

Abstract
We examined the uptake and distribution of an antisense phosphorothioated oligodeoxynucleotide (s-ODN) to c-fos, rncfosr115, infused into the left cerebral ventricle of male Long–Evans rats and the effect of this s-ODN on subsequent Fos, NGF, neurotrophin-3 (NT-3), and actin expression. To establish the uptake and turnover of s-ODN in the brain, we studied the copurification of the immunoreactivity of biotin with biotinylated s-ODN that was recovered from different regions of the brain. A time-dependent diffusion and the localization of s-ODN were further demonstrated by labeling the 3′-OH terminus of s-ODN in situ with digoxigenin-dUTP using terminal transferase and detection using anti-digoxigenin IgG–FITC. Cellular uptake of the s-ODN was evident in both the hippocampal and cortical regions, consistent with a gradient originating at the ventricular surface. Degradation of the s-ODN was observed beginning 48 hr after delivery. The effectiveness of c-fos antisense s-ODN was demonstrated by its suppression of postischemic Fos expression, which was accompanied by an inhibition of ischemia-induced NGF mRNA expression in the dentate gyrus. Infusion of saline, the sense s-ODN, or a mismatch antisense s-ODN did not suppress Fos expression. That this effect of c-fos antisense s-ODN was specific to NGF was demonstrated by its lack of effect on the postischemic expression of the NT-3 and β-actin genes. Our results demonstrate that c-fos antisense s-ODN blocks selected downstream events and support the contention that postischemic Fos regulates the subsequent expression of the NGF gene and that Fos expression may have a functional component in neuroregeneration after focal cerebral ischemia-reperfusion.
Keywords: antisense DNA, experimental cerebral ischemia, c-fos, drug target validation, gene regulation, gene function analysis, immediate early genes, intracerebroventricular delivery, neurotrophin, NGF, oligodeoxynucleotide, transfection, stroke
The expression of the immediate early genes (IEGs) (such as c-fos and c-jun), followed by the expression of the late effector genes, such as the neurotrophin genes (NTGs), in the ischemic brain raises the possibility that IEG expression may play a role in the subsequent expression of NTGs (Sharp, 1994). The expression of NTGs [e.g., nerve growth factor (NGF) and brain-derived neurotrophic factor] may be important in regenerative processes and functional recovery after stroke (Lindvall et al., 1992; Ghosh et al., 1994). However, the pathophysiological significance of the postischemic expression of the IEGs remains to be fully elucidated (Sagar, 1993; Akins et al., 1996). In eukaryocytes, gene expression is regulated primarily through sequence-specific transcription factors. IEG products, such as Fos and Jun, form heterodimers that bind specifically to the AP-1 site and are nuclear regulatory proteins. The AP-1 binding sequence has been identified in the promoter region of a number of genes, including NGF (Zheng and Heinrich, 1988). A causal link between the activation of c-fos and the subsequent expression of NGF has been foundin vitro (Hengerer et al., 1990), although the reverse sequence of events had been reported earlier (Kruijer et al., 1985). Further clarification of the relationship between c-fos and NGF expression in the brain is needed.
We have shown that in vivo Fos–AP-1 knockdown is feasible using a c-fos antisense phosphorothioated oligodeoxynucleotide (s-ODN), which selectively blocks the translation of the target c-fos mRNA (Liu et al., 1994). The commonly used antisense s-ODNs for manipulation of gene expression are single-stranded s-ODN, limited to 15 and 30 nucleotides in length for an effective uptake by endocytosis (Yakubov et al., 1989). Although the stability, disposition, and clearance of ODNs within the CSF of the rat have been described (Whitesell et al., 1993), the parenchymal uptake and distribution of ODNs within the brain are primarily unexplored. Our decision to use an s-ODN to c-fos to explore the regulation of gene expression in the brain was based on several factors: (1) the possible involvement of AP-1 in apoptosis initiated by tumor necrosis factor-α (Roulston et al., 1998); (2) the robust expression of c-fos after ischemic or traumatic brain injury (An et al., 1993); (3) the well established methodology for a detection of regional expression of Fos in the brain (Sagar et al., 1988; Sharp et al., 1991); (4) the effective selection of ODN sequences usingin vitro translation and immunoprecipitation (Liu et al., 1994); and (5) the ability to assess in vivo Fos function based on its AP-1 binding activity in the brain extract. Moreover, many laboratories have successfully applied c-fos antisense ODNs in animal models (Funato et al., 1992; Chiasson et al., 1992, 1994;Heilig et al., 1993; Dragunow et al., 1994; Hooper et al., 1994;Schlingensiepen et al., 1994; Simonson, 1994; Cirelli et al., 1995a; Quercia and Chang, 1996; Yu et al., 1996), which will allow us to compare our results with those of these other laboratories. In this study, we demonstrate the location of s-ODN and study the effects of s-ODNs.
MATERIALS AND METHODS
s-ODN and preparation of biotinylated s-ODN
The s-ODNs to c-fos, including the following, were custom made by National Biosciences, Inc. (Plymouth, MN): sense orientation (sense-rncfosr115) 5′-ggtttgcccaaaccacgaccatgatg-3′-OH; the antisense orientation (antisense-rncfosr115) 5′-catcatggtcgtggtttgggcaaacc-3′-OH; the same antisense-rncfosr115 with the 3′-OH terminus blocked with one dideoxynucleotide (s-ODN-ddC); and the mismatch MA17 s-ODN 5′-gaacatcatCgtGgCgg-3′-OH. The mismatch MA17 s-ODN had three mismatching nucleotides and contained only 14 matching nucleotides to the c-fos gene. Unless otherwise specified, the s-ODNs and biotinylated s-ODN (bio-s-ODNs) we used in this study were in the antisense orientation. The sense s-ODNs and the mismatch s-ODN to c-fos were used only in selected control experiments to confirm the specific effects of the antisense s-ODN. The s-ODN stock was diluted to a final concentration of 1 nmol/μl saline. The antisense s-ODN to c-fos was labeled using photobiotin acetate according to the protocol described by Life Technologies (Gaithersburg, MD). The resultant bio-s-ODN was purified using a Sephadex G-25 column (Boehringer Mannheim, Indianapolis, IN) to remove the unconjugated photobiotin acetate and stored at −20°C.
To verify that the length of the purified bio-s-ODN was the same as the original c-fos antisense s-ODN (26 nucleotides), the bio-s-ODN was labeled with [γ32P]ATP (New England Nuclear, Boston, MA) using T4 polynucleotide kinase (Promega, Madison, WI) and then resolved using PAGE (16%; Bio-Rad, Richmond, CA). The concentration of the bio-s-ODN was measured using a colormetric detection kit and the protocol specified by the manufacturer (Life Technologies). The pixel values of the biotin spots on the blot were quantified using an image analyzer (IS2000; Alpha Innotech Corp., San Leandro, CA).
Delivery of s-ODNs
One hundred ninety-one Long–Evans hooded male rats (body weight 300 ± 30 gm) from Harlan (Indianapolis, IN) were used in the entire study. Housing and anesthesia concurred with guidelines established by the Institutional Animal Welfare Committee in accordance with the Public Health Service Guide for the Care and Use of Laboratory Animals, United States Department of Agriculture regulations, and the American Veterinary Medical Association Panel on Euthanasia guidelines. Anesthesia was induced with ketamine (100 mg/kg, i.p.) plus xylazine (10 mg/kg, i.p.). A total of 40 μl of artificial CSF (aCSF) containing lipofectin (10 μg, unless otherwise indicated) and one of various s-ODNs (as specified in the text) was infused over 30 sec into the left lateral ventricle guided by a stereotaxic device (Liu et al., 1994). In lipofectin controls, lipofectin without s-ODNs (n = 3) in 40 μl of aCSF was infused in the same manner. In other controls, saline (n = 16), s-ODN alone in aCSF (n = 9), or animals without surgery (n = 5) (saline, S-ODN minus lipofectin, or normal controls; respectively) was delivered. At specified times after delivery, the animals were anesthetized and tissue fixation (s-ODN uptake and recovery studies) or for the surgery to produce experimental stroke (Fos and neurotrophin gene expression studies). All experiments were performed in a blinded manner, with the rats and s-ODN or control treatments randomly administered to minimize any selection, treatment, or observation bias on the part of investigators. Unless specified, at least three animals were used for each treatment in the rat brain.
Tissue preparations
The animals were anesthetized if needed at the time of death using transcardial perfusion with 150 ml of saline at a rate of 40 ml/min, followed by 300 ml of 4% paraformaldehyde in 0.1 mphosphate buffer (PB), pH 7.4, at a rate of 20 ml/min. The brain was removed, and 3 mm coronal sections were prepared. The brain slices were incubated twice in 0.01 m PB for 1 hr and then in a series of increasing concentrations of ethanol (70–100%) in 0.01m PB before being embedded in paraffin. For in situ 3′ end labeling of s-ODN, coronal brain sections (5 μm) from paraffin-embedded brains were treated with xylene and then with chloroform plus xylene. For immunohistochemistry and in situ hybridization (ISH), the brain tissue was not embedded in paraffin but was stored as frozen sections (20-μm-thick on poly-l-lysine-coated slides). In all examinations, two to four brain slices from each animal were mounted on a poly-l-lysine-coated slide, and each slice was obtained ∼0.2 mm apart, located between 3.6–4.8 mm from bregma. For dot blot analysis of s-ODN recovery or for gene expression using reverse transcription-PCR (RT-PCR), the brains without perfusion were removed at the designated times. The cortex, hippocampus, and cerebellum were separated, flash-frozen in liquid nitrogen, and stored at −75°C (Liu et al., 1994).
In situ uptake and distribution of s-ODNs
In our studies described here, three complementary assays were used for the pharmacokinetic study of s-ODN uptake in the right hemisphere. These include a direct immunohistochemical staining of the bio-s-ODNs and an in situ 3′-OH-terminus labeling of s-ODNs. Moreover, the length of the recovered bio-s-ODN was purified using Sephadex G-25 column-recovered bio-s-ODNs and Centricon-10 (Amicon, Beverly, MA).
Direct detection using the immunohistochemical method. The objective of this study was to determine the optimal time during which the brain took up bio-s-ODNs in 14 animals that received the antisense bio-s-ODN. The presence of bio-s-ODN in the brain tissue was detected using the primary antibody (goat anti-biotin IgG; Sigma, St. Louis, MO), which was recognized using a secondary antibody (rabbit anti-goat IgG-FITC; Sigma). Briefly, the brain slices were incubated with the primary antibody (1:800) for 16 hr at 4°C, washed with PBS three times, and then incubated with the secondary antibody (1:1600) for 2 hr at room temperature. After washing in PBS, the DNA in the nuclei was stained using propidium iodide (PI) at a concentration of 0.5 μg/ml in the presence of heat-inactivated RNase A (0.5 μg/ml; Stratagene, La Jolla, CA) for 5 min. The orange–red coloration of the DNA caused by PI staining serves as an indicator for the location of the nuclei. Each brain slice was examined for the antibody FITC under a microscope using a Leica (Nussloch, Germany) I3 filter (450–510 nm) and a mercury light source. Photographs of the right cerebral hemisphere were acquired using either a regular camera with Kodak type film (Eastman Kodak, Rochester, NY) or a cooled color digital camera (the SPOT camera; Diagnostic Instruments, Sterling Heights, MI), with the digitized image stored on a computer diskette.
The stability of biotin signals on bio-s-ODNs was determined by detecting biotin label in the bio-s-ODN from the brains of eight animals at 29, 48, and 72 hr after delivery. The assay was designed to measure biotin on the recovered s-ODNs. Nuclear DNA from cerebral cortex, hippocampus, and cerebellum was extracted (Liu et al., 1994). One-fifth of each DNA sample (or 100 μl) was processed through the Sephadex G-25 Quick-Spin column to recover the bio-s-ODN of greater than 20 bases. Further dialysis of the column-recovered DNA in a Centricon-10 membrane (Amicon) detected no residual bio-s-ODN in the flow-through, indicating that the bio-s-ODN was 26 bases. The Sephadex G-25 column-purified bio-s-ODN was then vacuum-dried and resuspended in 10 μl of STE buffer [(in mm) 50 NaCl, 10 Tris-HCl, and 1 EDTA], and one-fifth (2 of 10 μl) of the column-purified fraction was applied onto blotting paper for the measurement of biotin signal. Of the recovered bio-s-ODN, a set of 2–10 pg bio-s-ODN standards (before column purification) and a set of recovered bio-s-ODN after column purification were also applied onto the blotting paper in the second row. The amount of bio-s-ODN was measured using the colormetric detection kit stated above. The detection limit using this method was 250 pg of bio-s-ODN from the brain sample.
In situ labeling of 3′-OH terminus of the s-ODN. The objective of this study was to determine the distribution of s-ODN in the brain tissue. Seventy-nine animals were studied (n= 3 each that received 1 and 50 nmol, and n = 2 in 100 nmol antisense c-fos s-ODN; n = 19 in the group that received 10 nmol of antisense c-fos s-ODN;n = 20 for s-ODN-ddC; and n = 15 for those that received sense c-fos s-ODN). The 3′-OH terminus on s-ODN in the brain can be labeled using terminal transferase (TdT) in the presence of digoxigenin (dig)-dUTP, followed by the antibody against digoxigenin with FITC conjugates. TdT synthesizes a DNA chain by 5′ to 3′ polymerization using 5′-dNTP (or dig-dUTP). This reaction requires a three nucleotide or longer primer with a free 3′-hydroxyl group to serve as a primer terminus for extension (Kornberg, 1980). TdT will incorporate dNTP without the nucleotide sequence dictated by a template. This technique allows multiple incorporation of dig-dUTP onto the 3′-OH terminus of the s-ODN (3′ end labeling), and the s-ODN signal can be amplified. For controls, saline or s-ODN-ddC (10 nmol) plus lipofectin in aCSF were delivered. We expected no signal derived from 3′ end labeling in animals pretreated with the control s-ODN-ddC, which contained no 3′-OH terminus and, therefore, produced no TdT-mediated signal. The incorporation of dig-dUTP onto s-ODN by TdT and the detection of the incorporated dig-dMP on s-ODN using fluorescein conjugate of antibody against digoxigenin were performed as described in the Apop Taq kit (Oncor, Inc., Gaithersburg, MD). The brain slices were examined under a microscope as described previously (Liu et al., 1994).
Experimental cerebral ischemia
The effect of c-fos antisense s-ODN on the expression of Fos, NGF, neurotrophin-3 (NT-3), and actin was examined. To determine whether Fos expression was inhibited by the antisense strategy described, we compared ischemic Fos expression using a well established focal cerebral ischemia-reperfusion (FCIR) model in two groups of animals that received c-fos antisense (n = 22; group A) and sense (n = 16; group S) s-ODN, respectively. Control animals received one of the following: (1) aCSF and no FCIR (n = 17; the sham control or group N); (2) FCIR (n = 15; the positive control or group P); or (3) mismatch M17 s-ODN (n = 4) in aCSF and FCIR. Briefly, 16 hr after s-ODN delivery, the animal was anesthetized, and the right middle cerebral artery and both common carotid arteries were occluded for 90 min. Reperfusion was initiated by releasing the occlusion and continued for 90 min. While still under anesthesia, the animal was killed for tissue preparations as described earlier.
The effect of s-ODN on the expression of ischemic Fos, NGF, NT-3, and actin
The detection of ischemic Fos expression. Coronal sections (20-μm-thick) from 20 animals were incubated in proteinase K (10 μg/ml) for digestion at 37°C for 30 min, then in PBS containing bovine serum albumin (BSA) (10 mg/ml) and 0.1% NaN3 for 30 min, followed by washing in PBS–BSA five times. The sections were then incubated with Ab-2 antibody (Oncogene Science, Manhattan, NY) in PBS–BSA at a suitable dilution (predetermined for its lot number) at 4°C overnight. We have found that a 1:400 to 1:500 dilution of Ab-2 gives the best result. After washes in PBS–BSA, the brain sections were incubated with a secondary antibody (goat anti-rabbit IgG conjugated with biotin at 1:400 dilution) at room temperature for 45 min. The antigen–IgG complex was incubated with avidin–horseradish peroxidase (1:100) for 30 min and then stained with diaminobenzidine using a Vectastain ABC kit (Vector Laboratories, Burlingame, CA). The specificity of the Fos signal was tested using negative controls (the animal samples from positive controls that underwent the same procedures in the absence of the primary antibody or preadsorption of the primary antibody with Fos antigen before immunostaining) in each determination.
Neurotrophin gene expression using ISH. We analyzed the expression of NGF, NT-3, and β-actin mRNA in the brain after FCIR in animals previously treated with aCSF or s-ODN (sense-rncfosr115 and anti-rncfosr115). Because the activation of NGF mRNA is limited to neurons in the dentate gyrus (Lindvall et al., 1992), we used ISH to determine NGF mRNA expression in the brain tissue as described previously (An et al., 1993), except that the cRNA transcript was labeled with [α-32P]UTP (107 cpm/ml). After hybridization, the brain slices were digested using RNase A at 37°C for 30 min to remove nonspecific hybrids. The specificity of the mRNA signal was tested in animal samples from group P for their inability to produce any signal using the [32P]mRNA (the sense) transcript of the NGF gene.
RT-PCR. Because the background level of NGF mRNA in the brain is ∼1% of that found in the actin mRNA, we sought to amplify the hippocampal NGF and β-actin genes using the RT-PCR method. We isolated total RNA from the right (ipsilateral or ischemic) and left (contralateral or nonischemic) hippocampus from 19 animals. The cDNA preparation from 5 μg of total RNA was performed using 200 U of SuperScript II (Life Technologies) as described previously (Liu, 1993). The cDNA was extracted with phenol and then with chloroform, followed by precipitation with ethanol. The cDNA was dissolved in 100 μl of TE buffer (Tris-HCl 10 mm and EDTA 1 mm, pH 8.0). One microliter of cDNA was calculated to be equivalent to that transcribed from 1.5 ng of mRNA. The criteria of an acceptable cDNA preparation were the expression of the β-actin gene in the cDNA sample; the β-actin cDNA levels from the RNA samples of 16 animals were not affected by FCIR, and there was no significant difference between samples derived from the right and left hippocampi using the hot-start PCR. To minimize the technical feasibility of running more than 16 samples simultaneously on one agarose gel, we pooled the cDNA from all four animals in each treatment. Amplification of β-actin in the pooled samples showed no change in the amount of PCR product.
The primers (forward and backward primers, respectively) used for PCR for the β-actin mRNA were 5′-ccttcctgggcatggagtcctg-3′ and 5′-ggagcaatgatcttgatcttc-3′ (Colotta et al., 1992); for the NGF mRNA, they were 5′-ggcaagtcagcctcttcttgcagcc-3′ and 5′-gtaatgtccatgttgttctacactctg-3′; and for the NT-3 mRNA, they were 5′-gcagagcataagagtcaccg-3′ and 5′-ccgatttttcttgacaaggc-3′ (Maisonpierre et al., 1991). Several technical factors can affect the amplification result by PCR. In the preliminary studies, we found that the optimal conditions for PCR amplification were 10 pmol of primers, 2–6 mm Mg2+ in the reaction solution, and an annealing temperature of 55°C. In addition, we found that the PCR product from the 3 μl cDNA sample increased linearly with increasing numbers of cycles from 18 to 32. We used 24 cycles for the β-actin and 28 cycles for NTG amplification. For quantitative purposes, one of the primers was 5′ end-labeled with [γ-32P]ATP, and PCR amplification was performed for 18 cycles. Finally, the PCR products of the β-actin, NGF, and NT-3 genes were gel-purified, and their sequences were determined diagnostically. We found that each PCR product contained its target sequence. Moreover, the primers specifically amplified the target cDNA, even in the presence of other cDNA that may share partial homology to the target gene under the PCR conditions we used.
For PCR, the 39 μl reaction mixture containing 3 μl of cDNA and 20 pmol of primers was heated at 95°C for 5 min and then cooled to 65°C. An 11 μl mixture that contained dNTPs, buffer, and 1.5 U ofTaq polymerase was added to the reaction, which was in a thermocycler block at 65°C (Perkin-Elmer, Emeryville, CA). Amplification was performed for no more than 30 cycles at 94°C for 45 sec, 55°C for 30 sec, and 72°C for 1 min and 30 sec. The reaction was terminated by a 10 min extension at 70°C. Ten microliters of each amplification reaction were resolved on a 2% agarose gel or a 6% PAGE.
RESULTS
In situ uptake and distribution of s-ODNs in rat brain
Direct detection of bio-s-ODNs using the immunohistochemical method
To study the correlation between the distribution and the effect of c-fos antisense s-ODN after FCIR, we first determined the uptake and distribution of c-fos antisense s-ODN in the right hemisphere of rat brains. After the delivery of the bio-s-ODNs into the left lateral ventricle, we detected no significant bio-s-ODN signal (green or yellow–green fluorescence) in the cortex within 6 hr (n = 3) after delivery (Fig.1A), except in a few nuclei in the CA neurons of the hippocampus (Fig.1D). The intensity of bio-s-ODN signals, as shown by anti-biotin antibodies, located around the nucleus and increased with time. The signal was noted in the cerebral cortex 2 d (n = 2) after receiving the antisense s-ODN (Fig.1B), whereas the signal in the hippocampal CA1 region was most intense 1 d (n = 5) after infusion (Fig.1E). The signal in the cortex and hippocampus lasted for at least 4 d (n = 4) (Fig.1C,F).
Fig. 1.

The presence of bio-s-ODN in the rat brain. Bio-s-ODN (1 nmol) with lipofectin (0.01 μg) was delivered via intracerebroventricular route as described previously (Liu et al., 1994). The presence of bio-s-ODN in the brain was detected by using goat IgG against biotin and rabbit anti-goat IgG-FITC conjugates. The nuclei were counterstained with PI and appeared red. Representative areas in layer II of the cortex (A,B, and C at 6, 48, and 96 hr after delivery, respectively) and in CA1 of the hippocampus (D, E, and F at 6, 24, and 96 hr after delivery, respectively) in the right hemisphere are shown. The magnification in all panels is the same. Scale bar, 20 μm. The image was acquired using double exposure and Kodak photographic film for FITC and PI signals. Because of the double staining of FITC and PI, the biotin signal appearedgreen (F) or yellow(a blend of red and green;B, C, E). The apparent larger sizes of the nuclei in B–F were attributable to the visualization of bio-s-ODNs in cell bodies in contrast to nuclear stain only by PI in A.
To determine whether biotin signal remained with the bio-s-ODNs in different regions of the brain, bio-s-ODNs were recovered from the cortex, hippocampus, and cerebellum at 1 (n = 3), 2 (n = 3), and 3 (n = 2) d after infusion. At 3 d after delivery, we did not recover measurable bio-s-ODN from the hippocampus or the cerebellum using dot blot analyses. Because bio-s-ODN of 21–30 bases in length would be recovered, the data indicate that the infused bio-s-ODN remained as intact s-ODN for 2 d after delivery. The disappearance of brain bio-s-ODN over time suggests degradation of the bio-s-ODN to <20 bases in the brain.
Uptake and distribution of s-ODN using in situ labeling of s-ODN 3′-OH terminus
Using immunohistochemical detection of bio-s-ODN, our results indicated that some of the infused bio-s-ODNs could be observed in nuclei of the hippocampus beginning at ∼6 hr, and the intensity of bio-s-ODN increased with time and lasted until ∼4 d after infusion (Fig. 1). However, dot blot analysis demonstrated that intact s-ODNs could not be recovered beyond 2 d after delivery. Therefore, a direct detection of labeled s-ODN was not adequate to determine the localization of s-ODN. To further confirm the uptake of the infused s-ODN in the brain, we labeled the 3′-OH terminus of the s-ODN in situ. As early as 4 hr after delivery of 1 nmol of s-ODN (n = 3), fluorescent signals were detected in the corpus callosum and adjacent hippocampus in all three animals that received the s-ODN (Fig.2B), whereas no fluorescent signal was observed in animals without surgery (the normal control; n = 5) and in animals that received saline (n = 2) (Fig. 2A). The intensity of the signal indicating 3′-OH termini increased with the amount of s-ODN delivered (10 nmol) (Fig. 2C). Larger doses of s-ODN (e.g., ≥50 nmol) led to the distribution of the signal in a wider region beyond the lateral ventricles (data not shown). More intense and uniform staining of ependymal hippocampal cell bodies, diffuse staining of the hippocampal neurites, and the endothelial cell bodies of the vessel walls were observed at 1 hr after infusion in animals that received 50 (n = 3) or 100 (n = 2) nmol of s-ODN. The appearance of fluorescence in the neurites distinguished the s-ODNs from the intranuclear 3′-OH labeling commonly associated with DNA fragmentation in apoptosis. The animals that received >50 nmol of s-ODN gave excellent signals. It appeared that the infused s-ODNs were taken up indiscriminately. However, all of the rats that received 100 nmol showed impaired neurological function with signs of shallow breathing and died within the first hour of s-ODN infusion. The animals that received 50 nmol of s-ODN began to show signs of shallow breathing at 1 hr after s-ODN delivery. Therefore, we chose to use 10 nmol of s-ODN, which did not appear to have any adverse effects.
Fig. 2.

The uptake of s-ODN in the ependyma of the lateral ventricle and beyond. The presence of s-ODNs was detected using 3′-OH-terminus labeling of TdT-mediated dig-dUTP incorporation, followed by immunohistochemical staining using anti-dig-IgG-FITC conjugates (Oncor, Inc.). The images were acquired using the SPOT digital camera with a single exposure using the green (FITC) spectrum. The anti-dig-FITC–dig-dUTP complex is shown as whitefluorescence. Four different doses of c-fos antisense s-ODNs, each administered with 10 μg of lipofectin (1 of the 3 animals from each group is shown) in 40 μl of aCSF (A, saline with no s-ODN; B, 1 nmol; C, 10 nmol). One of the three animals from each dosage is shown. The uptake time was 4 hr after infusion.
In rats that received 10 nmol of s-ODN, we examined the presence of s-ODN at 4 hr (n = 3), 16 hr (n = 4), 1 d (n = 4), and 2 d (n = 8) after delivery. Again, the signal in animals with 10 nmol of s-ODN at 4 hr was the same as that observed in Figure 2C and increased with time. There was no reduction in the fluorescent signal between 16 hr (Fig. 3B) and 2 d (Fig. 4) after infusion. The labeled s-ODNs could be observed in the nuclei of the dentate gyrus (Fig.3B) and the CA1 neurons (Fig. 4) in the hippocampus, the hypothalamus, and the various layers of the cortex (Fig.4I–VI). The s-ODNs appeared migrating from the ependyma of the lateral ventricle into the adjacent brain parenchyma within 1 d of delivery. Within the cells, the s-ODNs were found in the nuclei (Fig. 3B) and as aggregates in the perinuclear region (Fig. 4). Similar s-ODN uptake and distribution were observed when the sense s-ODN to c-fos was delivered (10 nmol: n = 5 for 4 hr; n = 2 for 6 hr;n = 5 for 1 d; and n = 3 for 2 d). Delivery of s-ODNs without lipofectin (n = 9) showed substantially no aggregates. No signal was observed in the control animals [saline (n = 14) or lipofectin alone in aCSF (n = 3)], and the animals received antisense s-ODN-ddC (10 nmol: n = 6 each at 4 hr, 1 d, and 2 d; and 50 nmol: n = 2 at 4 hr uptake times) or in the animals that received s-ODN but without adding TdT during 3′-OH end labeling (Fig. 3A). No 3′-OH in s-ODNs-ddC was available for polymerization by TdT up to 2 d after infusion. These data confirmed that the signal came from the incorporation of dig-dUTP onto 3′-OH termini of s-ODNs.
Fig. 3.

Representative digitized images of brain slices showing the uptake of s-ODN. The presence of s-ODNs in the dentate gyrus was detected using 3′ end labeling as in Figure 2, except that 10 nmol of c-fos antisense s-ODN with 10 μg of lipofectin in 40 μl of aCSF was delivered, and the images were acquired as in Figure 2. The animals were killed at 16 hr after infusion. The anti-dig-FITC–dig-dUTP complex is shown as bright whitefluorescence. Dark dots appear in the dentate gyrus of the animals that received s-ODN but without TdT for dUTP incorporation during the 3′-OH end labeling assay (A). Scale bar, 20 μm.
Fig. 4.
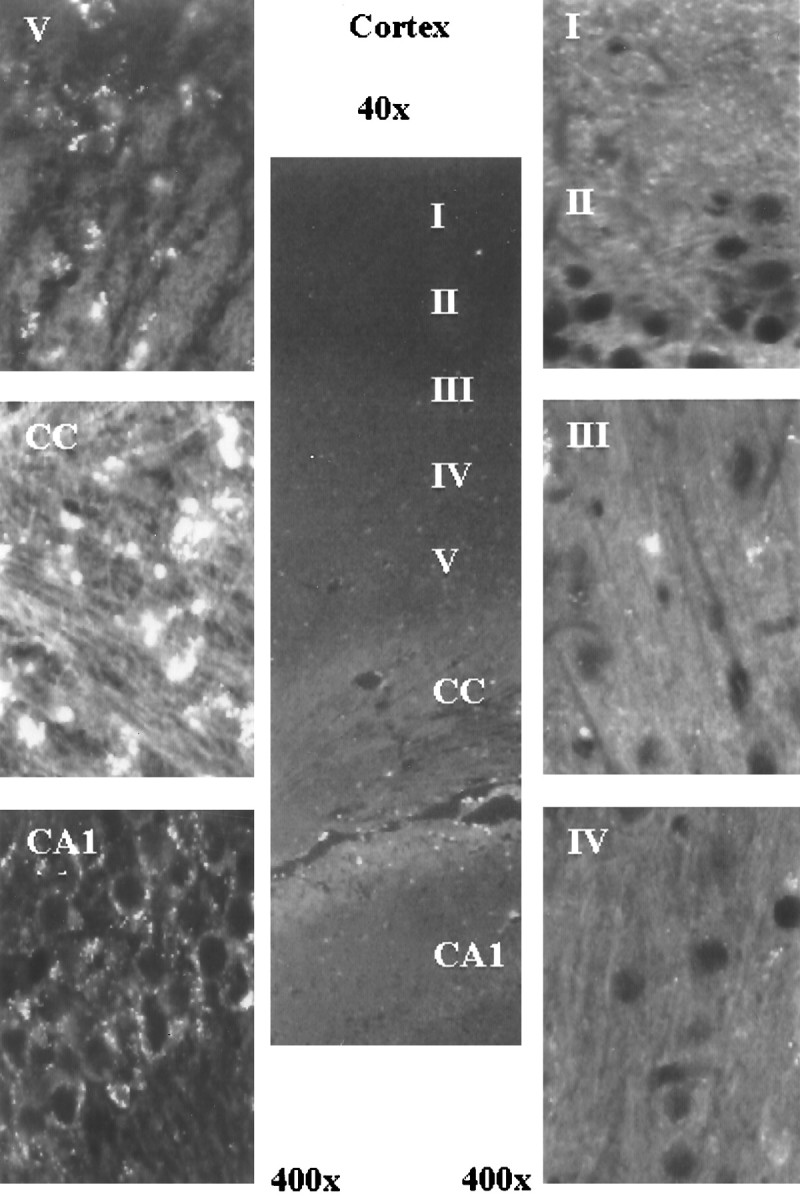
The presence of s-ODN 48 hr after delivery. The presence of infused s-ODNs appeared as bright whitefluorescence in one representative rat brain. Original magnifications (40× or 400×) were as indicated. Labels indicate the approximate cortical layers. CC, Corpus callosum. Images were acquired as in Figure 2. Because of single staining, the nuclei appeared as black holes.
The effect of s-ODN on the expression of postischemic Fos, NGF, NT-3, and actin expression
The detection of postischemic Fos expression
Very weak Fos expression was observed in the cerebral cortex and in the dentate gyrus in all of the sham-operated controls (Fig.5). Robust Fos expression was observed in the cortex and hippocampus in all of animals that received saline in the positive control (aCSF and FCIR) and in all of the animals that received the sense s-ODN to c-fos(sense-rncfosr115 and FCIR) (Fig. 5). Using mismatch MA17 s-ODN, we observed a slight reduction of Fos expression in the cortical neurons (Fig. 5), indicating a loss of stringency on the MA17 s-ODN. An inhibition of Fos expression, however, was observed in all FCIR animals that received the antisense s-ODN to c-fos(anti-rncfosr115). The knockdown of postischemic Fos expression in the dentate gyrus and the cortex by the antisense s-ODN to c-fos is in agreement with the robust uptake of the same s-ODN or bio-s-ODN in the same regions noted above (Figs.3B, 4, respectively).
Fig. 5.

The knockdown of Fos-expression after FCIR by c-fos antisense s-ODN. All samples shown were from the ischemic or the right cerebral cortex or hippocampus. A low basal level of Fos immunoreactivity was noted in the cortices without ischemia (sham controls). Significantly increased Fos expression was present in the cerebral cortical or hippocampal neurons in FCIR animals that received saline (positive controls), c-fos sense s-ODN (sense-rncfosr115), or mismatch MA17. Fos immunoreactivity was virtually absent in FCIR animals with c-fos antisense s-ODN (anti-rncfosr115).
Neurotrophin gene expression using ISH
Increased postischemic Fos expression is associated with an increase in AP-1 binding activity (An et al., 1993). The purpose of this study was to determine whether the expression of late effector genes was affected. Thirty-five animals were studied in two groups of animals that underwent one of four infusion treatments and FCIR: group N (n = 6), group P (n = 7), group S (n = 8), and group A (n = 14). Using ISH, we found a significant increase in postischemic NGF expression in the FCIR-treated right (ipsilateral) dentate gyrus in all of the positive controls (Fig. 6, P) and five of eight animals treated with c-fos sense s-ODN (Fig. 6, S) compared with the left (contralateral) hippocampus. In all of the sham-operated animals without ischemia (Fig.6, N), no difference between the expression of NGF mRNA in the right and left dentate gyri was noted. The increase in the NGF expression in the ipsilateral dentate gyrus of the positive and sense s-ODN controls was significant compared with the sham-operated controls (Fisher’s exact test; p < 0.05). In all FCIR animals that received c-fos antisense s-ODN (Fig. 6,A), NGF mRNA expression in the dentate gyrus (arrow) was not statistically different from that in the sham-operated group but was noticeably less than that observed in all of the animals with FCIR and in five of eight that received c-fos sense s-ODN with FCIR (Fisher’s exact test;p < 0.05). The knockdown of NGF mRNA localized to the dentate gyrus region, where there was s-ODN uptake (Fig. 3B) and no expression of the Fos peptide (Fig. 5).
Fig. 6.
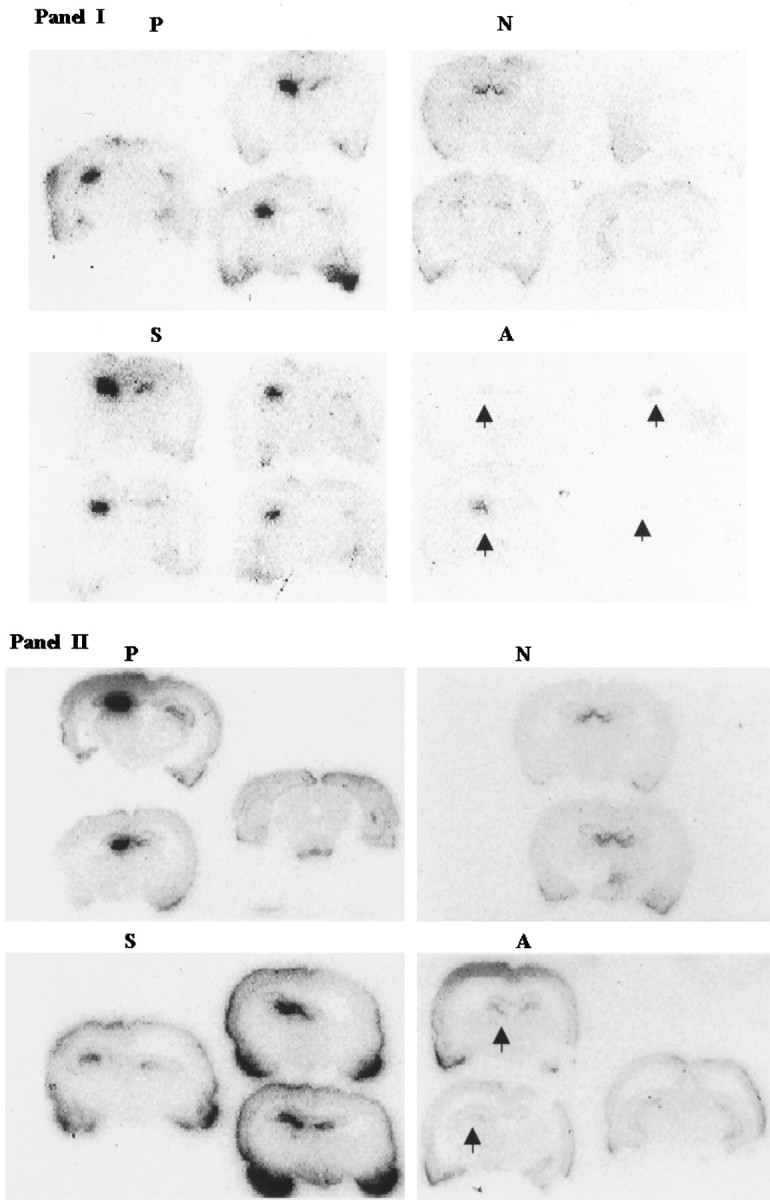
Suppression of NGF mRNA expression after FCIR by c-fos antisense s-ODN. NGF mRNA was detected in dentate gyri using a [32P]cRNA probe from the NGF gene. The autoradiograms show coronal brain sections (rostral to caudal orientation) of two representative groups of animals (sets one and two as shown in I and II, respectively) that underwent one of four infusion treatments and FCIR. P, Positive controls (saline and FCIR); N, sham controls (no FCIR); S, c-fos sense s-ODN;A, c-fos antisense s-ODN. Thearrows in A indicate FCIR-treated right dentate gyrus (see also Fig. 3B, s-ODN uptake; Fig. 5, Fos knockdown). Tissue from the sense-rncfosr115 group was hybridized with a [32P]mRNA probe; no signal was observed (data not shown).
Differential expression of NGF and NT-3 mRNA after FCIR
The expression of the hippocampal β-actin mRNA among the four groups of animals did not differ more than threefold (Fig.7); therefore, the expression of the β-actin mRNA was used as the control to determine the relative expression of hippocampal NGF mRNA. Using the coamplification RT-PCR (Fig. 7), the amount of hippocampal NGF mRNA (lane A) from the FCIR animals with the antisense s-ODN was no more than that in the non-FCIR control (lane N) but was less than that in the positive and sense controls (lanes P,S). Because there could be an interference of various substrates that might reduce the amplification of NGF mRNA from the animals that received c-fos antisense s-ODN during the co-amplification, we tested single gene amplification (Fig.8). Furthermore, the NGF mRNA was quantitatively examined in each set of RT-PCR experiments. The amount of NGF products from three hippocampi using RT-PCR gave a linear response curve (r = 0.95) using different amounts of cDNA that were transcribed from 0.75–9 ng of hippocampal mRNA. From this standard curve, we found that a greater than fivefold increase in the intensity of NGF products (from 200 to 900 cpm amplified from 0.75 to 3 ng, respectively) using this method was highly significant byt test (p < 0.01). On the other hand, a less than threefold increase in the intensity (from 900 to 2400 cpm amplified from 3 to 9 ng of mRNA, respectively) was not statistically significant. Figure 8A shows that postischemic NGF expression in the hippocampus significantly increased by eightfold (t test; p < 0.01) in FCIR animals receiving aCSF (n = 4; lane P) or the sense s-ODN (n = 4; lane S) compared with the sham controls without FCIR (n = 4;lane N). The hippocampal NGF gene expression in the animals that received the antisense s-ODN (n = 4;lane A) was not significantly different from that observed in the sham controls (lane N). The result was similar to that observed using coamplification in Figure 7. To determine that the observed NGF suppression after c-fosantisense s-ODN infusion was not secondary to nonspecific toxicity of the s-ODN, we also assessed the effect of this antisense s-ODN on postischemic expression of the hippocampal NT-3 gene in the same cDNA samples. Figure 8B shows that there was a twofold increase in the hippocampal NT-3 mRNA in the positive (lane P) and sense (lane S) controls compared with those in the antisense (lane A) and sham (lane N) controls. The antisense s-ODN to c-fos did not affect postischemic NT-3 expression. We concluded that there was no induction of NGF mRNA in the dentate gyrus of the animals that received c-fos antisense s-ODN after FCIR.
Fig. 7.

Differential display of hippocampal NGF and actin mRNA after FCIR by c-fos antisense s-ODN. The starting cDNA in each lane is from a mixture of all right FCIR hippocampi in each of the four groups (4 animals in each group). The PCR products were analyzed using nondenaturing PAGE (6%). The treatment groups are described in Figure 6. Lane M is the DNA size marker.
Fig. 8.
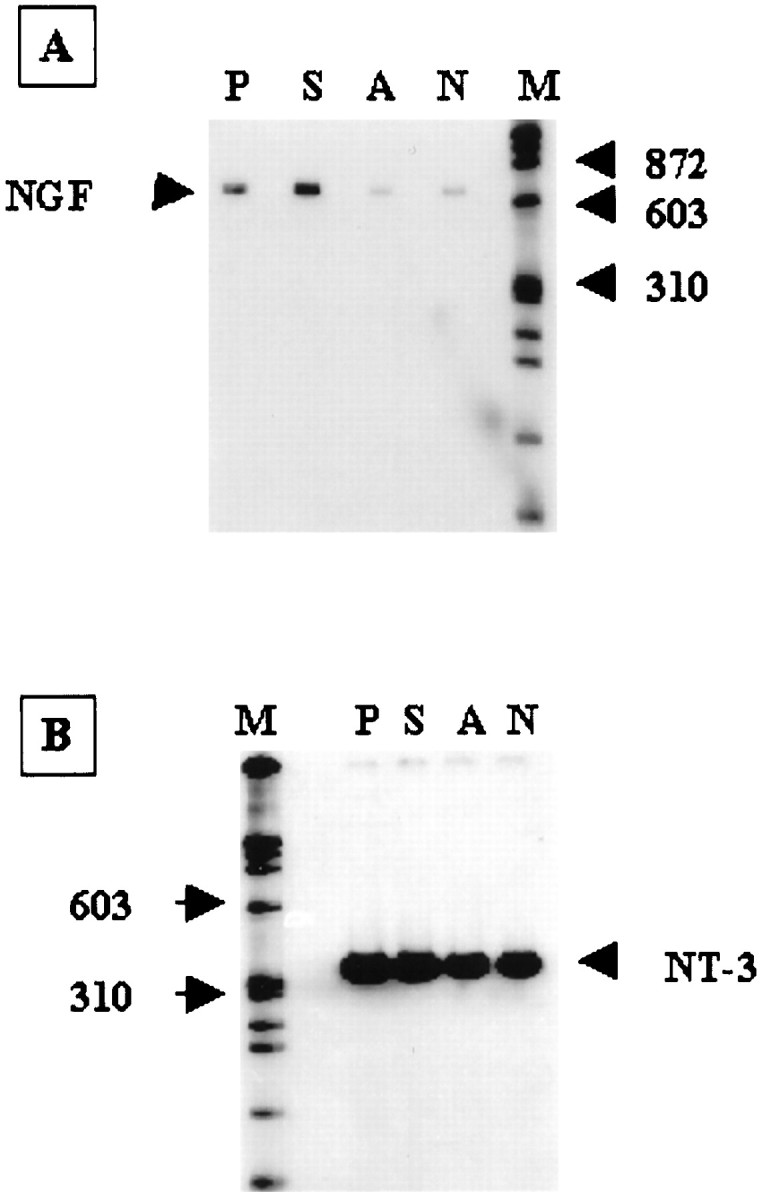
Postischemic expression of hippocampal NGF and NT-3 mRNA. NGF (A) and NT-3 (B) genes were individually amplified using 18 PCR cycles with a fixed amount of primer (10 pmol, one of which was labeled with 32P). The PCR product was resolved in nondenaturing 6% PAGE and was measured using the Betagen (Waltham, MA) Betascope blot analyzer. Each lane contains a mixture of cDNA from four animals that received the same treatment as those in Figures5-7.
DISCUSSION
A consequence of cerebral uptake of c-fos antisense s-ODN was the suppression of postischemic Fos expression after FCIR insult. The Fos expression is similar to that experienced in stroke or traumatic head injury. In addition, NGF mRNA expression in the dentate gyrus after FCIR was suppressed in animals treated with c-fos antisense s-ODN, whereas the expression of hippocampal β-actin and NT-3 genes was not affected. The gene products of several IEGs, including Fos, JunB, Jun, and cAMP response element-binding protein (Curran et al., 1984; Sagar et al., 1988; Sheng et al., 1990), are induced after cerebral ischemia (An et al., 1993; Sharp, 1994; Salminen et al., 1995; for review, see Akins et al., 1996). These proteins form activation signals to regulate the expression of various late effector genes (Sagar et al., 1988; Sharp et al., 1991; Gillardon et al., 1996). For example, Fos and Jun, the gene products of c-fos and c-jun, respectively, form a heterodimer via a leucine zipper (Halazonetis et al., 1988; Macgregor et al., 1990). This heterodimer binds the AP-1 DNA sequence and is presumed to regulate the expression of >900 genes (Sharp, 1994), including those coding for NGF (Zheng and Heinrich, 1988; Hengerer et al., 1990) and for DNA repair enzymes (Scanlon et al., 1991). A number of methods, including homologous recombination (Field et al., 1992) and RNA excision by ribozymes (Funato et al., 1992), can successfully inhibit c-fos gene expression. Antisense ODNs to c-fosgene have been used to regulate the expression of its mRNA in cell culture systems (Holt et al., 1986; Mercola et al., 1987; Nishikura and Murray, 1987; Edwards et al., 1988; Kerr et al., 1988; Levi and Ozato, 1988; Levi et al., 1988; Schönthal et al., 1988; Colotta et al., 1992) and in the rat brain (Chiasson et al., 1992, 1994; Heilig et al., 1993; Dragunow et al., 1994; Hooper et al., 1994; Schlingensiepen et al., 1994; Cirelli et al., 1995b; Quercia and Chang, 1996). Based on these works, we used in vivo transfection of c-fos antisense s-ODN to demonstrate the suppression of FCIR-induced Fos–AP-1 activities (Liu et al., 1994).
The neurotrophic genes support the survival of specific sets of neurons (Phillips et al., 1990; Wetmore et al., 1990; Snider, 1994). Expression of hippocampal NGF mRNA has been shown to increase significantly after cerebral ischemia (Lindvall et al., 1992), and we observed the same. In a motor neuron injury model, Gu and colleagues (1997) demonstrated that the increase in c-fos expression occurs before the induction of NGF mRNA. The selective suppression of hippocampal NGF, but not NT-3, mRNA after the administration of c-fos antisense s-ODN suggests that Fos–AP-1 activities may have different effects on various neurotrophins. The deletion of a DNA binding motif (such as -tgagtca-) in the 5′ regulatory region of a gene can be used to suppress activation by its regulator (such as AP-1) but does not guarantee a specific effect. In our study, the knockdown of Fos expression by c-fos antisense s-ODN after FCIR and the subsequent suppression of AP-1 activities (Liu et al., 1994), and NGF mRNA expression in the right dentate gyrus correlated positively. The specific suppression of NGF mRNA presented in this study as a direct result of the inhibition of Fos induction may indicate an association between Fos–AP-1 activities and NGF mRNA expression in the dentate gyrus after FCIR. Alternatively, the suppression effect may be the result of other effects caused by s-ODNs not mediated by c-fos. This indirect effect could be rather specific on NGF mRNA expression, however, in view of the fact that antisense c-fos s-ODN has no effect on the suppression of actin and NT-3 mRNA and that the sense and mismatch s-ODN have no effect on Fos peptide and NGF mRNA activation. Regardless, that c-fosantisense s-ODN suppresses NGF mRNA, directly or indirectly, suggests that this antisense technique could be applied to in vivoanti-NGF model in which a suppression of NGF is desirable. Moreover, the possible involvement of AP-1 in the signal cascade of apoptosis induced by tumor necrosis factor-α may permit c-fosantisense s-ODN a therapeutic potential to suppress AP-1 and delay or alter cell death (Roulston et al., 1998). Specific knockdown of peptide translation using the antisense strategy offers an alternative method for analyses of gene activation and regulation and for drug target discovery and gene function analysis in whole animals in which gene knockout technology is restricted.
The knockdown effect of c-fos antisense s-ODN on Fos–AP-1 activities and NGF mRNA demonstrated in our study correlated with the time-dependent uptake of transfected c-fos antisense s-ODN in the rat brain. The regional distribution of the s-ODN appears to follow a diffusion pattern from the periventricular wall toward the brain parenchyma. The s-ODNs appeared to be primarily confined to the ependymal layer of lateral ventricle 4 hr after delivery. The uptake of s-ODN in the parenchyma does not preclude the possibility that the actively dividing stem cells in the ependymal cell layer in the ventricle take up the s-ODNs and migrate to cell layers away from the ventricle (Rosario et al., 1997; Snyder et al., 1997). On the other hand, the direct detection of bio-s-ODN using the immunohistochemical method showed a time-dependent diffusion of biotin signal from the CSF space into the brain parenchyma for up to 96 hr after delivery. Our findings using this method are in agreement with those reported byWhitesell et al. (1993). The fluorescent signal detected by this method, however, does not distinguish between the degraded products and those attached to the s-ODN (Fig. 1). Therefore, we used the dot blot method to assess the content of intact bio-s-ODN after Sephadex G-25 column filtration. We observed that the cerebral bio-s-ODN uptake appeared to peak at 1 d and remained elevated up to 2 d after infusion. The decline in bio-s-ODN content in the brain beyond 2 d, especially in the hippocampus and cerebellum, suggests a probable degradation of the s-ODNs. The uptake of s-ODN was further confirmed byin situ 3′-OH-terminus labeling.
Our antisense s-ODN was designed to block protein translation by hybridizing to mRNA. We demonstrated previously that c-fosantisense s-ODN is hybridized to cellular RNA between 6 and 41 hr after delivery (Liu et al., 1994). The perinuclear localization of c-fos antisense s-ODN supports these observations and suggests the possible localization of c-fos mRNA. The perinuclear localization we observed is consistent with the observations of Agrawal et al. (1992) in their cell culture experiments but is not consistent with the results observed by Whitesell et al. (1993) in an animal study. In the latter study, the investigators delivered a large quantity of s-ODNs to the CSF space by continuous infusion for a minimum of 7 d using a mini-osmotic pump. Then, a marked uptake of the s-ODNs by the astrocytes, with the majority located in the nuclei, was noted (Whitesell et al., 1993). Differences between our study and that by Whitesell et al. (1993) include the method of infusion and the total amount of s-ODNs infused. Endocytosis may mediate the uptake of ODNs in cell culture conditions (Yakubov et al., 1989). An in vivo pharmacokinetics study using3H-ODN (12-mer) revealed that 3H-ODN given via tail vein injection showed no cerebral uptake in rats (Mirabelli and Crooke, 1993), suggesting that ODNs do not cross the blood–brain barrier when given intravenously. In our study, the s-ODN appeared in the corpus callosum and hippocampus before they appeared in the cerebral cortex. The s-ODN signal was detectable in the hippocampus beginning 4 hr after delivery. These findings suggest that cells closer to the ventricular site of infusion accumulate the s-ODN earlier and in greater quantities than do neurons more distal to the ventricle.
In this report, we have summarized our findings from interrelated experiments that explore the mechanisms of s-ODN uptake in the brain, with the focus on using antisense s-ODNs to regulate gene expression after FCIR. These include the following: (1) brain cells are able to take up s-ODNs; (2) it is possible to trace the distribution of specific s-ODNs by various labeling techniques; (3) antisense s-ODNs can affect target genes within 24 hr of delivery; and (4) the limited presence (3 d) of s-ODNs allows a short-term control of their effects in the CNS. The use of targeted s-ODN could facilitate a new avenue to study gene regulation in the CNS in vivo and to promote manipulation of genes under pathophysiological conditions in which permanent gene displacement may not be desirable. Although additional research is needed, the NGF-suppression model described here might be used to study the in vivo association of hippocampal NGF expression and neuronal regeneration (Hefti et al., 1989; Phillips et al., 1991; D’Mello et al., 1992). In conclusion, we show several lines of evidence that suggest that post-FCIR Fos knockdown could be used to suppress the subsequent NGF mRNA expression.
Footnotes
This work was completed during the term of Established Investigator Award 96N40202N to P.K.L. from the American Heart Association. This work was supported by National Institutes of Health Grants NS34810 to P.K.L. and NS25545, NS28995, and NS32636 to C.Y.H., and by Office of Naval Research Grant C4114503 to C.Y.H. We thank Y. Y. He, J. J. Xue, R. Speck, J. Wolff, A. Chen, and B. Chen for excellent technical assistance, and Dr. W. J. Hamilton (Baylor College of Medicine) for assistance in editing.
Correspondence should be addressed to Dr. Philip K. Liu, Department of Neurosurgery, Baylor College of Medicine, 6560 Fannin Street, Suite 944, Houston, TX 77030.
REFERENCES
- 1.Agrawal S, Sarin PS, Zamecnik M, Zamecnik PC. Cellular uptake and anti-HIV activity of oligonucleotides and their analogs. In: Erickson RP, des Izant JG, editors. Gene regulation: biology of antisense RNA and DNA. Raven; New York: 1992. pp. 273–228. [Google Scholar]
- 2.Akins PT, Liu PK, Hsu CY. Immediate early gene expression in response to cerebral ischemia: friend or foe? Stroke. 1996;27:1682–1687. doi: 10.1161/01.str.27.9.1682. [DOI] [PubMed] [Google Scholar]
- 3.An G, Lin T-N, Liu J-S, Xue J-J, He Y-Y, Hsu CY. Expression of c-fos and c-jun family genes after focal cerebral ischemia. Ann Neurol. 1993;33:457–464. doi: 10.1002/ana.410330508. [DOI] [PubMed] [Google Scholar]
- 4.Chiasson BJ, Hooper ML, Murphy PR, Robertson HA. Antisense oligonucleotide eliminates in vivo expression of c-fos in mammalian brain. Eur J Pharmacol. 1992;227:451–453. doi: 10.1016/0922-4106(92)90167-t. [DOI] [PubMed] [Google Scholar]
- 5.Chiasson BJ, Armstrong JN, Hooper ML, Murphy PR, Robertson HA. The application of antisense oligonucleotide technology to the brain: some pitfalls. Cell Mol Neurobiol. 1994;14:507–521. doi: 10.1007/BF02088834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cirelli C, Pompeiano M, Tononi G. Sleep-waking changes after c-fos antisense injections in the medial preoptic area. NeuroReport. 1995a;6:801–805. doi: 10.1097/00001756-199503270-00024. [DOI] [PubMed] [Google Scholar]
- 7.Cirelli C, Pompeiano M, Tononi G. In vivo antisense approaches to the role of immediate early gene expression in the brain. Regul Pept. 1995b;59:1551–1621. doi: 10.1016/0167-0115(95)00097-u. [DOI] [PubMed] [Google Scholar]
- 8.Colotta F, Polentarutti N, Sironi M, Mantovani A. Expression and involvement of c-fos and c-jun protooncogenes in programmed cell death induced by growth factor deprivation in lymphoid cell lines. J Biol Chem. 1992;267:18278–18283. [PubMed] [Google Scholar]
- 9.Curran T, Miller AD, Zokas L, Verma IM. Spiral and cellular Fos protein: a comparative analysis. Cell. 1984;36:259–268. doi: 10.1016/0092-8674(84)90219-8. [DOI] [PubMed] [Google Scholar]
- 10.D’Mello SR, Jiang C, Lamberti C, Martin SC, Heinrich G. Differential regulation of the nerve growth factor and brain-derived neurotrophic factor genes in L929 mouse fibroblasts. J Neurosci Res. 1992;33:519–526. doi: 10.1002/jnr.490330404. [DOI] [PubMed] [Google Scholar]
- 11.Dragunow M, Tse C, Glass M, Lawlor P. c-fos antisense reduces expression of Krox 24 in rat caudate and neocortex. Cell Mol Neurobiol. 1994;14:395–405. doi: 10.1007/BF02088826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards SA, Rundell YK, Adamson ED. Expression of c-fos antisense RNA inhibits the differentiation of F9 cells to parietal endoderm. Dev Biol. 1988;129:91–102. doi: 10.1016/0012-1606(88)90164-9. [DOI] [PubMed] [Google Scholar]
- 13.Field SJ, Johnson RS, Mortensen RM, Papaioannou VE, Spiegelman BM, Greenberg ME. Growth and differentiation of embryonic stem cells that lack an intact c-fos gene. Proc Natl Acad Sci USA. 1992;89:9306–9310. doi: 10.1073/pnas.89.19.9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Funato T, Yoshida E, Jiao L, Tone T, Kashani-Sabet M, Scanlon KJ. The utility of an anti-fos ribozyme in reversing cisplatin resistance in human carcinomas. Adv Enzyme Regul. 1992;32:195–209. doi: 10.1016/0065-2571(92)90017-t. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 16.Gillardon F, Shutella T, Uhlmann E, Holsboer F, Zimmerman M, Behl C. Activation of c-Fos contributes to amyloid b-peptide-induced neurotoxicity. Brain Res. 1996;706:169–172. doi: 10.1016/0006-8993(95)01332-6. [DOI] [PubMed] [Google Scholar]
- 17.Gu ZZ, Pan C, Cui JK, Klebec M, Shenaq S, Liu PK. Gene expression and apoptosis in the spinal cord neurons after peripheral nerve damage. Neurochem Int. 1997;30:417–426. doi: 10.1016/s0197-0186(96)00077-0. [DOI] [PubMed] [Google Scholar]
- 18.Halazonetis TD, Georgopoulos K, Greenberg ME, Leder P. c-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell. 1988;55:917–924. doi: 10.1016/0092-8674(88)90147-x. [DOI] [PubMed] [Google Scholar]
- 19.Hefti F, Hartikka J, Knusel B. Function of neurotrophic factors in the adult and aging brain and their possible use in the treatment of neurodegenerative diseases. Neurobiol Aging. 1989;10:515–533. doi: 10.1016/0197-4580(89)90118-8. [DOI] [PubMed] [Google Scholar]
- 20.Heilig M, Engel JA, Soderpalm B. c-fos antisense in the nucleus accumbens blocks the locomotor stimulant action of cocaine. Eur J Pharmacol. 1993;236:339–340. doi: 10.1016/0014-2999(93)90610-t. [DOI] [PubMed] [Google Scholar]
- 21.Hengerer B, Lindholm D, Heumann R, Ruther U, Wagner EF, Thoenen H. Lesion-induced increase in nerve growth factor mRNA is mediated by c-fos. Proc Natl Acad Sci USA. 1990;87:3899–3903. doi: 10.1073/pnas.87.10.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holt JT, Gopal TV, Moulton AD, Nienhuis AW. Inducible production of c-fos antisense RNA inhibits 3T3 cell proliferation. Proc Natl Acad Sci USA. 1986;83:4794–4798. doi: 10.1073/pnas.83.13.4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hooper ML, Chiasson BJ, Robertson HA. Infusion into the brain of an antisense oligonucleotide to the immediate-early gene c-fos suppresses production of fos and produces a behavioral effect. Neuroscience. 1994;63:917–924. doi: 10.1016/0306-4522(94)90559-2. [DOI] [PubMed] [Google Scholar]
- 24.Kerr LD, Holt JT, Matrisian LM. Growth factors regulate transin gene expression by c-fos-dependent and c-fos-independent pathways. Science. 1988;242:1424–1427. doi: 10.1126/science.2462278. [DOI] [PubMed] [Google Scholar]
- 25.Kornberg A. DNA replication, pp 225–229. Freeman; San Francisco: 1980. Terminal deoxynucleotidyl transferase. [Google Scholar]
- 26.Kruijer W, Schubert D, Verma IM. Induction of the proto-oncogene fos by nerve growth factor. Proc Natl Acad Sci USA. 1985;82:7330–7334. doi: 10.1073/pnas.82.21.7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levi BZ, Ozato K. Constitutive expression of c-fos antisense RNA blocks c-fos gene induction by interferon and by phorbol ester and reduces c-myc expression in F9 embryonal carcinoma cells. Genes Dev. 1988;2:554–566. doi: 10.1101/gad.2.5.554. [DOI] [PubMed] [Google Scholar]
- 28.Levi BZ, Kasik JW, Ozato K. c-fos antisense RNA blocks expression of c-fos gene in F9 embryonal carcinoma cells. Cell Differ Dev. 1988;25S:95–101. doi: 10.1016/0922-3371(88)90105-0. [DOI] [PubMed] [Google Scholar]
- 29.Lindvall O, Ernfors P, Bengzon J, Kokaia Z, Smith ML, Siesjo BK, Persson H. Differential regulation of mRNAs for nerve growth factor, brain-derived neurotrophic factor, and neurotrophin 3 in the adult rat brain following cerebral ischemia and hypoglycemic coma. Proc Natl Acad Sci USA. 1992;89:648–652. doi: 10.1073/pnas.89.2.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu PK. Enhanced expression in the α-type DNA polymerase genes reduces AZT-cytotoxicity in hamster tr5 cells. Somatic Cell Mol Genet. 1993;19:211–220. doi: 10.1007/BF01233069. [DOI] [PubMed] [Google Scholar]
- 31.Liu PK, Salminen A, He YY, Jiang MH, Xue JJ, Liu JS, Hsu CY. Suppression of ischemia-induced Fos expression and AP-1 activity by an antisense oligodeoxynucleotide to c-fos mRNA. Ann Neurol. 1994;36:566–576. doi: 10.1002/ana.410360405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macgregor PF, Abate C, Curran T. Direct cloning of leucine zipper proteins: Jun binds cooperatively to the CRE with CRE-BP1. Oncogene. 1990;5:451–458. [PubMed] [Google Scholar]
- 33.Maisonpierre PC, Le Beau MM, Espinosa R, III, Ip NY, Belluscio L, de la Monte SM, Squinto S, Furth ME, Yancopoulos GD. Human and rat brain-derived neurotrophic factor and neurotrophin-3: gene structures, distributions, and chromosomal localizations. Genomics. 1991;10:558–568. doi: 10.1016/0888-7543(91)90436-i. [DOI] [PubMed] [Google Scholar]
- 34.Mercola D, Rundell A, Westwick J, Edwards SA. Antisense RNA to the c-fos gene: restoration of density-dependent growth arrest in a transformed cell line. Biochem Biophys Res Commun. 1987;147:288–294. doi: 10.1016/s0006-291x(87)80119-5. [DOI] [PubMed] [Google Scholar]
- 35.Mirabelli CK, Crooke ST. Antisense oligonucleotides in the context of modern molecular drug discovery and development. In: Crooke ET, Lebleu B, editors. Antisense research and applications. CRC; Boca Raton, FL: 1993. pp. 7–35. [Google Scholar]
- 36.Nishikura K, Murray HM. Antisense RNA of protooncogene c-fos blocks renewed growth of quiescent 3T3 cells. Mol Cell Biol. 1987;7:637–649. doi: 10.1128/mcb.7.2.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phillips HS, Hains JM, Laramee GR, Rosenthal A, Winslow JW. Wide spread expression of BDNF but not NT3 by target areas of basal forebrain cholinergic neurons. Science. 1990;250:290–294. doi: 10.1126/science.1688328. [DOI] [PubMed] [Google Scholar]
- 38.Phillips HS, Haines JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron. 1991;7:695–702. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- 39.Quercia S, Chang SL. Antisense oligodeoxynucleotide attenuates in vivo expression of c-fos in the paraventricular hypothalamic nucleus of the rat brain. Neurosci Lett. 1996;209:89–92. doi: 10.1016/0304-3940(96)12627-6. [DOI] [PubMed] [Google Scholar]
- 40.Rosario CM, Yandava BD, Kosaras B, Zurakowski D, Sidman RL, Snyder EY. Differentiation of engrafted multipotent neural progenitors towards replacement of missing granule neurons in meander tail cerebellum may help determine the locus of mutant gene action. Development. 1997;124:4213–4224. doi: 10.1242/dev.124.21.4213. [DOI] [PubMed] [Google Scholar]
- 41.Roulston A, Reinhard C, Amiri P, Williams LT. Early activation of c-Jun N-terminal kinase and p38 kinase regulate cell survival in response to tumor necrosis factor alpha. J Biol Chem. 1998;273:10232–10239. doi: 10.1074/jbc.273.17.10232. [DOI] [PubMed] [Google Scholar]
- 42.Sagar SM. Expression of c-fos protein in brain: metabolic mapping at the cellular level. Ann Neurol. 1993;33:437–438. doi: 10.1126/science.3131879. [DOI] [PubMed] [Google Scholar]
- 43.Sagar SM, Sharp FR, Curran T. Expression of c-fos protein in brain: metabolic mapping at the cellular level. Science. 1988;240:1328–1331. doi: 10.1126/science.3131879. [DOI] [PubMed] [Google Scholar]
- 44.Salminen A, Liu PK, Hsu CY. Alteration of transcription factor binding activities in the ischemic rat brain. Biochem Biophys Res Commun. 1995;212:939–944. doi: 10.1006/bbrc.1995.2060. [DOI] [PubMed] [Google Scholar]
- 45.Scanlon KJ, Jiao L, Funato T, Wang W, Tine T, Rossi JJ, Kashani-Sabet M. Ribozyme-mediated cleavage of c-fos mRNA reduces gene expression of DNA synthesis enzymes and metallothionein. Proc Natl Acad Sci USA. 1991;88:10591–10595. doi: 10.1073/pnas.88.23.10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schlingensiepen KH, Wollnik F, Kunst M, Schlingensiepen R, Herdegen T, Brysch W. The role of Jun transcription factor expression and phosphorylation in neuronal differentiation, neuronal cell death, and plastic adaptations in vivo. Cell Mol Neurobiol. 1994;14:487–505. doi: 10.1007/BF02088833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schönthal A, Herrlic P, Rahmsdorf HJ, Ponta H. Requirement for fos gene expression in the transcriptional activation of collagenase by other oncogene and phorbol esters. Cell. 1988;54:325–334. doi: 10.1016/0092-8674(88)90195-x. [DOI] [PubMed] [Google Scholar]
- 48.Sharp FR. The sense of antisense fos oligonucleotides. Ann Neurol. 1994;36:566–576. doi: 10.1002/ana.410360403. [DOI] [PubMed] [Google Scholar]
- 49.Sharp JW, Sagar SM, Hisanaga K, Jasper P, Sharp FR. The NMDA receptor mediates cortical induction of fos and fos-related antigens following cortical injury. Exp Neurol. 1991;109:323–332. doi: 10.1016/s0014-4886(05)80023-8. [DOI] [PubMed] [Google Scholar]
- 50.Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- 51.Simonson MS. Anti-AP-1 activity of all-trans retinoic acid in glomerular mesangial cells. Am J Physiol. 1994;267:F805–815. doi: 10.1152/ajprenal.1994.267.5.F805. [DOI] [PubMed] [Google Scholar]
- 52.Snider WD. Functions of the neurotrophins during nervous system development: what the knockdowns are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 53.Snyder EY, Park KI, Flax JD, Liu S, Rosario CM, Yandava BD, Aurora S. Potential of neural “stem-like” cells for gene therapy and repair of the degenerating central nervous system. Adv Neurol. 1997;72:121–132. [PubMed] [Google Scholar]
- 54.Wetmore C, Ernfors P, Persson H, Olson L. Localization of brain-derived neurotrophic factor mRNA to neurons in the brain by in situ hybridization. Exp Neurol. 1990;109:141–152. doi: 10.1016/0014-4886(90)90068-4. [DOI] [PubMed] [Google Scholar]
- 55.Whitesell L, Geselowitz D, Chavany C, Fahmy B, Walbridge S, Alger JR, Neckers LM. Stability, clearance, and disposition of intraventricularly administered oligodeoxynucleotides: implications for therapeutic application within the central nervous system. Proc Natl Acad Sci USA. 1993;90:4665–4669. doi: 10.1073/pnas.90.10.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yakubov LA, Deeva EA, Zarytova VF, Ivanova EM, Ryte AS, Yurchenko LV, Vlassovm VV. Mechanism of oligonucleotide uptake by cells: involvement of specific receptors? Proc Natl Acad Sci USA. 1989;86:6454–6458. doi: 10.1073/pnas.86.17.6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu K, Lu D, Rowland NE, Raizada MK. Angitensin II regulation of tyrosine hydroxylase gene expression in the neuronal cultures of normotensive and spontaneously hypertensive rats. Endocrinology. 1996;137:3566–3576. doi: 10.1210/endo.137.8.8754788. [DOI] [PubMed] [Google Scholar]
- 58.Zheng M, Heinrich G. Structural and functional analysis of the promoter region of the nerve growth factor gene. Brain Res. 1988;427:133–140. doi: 10.1016/0169-328x(88)90058-7. [DOI] [PubMed] [Google Scholar]