

Abstract
Chronic stimulation of norepinephrine (NE) neuromodulation by angiotensin II (Ang II) involves activation of the Ras—Raf—MAP kinase signal transduction pathway in Wistar Kyoto (WKY) rat brain neurons. This pathway is only partially responsible for this heightened action of Ang II in the spontaneously hypertensive rat (SHR) brain neurons. In this study, we demonstrate that the MAP kinase-independent signaling pathway in the SHR neuron involves activation of PI3-kinase and protein kinase B (PKB/Akt). Ang II stimulated PI3-kinase activity in both WKY and SHR brain neurons and was accompanied by its translocation from the cytoplasmic to the nuclear compartment. Although the magnitude of stimulation by Ang II was comparable, the stimulation was more persistent in the SHR neuron compared with the WKY rat neuron. Inhibition of PI3-kinase had no significant effect in the WKY rat neuron. However, it caused a 40–50% attenuation of the Ang II-induced increase in norepinephrine transporter (NET) and tyrosine hydroxylase (TH) mRNAs and [3H]-NE uptake in the SHR neuron. In contrast, inhibition of MAP kinase completely attenuated Ang II stimulation of NET and TH mRNA levels in the WKY rat neuron, whereas it caused only a 45% decrease in the SHR neuron. However, an additive attenuation was observed when both kinases of the SHR neurons were inhibited. Ang II also stimulated PKB/Akt activity in both WKY and SHR neurons. This stimulation was 30% higher and lasted longer in the SHR neuron compared with the WKY rat neuron. In conclusion, these observations demonstrate an exclusive involvement of PI3-kinase–PKB-dependent signaling pathway in a heightened NE neuromodulatory action of Ang II in the SHR neuron. Thus, this study offers an excellent potential for the development of new therapies for the treatment of centrally mediated hypertension.
Keywords: PI3-kinase, protein kinase B, angiotensin, neuromodulation, spontaneously hypertensive rat, neuron
The key in the central actions of angiotensin II (Ang II) in the control of blood pressure (BP) is the interaction of Ang II with the neuronal AT1 receptor subtype in the cardioregulatory-relevant brain areas, such as hypothalamic and brainstem nuclei (Saavedra 1992; Steckelings et al., 1992; Timmermans et al., 1993; Raizada et al., 1994, 1998; Wright and Harding, 1994). Physiological mechanisms associated with this action are well understood and involve activation of sympathetic pathways, increased vasopressin secretion, and dampening of baroreceptor reflexes (Phillips, 1987; Saavedra, 1992; Steckelings et al., 1992; Sumners and Raizada, 1993; Timmermans et al., 1993; Raizada et al., 1994, 1998; Wright and Harding, 1994; Sumners et al., 1995). Despite this, the cellular and molecular bases of these physiological actions of brain Ang II in BP control are not clearly established.
Recent attempts to link Ang II activation of sympathetic pathways with the stimulation of norepinephrine transporter (NET) activity and transcriptional control of the synthesis and release of norepinephrine (NE) have provided a major step forward toward our understanding of the brain Ang II at a molecular level (Sumners and Raizada, 1993; Sumners et al., 1995; Lu et al., 1996a; Yu et al., 1996a; Raizada et al., 1998). These studies have established that enhanced or chronic stimulation of NE neuromodulatory activity is a result of transcriptional regulation of genes for NET, tyrosine hydroxylase (TH), and dopamine β-hydroxylase (DβH) in normotensive Wistar Kyoto (WKY) rat neuron (Lu et al., 1996a; Yu et al., 1996a; Gelband et al., 1998). This involves activation of the Ras–Raf–MAP kinase signaling pathway (Lu et al., 1996b; Yang et al., 1996a; Gelband et al., 1998). Activation of this cascade of kinases leads to an increased serum response element (SRE) and activator protein-1 (AP-1) binding activities that are associated with the stimulation of genes relevant to the synthesis and release of catecholamines (Lu et al., 1996b;Gelband et al., 1998).
Recent evidence implicates the brain Ang system in the development and establishment of hypertension (Raizada et al., 1994, 1998; Sumners et al., 1995). Thus, the cellular basis of the physiological actions of brain Ang II could be important in the design of novel therapies for treatment of the hypertensive state. This view is strengthened by the studies with a genetic model for primary hypertension in humans, spontaneously hypertensive rat (SHR), which have shown that the brain Ang system is hyperactive as a result of an increased expression of AT1 receptors (Phillips, 1987; Raizada et al., 1994, 1998;Sumners et al., 1995). This is associated with an increased turnover and synthesis of CA by Ang II in the SHR brain (Ding et al., 1990; Yang et al., 1991). A similar hyperactivity of the brain Ang system has been reported in a renin transgenic model of hypertension (Langheinrich et al., 1996). Finally, interruption in the expression of brain Ang hyperactivity by either pharmacological or genetic means normalizes BP in the SHR and not in normotensive, WKY rats (Berecek et al., 1983;Toney and Porter et al., 1993; Phillips et al., 1994). These studies have lead to the hypothesis that a hyperactive brain Ang system is a result of a heightened stimulation of signaling kinases by Ang II in the SHR neuron.
Our group has established an in vitro neuronal cell culture model from the hypothalamic–brainstem areas of WKY and SHR brains that provides evidence in support of this hypothesis (Sumners and Raizada, 1993; Raizada et al., 1994, 1998; Sumners et al., 1995). These studies have demonstrated that the in vivo expression of a hyperactive brain Ang system is maintained in the neuronal cultures of the SHR. This includes increased expression of AT1receptors and increased Ang II stimulation of NE neuromodulation in the SHR neuron compared with the WKY rat neuron (Raizada et al., 1993; Lu et al., 1996a; Gelband et al., 1998). These alterations are specific for the AT1 receptor system, because the AT2receptor and its signaling pathway are not altered (Sumners et al., 1995; Raizada et al., 1998). Thus, the in vitro neuronal cell culture system provides an excellent model for comparing the AT1 receptor-mediated signal transduction mechanism and determining the basis for heightened neuromodulatory actions of Ang II in the SHR neuron.
In previous studies we focused our attention on the Ras—Raf—MAP kinase pathway and postulated that an increased NE neuromodulation would be attributable to increased Ang II stimulation of this signaling system in the SHR neuron. However, our data demonstrated that although completely responsible for Ang II action in the WKY rat neuron, the Ras—Raf—MAP kinase pathway is only a partial player in the SHR neuron (Yang and Raizada, 1998). These observations led us to propose the presence of a MAP kinase-independent signaling pathway that must account for an increased NE neuromodulation effect of Ang II in the SHR neuron (Yang and Raizada, 1998). We hypothesized that the PI3-kinase signaling system could be such a pathway in the SHR. The rationale for this conclusion included the following: calcineurin/nuclear factor of activated T cells or protein kinase A and protein tyrosine kinase did not appear to participate in this signaling (Yang and Raizada, 1998); some G-protein-coupled receptors are capable of coupling to the PI3-kinase signaling system (Lopez-Ilasaca et al., 1997; Hazeki et al., 1998; Leopoldt et al., 1998); and Ang II has recently been shown to stimulate PI3-kinase activity in vascular smooth muscle cells (Saward and Zahradka, 1997). These cells are one of the major targets of peripheral actions of Ang II (Griendling et al., 1993). Because neurons are the primary target for the brain actions of Ang II, it was of obvious significance to explore this hypothesis.
Observations presented in this study demonstrate that although Ang II stimulates PI3-kinase in both WKY and SHR neurons, its effect on the SHR neuron is more persistent and longer lasting. The activation accounts for MAP kinase-independent signaling in Ang II stimulation of NE neuromodulation in the SHR. Our data also establish that this effect involves activation of PKB/Akt, whose stimulation by Ang II is more prolonged and significantly higher in the SHR neuron.
MATERIALS AND METHODS
Materials
WKY rats and SHRs (1 d old) were obtained from our breeding colony, which originated from Harlan Sprague Dawley (Indianapolis, IN). Blood pressure for breeder WKY rats was an average of 117 ± 5 mmHg, whereas it was 205 ± 8 mmHg for the SHRs. DMEM, plasma-derived horse serum (PDHS), and 1× crystallized trypsin were from Central Biomedia (Irwin, MO). [γ-32P]-ATP (3000 Ci/mmol), [γ-32P]-dCTP (3000 Ci/mmol), [3H]-NE, and chemiluminescence assay reagents were from DuPont/NEN (Boston, MA). Nitrocellulose membranes were from Micron Separations (Westboro, MA). Ang II, Wortmannin, and polyethyleneimine-cellulose thin-layer plates were purchased from Sigma (St. Louis, MO). Losartan potassium was a gift from DuPont/Merck (Wilmington, DE). PD123319 was from RBI (Natick, MA). Superscript Rnase H− reverse transcriptase and deoxynucleotide mixture were from Life Technologies (Grant Island, NY). Dynal beads and other reagents for poly(A+) RNA isolation were from Dynal (Lake Success, NY). Oligo-dT and Taq DNA polymerase were purchased from Promega (Madison, WI). Anti-PI3-kinase p85α and PKB/Akt were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All other reagents were purchased from Fisher Scientific (Pittsburgh, PA) and were the highest quality available.
Primers for TH, NET, and β-actin were synthesized in the DNA synthesis facility of the Interdisciplinary Center for Biotechnology for Research, University of Florida (Gainesville, FL). The sequences of these primers have been published previously (Lu et al., 1996a; Yu et al., 1996a).
Methods
Preparation of hypothalamus–brainstem neuronal cultures from WKY and SHR brains. Neuronal cultures were prepared essentially as described previously (Raizada et al., 1984, 1993, 1994). Briefly, hypothalamus–brainstem of 1-d-old WKY and SHR brains, which contained the paraventricular nucleus, the supraoptic, anterior, lateral, posterior dorsomedial and ventromedial nuclei, the medulla oblongata, and the pons, were dissected, and brain cells were dissociated by trypsin. Cells were plated on poly-l-lysine-precoated tissue-culture dishes in DMEM containing 10% PDHS. Culture dishes of 35 mm diameter (3 × 106 cells) or 100 mm diameter (1.5 × 107 cells) were prepared for experiments. Cultures were treated with 1% cytosine arabinoside for 3 d followed by establishment of cultures for 10 d before their use in the experiments. These cultures have been shown to contain 90–95% neuronal cells and 5–10% astroglial cells (Raizada et al., 1984,1993, 1994). Properties of neuronal cultures from the WKY brain are comparable with the SHR brain in regard to neuronal cells per dish, total protein per dish, catecholamines present, and many other biochemical criteria. Thus, they have been used extensively by ourin vitro model to study hyperactivity of the brain AT1 receptor function and its interaction with catecholamines in the SHR brain (Raizada et al., 1984, 1993, 1994).
PI3-kinase assay. PI3-kinase activity in extracts from neuronal cultures and in hypothalamic brain areas was measured essentially as described previously (Kaplan et al., 1986, 1987). Briefly, neuronal cell lysates were prepared in lysis buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 50 mm NaF, 1% NP-40, 1 mm EDTA, 1 mmNa3VO4, 1 mm PMSF, 2 μg/ml aprotinin, 2 μg/ml leupeptin). After centrifugation at 12,000 ×g for 10 min at 4°C, lysates were used for immunoprecipitation with anti-PI3-kinase p85α antibody essentially as described previously (Kaplan et al., 1986, 1987). Immunoprecipitates were washed twice in lysis buffer and twice in 0.5 m LiCl, 100 mm HEPES, pH 7.5, and once with 10 mmTris-HCl, pH 7.5, 100 mm NaCl, 1 mm EDTA. They were then incubated in PI3-kinase reaction buffer [20 mmHEPES, pH 7.4, 30 mm MgCl2, 50 μm ATP, 200 μm adenosine, 200 μg of PI, 10 μCi of [γ-32P] ATP (3000 Ci/mmol)] for 20 min at room temperature. Reactions were stopped by the addition of 100 μl of 1.0N HCl and 200 μl of a 1:1 mixture of chloroform/methanol, and lipids in the organic phase were resolved on TLC plates developed in chloroform/methanol/H2O/25% NH4OH (43:38:7:5). Radioactive phosphatidylinositol 3-monophosphate (PIP) products were visualized by autoradiography and quantitated (Lu et al., 1996b; Yang et al., 1996a).
Measurement of protein kinase B (PKB/Akt) activity. PKB activity was measured by a previously published method (Franke et al., 1995; Stokoe et al., 1997). In brief, the protocol was as follows. Neuronal cultures were extracted in lysis buffer containing 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10% glycerol, 1% NP-40, 10 mm NaF, 1 mmNa3VO4, 1 mmNa4O7P2, 2 μmleupeptin, 2 μm aprotinin, and 1 mm PMSF. Lysates were centrifuged for 10 min at 12,000 × g. The PKB protein was immunoprecipitated from cell-free extracts (200 μg protein) with the use of PKB-specific polyclonal antibody. Immune complexes were collected with protein A/G plus-agarose and washed three times with lysis buffer, and once with kinase assay buffer (20 mm HEPES, pH 7.4, 1 mm DTT, 10 mmMnCl2, 10 mm MgCl2).In vitro kinase assays were performed at 30°C in a 50 μl reaction volume of kinase assay buffer containing 5 μmATP, 100 μg/ml histone 2B (H2B), and 10 μCi of [γ-32P] ATP per sample. The reactions were stopped with 5× SDS-PAGE sample buffer, and reaction products were separated by SDS-PAGE gel and dried for autoradiography. Phosphorylated H2B was quantitated by ultraviolet products Imagestore 5000 system (Lu et al., 1996b; Yang et al., 1996a).
Western blotting for PI3-kinase and PKB/Akt. Neuronal cell-free lysates were prepared as described above for the enzyme assay. Lysates were electrophoresed on 10% SDS/PAGE, and proteins were transferred to nitrocellulose membranes. Membranes were blocked by using 5% nonfat dry milk in TBST (20 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 0.05% Tween 20) for 1 hr followed by incubation for 1 hr at room temperature with anti-PI3-kinase p85α antibody or anti-PKB antibody as described previously (Yang et al., 1996a). Protein-bound primary antibody was detected by using horseradish peroxidase-labeled secondary antibody and enhanced by chemiluminescence assay reagent (Lu et al., 1996b; Yang et al., 1996a). The bands recognized by the primary antibody were visualized by autoradiography and quantitated essentially as described previously (Lu et al., 1996b; Yang et al., 1996a).
Preparation and cytoplasmic and nuclear extracts from neuronal cultures. Neuronal cultures from WKY and SHR that had been established on 100-mm-diameter culture dishes were used to prepare cytoplasmic and nuclear fractions to determine Ang II-induced redistribution of PI3-kinase and PKB immunoreactivity. The protocol was established previously (Lu et al., 1996b; Yang et al., 1997).
Measurement of AP-1 binding activity by gel shift assay. The protocol was essentially the same as described previously (Lu et al., 1996b). In brief, 3 pmol of each oligonucleotide was first labeled with [γ-32P] ATP at the 5′ end by 10 min incubation at 37°C with 4 μ of T4-polynucleotide kinase in 10 μl solution containing 70 mm Tris-HCl, pH 7.6, 10 mm MgCl2, 5 mm DTT, and 10 μCi of [γ-32P] ATP. The reaction was stopped by the addition of 1 μl of 0.5 m EDTA, and the volume was expanded to 100 μl with 1× gel-shift assay buffer. [32P]-labeled oligonucleotides (0.1 μCi) were mixed with 5 μg of nuclear extract protein containing 1× gel-shift assay buffer, 100 mg of salmon sperm DNA, 2 μg of DNA duplex poly (dl-dc), and 10 μg of BSA in a final volume of 10 μl. The mixture was incubated at room temperature for 15 min and electrophoresed on a 5% PAGE gel in 20 mm Tris-acetate buffer containing 5 nm EDTA, pH 8.0, for 90 min at a constant 200 V. The gel was decasted, wrapped in a plastic bag, and exposed and analyzed by an Imagestore system (Ultra Violet Products, San Gabriel, CA). The densities of shifted bands on the gel representing the oligonucleotide–protein complexes were quantitated by the SW 5000 Gel Analysis program (Lu et al., 1996b; Yang et al., 1996a).
Measurements of norepinephrine neuromodulation activity.Uptake of [3H]-NE and levels of NET and TH mRNAs were used as parameters of the effects of Ang II on NE neuromodulation. The effect of Ang II on specific uptake of [3H]-NE by neuronal cells was determined as described previously (Lu et al., 1996a). Levels of NET, TH, and β-actin mRNAs were measured and quantitated essentially as described elsewhere (Lu et al., 1996b, 1998;Yang et al., 1996a).
Measurement of the effects of Ang II on PI3-kinase in vivo. Adult male WKY rats and SHRs were housed singly after being fitted with an indwelling cannula (10 mm long, 23 gauge stainless steel) stereotaxically aimed to end in or just above the lumen of the right lateral ventricle and firmly fixed to the skull with jeweler’s screws and dental acrylic (Yang et al., 1996b). Rats were allowed to recover for 1 week before the experiments. Injections of Ang II were made through an 11 mm gauge injector needle attached to a 35 mm syringe. Five microliters of either PBS, pH 7.4, or PBS containing 10 ng of Ang II were injected into each rat. Hypothalami were dissected, and tissues were processed for the measurement of PI3-kinase activity as described above.
Experimental groups and data analysis. Each experiment for the effect of Ang II on the activation of PI3-kinase was conducted in triplicate culture dishes, and cells in each dish were derived from multiple brains of 1-d-old rats. WKY and SHR brain neurons were used in parallel for each experiment, and both samples were run on the same gel to minimize experimental variations. Triplicate hypothalami were used to determine the effect of Ang II on PI3-kinase in vivo. Each experiment was repeated at least three times, unless indicated otherwise. [3H]-NE uptake and release were measured with the use of the same number of cultures in triplicate dishes. For the analysis of mRNA levels, triplicate culture dishes were used for each data point, and poly(A+) RNA was pooled. Each experiment was repeated three times unless indicated otherwise. Densities of PCR bands were quantified, and data were presented as relative absorbance of the mean ± SE derived from normalization with β-actin for equal loading (Lu et al., 1996b, 1998;Yang et al., 1996a). Comparisons between the control and experimental groups were made using t test with the use of statistical software.
RESULTS
Effects of Ang II on PI3-kinase in WKY and SHR neurons
A time-dependent stimulation of PI3-kinase activity was observed when WKY rat brain neurons were incubated with 100 nm Ang II. The increase was transient. A significant stimulation was seen as early as 1 min; it reached maximal levels of ∼3.6-fold within 5 min. By 30 min the activity returned to control levels (Fig.1A). The stimulation was mediated by the interaction of Ang II with the AT1receptor subtype, because it was completely blocked by 10 μm losartan but not by PD 123319, an AT2receptor subtype antagonist (Fig. 1B). The stimulation was also Ang II concentration-dependent, and 100 nm of Ang II provided a maximal stimulation of 3.8-fold (Fig. 1C). The effect of Ang II on PI3-kinase activity between WKY and SHR neurons was compared, because neurons from SHR express two- to fourfold higher levels of AT1 receptors and a parallel increase in NE neuromodulation (Lu et al., 1996a; Yu et al., 1996a; Raizada et al., 1998). Despite these increases, the stimulation of PI3-kinase by Ang II was comparable (∼3.6-fold) in both strains of neurons (Fig. 1). However, the time course of inactivation after Ang II stimulation was significantly delayed in the SHR neuron. As a result, PI3-kinase activity remained maximally stimulated at 30 min in the SHR, whereas it returned to control levels in the WKY rat neuron (Fig.1A). In fact, the enzyme activity was approximately twofold higher even at 60 min in the SHR neuron compared with the WKY rat neuron.
Fig. 1.

A, Effect of Ang II on PI3-kinase activity in WKY and SHR brain neurons. Time course: neuronal cultures of WKY and SHR were incubated with 100 nm Ang II for the indicated time periods. PI3 kinase activity was measured after immunoprecipitation with the PI3-kinase p85 subunit-specific antibody essentially as described in Materials and Methods. Top, Representative autoradiogram of a TLC plate. Bottom, Quantitation of radioactive spots corresponding to PIP, mean ± SE (n = 3 experiments). * Significantly different from zero time (p < 0.01). # Significantly different from Ang II-treated WKY rat neuron (p < 0.01). B, Effect of Ang receptor antagonists. WKY and SHR neuronal cultures were incubated without (Control) or with (2, 4, 6) 100 nm Ang II for 10 min in the absence or presence of 10 μm losartan (Los) or PD 123319 (PD). PI3-kinase activity was determined as described in Materials and Methods. Top, Representative autoradiogram. Bottom, Quantitation of radioactive spot, mean ± SE (n = 3). * Significantly different from control (p < 0.0l). C, Ang II concentration. Incubation and assay conditions with indicated concentration of Ang II for 10 min were essentially as described inA. Top, Representative autoradiogram.Bottom, Data from three experiments ± SE. * Significantly different from no Ang II (p< 0.05).
The effect of Ang II on PI3-kinase activity was performed in intact hypothalamus from adult WKY rats and SHRs to validate that the effects observed in primary neuronal cultures are not an artifact of culture conditions. Figure 2 shows that similar to neuronal cultures, Ang II caused a three- to fourfold stimulation of PI3-kinase in 15 min, and the levels of this stimulation were comparable in WKY and SHR hypothalamus. In addition, the stimulation was persistent even at 30 min in the SHR, whereas it was completely reversed in the WKY rat hypothalamus (Fig. 2). Ang II stimulation of PI3-kinase activity in neuronal cultures as well as in intact hypothalamus of both WKY and SHR did not alter the immunoreactive PI3-kinase levels (Fig. 3).
Fig. 2.
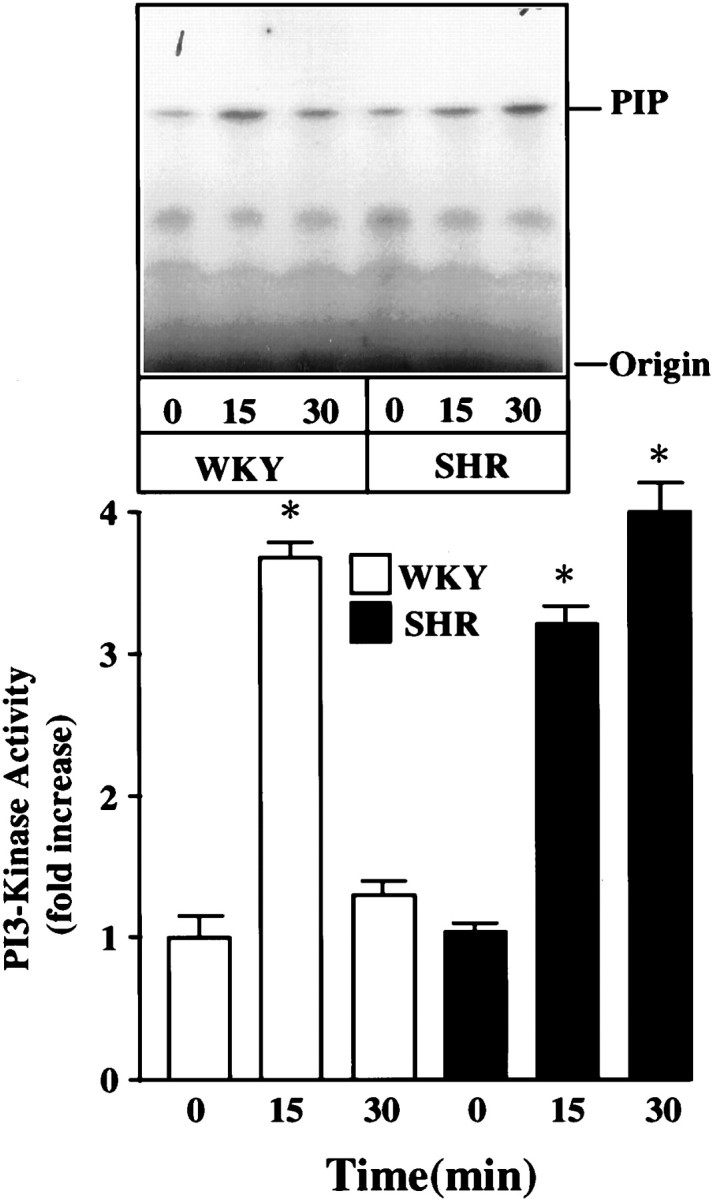
Effect of Ang II on PI3-kinase activity in hypothalami of adult WKY rats and SHRs. WKY rats and SHRs were cannulated and subjected to Ang II injection and isolation of hypothalamic blocks. Tissues were homogenized and used for the measurement of PI3-kinase activity as described in Materials and Methods. Top, Representative autoradiogram.Bottom, Quantitation of PIP spots from three animals. Data are mean ± SE (n = 3). * Significantly different from zero time (p < 0.01).
Fig. 3.

Effect of Ang II on immunoreactive PI3-kinase in WKY and SHR brain neurons (in vitro) and in hypothalami (in vivo). Lysates from neuronal cultures and hypothalamic blocks of adult WKY and SHR neurons were prepared, and PI3-kinase immunoreactivity was precipitated with the use of p85 PI3-kinase-specific antibody. Western blotting was performed as described in Materials and Methods.
Intracellular distribution of PI3 kinase was studied with the use of immunoblot analysis in Ang II-treated neurons, and nuclear and cytoplasmic fractions were isolated from Ang II-treated neurons and subjected to immunoblotting with the use of p85α antibody. Ang II caused a time-dependent decrease in immunoreactive PI3-kinase in cytoplasmic fractions of both WKY and SHR neurons (Fig.4A). A 54% decrease was observed in 30 min. This was associated with a time-dependent increase in the immunoreactivity in the nuclear functions of Ang II-treated WKY and SHR neurons (Fig. 4B).
Fig. 4.
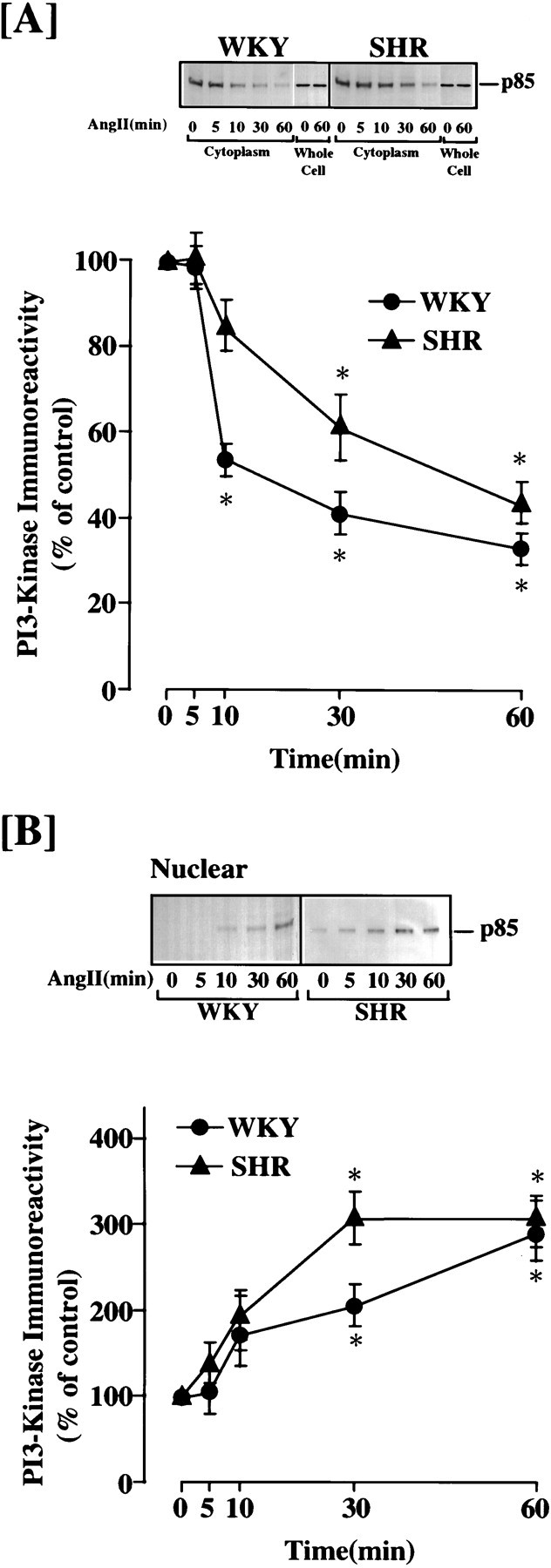
Ang II-induced nuclear translocation of PI3-kinase in WKY and SHR neurons. Neuronal cultures of WKY rats and SHRs were incubated without or with 100 nm Ang II for indicated time periods. Cytoplasmic and nuclear fractions were isolated and subjected to Western analysis as described in Materials and Methods: (A) cytoplasmic fractions and (B) nuclear fractions. Whole-cell lysates from control and Ang II-treated cells were also run in separate experiments to determine levels of the enzyme. A, B,Top, Representative autoradiograms;bottom, quantitation of immunoreactive bands. Mean ± SE (n = 3). * Significantly different from control (p < 0.05).
Role of PI3-kinase in Ang II regulation of NE neuromodulation
We studied the effect of PI3-kinase inhibition by Wortmannin on Ang II stimulation of NE neuromodulation in WKY and SHR neurons. We postulated that MAP kinase-independent NE neuromodulation may involve PI3-kinase in the SHR neurons. Neuronal cultures were preincubated with 100 nm Wortmannin for 30 min. This resulted in a complete inhibition of PI3-kinase activity in both strain of neurons (Fig. 5). Ang II caused threefold and sixfold increases in NET mRNA levels in the WKY and the SHR neurons, respectively (Fig. 6A). This observation is consistent with our earlier report demonstrating an increase of AT1 receptors in this strain (Raizada et al., 1993, 1994). Treatment of the WKY neuron with 100 nmWortmannin had no effect on Ang II stimulation of NET mRNA levels. In contrast, Wortmannin caused a 50% inhibition of NET mRNA in the SHR neuron (Fig. 6A). Interestingly, the level of stimulation of PI3-kinase by Ang II in Wortmannin-treated SHR neurons was comparable to the Wortmannin-treated WKY neurons. Similar observations were noted for the effects of Ang II and Wortmannin on TH mRNA and [3H]-NE uptake between WKY and SHR neurons (Fig. 6B,C). For example, Ang II stimulation of TH mRNA levels and [3H]-NE uptake were significantly higher in the SHR neuron. Also, Wortmannin partially inhibited Ang II stimulation of these activities in the SHR neuron and not in the WKY neuron. These data indicated that PI3-kinase plays a minimal role in Ang II regulation of NE neuromodulation in the WKY rat neuron. However, it is partially responsible for NE neuromodulatory effects of Ang II in the SHR neuron. This difference in the ability of Wortmannin to selectively inhibit Ang II stimulation of NE neuromodulation in the SHR neuron is not a result of a differential inhibition of PI3-kinase, because Wortmannin inhibition of this enzyme was comparable in both strains of neurons. These observations led us to hypothesize that the Ras—Raf—MAP kinase signaling pathway is involved in the response of Ang II on the WKY rat neuron, whereas both Ras—Raf—MAP kinase and PI3-kinase signaling pathways are responsible for the neuromodulatory actions of Ang II in the SHR neuron.
Fig. 5.
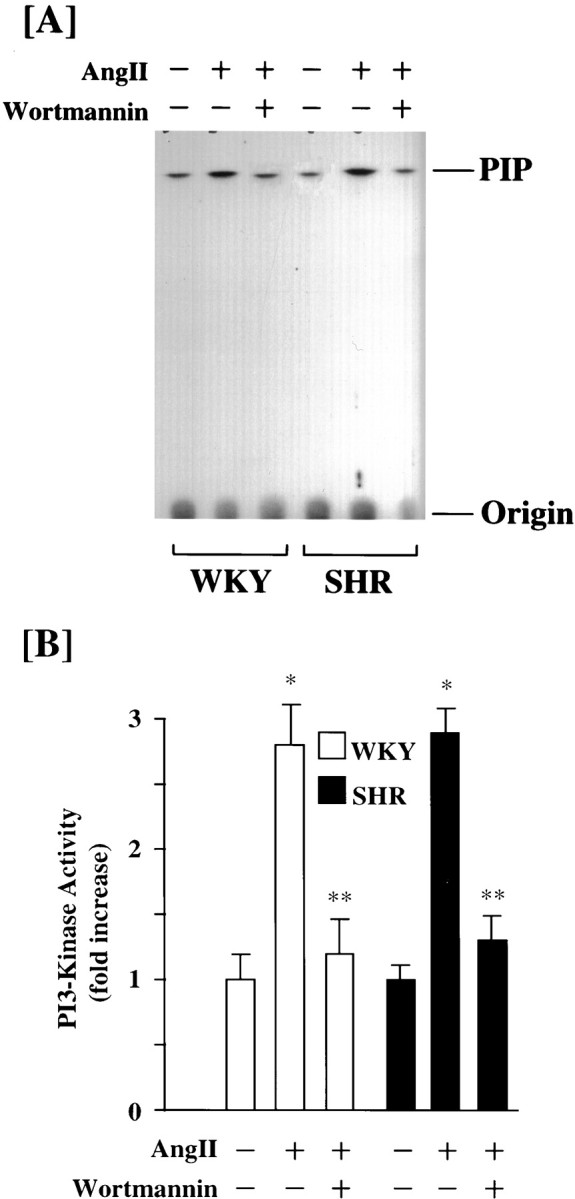
Effect of Wortmannin on Ang II stimulation of PI3-kinase activity in WKY and SHR neurons. Neuronal cultures of WKY rats and SHRs were incubated with 100 nm Ang II for 15 min in the absence and presence of 100 nm Wortmannin. PI3-kinase activity was determined as described in Materials and Methods. A, Representative autoradiogram.B, Mean ± SE (n = 3). * Significantly different from untreated cells (p < 0.01). ** Significantly different from Ang II-treated cells (p < 0.01).
Fig. 6.

Neuronal cultures of WKY rats and SHRs were incubated with 100 nm Ang II in the absence or presence of 100 nm Wortmannin for 4 hr at 37°C. Levels of NET mRNA (A), TH mRNA (B), and [3H]-NE uptake (C) were measured essentially as described in Materials and Methods. A, B, Top, Representative autoradiograms; bottom, mean data from three experiments ± SE. * Significantly different from control (p < 0.01). ** Significantly different from Ang II-treated neurons (p < 0.05). # Significantly different from Ang II-treated WKY rat neurons (p < 0.01).
Next, we performed experiments with the use of both Wortmannin and PD 98059, a MAP kinase-kinase inhibitor, to provide evidence in support of the above hypothesis. Previous studies have established that PD 98059, at 50 μm, caused a complete attenuation of the Ang II stimulation of NET and TH mRNAs in the WKY rat neuron, whereas a similar treatment caused only a 43–50% inhibition in the SHR neuron (Yang and Raizada, 1998). This was despite the fact that PD 98059 completely and equally attenuated Ang II-induced activation of MAP kinase in both strains of neurons (Yang and Raizada, 1998). In contrast to PD 98059, Wortmannin showed no effect in the WKY rat neuron and partially inhibited Ang II stimulation of NET and TH mRNA levels in the SHR neuron. Interestingly, inclusion of both PD 98059 and Wortmannin completely attenuated Ang II’s stimulatory effects on NET and TH mRNA levels in the SHR neuron (Fig. 7). This combination failed to further lower the stimulation significantly in the WKY rat neuron.
Fig. 7.
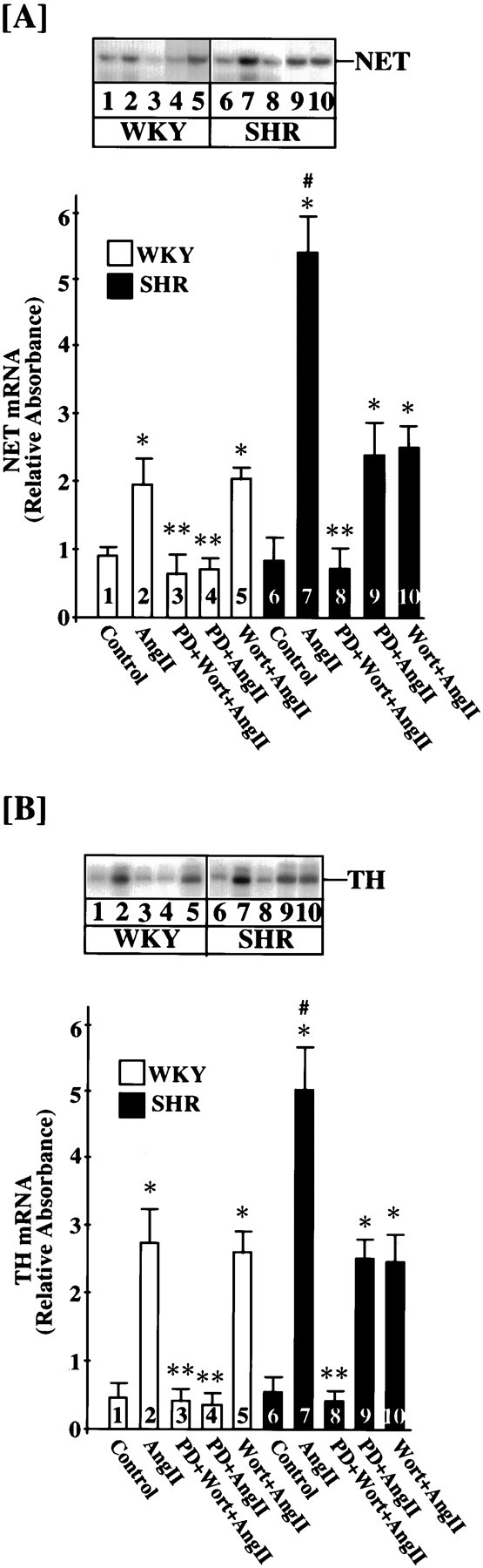
Effects of Wortmannin and PD 98059 on NE neuromodulation in WKY and SHR neurons. WKY rat and SHR neurons were treated without or with 100 nm Ang II (AngII) for 4 hr at 37°C in the absence or presence of 100 nm Wortmannin (Wort) or 50 μm PD 98059 (PD). mRNA levels for NET (A) and TH (B) were measured essentially as described in Materials and Methods.Top, Representative autoradiograms;bottom, mean ± SE (n = 3). * Significantly different from control (p< 0.01). ** Significantly different from Ang II-treated neurons (p < 0.05). # Significantly different from Ang II-treated WKY rat neurons (p < 0.01).
Our previous studies have demonstrated that enhancedneuromodulatory actions of Ang II are associated with the stimulation of MAP kinase, its translocation into the nucleus, and the activation of AP-1 binding activity, which is involved in the transcription of NET, TH, and DβH genes in the WKY rat neuron (Lu et al., 1996b). On the basis of this information and our above data on a selective effect of PI3-kinase in the SHR neuron, we decided to compare the effect of PI3-kinase inhibition on Ang II stimulation of AP-1 binding activity in both strains of neurons. Figure 8shows that Ang II caused a fivefold increase in the AP-1 binding activity in the nuclear fractions of the WKY rat neuron. The stimulation in the nuclear fractions of the Ang II-treated SHR neuron was 50% higher compared with WKY rat neuron. Wortmannin caused a 60% decrease in the AP-1 binding activity in the SHR neuron, whereas it had no effect on the WKY rat neuron.
Fig. 8.

Effect of Wortmannin on Ang II stimulation of AP-1 binding activity in the nuclear fractions of WKY and SHR neurons. Nuclear extracts of control, Ang II-treated (100 nm) or Ang II + Wortmannin-treated WKY rat and SHR neurons were subjected to gel shift analysis to determine the levels of AP-1 binding activity essentially as described in Materials and Methods.
Involvement of PI3-kinase in Ang II stimulation of PKB/Akt in WKY rat and SHR neurons
PKB/Akt is an established downstream signaling kinase in the propagation of PI3-kinase-mediated signals (Franke et al., 1997; Marte and Downward, 1997). Activation of PKB/Akt is associated with its translocation from the cytoplasmic to nuclear compartment, where it acts as a transcription factor to influence the expression of various genes (Andjelkovic et al., 1997; Alessi and Cohen, 1998). In view of our above observations, our next objective was to determine whether the differential effect of Ang II on the SHR neuron was caused by the effects of Ang II on PKB/Akt activation. Ang II caused a time-dependent increase in PKB/Akt activity in both strains of neurons (Fig. 9A). There were two differences in the nature of stimulation between the WKY and SHR neurons: (1) the stimulation persisted for longer time periods in the SHR neuron, and (2) maximal stimulation of PKB/AKt activity was 30–40% higher in the SHR neuron compared with the WKY rat neuron. The stimulation was Ang II concentration-dependent in both cell types, although the SHR neuron was 30% more responsive at each dose of Ang II (Fig. 9B). Losartan completely blocked the Ang II stimulation of PKB/Akt activity, whereas PD 123319 had little effect (Fig. 9C).
Fig. 9.

Effect of Ang II on protein kinase B activity in WKY and SHR neurons. A, Time course: neuronal culture of WKY rat and SHR were incubated with 100 nm Ang II for the indicated time at 37°C. PKB activity in neuronal extract was determined essentially as described in Materials and Methods.Top, Representative autoradiogram;bottom, mean ± SE (n = 3). * Significantly different from zero time (p< 0.05). # Significantly different from the WKY (p < 0.05). B, Ang II concentrations: neuronal cultures of WKY rat and SHR were incubated with the indicated concentrations of Ang II for 10 min at 37°C before determination of PKB activity. Top, Representative autoradiogram; bottom, mean ± SE (n = 3). * Significantly different from control (p < 0.05). # Significantly different from the WKY (p < 0.05). C, Effect of Ang receptor antagonists: experimental conditions were essentially as described in the legend to Figure10B. WKY rat and SHR neuronal cultures were incubated without (C) or with 100 nmAng II in the absence or presence of 10 μm losartan (Los) or PD123319 (PD).Top, Representative autoradiogram;bottom, mean ± SE (n = 3). * Significantly different from control (p< 0.05). ** Significantly different from Ang II treatment (p < 0.05).
The stimulation of PKB/Akt activity by Ang II appeared to be mediated by activation of PI3-kinase and not by MAP kinase. Incubation of WKY and SHR neurons with 100 nm Wortmannin completely attenuated Ang II stimulation of PKB/Akt activity, whereas 50 μm PD 98059 had little effect on this stimulation (Fig.10). The levels of PKB/Akt in the cytoplasmic and nuclear fractions were compared to determine whether Ang II treatment causes nuclear translocation of this enzyme. Stimulation with Ang II caused a time-dependent decrease in the cytoplasmic PKB/Akt. Its levels were 60% lower in the cytoplasmic fraction of the WKY rat neuron after 30 min of Ang II treatment (Fig.11A). This decrease was associated with increases in its levels in the nuclear fraction (Fig. 11B). An approximately twofold increase was seen 30 min after Ang II treatment in the WKY rat neuron. Comparison of data showed that the levels of PKB/Akt immunoreactivity in the nucleus and fold translocation by Ang II in the SHR neuron were comparable with the WKY rat neuron (Fig. 11). This would suggest that the translocated PKB/Akt activity in the nuclear fraction of the SHR neuron would be significantly higher than in the WKY rat neurons, because the activity of this enzyme per unit of protein is higher in this strain of cells.
Fig. 10.
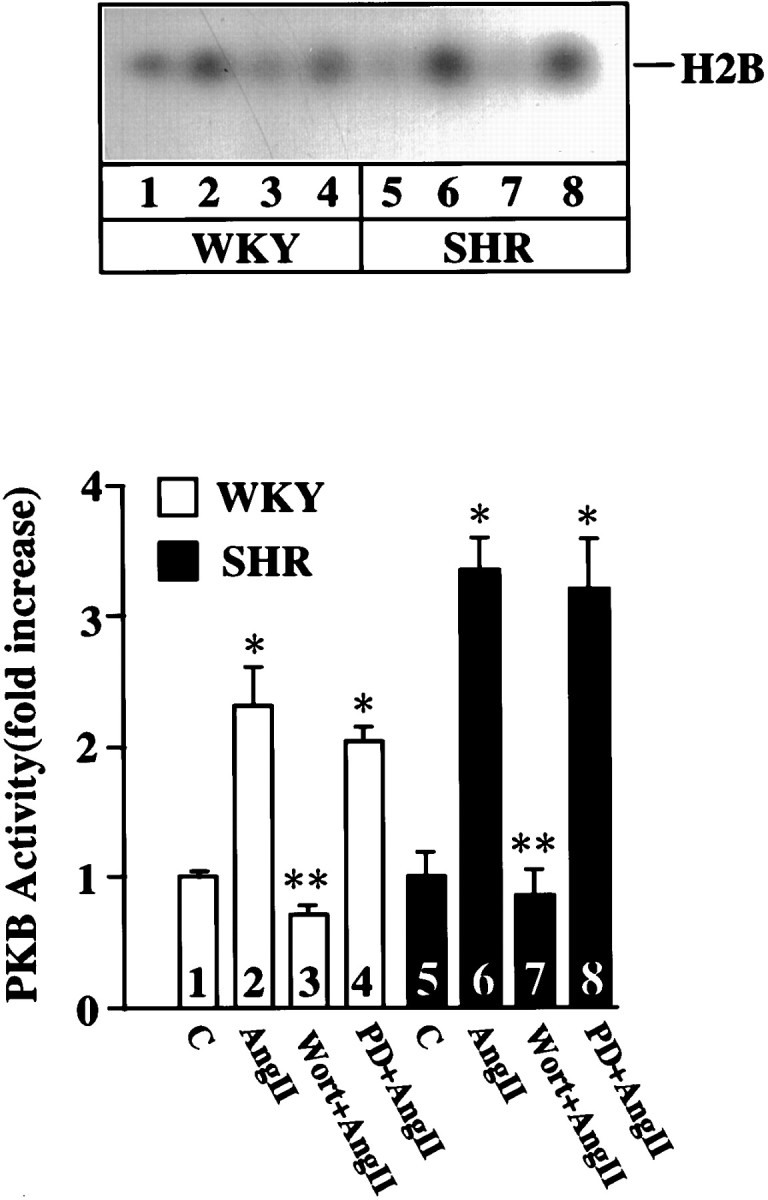
Effects of PD 98059 and Wortmannin on PKB activity in WKY and SHR neurons. WKY rat and SHR neurons were treated without or with 100 nm Ang II (AngII) for 10 min at 37°C in the absence or presence of 50 μmPD 98059 (PD) or 100 nm Wortmannin (Wort). Top, Representative autoradiogram; bottom, mean ± SE (n = 3) * Significantly different from control (p < 0.05). ** Significantly different from Ang II-treated neurons (p < 0.05).
Fig. 11.
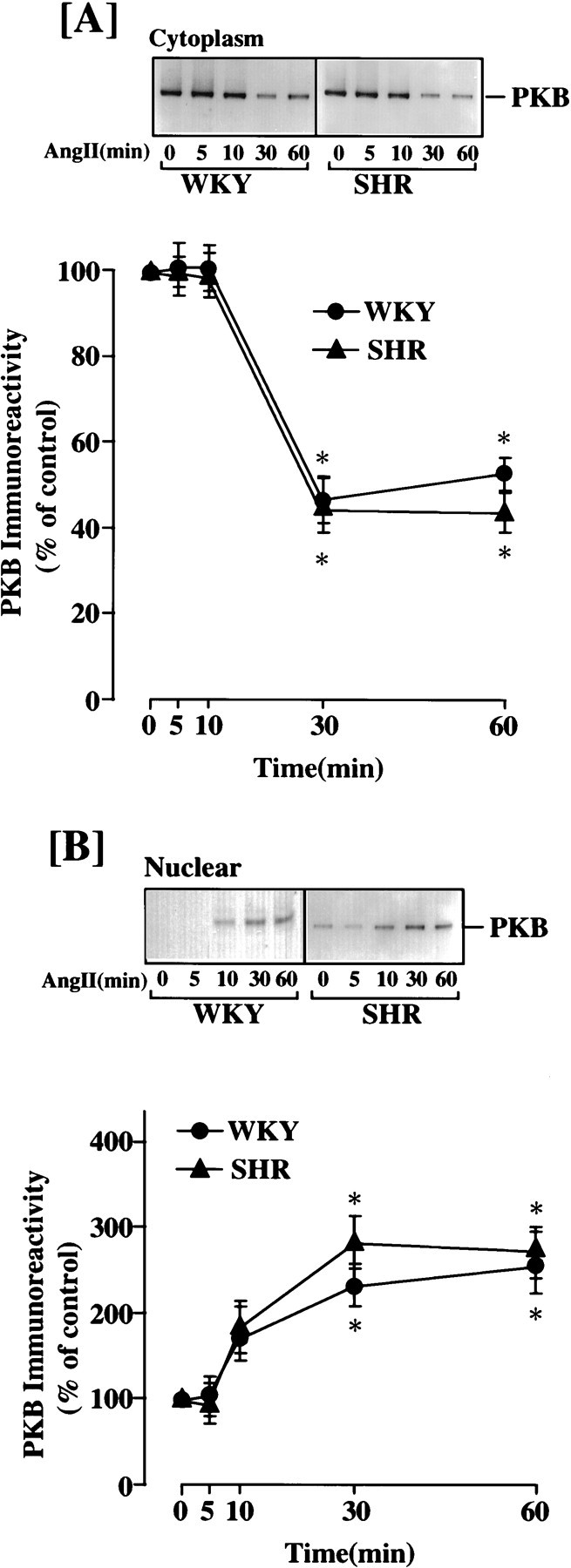
Ang II-induced nuclear translocation of PKB in WKY and SHR neurons. Neuronal cultures of WKY and SHR were incubated without or with 100 nm Ang II for the indicated time periods. Cytoplasmic and nuclear fractions were isolated and subjected to Western analysis as described in Materials and Methods.A, Cytoplasmic fractions; B, nuclear fractions. A, B, Top, Representative autoradiograms;bottom, quantitation of immunoreactive bands. Mean ± SE (n = 3). * Significantly different from control (p < 0.01).
DISCUSSION
The most significant observation of this study is the exclusive involvement of PI3-kinase and PKB/Akt activities in Ang II-induced regulation of NE neuromodulation in the SHR neuron. Thus, the PI3-kinase-PKB signaling system constitutes the MAP kinase-independent signal transduction mechanism for a heightened Ang II stimulation of NET, TH, and DβH gene transcription and [3H]-NE uptake in the SHR neuron previously postulated by us (Yang and Raizada, 1998). Diagram FD1 summarizes our observations. Ras—Raf—MAP kinase appears to be the sole signal transduction pathway for NE neuromodulatory action in the WKY rat neuron. This is primarily based on our earlier observations that inhibition of MAP kinase activity, either by PD 98059 or by MAP kinase-specific antisense oligonucleotides, completely attenuates Ang II stimulation of NET, TH, and DβH mRNAs and [3H]-NE uptake (Yang et al., 1996a; Gelband et al., 1998). In addition, a complete attenuation is seen in Ang II activation of SRE and AP-1 binding activities, a downstream regulatory step in the transcriptional control of CA-synthesizing genes (Lu et al., 1996b). AT1 receptor levels and its transcription are two- to fourfold higher in the SHR neuron compared with the WKY rat neuron, a normotensive control (Raizada et al., 1993, 1998). This increase is associated with a two- to fourfold heightened response of the SHR neuron to Ang II in the stimulation of NE neuromodulation (Lu et al., 1996a; Yu et al., 1996a;Yang and Raizada, 1998). Despite this, the MAP kinase signaling pathway is only partially responsible. The heightened response appears to be caused by the coupling of the SHR AT1 receptor to the PI3-kinase-PKB signaling pathway. Thus, neuromodulation mediated by AT1 receptors in the SHR neuron is linked to both MAP kinase and PI3-kinase signaling systems, whereas it is linked to only the MAP kinase system in the WKY rat neuron. This conclusion is further supported by our data, which indicate that PI3-kinase is not involved in the regulation of MAP kinase activity in brain neurons, in contrast to the evidence in other systems (Karnitz et al., 1995; Nishioka et al., 1995).
Fig. D1.

Summary of signaling mechanisms of AT1 receptor-mediated neuromodulation in WKY and SHR neurons.
These observations are highly significant because (1) they constitute the first example in which the PI3-kinase-PKB signaling system has been linked to the NE neuromodulatory activity. The involvement of PI3-kinase in the regulation of growth, differentiation, and cellular metabolic actions of many hormones is well established (Soltoff et al., 1992; Kapeller and Cantley, 1994), but its ability to regulate catecholamine turnover, synthesis, and release is a unique function and links this enzyme to the regulation of neurotransmission. The observations establish that (2) the AT1 receptor is coupled to the PI3-kinase-neuromodulation cascade only in the SHR neuron. This is highly relevant from a pathophysiological perspective and may turn out to have important pharmacological implications in the control of centrally regulated hypertension. For example, members of the PI3-kinase signaling cascade could serve as highly specific targets for pharmacological and/or gene therapy, because the pathway is only active in the SHR. Our data further support this implication. Hypothalamic nuclei in the adult brain contain functional AT1 receptors, which are predominantly responsible for Ang II-mediated NE neuromodulation leading to is effects on blood pressure (Saavedra, 1992; Steckelings et al., 1992; Timmermans et al., 1993). In addition, AT1 receptor expression in SHR hypothalamus is increased similar to that observed in vitro (Raizada et al., 1998). The present observation that Ang II stimulates PI3-kinase in the hypothalamus of both WKY and SHR, with more sustained stimulation in the SHR, is also consistent with the in vitro studies and their relevance to the in vivo situation. Despite a strong correlation between in vitro and in vivo data on the effects of Ang II on the NE system, some degree of caution must observed when be in vitro data are extrapolated to the adult animal. This is particularly relevant in view of the fact that neuronal phenotypes may be different in vitro and in situ. In addition, although in vitro effects of Ang II on catecholamic systems are fairly well studied (Raizada et al., 1998), the nature of these interactions in situ is not established and may be distinct.
This study raises an important question concerning the mechanism by which the PI3-kinase pathway is uniquely linked to neuromodulationonly in the SHR. Currently there is no direct evidence that examines this issue. However, we propose that a persistent stimulation of both PI3-kinase and PKB/Akt and a significantly higher stimulation of PKB/Akt in the SHR neuron may be sufficient to shift the signaling equilibrium toward this specificity. It is likely that increased activation of PKB/Akt by persistently stimulated PI3-kinase could lead to an increased nuclear translocation of PKB/Akt-specific activity. This increase in the nuclear PKB/Akt activity may participate in a transcriptional control of catecholamine-relevant genes. In the future, the availability of improved techniques to measure nuclear PKB/Akt activity in neuronal cultures would provide evidence to support or refute this view.
Many other possibilities concerning the mechanism, however, cannot be ruled out at the present time. For example, the signaling steps downstream from the PKB/Akt activation may be different between the two strains of neurons. This hypothesis would suggest that Ang II stimulation of PI3-kinase cascade in the WKY rat neuron may be involved in other distinct functions. Preliminary data, in fact, support this view. Ang II stimulates plasminogen activator inhibitor-1 (PAI-1) synthesis in brain neurons (Yu et al., 1996b). This stimulation, which is at the level of PAI-1 gene transcription, has been linked to the trophic actions of Ang II in neuronal cultures (Rydzewski et al., 1993). Ang II stimulation of PAI-1 mRNA was exclusively attenuated by Wortmannin in the WKY rat neuron. PI3-kinase inhibition did not have such an effect on the stimulation of PAI-1 in the SHR neuron. From these data, although preliminary, it is tempting to speculate that the PI3-kinase cascade is involved in trophic actions of Ang II in the WKY neuron.
The presence of two distinct AT1 receptor subtypes in the SHR neuron, one coupled to MAP kinase and the other to PI3-kinase, can also explain these observations. Although no pharmacological difference between the AT1 receptors of WKY and SHR neurons have been noted (Sumners and Raizada, 1993; Raizada et al., 1994, 1998; Sumners et al., 1995), the proposal needs a critical evaluation. This is especially relevant because the AT1 receptor gene expression is higher in the SHR neuron and because two subtypes of the AT1 receptor, AT1A and AT1B, have been reported in the neurons (Sumners and Raizada, 1993). Finally, coupling of the AT1 receptor to G-proteins may be different in the two strains of neurons. We have proposed that Gβγ subunits of Gq may be involved in the NE neuromodulatory actions of the AT1 receptor (Gelband et al., 1998). This is consistent with the evidence that Gβγ subunits play an important role in the regulation of this enzyme (Hazeki et al., 1998; Leopoldt et al., 1998). Whatever the precise mechanism for this diversity of Ang II actions may be, the observations pinpoint a unique signaling cascade involving PI3-kinase in NE neuromodulatory actions of Ang II in the SHR neuron.
Footnotes
This study was supported by National Institutes of Health Grant HL33610. Hong Yang is a postdoctoral fellow of the American Heart Association, Florida Affiliate. We thank Ling Liu and Hung Dang for technical assistance.
Correspondence should be addressed to Dr. Mohan K. Raizada, Department of Physiology, College of Medicine, University of Florida, P.O. Box 100274, Gainesville, FL 32610.
REFERENCES
- 1.Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. Curr Opin Genet Dev. 1998;8:55–62. doi: 10.1016/s0959-437x(98)80062-2. [DOI] [PubMed] [Google Scholar]
- 2.Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 3.Berecek KH, Okuno T, Nagahama T, Oparil S. Altered vascular reactivity and baroreflex sensitivity induced by chronic central administration of captopril in the spontaneously hypertensive rat. Hypertension. 1983;5:687–700. doi: 10.1161/01.hyp.5.5.689. [DOI] [PubMed] [Google Scholar]
- 4.Ding H, Zhou Q, Deng J, Lao HY, Yang K. Effect of the overactivated central renin-angiotensin system on the concentration of brain norepinephrine and epinephrine in stroke-prone spontaneously hypertensive rats and its significance. Sheng Li Hsueh Pao. 1990;42:379–384. [PubMed] [Google Scholar]
- 5.Franke TF, Yang SI, Chan TO, Datta CK, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the AKT proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3 kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 6.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 7.Gelband CH, Sumners C, Lu D, Raizada MK. Angiotensin receptors and norepinephrine neuromodulation: implications of functional coupling. Regul Pept. 1998;73:141–147. doi: 10.1016/s0167-0115(97)11050-3. [DOI] [PubMed] [Google Scholar]
- 8.Griendling KK, Murphy TJ, Alexander RW. Molecular biology of the renin-angiotensin system. Circulation. 1993;87:1816–1828. doi: 10.1161/01.cir.87.6.1816. [DOI] [PubMed] [Google Scholar]
- 9.Hazeki O, Okada T, Kutosu H, Takasuga S, Suzuki T, Katada T. Activation of PI 3-kinase by G protein βγ subunits. Life Sci. 1998;62:1555–1559. doi: 10.1016/s0024-3205(98)00106-4. [DOI] [PubMed] [Google Scholar]
- 10.Kapeller R, Cantley LC. Phosphatidylinositol 3 kinase. Bioessays. 1994;16:565–576. doi: 10.1002/bies.950160810. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan DR, Whitman M, Schaffhausen B, Pallas DC, White M, Cantley L, Roberts TM. Phosphatidylinositol metabolism and polyoma-mediated transformation. Proc Natl Acad Sci USA. 1986;83:362–364. doi: 10.1073/pnas.83.11.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaplan DR, Whitman M, Schafthausen B, Pallas DC, White M, Cantley L, Roberts TM. Common elements in growth factor stimulation and oncogene transformation: 85kd phosphoprotein and phosphatidylinositol kinase activity. Cell. 1987;50:1021–1029. doi: 10.1016/0092-8674(87)90168-1. [DOI] [PubMed] [Google Scholar]
- 13.Karnitz LM, Burnsx LA, Sutor SL, Blenis J, Abraham RT. Interleukin-2 triggers a novel phosphatidylinositol 3-kinase-dependent MEK activation pathway. Mol Cell Biol. 1995;15:3049–3057. doi: 10.1128/mcb.15.6.3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langheinrich M, Lee MA, Bohn M, Pinto YM, Ganten D, Paul M. The hypertensive Ren-2 transgenic rat TGR (mREN2)27 in hypertension research. Characteristics and functional aspects. Am J Hypertens. 1996;9:506–512. doi: 10.1016/0895-7061(95)00400-9. [DOI] [PubMed] [Google Scholar]
- 15.Leopoldt D, Hanck T, Exner T, Maier V, Wetzker R, Nurnkeg B. Gβγ stimulates phosphoinositide 3-kinase-γ by direct interaction with two domains of the catalytic p110 subunit. J Biol Chem. 1998;273:7024–7029. doi: 10.1074/jbc.273.12.7024. [DOI] [PubMed] [Google Scholar]
- 16.Lopez-llasaca M, Crespo P, Pellici PG, Gulkind JS, Wetzker R. Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI3-kinase γ. Science. 1997;265:394–396. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- 17.Lu D, Yu K, Paddy MR, Rowland NE, Raizada MK. Regulation of norepinephrine transport system by angiotensin II in neuronal cultures of normotensive and spontaneously hypertensive rat brains. Endocrinology. 1996a;137:763–772. doi: 10.1210/endo.137.2.8593828. [DOI] [PubMed] [Google Scholar]
- 18.Lu D, Yang H, Raizada MK. Angiotensin II regulation of neuromodulation: downstream signaling mechanism from activation of mitogen activated protein kinase. J Cell Biol. 1996b;135:1609–1617. doi: 10.1083/jcb.135.6.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu D, Yang H, Lenox RH, Raizada MK. Regulation of angiotensin II-induced neuromodulation by MARCKS in brain neurons. J Cell Biol. 1998;142:217–227. doi: 10.1083/jcb.142.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marte BM, Downward J. PKB/Akt: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biol Sci. 1997;22:355–358. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- 21.Nishioka N, Hirai S, Mizuno K, Osada S, Suzuki A, Kosaka K, Ohno S. Wortmannin inhibits the activation of MAP kinase following vasopressin V1 receptor stimulation. FEBS Lett. 1995;377:393–398. doi: 10.1016/0014-5793(95)01382-2. [DOI] [PubMed] [Google Scholar]
- 22.Phillips MI. Brain angiotensin. In: Gross P, editor. Circumventricular organs and body fluids, Vol III. CRC; Boca Raton, FL: 1987. pp. 163–182. [Google Scholar]
- 23.Phillips MI, Wielbo D, Gyuoko R. Antisense inhibition of hypertension: a new strategy for renin-angiotensin candidate gene. Kidney Int. 1994;46:1554–1556. doi: 10.1038/ki.1994.444. [DOI] [PubMed] [Google Scholar]
- 24.Raizada MK, Muther TF, Sumners C. Increased angiotensin II specific receptors in neuronal culture of spontaneously hypertensive rat brain. Am J Physiol. 1984;247:C364–C372. doi: 10.1152/ajpcell.1984.247.5.C364. [DOI] [PubMed] [Google Scholar]
- 25.Raizada MK, Lu D, Tang W, Kurian P, Sumners C. Increased angiotensin II type-1 receptor gene expression in neuronal cultures from spontaneously hypertensive rats. Endocrinology. 1993;132:1715–1722. doi: 10.1210/endo.132.4.8462471. [DOI] [PubMed] [Google Scholar]
- 26.Raizada MK, Lu D, Sumners C. AT1 receptors and angiotensin actions in the brain and neuronal cultures of normotensive and hypertensive rats. In: Mukhopadhyay A, Raizada MK, editors. Current concepts: tissue renin-angiotensin system as local regulators in reproductive and endocrine organs. Plenum; New York: 1994. pp. 331–348. [DOI] [PubMed] [Google Scholar]
- 27.Raizada MK, Lu D, Yang H, Richards EM, Gelband CH, Sumners C. Brain angiotensin receptor subtypes and their coupling to distinct signal transduction pathways. In: LeRoith D, editor. Advances in molecular and cellular endocrinology, Vol 3. JAI; Greenwich, CT: 1998. pp. 73–79. [Google Scholar]
- 28.Rydzewski B, Wozniak M, Sumners C, Raizada MK. The plasminogen activator system and its interactions with angiotensin II in the brain. In: Raizada MK, Phillips MI, Sumners C, editors. Cellular and molecular biology of the renin angiotensin system. CRC; Boca Raton, FL: 1993. pp. 485–512. [Google Scholar]
- 29.Saavedra JM. Brain and pituitary angiotensin. Endocr Rev. 1992;13:329–380. doi: 10.1210/edrv-13-2-329. [DOI] [PubMed] [Google Scholar]
- 30.Saward L, Zahradka P. Angiotensin II activates phosphatidylinositol 3-kinase in vascular smooth muscle cells. Circ Res. 1997;81:249–257. doi: 10.1161/01.res.81.2.249. [DOI] [PubMed] [Google Scholar]
- 31.Soltoff SP, Carpenter CL, Auger KR, Kapeller R, Schaffhausen B, Cantley LC. Phosphatidylinositol 3 kinase and growth regulation. Cold Spring Harb Symp Quant Biol. 1992;57:75–80. doi: 10.1101/sqb.1992.057.01.010. [DOI] [PubMed] [Google Scholar]
- 32.Steckelings U, Lebrun C, Quadri F, Veltman A, Unger T. Role of brain angiotensin in cardiovascular regulation. J Cardiovasc Pharmacol. 1992;19[Suppl 6]:S73–S79. doi: 10.1097/00005344-199219006-00012. [DOI] [PubMed] [Google Scholar]
- 33.Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 34.Sumners C, Raizada MK. Angiotensin II receptor subtypes in neuronal cells. In: Raizada MK, Phillips MI, Sumners C, editors. Cellular and molecular biology of the renin-angiotensin system. CRC; Boca Raton, FL: 1993. pp. 379–411. [Google Scholar]
- 35.Sumners C, Raizada MK, Kang J, Lu D, Posner P. Receptor mediated effects of angiotensin II on neurons. Front Neuroendocrinol. 1995;15:203–230. doi: 10.1006/frne.1994.1009. [DOI] [PubMed] [Google Scholar]
- 36.Timmermans PBMWM, Wang PC, Chiu AT, Herblin MF, Benfield P, Carini DJ, Lee RJ, Wexler RR, Saye JAM, Smith RD. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev. 1993;45:205–251. [PubMed] [Google Scholar]
- 37.Toney GM, Porter JP. Effects of blockade of AT1 and AT2 recep- tors in brain on the central angiotensin II pressor response in conscious spontaneously hypertensive rats. Neuropharmacology. 1993;32:581–589. doi: 10.1016/0028-3908(93)90054-7. [DOI] [PubMed] [Google Scholar]
- 38.Wright JW, Harding JW. Brain angiotensin receptor subtypes in the control of physiological and behavioral responses. Neurosci Biobehav Rev. 1994;18:21–53. doi: 10.1016/0149-7634(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 39.Yang H, Lu D, Yu K, Raizada MK. Regulation of neuromodulatory actions of angiotensin II in the brain neurons by the Ras-dependent mitogen-activated protein kinase pathway. J Neurosci. 1996a;16:4047–4058. doi: 10.1523/JNEUROSCI.16-13-04047.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang H, Lu D, Raizada MK. Lack of cross talk between α1-adrenergic and angiotensin type 1 receptors in neurons of spontaneously hypertensive rat brain. Hypertension. 1996b;27:1277–1283. doi: 10.1161/01.hyp.27.6.1277. [DOI] [PubMed] [Google Scholar]
- 41.Yang H, Lu D, Vinson GP, Raizada MK. Involvement of MAP kinase in angiotensin II-induced phosphorylation and intracellular targeting of neuronal AT1 receptors. J Neurosci. 1997;17:1660–1669. doi: 10.1523/JNEUROSCI.17-05-01660.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang H, Raizada MK. MAP kinase-independent signaling in angiotensin II regulation of neuromodulation in SHR neurons. Hypertension. 1998;32:473–481. doi: 10.1161/01.hyp.32.3.473. [DOI] [PubMed] [Google Scholar]
- 43.Yang K, Ding H, Zhou Q, Luo HY, Wu ZY. Central norepinephrine and angiotensin II contents in the brain regions of spontaneously hypertensive rats (SHR) and the interaction between them. Sheng Li Hsueh Pao. 1991;43:345–351. [PubMed] [Google Scholar]
- 44.Yu K, Lu D, Rowland NE, Raizada MK. Angiotensin II regulation of tyrosine hydroxylase gene expression in the neuronal cultures of normotensive and spontaneously hypertensive rats. Endocrinology. 1996a;137:3566–3576. doi: 10.1210/endo.137.8.8754788. [DOI] [PubMed] [Google Scholar]
- 45.Yu K, Lu D, Paddy MR, Lenk SE, Raizada MK. Angiotensin II regulation of plasminogen activator inhibitor-1 gene expression in neurons of normotensive and spontaneously hypertensive rat brains. Endocrinology. 1996b;137:2503–2513. doi: 10.1210/endo.137.6.8641204. [DOI] [PubMed] [Google Scholar]