Ginsberg HS, Moldawer LL, Sehgal PB, Redington M, Kilian PL, Chanock RM, Prince GA. A mouse model for investigating the molecular pathogenesis of adenovirus pneumonia. Proc Natl Acad Sci USA. 1991;88:1651–1655. doi: 10.1073/pnas.88.5.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hermens WT, Verhaagen J. Adenoviral vector-mediated gene expression in the nervous system of immunocompetent Wistar and T cell-deficient nude rats: preferential survival of transduced astroglial cells in nude rats. Hum Gene Ther. 1997;8:1049–1063. doi: 10.1089/hum.1997.8.9-1049. [DOI] [PubMed] [Google Scholar]
- 21.Klir JJ, Roth J, Szelenyi Z, McClellan JL, Kluger MJ. Role of hypothalamic interleukin-6 and tumor necrosis factor-α in LPS-fever in rat. Am J Physiol. 1993;265:R512–R517. doi: 10.1152/ajpregu.1993.265.3.R512. [DOI] [PubMed] [Google Scholar]
- 22.Klir JJ, McClellan JL, Kluger MJ. Interleukin-1β causes the increase in anterior hypothalamic interleukin-6 during LPS-induced fever in rats. Am J Physiol. 1994;266:R1845–R1848. doi: 10.1152/ajpregu.1994.266.6.R1845. [DOI] [PubMed] [Google Scholar]
- 23.Kluger MJ. Fever: the role of pyrogens and cryogens. Physiol Rev. 1991;71:93–127. doi: 10.1152/physrev.1991.71.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lieber A, He CY, Meuse L, Schowalter D, Kirillowva I, Winther B, Kay MA. The role of Kupffer cell activation and viral gene expression in early liver toxicity after infusion of recombinant adenovirus vectors. J Virol. 1997;71:8798–8807. doi: 10.1128/jvi.71.11.8798-8807.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Marino MW, Wong G, Grail D, Dunn A, Bettadapura J, Slavin AJ, Old L, Bernard CC. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat Med. 1998;4:78–83. doi: 10.1038/nm0198-078. [DOI] [PubMed] [Google Scholar]
- 26.Lowenstein PR, Wilkinson GWG, Castro MG, Shering AF, Fooks AR, Bain D. Non-neurotropic adenovirus: a vector for gene transfer to the brain and possible gene therapy of neurological disorders. In: Latchman DS, editor. Genetic manipulation of the nervous system. Academic; London: 1996. pp. 11–39. [Google Scholar]
- 27.Luheshi GN, Stefferl A, Turnbull AV, Dascombe MJ, Brouwer S, Hopkins SJ, Rothwell NJ. Febrile response to tissue inflammation involves both peripheral and brain IL-1 and TNF-α in the rat. Am J Physiol. 1997;272:R862–R868. doi: 10.1152/ajpregu.1997.272.3.R862. [DOI] [PubMed] [Google Scholar]
- 28.McCoy RD, Davidson BL, Roessler BJ, Huffnagle GB, Janich SL, Laing TJ, Simon RH. Pulmonary inflammation induced by incomplete or inactivated adenoviral particles. Hum Gene Ther. 1995;6:1553–1560. doi: 10.1089/hum.1995.6.12-1553. [DOI] [PubMed] [Google Scholar]
- 29.Morelli AE, Larregina AT, Smith-Arica J, Dewey RA, Southgate TD, Ambar B, Fontana A, Castro MG, Lowenstein PR (1999) Neuronal and glial cell-type specific promoters within adenovirus recombinants restrict the expression of the apoptosis inducing molecule Fas ligand to predetermined brain cell types, and abolish peripheral liver toxicity. J Gen Virol, in press. [DOI] [PubMed]
- 30.Paxinos G, Watson C. The rat brain in stereotaxic co-ordinates. Academic; Sydney: 1986. [Google Scholar]
- 31.Rees GS, Ball C, Ward HL, Gee CK, Tarrant G, Mistry Y, Poole S, Bristow AF (1998) Rat interleukin 6: expression in recombinantEscherichia coli, purification and development of a novel ELISA. Cytokine, in press. [DOI] [PubMed]
- 32.Safieh-Garabedian B, Poole S, Allchorne A, Winter J, Woolf CJ. Contribution of interleukin-1β to the inflammation-induced increase in nerve growth factor levels and inflammatory hyperalgesia. Br J Pharmacol. 1995;115:1265–1275. doi: 10.1111/j.1476-5381.1995.tb15035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shering AF, Bain D, Stewart S, Epstein AL, Castro MG, Wilkinson GWG, Lowenstein PR. Cell type-specific expression in brain cell cultures from a short human cytomegalovirus major immediate early promoter depends on whether it is inserted into herpesvirus or adenovirus vectors. J Gen Virol. 1997;78:445–459. doi: 10.1099/0022-1317-78-2-445. [DOI] [PubMed] [Google Scholar]
- 34.Stefferl A, Hopkins SJ, Rothwell NJ, Luheshi GN. The role of TNF-α in fever: opposing actions of human and murine TNF-α and interactions with IL-1β in the rat. Br J Pharmacol. 1996;118:1919–1924. doi: 10.1111/j.1476-5381.1996.tb15625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevenson PG, Freeman S, Bangham CR, Hawke S. Virus dissemination through the brain parenchyma without immunologic control. J Immunol. 1997a;159:1876–1884. [PubMed] [Google Scholar]
- 36.Stevenson PG, Hawke S, Sloan DJ, Bangham CR. The immunogenicity of intracerebral virus infection depends on anatomical site. J Virol. 1997b;71:145–151. doi: 10.1128/jvi.71.1.145-151.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takao T, Tracey DE, Mitchell WM, De Souza EB. Interleukin-1 receptors in mouse brain: characterisation and neuronal localization. Endocrinology. 1990;127:3070–3078. doi: 10.1210/endo-127-6-3070. [DOI] [PubMed] [Google Scholar]
- 38.Tringali G, Mancuso C, Mirtella A, Pozzoli G, Parente L, Preziosi P, Navarra P. Evidence for the neuronal origin of immunoreactive interleukin-1 beta released by rat hypothalamic explants. Neurosci Lett. 1996;219:143–146. doi: 10.1016/s0304-3940(96)13195-5. [DOI] [PubMed] [Google Scholar]
- 39.Wilkinson GW, Akrigg A. Constitutive and enhanced expression from the CMV major 1E promoter in a defective adenovirus vector. Nucleic Acids Res. 1992;20:2233–2239. doi: 10.1093/nar/20.9.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wood MJ, Charlton HM, Wood KJ, Kajiwara K, Byrnes AP. Immune responses to adenovirus vectors in the nervous system. Trends Neurosci. 1996;19:497–501. doi: 10.1016/S0166-2236(96)10060-6. [DOI] [PubMed] [Google Scholar]
- 41.Worgall S, Wolff G, Falck-Pedersen E, Crystal RG. Innate immune mechanisms dominate elimination of adenoviral vectors following in vivo administration. Hum Gene Ther. 1997;8:37–44. doi: 10.1089/hum.1997.8.1-37. [DOI] [PubMed] [Google Scholar]
- 42.Yabuuchi K, Minami M, Katsumata S, Satoh M. Localization of type I interleukin-1 receptor mRNA in the rat brain. Mol Brain Res. 1994;27:27–36. doi: 10.1016/0169-328x(94)90180-5. [DOI] [PubMed] [Google Scholar]
- 43.Zampronio AR, Souza GE, Silva CA, Cunha FQ, Ferreira SH. Interleukin-8 induces fever by a prostaglandin-independent mechanism. Am J Physiol. 1994;266:R1670–R1674. doi: 10.1152/ajpregu.1994.266.5.R1670. [DOI] [PubMed] [Google Scholar]
- 44.Zsengeller ZK, Wert SE, Hull WM, Hu X, Yei S, Trapnell BC, Whitsett JA. Persistence of replication-deficient adenovirus-mediated gene transfer in lungs of immune-deficient (nu/nu) mice. Hum Gene Ther. 1995;6:457–467. doi: 10.1089/hum.1995.6.4-457. [DOI] [PubMed] [Google Scholar]