

Abstract
Methamphetamine-induced toxicity has been shown to require striatal dopamine and to involve mechanisms associated with oxidative stress. Dopamine is a reactive molecule that can oxidize to form free radicals and reactive quinones. Although this has been suggested to contribute to the mechanism of toxicity, the oxidation of dopamine has never been directly measured after methamphetamine exposure. In this study we sought to determine whether methamphetamine-induced toxicity is associated with the oxidation of dopamine by measuring the binding of dopamine quinones to cysteinyl residues on protein. We observed that administration of neurotoxic doses of methamphetamine to rats resulted in a two- to threefold increase in protein cysteinyl-dopamine in the striatum 2, 4, and 8 hr after treatment. When methamphetamine was administered at an ambient temperature of 5°C, no increase in dopamine oxidation products was observed, and toxicity was prevented. Furthermore, as shown by striatal microdialysis, animals treated with methamphetamine at 5°C showed DA release identical to that of animals treated at room temperature. These data suggest that the toxicity of methamphetamine and the associated increase in dopamine oxidation are not exclusively the result of increases in extracellular dopamine. Because dopamine-induced modifications of protein structure and function may result in cellular toxicity, it is likely that dopamine oxidation contributes to methamphetamine-induced toxicity to dopamine terminals, adding support to the role of dopamine and the evidence of oxidative stress in this lesion model.
Keywords: cysteinyl-dopamine, dopamine quinone, dopamine oxidation, methamphetamine, neurotoxicity, oxidative stress
Administration of high doses of the indirect dopamine (DA) agonist methamphetamine (METH) has been shown to result in damage to both DA and serotonin (5-HT) terminals, and perhaps in cell loss, in the brains of rodents and nonhuman primates (Seiden et al., 1975; Hotchkiss and Gibb, 1980; Wagner et al., 1980; Ricaurte et al., 1982; Sonsalla et al., 1996). It has been firmly established that DA plays a role in the degeneration of both DA and 5-HT terminals. Pharmacological interventions that lower striatal DA content have proven protective against METH-induced toxicity (Gibb and Kogan, 1979;Wagner et al., 1983; Schmidt et al., 1985; Johnson et al., 1987). It has also been suggested that the magnitude of DA release after METH exposure is predictive of its long-term toxicity (O’Dell et al., 1991). These findings have clearly demonstrated a role for DA release and/or redistribution in METH-induced toxicity; however, the exact mechanism remains unclear.
Oxidative stress has also been implicated in METH-induced toxicity (for review, see Cadet and Brannock, 1998). Early studies showed that systemic administration of antioxidants such as ascorbate and vitamin E attenuates METH-induced toxicity (De Vito and Wagner, 1989). The formation of 6-hydroxydopamine, presumably via a reaction between DA and the hydroxyl radical, was observed after administration of a single high dose of METH (Seiden and Vosmer, 1984); however, this was not observed by others (Karoum et al., 1993). More recently, Giovanni et al. (1995) showed that after neurotoxic doses of METH, salicylate administration resulted in increased formation of 2,3-dihydroxybenzoic acid, which is also indicative of hydroxyl radical production. Transgenic mice over-expressing the antioxidant enzyme Cu/Zn superoxide dismutase showed an attenuated response to toxic doses of METH (Cadet et al., 1994; Hirata et al., 1995). An electron spin-trapping agent capable of inactivating various free radicals has also been shown to prevent METH-induced toxicity, as well as the toxicity of another amphetamine analog (Colado and Green, 1995; Schmidt and Taylor, 1995;Cappon et al., 1996). These reports have established the likelihood that oxidative stress plays a role in the toxicity of METH.
Catecholamines have been shown to be toxic both in vivo andin vitro via mechanisms of oxidative stress (Graham et al., 1978; Rosenberg, 1988; Michel and Hefti, 1990; Mena et al., 1992;Filloux and Townsend, 1993; Hastings et al., 1996). Under such conditions, DA may oxidize to form superoxide and hydrogen peroxide, which can then form the hydroxyl radical in the presence of transition metals (Graham, 1978). These are the same free radicals implicated in METH-induced toxicity. DA oxidation also results in the formation of DA quinone, which readily participates in nucleophilic addition reactions with sulfhydryl groups on free cysteine, glutathione, or cysteine found in protein (Graham et al., 1978; Fornstedt et al., 1986;Hastings and Zigmond, 1994). The reaction between DA quinone and cysteine results in the formation of 5-cysteinyl-DA (Fig.1). Because cysteinyl residues are often found at the active site of proteins, the covalent addition of the catechol moiety to cysteine may inhibit protein function and possibly lead to cellular damage and/or cell death. In addition, DA quinone is able to react with the sulfhydryl group of cysteine in glutathione, which may decrease levels of this important antioxidant. The reactive quinones and free radicals produced by the oxidation of DA may contribute to the oxidative stress associated with METH-induced toxicity.
Fig. 1.

The oxidation of DA to DA quinone and the resultant conjugation with cysteine (5-Cys-DA).
In this study, we investigated whether administration of a neurotoxic dose of METH to rats results in the formation of oxidative metabolites of DA in the striatum and, if so, whether an intervention that attenuates the toxicity of METH also prevents the oxidation of DA. We observed that increases in DA oxidation occurred only under conditions resulting in toxicity, suggesting that the oxidation of DA may contribute to the mechanism of METH-induced damage to dopaminergic terminals.
MATERIALS AND METHODS
Animals and methamphetamine administration. Male Sprague Dawley rats (Zivic-Miller, Allison Park, PA) weighing 325–375 gm were housed individually in suspended wire-mesh cages in a temperature- and humidity-controlled environment. Food and water were provided ad libitum, and animals were subject to a 12 hr light/dark cycle. All procedures and handling techniques used were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All materials were purchased from Sigma (St. Louis, MO) unless otherwise noted.
METH or saline was administered subcutaneously to rats (15 mg of free base/kg) for a total of four injections, with all injections spaced 2 hr apart. Core-body temperature was recorded in all animals at 30 min intervals by the use of a small-animal rectal probe (YSI, Yellow Springs, Ohio). Any animal that surpassed a threshold of 41.3°C was placed in a plastic container of ice for 15 min to prevent hyperthermia-related mortality. Animals to be treated with METH or saline at a lowered ambient temperature were moved into a refrigerated room, maintained at 5°C, 30 min before the first injection and remained there until 1 hr after the last injection (a total of 7.5 hr). They were then returned to the colony room, which was maintained at 23°C, and carefully monitored. Animals receiving METH or saline were killed 2, 4, or 8 hr or 7 d after the fourth injection. Striatal tissue collected at 2, 4, and 8 hr time points was analyzed for levels of DA, dihydroxyphenylacetic acid (DOPAC), and DA oxidation products. Striatal tissue collected at 7 d was analyzed for levels of DA, DOPAC, 5-HT, and 5-hydroxyindoleacetic acid.
Analysis of monoamines and DA oxidation products. Animals were killed by decapitation, and brains were rapidly dissected on ice. Whole striata were removed, weighed, and then immediately frozen on dry ice. Tissue was then stored at −80°C until analysis, at which time striatal tissue (∼24 mg) was homogenized in 1 ml of 0.1N perchloric acid containing 0.2 mm sodium bisulfite, 1 mmdithiothreitol, and 1 mm ascorbate. The homogenate was then centrifuged at 30,000 × g for 20 min. An aliquot (0.8 ml) of the resulting supernatant was extracted over alumina and analyzed for free cysteinyl-DA, free cysteinyl-DOPAC, and glutathione-DA by HPLC with electrochemical detection. The protein pellet was washed once in 0.1N perchloric acid and then hydrolyzed under vacuum in 6N HCl for 20 hr at 110°C. The hydrolysate (containing modified amino acid residues) was freeze-dried to remove the HCl and then redissolved in perchloric acid before extraction with alumina and final analysis on HPLC to determine the levels of protein cysteinyl-DA and cysteinyl-DOPAC (Ito et al., 1988; Hastings and Zigmond, 1994). For both free and protein cysteinyl-catechols, a Waters Associates (Milford, MA) 460 amperometric detector set at an oxidizing potential of +0.6 V was used to quantify samples. Analytes were separated with a Microsorb 86–200-C5 column (5 μm; 250 × 4.6 mm; Rainin, Ridgefield, NJ). Sample peaks were compared with synthesized standards (Rosengren et al., 1985). For the analysis of the parent catechols DA and DOPAC, an aliquot of the supernatant was extracted over alumina and injected onto an HPLC system equipped with an ESA (Chelmsford, MA) Coulochem II coulometric detector set at −200 and +280 mV. This system was also used to determine the tissue levels of DA, 5-HT, DOPAC, and 5-hydroxyindoleacetic acid 7 d after METH. For 7 d analyses, an aliquot of the supernatant was filtered (0.2 μm) and then injected directly onto the HPLC system. When possible, data from both striata were averaged together before statistical analysis. Student’s t tests confirmed that no hemispheric differences existed among the various analyses.
Striatal microdialysis. Concentric microdialysis probes were constructed as described previously (Abercrombie and Finlay, 1991). Rats were anesthetized with equithesin at 3 ml/kg [258 mmchloral hydrate, 20% (v/v) Nembutal, 86 mmMgSO4, and 25% (v/v) propylene glycol] before the stereotaxic implantation of a probe into the left striatum (+0.5 mm anteroposterior, +2.5 mm mediolateral from bregma, and −7.5 mm dorsoventral to dura). Probes were secured to the skull using stainless steel screws and dental acrylic and were continuously perfused with artificial CSF (145 mm NaCl, 2.7 mm KCl, 1.0 mm MgCl2, and 1.2 mmCaCl2) at a rate of 1.5 μl/min. A period of at least 18 hr elapsed between the implantation of the microdialysis probe and the start of the experiment. Dialysate was collected in 30 min fractions and frozen at −80°C until analysis by HPLC for levels of DA and DOPAC. Animals to be treated with METH at 5°C were placed into a temperature-controlled chamber, initially kept at 23°C, ∼1 hr before the collection of the first baseline sample. The chamber was rapidly brought down to 5°C 30 min before the first injection of METH. A minimum of three samples was taken to establish baseline levels of extracellular DA and DOPAC before the administration of four systemic injections of METH.
RESULTS
Neurotoxic potential of METH
Tissue levels of DA and 5-HT measured several days after exposure to METH have been shown to concur with the loss of striatal tyrosine and tryptophan hydroxylase activities and thus seem to represent the magnitude of monoaminergic terminal damage in the striatum (Ricaurte et al., 1980, 1982). Therefore, we measured the striatal content of DA and 5-HT at 7 d as an index of monoaminergic terminal loss after METH administration. When rats were treated at room temperature (23°C), METH caused significant depletions in striatal DA (−47%) and 5-HT (−61%) as compared with the levels in saline-injected controls (Fig.2). This degree of terminal damage is consistent with previous findings from our laboratory (Giovanni et al., 1995). 5-Hydroxyindoleacetic acid, the primary metabolite of 5-HT, was decreased (−36%) at 7 d; however, DOPAC levels were not different from control values, indicative of increased DA turnover as suggested previously after METH (Robinson et al., 1990). When METH was administered at a lowered ambient temperature (5°C), tissue levels of DA and 5-HT (Fig. 2), as well as their primary metabolites (data not shown), were not significantly different from control levels at 7 d, demonstrating protection against METH-induced toxicity.
Fig. 2.

Effect of METH on striatalDA and 5-HT levels. Striatal tissue levels of DA and 5-HT were measured 7 d after treatment with saline or METH (15 mg/kg × 4) at room temperature (23°C) or at 5°C. Values are expressed as the percent of control, as determined on a per experiment basis (mean ± SEM; n = 3–8). Control values forDA were 75.96 ± 12.1 nmol/gm of tissue (23°C) and 79.22 ± 3.95 nmol/gm of tissue (5°C). Control values for5-HT were 2.31 ± 0.56 nmol/gm of tissue (23°C) and 2.66 ± 0.33 nmol/gm of tissue (5°C). *, Significantly different from control values (p < 0.05).
Effects of METH on DA and reactive metabolites
Protein cysteinyl-DA and cysteinyl-DOPAC are stable oxidative metabolites of DA and DOPAC. Therefore, measurement of these compounds is not only an index of the oxidation of the parent catechols but also a direct measure of protein modification. Animals treated with METH at room temperature (23°C) showed significant increases in protein cysteinyl-DA above that in saline-injected controls at 2, 4, and 8 hr after the fourth injection of METH (Fig.3). Cysteinyl-DA levels were increased to 284% at 2 hr, 193% at 4 hr, and 208% at 8 hr, as compared with control levels. Protein cysteinyl-DOPAC, however, did not differ significantly from control levels at any time point (Fig. 3). Free cysteinyl-DA, free cysteinyl-DOPAC, and glutathione-DA were found in the acid-soluble supernatant of some METH-treated striata, but this was not a consistent observation. Control levels of these acid-soluble DA oxidation products in a single rat striatum are typically below the limit of detection by HPLC (Fornstedt et al., 1990; Hastings et al., 1996).
Fig. 3.

Effect of METH on the levels of proteincysteinyl-DA and cysteinyl-DOPAC. Levels of protein cysteinyl-DA andcysteinyl-DOPAC were examined in the striatum at 2, 4, and 8 hr after the last injection of METH (15 mg/kg × 4) at room temperature (23°C) and were compared with levels in saline-injected controls (mean ± SEM; n = 4–6). Control values of protein cysteinyl-DA orcysteinyl-DOPAC did not differ between time points and therefore were combined. An overall ANOVA of the proteincysteinyl-DA [F(3,18) = 5.8;p < 0.01] and the proteincysteinyl-DOPAC [F(3,18) = 1.2; p > 0.05] was followed by pairwise comparisons with the Student’s t test and layered Bonferroni correction. *, Significantly different from control (p < 0.05).
Striatal tissue DA and DOPAC levels also were analyzed in animals treated at room temperature at 2, 4, and 8 hr after the fourth injection of METH (Fig. 4). It was observed that METH caused a transient depletion of tissue DA (−58%) at 2 hr that returned to control levels by 8 hr. METH-treated animals showed a persistent decrease in striatal DOPAC levels, −85% at 2 hr and −68% at 8 hr after METH (Fig. 4).
Fig. 4.

Striatal DA andDOPAC immediately after METH. Striatal tissueDA and DOPAC levels were measured at 2, 4, and 8 hr after administration of METH (15 mg/kg × 4) at room temperature (23°C) and were compared with levels in saline-injected controls (mean ± SEM; n = 4–6). An overall ANOVA of the DA [F(3,18) = 4.8; p < 0.05] and DOPAC[F(3,18) = 51.1; p < 0.01] levels was followed by pairwise comparisons with the Student’st test and layered Bonferroni correction. *, Significantly different from control (p < 0.05).
Effect of reduced ambient temperature on the oxidation of DA
To determine whether DA oxidation only occurred during conditions resulting in toxicity, we analyzed protein cysteinyl-DA and cysteinyl-DOPAC levels in striatum 2 hr after administration of either METH or saline at 23 or 5°C. Again, we observed that animals receiving METH at room temperature showed increased protein cysteinyl-DA at 2 hr (263% of control) (Fig.5). However, animals administered METH at 5°C showed no change in the levels of cysteinyl-DA (Fig. 5). Protein cysteinyl-DOPAC levels were not different from control (0.201 ± 0.03 nmol/gm of tissue) in either group (data not shown).
Fig. 5.

Effect of temperature onMETH-induced DA oxidation. Proteincysteinyl-DA levels were examined in the striatum 2 hr after administration of METH (15 mg/kg × 4) at an ambient temperature of 5°C or at room temperature (23°C) and were compared with the levels in saline-injected controls (mean ± SEM;n = 4–7). Student’s t tests were used to compare the METH-treated striata with their appropriate controls. *, Significantly different from control (p < 0.05).
Animal core-body temperatures were recorded at 30 min intervals for the duration of the METH injection regimen (7 hr). The results showed that animals treated with METH at 23°C had a hyperthermic response to the drug (Fig. 6). However, animals that were treated with METH at a reduced ambient temperature (5°C) did not undergo hyperthermia but showed a severe hypothermic response to METH (Fig. 6). In fact the absolute magnitude of METH-induced hypothermia was several times greater than the magnitude of METH-induced hyperthermia.
Fig. 6.

Effect of ambient temperature on the thermoregulatory response to METH. Core-body temperature was recorded in animals every 30 min during the administration of METH (15 mg/kg × 4; the four injections of METH indicated byarrows) (mean ± SEM; n= 6–13). The dashed line represents the average core-body temperature of nontreated animals before administration of METH in both temperature conditions, because there was no difference in initial core-body temperature. Saline-treated animals did not exhibit changes in core-body temperature over time in either ambient temperature, and therefore, these data are not shown.
Temperature, however, did not influence the transient METH-induced depletion of striatal DA and DOPAC. At 2 hr after METH, tissue DA and DOPAC levels were decreased from control to a similar extent both in animals treated at 23°C (−64 and −51%, respectively) and in animals treated at 5°C (−71 and −54%, respectively) (Fig.7).
Fig. 7.

Effect of temperature on theMETH-induced depletion of DA andDOPAC. Striatal DA andDOPAC levels were measured 2 hr after administration ofMETH (15 mg/kg × 4) at 23 and 5°C (mean ± SEM; n = 4). Student’s t tests revealed significant depletions in tissue DA andDOPAC, as compared with respective control levels. No differences were observed across temperature conditions. *, Significantly different from control (p < 0.05).
Effect of ambient temperature on METH-induced DA release
Because the oxidation of DA may be influenced by the availability of DA in the low-antioxidant environment of the extracellular fluid, we used the technique of striatal microdialysis to compare the amount of DA released when METH was administered at an ambient temperature of 23 or 5°C. Results showed that METH caused significant changes in extracellular DA and DOPAC. Peak extracellular DA was increased 20-fold above baseline in animals treated at both 23 and 5°C (Fig.8A). No significant difference in extracellular DA was observed between these two groups of animals. Likewise, extracellular DOPAC was found to be decreased similarly under both conditions (Fig. 8B).
Fig. 8.
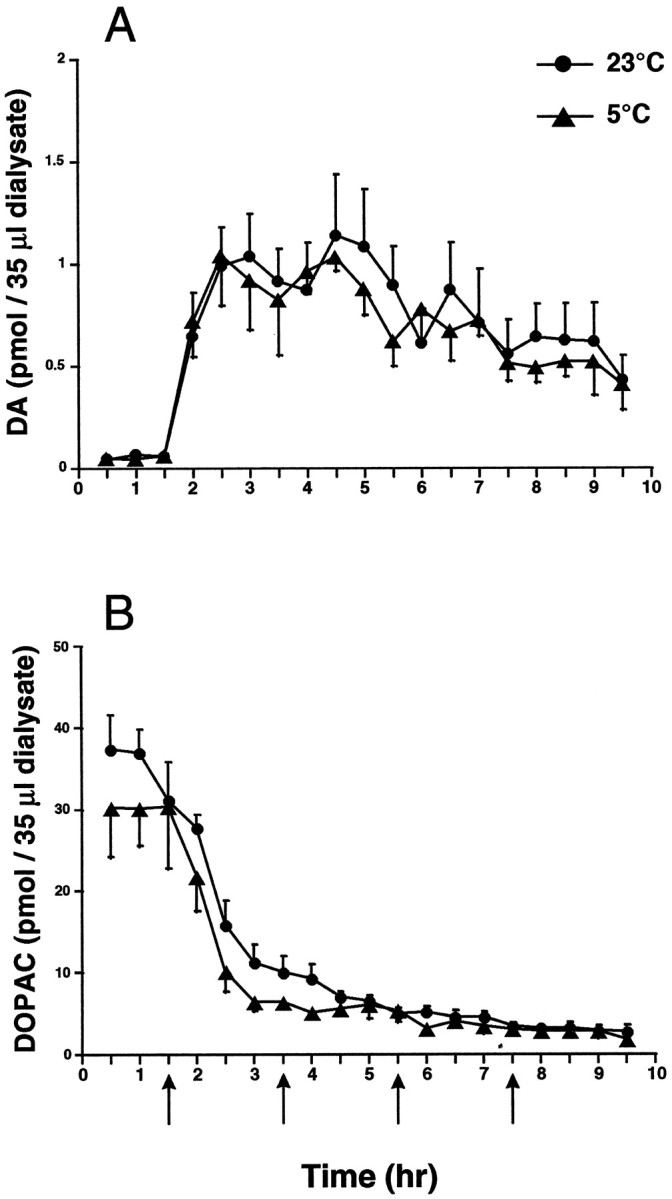
The effect of METH on extracellular levels of striatal DA and DOPAC. Extracellular levels of striatal DA (A) andDOPAC (B) were measured in the striatum by in vivo microdialysis during METH administration (15 mg/kg × 4) at both 5 and 23°C (mean ± SEM; n = 5). Arrows indicate each of the four injections of METH. A two-way, repeated-measures ANOVA did not reveal any differences between treatment conditions in either extracellular DA [F(1,18) = 0.37; p > 0.05] or DOPAC[F(1,18) = 1.98; p > 0.05].
DISCUSSION
In this study, we showed that four systemic injections of METH resulted in toxicity to DA and 5-HT terminals in rat striatum and that this toxicity was accompanied by a threefold increase in protein cysteinyl-DA, a stable oxidative metabolite of DA. This increase appeared greatest 2 hr after METH, but levels of protein cysteinyl-DA remained significantly elevated for at least 8 hr after drug administration. We observed that administration at 5°C prevented both the acute increase in protein cysteinyl-DA and the long-term toxicity of METH. These data suggest that a relationship exists between METH-induced toxicity and DA oxidation.
The increase in protein cysteinyl-DA that preceded METH-induced toxicity represents a direct measure of the covalent modification of cysteinyl residues on protein by DA quinones. Although suggested in previous studies (Schmidt et al., 1985; Sonsalla et al., 1986;Weihmuller et al., 1992; Stephans and Yamamoto, 1994), this is the first direct evidence of the formation of DA quinone in methamphetamine-induced toxicity. In fact, the generation of DA quinones and binding to protein has been implicated in other studies of protein inactivation and toxicity (Graham et al., 1978; Berman et al., 1996; Hastings et al., 1996). Because of the complex nature of METH-induced toxicity, there are likely many contributing factors. However, the observed oxidation of DA provides one possible explanation for the role of striatal DA in the long-term toxicity of METH.
The contribution of a threefold increase in protein cysteinyl-DA formation to the toxicity of METH would depend highly on the proteins targeted for modification. One cannot assume that the proteins modified by DA quinones under basal conditions (control levels of cysteinyl-DA) are the same proteins targeted under pathological conditions. METH causes a redistribution of DA and increased oxidative stress within DA cells (Cubells et al., 1994) that may allow for the covalent modification of proteins that are normally not exposed to DA or DA quinones. Therefore, a threefold increase in cysteinyl-DA may represent a very significant challenge to cell viability. Other investigators have reported similar increases in other indices of oxidative stress associated with neurotoxic METH exposure (Giovanni et al., 1995;Fleckenstein et al., 1997). The identification of proteins targeted for modification by DA quinones is the focus of ongoing experiments in the laboratory.
At present, we cannot directly conclude which proteins were attacked by DA quinones; however, several pieces of evidence suggest an intracellular oxidation of DA. We observed that METH caused an acute increase in protein cysteinyl-DA, whereas protein cysteinyl-DOPAC was unchanged. Because both DA and DOPAC are capable of oxidizing and binding to sulfhydryl groups on protein (Ito et al., 1988; Hastings and Zigmond, 1994), the selective increase in cysteinyl-DA may reflect the relative concentrations of DA and DOPAC at the site of oxidation. The selective increase in cysteinyl-DA suggests that the oxidation occurred in a compartment in which DA exists in higher concentrations than DOPAC, such as the cytoplasm of a DA terminal. Presumably, the intracellular concentration of DA is increased after METH administration by the vesicular release of DA into the cytoplasm (Cubells et al., 1994), and cytoplasmic DOPAC levels are decreased by inhibition of monoamine oxidase by metabolites of METH (Egashira et al., 1987). Therefore, the selective increase in cysteinyl-DA suggests that the oxidation of DA occurs in the intracellular compartment.
However, in response to this regimen of METH, extracellular DA levels increase considerably, which might promote DA oxidation in the extracellular compartment. Indeed, extracellular DA increases ∼2000%, and extracellular DOPAC levels decreased −83% from control (Fig. 8). Despite these changes, DOPAC levels are still 7.5-fold higher than the peak levels of extracellular DA. Thus, if the oxidation of DA occurred in the extracellular space, we would have expected increases in cysteinyl-DOPAC, as well. These observations suggest that in the case of METH-induced toxicity, the extracellular environment is not involved in the oxidation of DA.
It was also observed that the toxicity of METH could be prevented if administered at an ambient temperature of 5°C. This is in accordance with previous findings showing that reduced environmental temperatures prevent the severe hyperthermia necessary to generate the long-term toxicity of METH (Bowyer et al., 1992, 1994; Ali et al., 1994; Miller and O’Callaghan, 1994; Albers and Sonsalla, 1995). In addition to the prevention of METH-induced hyperthermia and toxicity, we also observed no increase in DA oxidation products when METH was administered at 5°C, suggesting a connection between increased DA oxidation and the mechanism of toxicity. Animals treated at 5°C actually showed a significant drug-induced hypothermia instead of hyperthermia (Fig. 6), resulting in a greater net change in core-body temperature than seen in animals treated at room temperature. As suggested previously (Yehuda and Wurtman, 1972), these data show that high doses of METH interfere with thermoregulation rather than cause hyperthermia.
Microdialysis experiments demonstrated that the protection observed by administration of METH at 5°C is not the result of decreased DA release. This novel observation is important to understanding the mechanism of METH-induced toxicity because it shows that reduced ambient temperature does not interfere with the pharmacological response to METH but merely prevents METH-induced hyperthermia. This is further supported by the observation that striatal tissue levels of DA and DOPAC are decreased to similar extents in both temperature conditions 2 hr after METH (Fig. 7). These data are in contrast to those in one previously reported study showing an attenuated release of DA in response to METH (5 mg/kg × 4) at 4°C; however, this dose did not result in toxicity (Bowyer et al., 1993). Our results also clearly show that elevations in extracellular DA alone are not sufficient to cause toxicity. Although extracellular DA may be important to the mechanism of METH-induced toxicity, as proposed previously (O’Dell et al., 1991), additional factors also must be involved.
Finally, our microdialysis data indicated that the rise in extracellular DA was not sufficient to cause an increase in DA oxidation products. Because the extracellular environment has lower antioxidant levels than the intracellular compartment (Schenk et al., 1982), it might be expected that large increases in extracellular DA would overload the antioxidant capacity, resulting in DA oxidation. However, animals treated at 5°C exhibited the same extracellular levels of DA as did animals treated at 23°C but did not show an increase in DA oxidation products. Overall, these results suggest that the oxidation of DA may be more predictive of toxicity than are changes in extracellular DA. However, it is clear that other factors such as METH-induced hyperthermia play a key role in both the oxidation of DA and the long-term toxicity of METH.
It has been well demonstrated that DA can oxidize and form cysteinyl-DA via an autoxidation mechanism that is facilitated by the presence of transition metals, such as iron (Graham, 1978). It is possible that the METH-induced hyperthermia causes the release of free iron, as occurs in liver (Skibba and Gwartney, 1997), thus promoting the oxidation of DA. DA may also be oxidized via enzymatic mechanisms. We and others have shown previously that prostaglandin H synthase has the ability to oxidize DA and increase the formation of protein cysteinyl-DA (Hastings, 1995; Mattammal et al., 1995). Other enzymes such as lipoxygenase, tyrosinase, and xanthine oxidase also will oxidize DA (Korytowski et al., 1987; Rosei et al., 1994; Foppoli et al., 1997). It is possible that severe hyperthermia might result in the activation or upregulation of proteins capable of oxidizing DA.
Pro-oxidants may also be formed in vivo that can react with DA and increase the formation of protein cysteinyl-DA. METH-induced toxicity has been shown to involve increases in extracellular glutamate (Nash and Yamamoto, 1992; Abekawa et al., 1994; Stephans and Yamamoto, 1994). The activation of glutamate receptors has been linked to the formation of reactive oxygen and nitrogen species, such as superoxide and nitric oxide (Lafon-Cazal et al., 1993; Gunasekar et al., 1995;Reynolds and Hastings, 1995). Superoxide has been implicated in METH-induced toxicity (Cadet et al., 1994; Hirata et al., 1995) and the oxidation of catechols (Ito and Fujita, 1982). Superoxide also may react with nitric oxide to produce peroxynitrite, another compound shown to oxidize DA (LaVoie and Hastings, 1997). In fact, it has been demonstrated that inhibition of neuronal nitric oxide synthase prevents METH-induced toxicity (Di Monte et al., 1996; Itzhak and Ali, 1996). Likewise, mice lacking the neuronal nitric oxide synthase gene are resistant to the toxic effects of METH (Itzhak et al., 1998). Therefore, additional work is needed to elucidate the possible interactions of hyperthermia, nitric oxide, and DA oxidation and their relevance to METH-induced toxicity.
DA also plays an important role in the toxicity of METH to 5-HT terminals (Gibb and Kogan, 1979; Wagner et al., 1983; Schmidt et al., 1985; Johnson et al., 1987), but the mechanism is unknown. It has been speculated that the METH-induced increase in extracellular DA results in DA uptake into 5-HT terminals, which may then oxidize and damage 5-HT terminals (Schmidt et al., 1985). Although evidence of the inactivation of tryptophan hydroxylase, an enzyme unique to 5-HT terminals, suggests an intracellular oxidative stress (Fleckenstein et al., 1997), DA uptake alone does not seem sufficient to explain METH-induced 5-HT terminal loss. Intrastriatal injections of high concentrations of DA have been shown to cause a selective lesion of DA terminals that correlates with the oxidation of catechols. These injections of DA result in far greater extracellular concentrations of DA than does METH administration, and yet 5-HT terminals are spared in this model (Hastings et al., 1996; Rabinovic et al., 1996). Thus, similar to the mechanism of toxicity to DA terminals, it seems that the METH-induced toxicity to 5-HT terminals likely involves a complex interaction of many factors.
This study has provided several lines of evidence suggesting that increased DA oxidation is associated with the neurotoxicity of METH. METH may either cause the generation of reactive species such as superoxide or peroxynitrite, which are capable of oxidizing DA to DA quinone, or may create an environment that favors the autoxidation or the enzyme-catalyzed oxidation of DA. Regardless of the sequence of events that leads to the oxidation of DA, generation of the DA quinone seems to be linked to the toxicity of METH and may contribute to the mechanism involved. These findings may be important not only to the understanding of METH-induced toxicity but to the selective vulnerability of monoaminergic terminals in neurodegeneration, as well.
Footnotes
This work was supported by National Institute on Drug Abuse Grants DA09601 (T.G.H.) and DA05811 (M.J.L.). We thank Dr. Alan F. Sved for helpful discussions and a critical review of this manuscript.
Correspondence should be addressed to Dr. Teresa G. Hastings, S-526 Biomedical Science Tower, Department of Neurology, University of Pittsburgh, Pittsburgh, PA 15261.
REFERENCES
- 1.Abekawa T, Ohmori T, Koyama T. Effects of repeated administration of a high dose of methamphetamine on dopamine and glutamate release in the rat striatum and nucleus accumbens. Brain Res. 1994;643:276–281. doi: 10.1016/0006-8993(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 2.Abercrombie ED, Finlay JM. Monitoring extracellular norepinephrine in brain using in vivo microdialysis and HPLC-EC. In: Robinson T, Justice J Jr, editors. Microdialysis in the neurosciences. Elsevier; Amsterdam: 1991. pp. 253–274. [Google Scholar]
- 3.Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and non-protective agents. J Pharmacol Exp Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- 4.Ali SF, Newport GD, Holson RR, Slikker W, Jr, Bowyer JF. Low environmental temperatures or pharmacologic agents that produce hypothermia decrease methamphetamine neurotoxicity in mice. Brain Res. 1994;658:33–38. doi: 10.1016/s0006-8993(09)90007-5. [DOI] [PubMed] [Google Scholar]
- 5.Berman SB, Zigmond MJ, Hastings TG. Modification of dopamine transporter function: effect of reactive oxygen species and dopamine. J Neurochem. 1996;67:593–600. doi: 10.1046/j.1471-4159.1996.67020593.x. [DOI] [PubMed] [Google Scholar]
- 6.Bowyer JF, Tank WA, Newport GD, Slikker W, Jr, Ali SF, Holson RR. The influence of environmental temperature on the transient effects of methamphetamine on dopamine levels and dopamine release in rat striatum. J Pharmacol Exp Ther. 1992;260:817–824. [PubMed] [Google Scholar]
- 7.Bowyer JF, Gough B, Slikker W, Jr, Lipe GW, Newport GD, Holson RR. Effects of a cold environment or age on methamphetamine-induced dopamine release in the caudate putamen of female rats. Pharmacol Biochem Behav. 1993;44:87–98. doi: 10.1016/0091-3057(93)90284-z. [DOI] [PubMed] [Google Scholar]
- 8.Bowyer JF, Davies DL, Schmued L, Broening HW, Newport GD, Slikker W, Jr, Holson RR. Further studies on the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- 9.Cadet JL, Brannock C. Free radicals and the pathology of brain dopamine systems. Neurochem Int. 1998;32:117–131. doi: 10.1016/s0197-0186(97)00031-4. [DOI] [PubMed] [Google Scholar]
- 10.Cadet JL, Sheng P, Ali SF, Rothman RB, Carlson E, Epstein C. Attenuation of methamphetamine-induced neurotoxicity in copper/zinc superoxide dismutase transgenic mice. J Neurochem. 1994;62:380–383. doi: 10.1046/j.1471-4159.1994.62010380.x. [DOI] [PubMed] [Google Scholar]
- 11.Cappon GD, Broening HW, Pu C, Morford L, Vorhees CV. Alpha-phenyl-n-tert-butyl nitrone attenuates methamphetamine-induced depletion of striatal dopamine without altering hyperthermia. Synapse. 1996;24:173–181. doi: 10.1002/(SICI)1098-2396(199610)24:2<173::AID-SYN9>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 12.Colado MI, Green AR. The spin trap α-phenyl-tert-butyl-nitrone prevents “ecstasy”-induced neurodegeneration of 5-hydroxytryptamine neurones. Eur J Pharmacol. 1995;280:343–346. doi: 10.1016/0014-2999(95)00298-y. [DOI] [PubMed] [Google Scholar]
- 13.Cubells JF, Rayport S, Rajendran G, Sulzer D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci. 1994;14:2260–2271. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Vito MJ, Wagner GC. Methamphetamine-induced neuronal damage: a possible role for free radicals. Neuropharmacology. 1989;28:1145–1150. doi: 10.1016/0028-3908(89)90130-5. [DOI] [PubMed] [Google Scholar]
- 15.Di Monte DA, Royland JE, Jakowec MW, Langston JW. Role of nitric oxide in methamphetamine-induced neurotoxicity: protection by 7-nitroindazole, an inhibitor of neuronal nitric oxide synthase. J Neurochem. 1996;67:2443–2450. doi: 10.1046/j.1471-4159.1996.67062443.x. [DOI] [PubMed] [Google Scholar]
- 16.Egashira T, Yamamoto T, Yamanaka Y. Effects of d-methamphetamine on monkey brain monoamine oxidase, in vivo and in vitro. Jpn J Pharmacol. 1987;45:79–88. doi: 10.1254/jjp.45.79. [DOI] [PubMed] [Google Scholar]
- 17.Filloux F, Townsend JJ. Pre- and postsynaptic neurotoxic effect of dopamine demonstrated by intrastriatal injection. Exp Neurol. 1993;119:79–88. doi: 10.1006/exnr.1993.1008. [DOI] [PubMed] [Google Scholar]
- 18.Fleckenstein AE, Wilkins DG, Gibb JW, Hanson GR. Interaction between hyperthermia and oxygen free radical formation in the 5-hydroxytryptaminergic response to a single methamphetamine administration. J Pharmacol Exp Ther. 1997;283:281–285. [PubMed] [Google Scholar]
- 19.Foppoli C, Coccia R, Cini C, Rosei MA. Catecholamines oxidation by xanthine oxidase. Biochim Biophys Acta. 1997;1334:200–206. doi: 10.1016/s0304-4165(96)00093-1. [DOI] [PubMed] [Google Scholar]
- 20.Fornstedt B, Rosengren E, Carlsson A. Occurrence and distribution of 5-S-cysteinyl derivatives of dopamine, dopa and dopac in the brains of eight mammalian species. Neuropharmacology. 1986;25:451–454. doi: 10.1016/0028-3908(86)90242-x. [DOI] [PubMed] [Google Scholar]
- 21.Fornstedt B, Bergh I, Rosengren E, Carlsson A. An improved HPLC-electrochemical detection method for measuring brain levels of 5-S-cysteinyldopamine, 5-S-cysteinyl-3,4-dihydroxyphenylalanine, and 5-S-cysteinyl-3,4-dihydroxyphenylacetic acid. J Neurochem. 1990;54:578–586. doi: 10.1111/j.1471-4159.1990.tb01910.x. [DOI] [PubMed] [Google Scholar]
- 22.Gibb JW, Kogan FJ. Influence of dopamine synthesis on methamphetamine-induced changes in striatal and adrenal tyrosine hydroxylase activity. Naunyn Schmiedebergs Arch Pharmacol. 1979;310:185–187. doi: 10.1007/BF00500283. [DOI] [PubMed] [Google Scholar]
- 23.Giovanni A, Liang LP, Hastings TG, Zigmond MJ. Estimating hydroxyl radical content in rat brain using systemic and intraventricular salicylate: impact of methamphetamine. J Neurochem. 1995;64:1819–1825. doi: 10.1046/j.1471-4159.1995.64041819.x. [DOI] [PubMed] [Google Scholar]
- 24.Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- 25.Graham DG, Tiffany SM, Bell WR, Jr, Gutknecht WF. Autoxidation versus covalent binding of quinones as a mechanism of toxicity of dopamine, 6-hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol Pharmacol. 1978;14:644–653. [PubMed] [Google Scholar]
- 26.Gunasekar PG, Kanthasamy AG, Borowitz JL, Isom GE. NMDA receptor activation produces concurrent generation of nitric oxide and reactive oxygen species: implications for cell death. J Neurochem. 1995;65:2016–2021. doi: 10.1046/j.1471-4159.1995.65052016.x. [DOI] [PubMed] [Google Scholar]
- 27.Hastings TG. Enzymatic oxidation of dopamine: the role of prostaglandin H synthase. J Neurochem. 1995;64:919–924. doi: 10.1046/j.1471-4159.1995.64020919.x. [DOI] [PubMed] [Google Scholar]
- 28.Hastings TG, Zigmond MJ. Identification of catechol-protein conjugates in neostriatal slices incubated with [3H]-dopamine: impact of ascorbic acid and glutathione. J Neurochem. 1994;63:1126–1132. doi: 10.1046/j.1471-4159.1994.63031126.x. [DOI] [PubMed] [Google Scholar]
- 29.Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci USA. 1996;93:1956–1961. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirata H, Ladenheim B, Rothman RB, Epstein C, Cadet JL. Methamphetamine-induced neurotoxicity is mediated by superoxide radicals. Brain Res. 1995;667:345–347. doi: 10.1016/0006-8993(95)00218-f. [DOI] [PubMed] [Google Scholar]
- 31.Hotchkiss AJ, Gibb JW. Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J Pharmacol Exp Ther. 1980;214:257–262. [PubMed] [Google Scholar]
- 32.Ito S, Fujita K. Conjugation of DOPA and 5-S-cysteinyldopa with cysteine mediated by superoxide radical. Biochem Pharmacol. 1982;31:2887–2889. doi: 10.1016/0006-2952(82)90259-3. [DOI] [PubMed] [Google Scholar]
- 33.Ito S, Kato T, Fujita K. Covalent binding of catechols to proteins through the sulphydryl group. Biochem Pharmacol. 1988;37:1707–1710. doi: 10.1016/0006-2952(88)90432-7. [DOI] [PubMed] [Google Scholar]
- 34.Itzhak Y, Ali SF. The neuronal nitric oxide synthase inhibitor, 7-nitroindazole, protects against methamphetamine-induced neurotoxicity in vivo. J Neurochem. 1996;67:1770–1773. doi: 10.1046/j.1471-4159.1996.67041770.x. [DOI] [PubMed] [Google Scholar]
- 35.Itzhak Y, Gandia C, Huang PL, Ali SF. Resistance of neuronal nitric oxide synthase-deficient mice to methamphetamine-induced dopaminergic neurotoxicity. J Pharmacol Exp Ther. 1998;284:1040–1047. [PubMed] [Google Scholar]
- 36.Johnson M, Stone DM, Hanson GR, Gibb JW. Role of the dopaminergic nigrostriatal pathway in methamphetamine-induced depression of the neostriatal serotonergic system. Eur J Pharmacol. 1987;135:231–234. doi: 10.1016/0014-2999(87)90616-9. [DOI] [PubMed] [Google Scholar]
- 37.Karoum F, Chrapusta SJ, Egan MF, Wyatt RJ. Absence of 6-hydroxydopamine in the rat brain after treatment with stimulants and other dopaminergic agents: a mass fragmentographic study. J Neurochem. 1993;61:1369–1375. doi: 10.1111/j.1471-4159.1993.tb13630.x. [DOI] [PubMed] [Google Scholar]
- 38.Korytowski W, Sarna T, Kalyanaraman B, Sealy RC. Tyrosinase-catalyzed oxidation of dopa and related catechol(amine)s: a kinetic electron spin resonance investigation using spin-stabilization and spin label oximetry. Biochim Biophys Acta. 1987;924:383–392. doi: 10.1016/0304-4165(87)90152-8. [DOI] [PubMed] [Google Scholar]
- 39.Lafon-Cazal M, Pietrl MC, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 40.LaVoie MJ, Hastings TG. Peroxynitrite potentiates the oxidation of dopamine in vitro: implications for MPTP- and methamphetamine-induced toxicity. Soc Neurosci Abstr. 1997;23:1371. [Google Scholar]
- 41.Mattammal MB, Strong R, Lakshmi VM, Chung HD, Stephanson AH. Prostaglandin H synthase-mediated metabolism of dopamine: implications for Parkinson’s disease. J Neurochem. 1995;64:1645–1654. doi: 10.1046/j.1471-4159.1995.64041645.x. [DOI] [PubMed] [Google Scholar]
- 42.Mena MA, Pardo B, Caserejos MJ, Fahn S, De Yebenes JG. Neurotoxicity of levodopa on catecholamine-rich neurons. Mov Disord. 1992;7:23–51. doi: 10.1002/mds.870070105. [DOI] [PubMed] [Google Scholar]
- 43.Michel PP, Hefti F. Toxicity of 6-hydroxydopamine and dopamine for dopaminergic neurons in culture. J Neurosci Res. 1990;26:428–435. doi: 10.1002/jnr.490260405. [DOI] [PubMed] [Google Scholar]
- 44.Miller DB, O’Callaghan JP. Environment-, drug- and stress-induced alterations in body temperature affect the neurotoxicity of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:752–760. [PubMed] [Google Scholar]
- 45.Nash JF, Yamamoto BK. Methamphetamine neurotoxicity and striatal glutamate release: comparison to 3,4-methylenedioxymethamphetamine. Brain Res. 1992;581:237–243. doi: 10.1016/0006-8993(92)90713-j. [DOI] [PubMed] [Google Scholar]
- 46.O’Dell SJ, Weihmuller FB, Marshall JF. Multiple methamphetamine injections induce marked increases in extracellular striatal dopamine which correlate with subsequent neurotoxicity. Brain Res. 1991;564:256–260. doi: 10.1016/0006-8993(91)91461-9. [DOI] [PubMed] [Google Scholar]
- 47.Rabinovic AD, Zigmond MJ, Hastings TG. Early indices of oxidative stress: immediate response to neurotoxic levels of exogenous dopamine. Soc Neurosci Abstr. 1996;22:221. [Google Scholar]
- 48.Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- 50.Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- 51.Robinson TE, Yew J, Paulson PE, Camp DM. The long-term effects of neurotoxic doses of methamphetamine on the extracellular concentration of dopamine measured with microdialysis in striatum. Neurosci Lett. 1990;110:193–198. doi: 10.1016/0304-3940(90)90810-v. [DOI] [PubMed] [Google Scholar]
- 52.Rosenberg PA. Catecholamine toxicity in cerebral cortex in dissociated cell culture. J Neurosci. 1988;8:2887–2894. doi: 10.1523/JNEUROSCI.08-08-02887.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosengren E, Linder-Eliasson E, Carlsson A. Detection of 5-S-cysteinyldopamine in human brain. J Neural Transm. 1985;63:247–253. doi: 10.1007/BF01252029. [DOI] [PubMed] [Google Scholar]
- 54.Rosei MA, Blarzino C, Foppoli C, Mosca L, Coccia R. Lipoxygenase-catalyzed oxidation of catecholamines. Biochem Biophys Res Commun. 1994;200:344–350. doi: 10.1006/bbrc.1994.1454. [DOI] [PubMed] [Google Scholar]
- 55.Schenk JO, Miller E, Gaddis R, Adams RN. Homeostatic control of ascorbate concentration in CNS extracellular fluid. Brain Res. 1982;253:353–356. doi: 10.1016/0006-8993(82)90709-0. [DOI] [PubMed] [Google Scholar]
- 56.Schmidt CJ, Taylor VL. The dopaminergic neurotoxicity of methamphetamine is prevented by nitrone spin trapping agents. Soc Neurosci Abstr. 1995;21:1246. [Google Scholar]
- 57.Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 1985;233:539–544. [PubMed] [Google Scholar]
- 58.Seiden LS, Vosmer G. Formation of 6-hydroxydopamine in caudate nucleus of the rat brain after a single large dose of methylamphetamine. Pharmacol Biochem Behav. 1984;21:29–31. doi: 10.1016/0091-3057(84)90125-4. [DOI] [PubMed] [Google Scholar]
- 59.Seiden LS, Fischman MW, Schuster CR. Long-term methamphetamine induced changes in brain catecholamines in tolerant rhesus monkeys. Drug Alcohol Depend. 1975;1:215–219. doi: 10.1016/0376-8716(76)90030-2. [DOI] [PubMed] [Google Scholar]
- 60.Skibba JL, Gwartney EA. Liver hyperthermia and oxidative stress: role of iron and aldehyde production. Int J Hyperthermia. 1997;13:215–226. doi: 10.3109/02656739709012384. [DOI] [PubMed] [Google Scholar]
- 61.Sonsalla PK, Gibb JW, Hanson GR. Role of D1 and D2 dopamine receptor subtypes in mediating the methamphetamine-induced changes in monoamine systems. J Pharmacol Exp Ther. 1986;238:932–937. [PubMed] [Google Scholar]
- 62.Sonsalla PK, Jochnowitz ND, Zeevalk GD, Oostveen JA, Hall ED. Treatment of mice with methamphetamine produces cell loss in the substantia nigra. Brain Res. 1996;738:172–175. doi: 10.1016/0006-8993(96)00995-x. [DOI] [PubMed] [Google Scholar]
- 63.Stephans SE, Yamamoto BK. Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse. 1994;17:203–209. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- 64.Wagner GC, Ricaurte GA, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- 65.Wagner GC, Lucot JB, Schuster CR, Seiden LS. Alpha-methyltyrosine attenuates and reserpine increases methamphetamine-induced neuronal changes. Brain Res. 1983;270:285–288. doi: 10.1016/0006-8993(83)90602-9. [DOI] [PubMed] [Google Scholar]
- 66.Weihmuller FB, O’Dell SJ, Marshall JF. MK-801 protection against methamphetamine-induced striatal dopamine terminal injury is associated with attenuated dopamine overflow. Synapse. 1992;11:155–163. doi: 10.1002/syn.890110209. [DOI] [PubMed] [Google Scholar]
- 67.Yehuda S, Wurtman RJ. The effects of d-amphetamine and related drugs on colonic temperatures of rats kept at various ambient temperatures. Life Sci. 1972;11:851–859. doi: 10.1016/0024-3205(72)90101-4. [DOI] [PubMed] [Google Scholar]