

Abstract
Methamphetamine (METH) is a powerful psychostimulant that is increasingly abused worldwide. Although it is commonly accepted that the dopaminergic system and oxidation of dopamine (DA) play pivotal roles in the neurotoxicity produced by this phenylethylamine, the primary source of DA responsible for this effect has remained elusive. In this study, we used mice heterozygous for vesicular monoamine transporter 2 (VMAT2 +/− mice) to determine whether impaired vesicular function alters the effects of METH. METH-induced dopaminergic neurotoxicity was increased in striatum of VMAT2 +/− mice compared with wild-type mice as revealed by a more consistent DA and metabolite depletion and a greater decrease in dopamine transporter expression. Interestingly, increased METH neurotoxicity in VMAT2 +/− mice was accompanied by less pronounced increase in extracellular DA and indices of free radical formation compared with wild-type mice. These results indicate that disruption of vesicular monoamine transport potentiates METH-induced neurotoxicity in vivo and point, albeit indirectly, to a greater contribution of intraneuronal DA redistribution rather than extraneuronal overflow on mediating this effect.
Keywords: vesicular monoamine transporter, methamphetamine, dopamine, microdialysis, GFAP, free radicals
Administration of the psychostimulant methamphetamine (METH) to experimental animals results in prolonged reduction of dopamine (DA) and serotonin levels, depressed activity of tyrosine hydroxylase (TH) and tryptophan hydroxylase, and decreased dopamine transporter (DAT) and serotonin transporter binding sites in the brain, alterations that are commonly considered to be indices of METH neurotoxic action (Gibb and Kogan, 1979; Hotchkiss and Gibb, 1980; Ricaurte et al., 1980; Wagner et al., 1980; Seiden and Ricaurte, 1987; Pu and Vorhees, 1993; Seiden and Sabol, 1995). The mechanisms underlying METH neurotoxicity have been studied primarily with respect to the dopaminergic system and suggest that DA contributes to the neurotoxic effects of METH. In fact, α-methyl-p-tyrosine (αMPT), a catecholamine synthesis inhibitor, prevents the toxic effects of METH (Wagner et al., 1983;Schmidt et al., 1985; Axt et al., 1990), whereasl-3,4-dihydroxyphenylalanine administration reverses the protective effects of αMPT (Schmidt, 1992; Weihmuller et al., 1993). Moreover, it has been suggested that the interaction of METH with DAT alters the homeostasis of the dopaminergic neuron, resulting in neurotoxicity (Schmidt and Gibb, 1985; Marek et al., 1990a,b). In view of this hypothesis, we have recently demonstrated that mice lacking the DAT are protected against the toxic effects of METH (Fumagalli et al., 1998), highlighting the essential role DAT plays in METH-induced dopaminergic neurotoxicity.
Two major hypotheses have been proposed to explain METH-induced neurotoxicity. First, it has been suggested that the ability of the drug to mobilize DA from intraneuronal pools to the extracellular space by outward transport through DAT may allow extraneuronal DA oxidation to highly reactive molecules, resulting in subsequent neurotoxicity (Seiden and Vosmer, 1984; De Vito and Wagner, 1989; Axt et al., 1990;Marek et al., 1990a,b; O’Dell et al., 1991, 1993). Alternatively, redistribution of DA from synaptic vesicles to cytoplasmic compartments and consequent elevation of oxidizable DA concentrations has been postulated to be primarily responsible for dopamine terminal injury by amphetamines (Cubells et al., 1994; Liu and Edwards, 1997; Wrona et al., 1997; Uhl, 1998). Thus, although DA clearly plays a role in METH neurotoxicity, the DA pool responsible for this toxicity remains unclear. However, both hypotheses suggest that disruption of the delicate balance that exists among vesicular, cytoplasmic, and extracellular DA pools may cause the neurotoxic effect of METH. Hence, synaptic processes regulated by vesicular and plasmalemmal transporters might be determinants of the cell vulnerability to METH action.
In the CNS, DA, serotonin, and norepinephrine are packaged into specialized secretory vesicles by vesicular monoamine transporter 2 (VMAT2) (Rudnick and Clark, 1993; Schuldiner et al., 1995; Lesch et al., 1996; Fon et al., 1997; Liu and Edwards, 1997; Takahashi et al., 1997; Varoqui and Erickson, 1997; Wang et al., 1997). It has been suggested that amphetamines, through interaction with VMAT2, cause monoamine displacement from these vesicles, resulting in increased cytoplasmic DA levels (Pifl et al., 1995; Sulzer et al., 1995; Fon et al., 1997; Wang et al., 1997; Jones et al., 1998b). The recent development of genetically altered mice heterozygous for VMAT2 (VMAT2 +/− mice) (Fon et al., 1997; Takahashi et al., 1997; Wang et al., 1997) provides a unique opportunity to gain insight into the neuronal mechanisms participating in the neurotoxicity of METH. Thus, to define the role played by vesicular monoamine uptake and storage in METH-induced dopaminergic neurotoxicity, we have examined the effects of METH in VMAT2 +/− mice.
MATERIALS AND METHODS
Animals. The heterozygous mice (VMAT2 +/− mice) were generated as described previously (Wang et al., 1997). Whereas homozygous mice (VMAT2 −/− mice) die within 1 week of birth, VMAT2 +/− mice are viable into adulthood. Mice were genotyped by Southern blot analysis of tail biopsies (Wang et al., 1997). Male wild-type (VMAT2 +/+) and VMAT2 +/− C57BL/129SvJ mice (3 months old) were used for all experiments. Animals were separated into different cages according to sex and genotype and maintained under standard housing conditions. Food and water were provided ad libitum. Animal care was in accordance with the Guide for Care and Use of Laboratory Animals (National Institutes of Health publication 865–23, Bethesda, MD) and approved by the Institutional Animal Care and Use Committee. Rectal body temperature was determined using a digital thermometer (Physitemp, Clifton, NJ).
Drugs. Methamphetamine (Sigma, St. Louis, MO) or vehicle (0.9% NaCl) were administered as either four subcutaneous injections (15 mg/kg, s.c.), each given 2 hr apart, or a single dose (15 or 30 mg/kg, s.c.). Reserpine and tetrabenazine were dissolved in acetic acid, diluted and administered at 5 mg/kg intraperitoneally. α-Methyl-p-tyrosine (Research Biochemicals, Natick, MA) was suspended in Tween 20 and administered at 250 mg/kg intraperitoneally.
Measurements of DA and metabolites. Dissected striata were homogenized in 0.1 m perchloric acid containing 100 ng/ml 3,4-dihydroxybenzylamine (DHBA) as an internal standard. Homogenates were centrifuged for 10 min at 10,000 × g. Supernatants were filtered through 0.22 μm filters and analyzed for levels of DA, 3,4-dihydroxyphenilacetic acid (DOPAC), and homovanillic acid (HVA), using HPLC with electrochemical detection (HPLC-EC). DA and metabolites were separated on a microbore reversed-phase column (C-18; 5 μm; 1 × 150 mm; Unijet; BAS, West Lafayette, IN) using a mobile phase consisting of 0.03 m citrate phosphate buffer with 2.1 mm octanesulfonic acid, 0.1 mmEDTA, and 17% methanol, pH 3.6, at a flow rate of 90 μl/min and detected by a 3 mm glassy carbon Unijet electrode (BAS) set at +0.85 V (Gainetdinov et al., 1997).
Western blotting. Analysis of DAT protein levels in the striata of mutant and wild-type mice was performed by Western blotting (Gainetdinov et al., 1998a). Dissected striata were homogenized in a buffer containing 320 mm sucrose, 5 mm HEPES, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 μg/ml pepstatin. Homogenates were centrifuged, and the resulting pellet was resuspended in sample buffer (62.5 mm Tris-HCl, 20% glycerol, 2% SDS, 0.01% bromphenol blue, and 1 mmdithiothreitol) and subjected to SDS-PAGE (10%). Proteins were electrophoretically transferred to a polyvinylidene difluoride membrane, and nonspecific sites were blocked in 5% nonfat dry milk in Tris-buffered saline (135 mm NaCl, 2.5 mm KCl, 50 mm Tris, and 0.1% Tween 20, pH 7.4). Membranes were then incubated in the presence of a monoclonal antibody to the N terminus of DAT [DAT-Nt (Miller et al., 1997)] in Tris-buffered saline with 2% nonfat dry milk. DAT antibody binding was detected using a sheep anti-rat horseradish peroxidase secondary antibody (ICN Biochemicals, Costa Mesa, CA) and enhanced chemiluminescence (Pierce, Rockford, IL). Densitometric analysis was performed and calibrated to coblotted dilutional standards of control striatum. Blots were then stripped for 20 min at 80°C (8m urea, 100 mm 2-mercaptoethanol, and 62.5 mm Tris, pH 6.8) and reprobed with an antibody to α-tubulin (Sigma).
In vivo microdialysis. In vivo microdialysis in freely moving mice was performed as described previously (Gainetdinov et al., 1997; Wang et al., 1997; Fumagalli et al., 1998). Mice were anesthetized with chloral hydrate (400 mg/kg, i.p.) and placed in a stereotaxic apparatus. Dialysis probes (2 mm membrane length; 0.24 mm outer diameter; cutoff of 6000 Da; Cuprophane; CMA/Microdialysis, Solna, Sweden) with CMA-11 guide cannulas implanted into the right striatum. The stereotaxic coordinates for implantation of microdialysis probes were as follows: anteroposterior, 0.0; dorsoventral, 4.4; and lateral, 2.5 for both wild-type and VMAT2 +/− mice relative to bregma (Franklin and Paxinos, 1996). Placement of the probe was verified by subsequent histological examination.
After surgery, animals were returned to their home cages with access to food and water ad libitum. Twenty-four hours after surgery, the dialysis probe was connected to a syringe pump (A-99; Razel Scientific Instruments, Stamford, CT) and perfused at 1 μl/min with artificial CSF (in mm): Na+ 150; K+ 3.0; Ca2+ 1.4; Mg2+ 0.8; PO4− 31.0; and Cl− 155, pH 7.3 (ESA Inc., Bedford, MA). After a 1 hr equilibration period, the perfusate was collected every 20 min into tubes containing 1 μl of 2 m perchloric acid. At least four control samples were taken before METH was administered. Perfusate samples were assayed for DA using HPLC-EC by the same chromatographic conditions described above. The sensitivity of the method permitted detection of ∼3 fmol of DA.
Analysis of free radical formation. To evaluate the level of hydroxyl radical formation, the “salicylate” microdialysis technique on freely moving mice was used (Obata and Chiueh, 1992;Chiueh et al., 1993; Fumagalli et al., 1998). The microdialysis procedure was performed as above. For trapping hydroxyl radicals, the striatum was perfused with 5 mm sodium salicylate in Ringer’s solution. Brain dialysate (1 μl/min) was collected every 20 min and assessed immediately for 2,3-DHBA and 2,5-DHBA concentrations using HPLC-EC under the chromatographic conditions described previously (Fumagalli et al., 1998).
Data analysis. The data are presented as means ± SEM and were analyzed statistically using ANOVA with Fisher’s protected least significant difference test.
RESULTS
Pharmacological characterization of DA pools in VMAT2 +/− mice
Previous studies demonstrated that reduced levels of VMAT2 protein in VMAT2 heterozygote knock-out mice result in a decrease in the tissue content and release of striatal DA (Fon et al., 1997; Wang et al., 1997; Gainetdinov et al., 1998a). To determine whether the impaired vesicular transport can affect intracellular compartmentalization of DA (i.e., portion of cytoplasmic vs vesicular storage) in VMAT2 +/− mice, the effects of drugs interacting with monoamine vesicular transport or synthesis of DA were assessed in the striatum and frontal cortex of wild-type (VMAT2 +/+) and VMAT2 +/− mice. Reserpine and tetrabenazine, irreversible and short-acting inhibitors of vesicular transport, respectively, were administered at 5 mg/kg, and the content of DA and its metabolites, DOPAC and HVA, were determined. Both drugs caused modest to marked depressions in the striatal and cortical DA content of the two genotypes at the time points examined, with reserpine displaying more pronounced effects (Fig.1A,B). In fact, the levels of DA in frontal cortex of reserpine-treated animals were below the limits of detection in our assay system. In contrast, DA metabolite levels were markedly increased (1.5-fold to sixfold) in both brain regions and in both genotypes, with the exception of reserpine-treated VMAT2 +/− striatum. In this case, no elevation was detected in either DOPAC or HVA levels. These observations (i.e., depression of tissue DA content and elevation of metabolite levels) represent characteristic responses to vesicular transport inhibitors and are believed to reflect monoamine redistribution from vesicles to cytoplasm and subsequent metabolism by monoamine oxidase (MAO) (Brodie et al., 1955; Carlsson, 1987; Callaway et al., 1989; Fairbrother et al., 1990; Colzi et al., 1993). It should be noted that, consistent with these pharmacological observations, the steady state tissue levels of DA are reduced and DOPAC levels are elevated in the striatum of VMAT2 +/− mice (Wang et al., 1997) (Figs.1, 2). Importantly, the reduced ability of the drugs at elevating DOPAC, the intraneuronal product of cytosolic DA metabolism by MAO (Zetterstrom et al., 1988), in VMAT2 +/− compared with wild-type mice suggests that less DA has been mobilized from the vesicles to the cytoplasm.
Fig. 1.
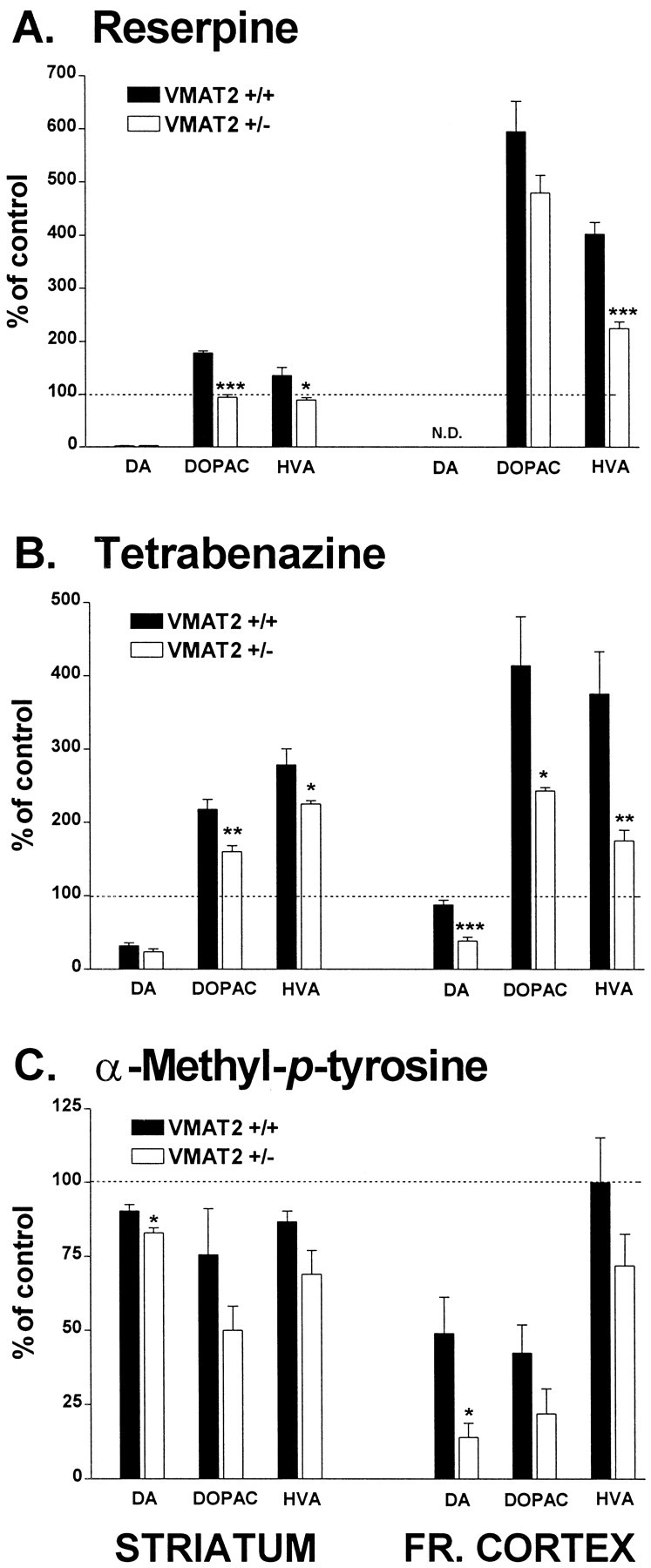
Effect of reserpine (A), tetrabenazine (B), and α-methyl-p-tyrosine (C) on DA and metabolite content in striatum and frontal cortex. The data are presented as percentages of DA, DOPAC, and HVA levels in saline-treated controls, respectively, which were as follows: striatum, 20.7 ± 2.3, 1.3 ± 0.05, and 1.6 ± 0.1 ng/mg wet tissue in wild-type mice; 15.2 ± 0.4, 1.7 ± 0.1, and 1.4 ± 0.05 ng/mg wet tissue in VMAT2 +/− mice; frontal cortex, 0.05 ± 0.03, 0.02 ± 0.01, and 0.08 ± 0.01 ng/mg wet tissue in wild-type mice; 0.06 ± 0.005, 0.03 ± 0.04, and 0.1 ± 0.005 ng/mg wet tissue in VMAT2 +/− mice. Animals treated with reserpine or tetrabenazine (5 mg/kg, i.p) were killed 6 and 1 hr, respectively, after injection. Animals treated with αMPT (250 mg/kg) were killed 1 hr after administration. Dopamine and metabolite levels were determined using HPLC-EC as described in Materials and Methods. Values represent the mean ± SEM of four to five independent determinations. ***p < 0.001; **p< 0.01; and *p < 0.05 versus treated wild-type animals.
Fig. 2.
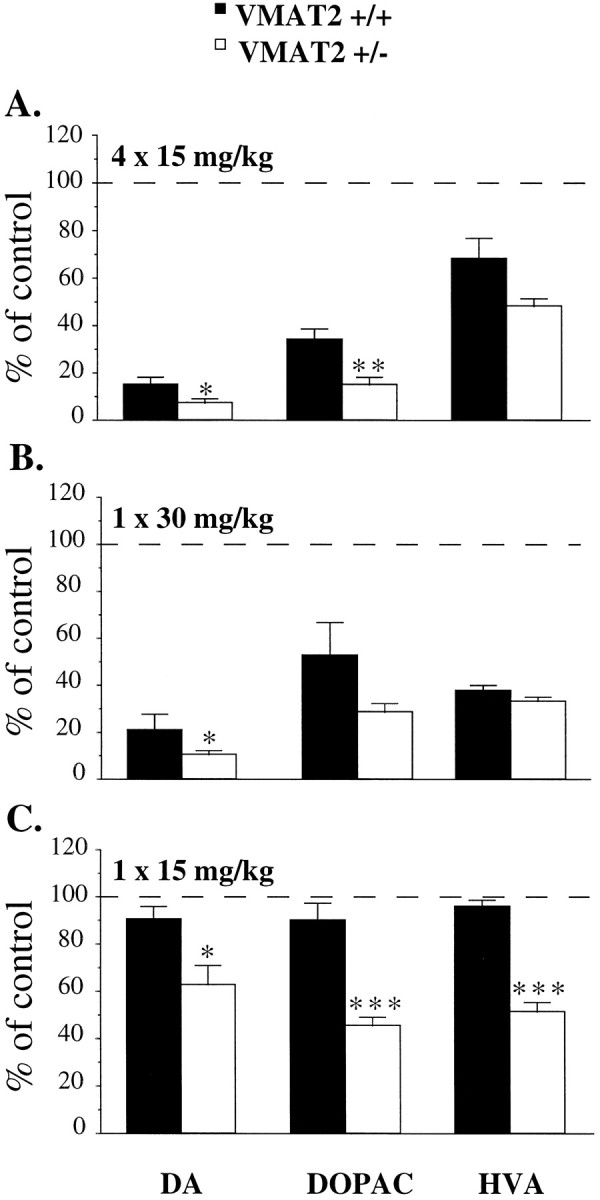
Effects of METH on striatal DA and metabolite content after different paradigms of administration and dosing regimens. A, Two-day post-METH administration. Animals were treated with METH (4 times at 15 mg/kg, s.c., each given 2 hr apart) and killed 2 d after the last injection. The data are presented as percentages of saline-treated controls, which were as follows: 15.6 ± 1.4, 1.08 ± 0.01, and 1.39 ± 0.1 ng/mg wet tissue in wild-type mice; 14.3 ± 1.2, 1.4 ± 0.3, and 1.47 ± 0.2 ng/mg wet tissue in VMAT2 +/− mice for striatal DA, DOPAC, and HVA, respectively. B, C, Seven-day post-METH administration. Animals were treated with a single subcutaneous injection of METH at 30 (B) or 15 (C) mg/kg and killed 7 d after injection. The data are presented as percentages of saline-treated controls, which were as follows: 20.7 ± 0.8, 1.2 ± 0.1, and 1.4 ± 0.1 ng/mg wet tissue in wild-type mice; 15.6 ± 1.2, 1.5 ± 0.1, and 1.5 ± 0.1 ng/mg wet tissue in VMAT2 +/− mice for striatal DA, DOPAC, and HVA, respectively. DA and metabolite levels were determined using HPLC-EC as described in Materials and Methods. Values represent the mean ± SEM of four to five independent determinations. **p < 0.01; *p< 0.05 versus METH-treated wild-type animals.
A quite different picture was revealed when the effect of catecholamine synthesis blockade on DA and metabolite levels was examined. Short-term treatment (1 hr) with 250 mg/kg intraperitoneal αMPT, a potent TH inhibitor (Javoy and Glowinsky, 1971; Bannon et al., 1981; Mignot and Laude, 1985; Fairbrother et al., 1990), modestly reduced striatal DA levels in both wild-type and VMAT2 +/− mice, with a slight but significant increase in the response in the latter animals (Fig.1C). Similarly, the greater reductions in DA levels were found in frontal cortex of VMAT2 +/− mice. The increased sensitivity to TH inhibition in frontal cortex compared with striatum confirms a higher DA turnover rate of mesocortical DA neurons (Bannon et al., 1981). Moreover, the heightened dopaminergic sensitivity to catecholamine synthesis inhibition in both brain regions of VMAT2 +/− mice suggests that, under basal conditions, the portion of newly synthesized DA, which is presumably cytoplasmic (Fairbrother et al., 1990; Seiden et al., 1993), is higher in heterozygous animals.
Effect of METH on DA content in striatum
The effects of METH on the dopaminergic parameters in the striatum of wild-type and VMAT2 +/− mice were measured by HPLC-EC as an index of neurotoxicity. Multiple injections of METH (four times at 15 mg/kg, s.c., each given 2 hr apart) produced an 85% decrease in DA levels in the striatum of wild-type animals 2 d after treatment (Fig.2A). However, METH caused a slightly greater reduction (−93%) in DA levels of VMAT2 +/− mice (Fig.2A). DOPAC and HVA levels also tended to be more depressed in VMAT2 +/− compared with wild-type animals (Fig.2A). The apparent greater reductions in DA and metabolite content of VMAT2 +/− mice compared with wild-type mice are consistent with an increased susceptibility of the dopaminergic system to the neurotoxic action of METH in mutant animals.
The long-term effects of METH toxicity were also tested 7 d after administration using two dosing regimens. First, a single injection of a high dose of METH (30 mg/kg, s.c.) was administered and found to produce substantial decreases in striatal DA, DOPAC, and HVA levels in both genotypes, although reduction in DA level was more pronounced in VMAT2 +/− mice (Fig. 2B). Whereas we previously used this same paradigm and clearly demonstrated striatal DA depletion in wild-type mice 7 d after METH administration (Fumagalli et al., 1998), in this study, we used a lower dose (15 mg/kg, s.c.) to better discriminate the decrease in DA content between the two genotypes. In this case, no significant decrease in DA or metabolite levels was observed in wild-type mice, whereas DA, DOPAC, and HVA levels were decreased 37, 54, and 49%, respectively, in VMAT2 +/− mice 7 d after METH injection (Fig. 2C). These results further support the contention that METH induces more pronounced changes in DA and metabolites in VMAT2 +/− than wild-type mice.
Effect of METH on striatal DAT protein levels
To test whether the more pronounced reductions in DA levels in VMAT2 +/− mice after METH reflect neurodegeneration rather than enhanced ability of the drug to deplete DA in these animals (Wilson et al., 1996a; Gainetdinov et al., 1998b; Fumagalli et al., 1998), we analyzed DAT protein levels as an index of nigrostriatal dopamine terminal integrity. Recent studies suggested that striatal DAT protein levels can reliably serve as a marker of DA terminal viability (Wilson et al., 1996b, Miller et al., 1997; Gainetdinov et al., 1998a). Western blot analysis of DAT protein was performed to assess the neurotoxic effects of METH in the mice. Administration of METH (15 mg/kg, s.c.) reduced DAT protein expression by 32.1 ± 5% in wild-type and 61 ± 11.4% in VMAT2 +/− mouse striatum, demonstrating a significant increase in the nigrostriatal damage in VMAT2 +/− mice (Fig. 3). No differences were observed, however, between VMAT2 +/− and wild-type mice in basal striatal DAT protein levels or synaptosomal uptake of DA (Gainetdinov et al., 1998b), suggesting that the increased neurotoxicity in heterozygous mice is not caused by alterations in DAT-mediated transport of DA and/or METH.
Fig. 3.

Effect of METH on striatal DAT-Nt immunoreactivity. Animals were treated with METH (15 mg/kg, s.c.), and striata were harvested 7 d after the drug administration. Densitometric analysis of DAT-Nt immunoreactivity was performed in both samples from individual animals (7 mice per group) and pooled samples for each group. Data are presented as a representative blot using pooled samples from each group. Immunoreactive bands of 85 kDa for DAT-Nt and 55 kDA for α-tubulin were observed. Lanes 1–5, Standard curve from wild-type mouse striatum (1.25, 2.5, 5, 10, and 20 μg, respectively). Lane 6, Wild-type mice, saline-treated (10 μg). Lane 7, Wild-type mice, METH-treated (10 μg). Lane 8, VMAT2+/− mice, saline-treated (10 μg). Lane 9, VMAT2+/− mice, METH-treated (10 μg). Individual measurements revealed a 32.1 ± 5% reduction in wild-type versus 61 ± 11.4% reduction in the VMAT2 +/− mice after METH.p < 0.05, between genotypes.
Effect of METH on striatal extracellular DA levels
In vivo microdialysis was used to measure extracellular levels of striatal DA in freely moving mice. METH administration (30 mg/kg, s.c.) induced 14-fold and sixfold increases in extracellular DA levels in wild-type and VMAT2 +/− mice, respectively (Fig.4A). These observations agree with previous reports, which demonstrate a reduced ability of amphetamine to release DA in VMAT2 +/− mice (Fon et al., 1997; Wang et al., 1997). It should also be noted that the basal extracellular DA levels are 40% lower in VMAT2 +/− mouse striatum compared with wild-type mice (Wang et al., 1997); dialysate concentrations of DA under the present conditions were 95.1 ± 28.3 (n= 5) and 61.4 ± 18.2 (n = 7) fmol/20 μl for wild-type and VMAT2 +/− mice, respectively. In addition, dialysate levels of DOPAC after METH were depressed similarly (∼50%) in both genotypes (data not shown), confirming that METH directly inhibits MAO (Seiden et al., 1993; Fumagalli et al., 1998) and/or deprives MAO of substrate via reverse DAT-mediated DA transport from cytoplasmic to extracellular compartments (Zetterstrom et al., 1988).
Fig. 4.
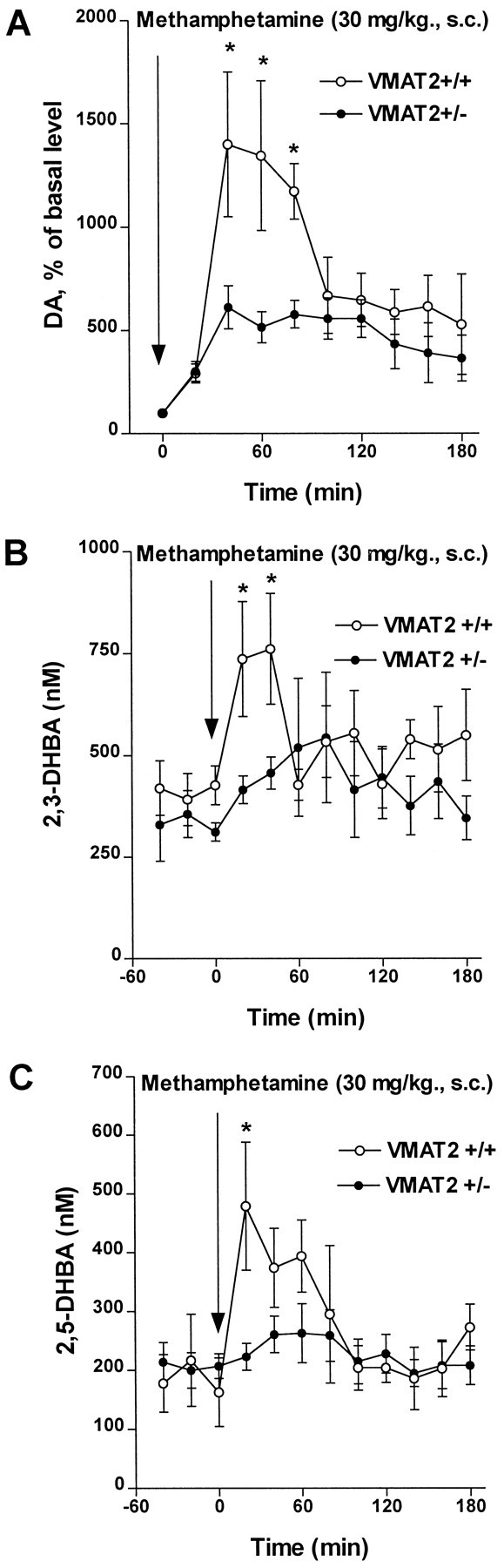
Effect of METH (30 mg/kg, s.c.) on extracellular striatal DA and indices of free radical formation. A, Microdialysis was used to measure extracellular monoamine levels in freely moving mice as described in Materials and Methods. The data are expressed as percentage of average values of at least three basal values before drug administration, each mouse used as its own control. Means ± SEM are shown (n = 5–7). All points representing the effect of METH in both wild-type and VMAT2+/− mice were significantly different (p < 0.05) from saline-treated controls (data not shown). *p < 0.05 versus VMAT2+/− mice.B, C, Effect of METH on the in vivo indices of hydroxyl radical formation in the striatum. 2,3-DHBA (B) and 2,5-DHBA (C) dialysate concentrations were measured during infusion of 5 mm salicylate into the striatum of freely moving mice as described in Materials and Methods. Values represent the mean ± SEM of four to six experiments. *p < 0.05 versus VMAT2+/− mice.
Effect of METH on the indices of hydroxyl radical formationin vivo
Formation of oxygen radicals has been suggested to underlie METH-induced neurotoxicity, most likely through intraneuronal and/or extraneuronal accumulation of DA and its consequent oxidation (De Vito and Wagner, 1989; Chiueh et al., 1993; Cadet et al., 1994;Cubells et al., 1994; Fleckenstein et al., 1997). Microdialysis and HPLC-EC were used to measure the effect of METH on the markers of hydroxyl radical formation, the levels of 2,3-DHBA and 2,5-DHBA, during intrastriatal infusion of salicylate (Obata and Chiueh, 1992; Coudray et al., 1995). In agreement with our previous study (Fumagalli et al., 1998), METH produced a transient twofold to threefold increase in both 2,3-DHBA and 2,5-DHBA levels in striatum of wild-type mice, whereas only a marginal increase was observed in VMAT2 +/− mice (Fig.4B,C). It is noteworthy that basal dialysate levels of 2,3-DHBA and 2,5-DHBA were found to be similar in mutant and wild-type mice.
Effect of METH on body temperature
Hyperthermia has been reported to directly correlate with dopaminergic neurotoxicity in mice (Bowyer et al., 1992, 1994; Albers and Sonsalla, 1995). To examine whether differences in hyperthermia might correlate with the increased susceptibility of VMAT2 +/− mice to METH administration, rectal temperature was monitored in wild-type and VMAT2 +/− mice before and after METH injection (15 and 30 mg/kg, s.c.). No differences were observed between the two genotypes in the basal core temperatures (37.1 ± 0.1 and 37.0 ± 0.1°C in wild-type and VMAT2 +/− mice, respectively) (n = 13). One hour after the injection of 15 mg/kg METH, body temperatures rose to 39.7 ± 0.2 (n = 7) and 39.4 ± 0.3°C (n = 7) and after 30 mg/kg METH to 39.0 ± 0.1 (n = 6) and 39.4 ± 0.2°C (n = 6) for wild-type and VMAT2 +/− mice, respectively. Thus, it is unlikely that a more pronounced hyperthermia could be a cause for the heightened sensitivity of VMAT2 +/− mice to METH-induced neurotoxicity.
DISCUSSION
In this study, we provide evidence indicating that an impairment in vesicular uptake increases METH-induced dopaminergic neurotoxicityin vivo. First, METH produced a greater depletion of DA and metabolite content in the striatum of VMAT2 +/− compared with wild-type mice. Second, DAT expression was reduced more in the striatum of VMAT2 +/− mice after treatment. Most importantly, however, our results are most consistent with intraneuronal DA redistribution making a greater contribution than extraneuronal overflow in mechanisms of METH-induced dopaminergic neurotoxicity. Indeed, despite the attenuated increase in extracellular DA and indices of hydroxyl radical formation in VMAT2 +/− mice in response to METH, a more pronounced METH-induced dopaminergic toxicity was found in VMAT2 +/− compared with wild-type mice.
Vesicular transporters, by modulating transport of neurotransmitters and/or neurotoxins into vesicles, represent potential sites for the regulation of synaptic function, as well as protection against neurotoxicity (Reinhard et al., 1988; Liu and Edwards, 1997; Takahashi et al., 1997; Varoqui and Erickson, 1997; Gainetdinov et al., 1998a). In fact, VMAT2 +/− mice are more susceptible to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) toxicity, demonstrating the role of vesicles in sequestration of MPP+, the toxic metabolite of MPTP (Takahashi et al., 1997; Gainetdinov et al., 1998a). The findings that diminished vesicular transport potentiates the damage caused by neurotoxins with different mechanisms of action (i.e., MPTP and METH) emphasizes the notion of vesicular stores as important defense systems against neurotoxicity. In the case of METH, perturbation of vesicular loading might alter the susceptibility of the neurons to degeneration, perhaps as a result of redistribution of neurotransmitters between storage vesicles and cytoplasm. Furthermore, these data, together with our previous results showing the essential role that DAT plays in METH-induced neurotoxicity (Fumagalli et al., 1998), suggest the importance of the balance between DAT- and VMAT2-mediated transport as a determinant of cell vulnerability to METH.
There is an extensive literature regarding the role played by DA as a mediator of the neurotoxic effects of METH. However, the question as to whether intraneuronal or released DA mediates METH-induced dopaminergic neurotoxicity has remained unanswered. The majority of the supporting data are consistent with neurotoxicity requiring DA release and extracellular reactive species (Seiden and Vosmer, 1984; Schmidt et al., 1985; Wagner et al., 1985; De Vito and Wagner, 1989; Marek et al., 1990b; O’Dell et al., 1991, 1993; Cadet et al., 1994). In the present study, in agreement with previous observations with amphetamine (Fon et al., 1997; Wang et al., 1997), we found that VMAT2 +/− mice display an attenuated striatal extracellular DA overflow after METH treatment compared with wild-type mice. In addition, indices of hydroxyl radical formation were elevated by METH markedly less in VMAT2 +/− mice than in wild-type animals. Nevertheless, more prominent DA and metabolite depletion and decrease in DAT expression were observed in heterozygous mice. These results suggest a dissociation between the ability of the drug to modulate extraneuronal DA dynamics and the degree of METH-induced neurotoxicity in VMAT2 +/− mice. Furthermore, these data suggest that alterations in intraneuronal DA compartmentalization rather than elevation in extraneuronal levels may represent the primary cause for the increased vulnerability of the cell to the neurotoxic action of METH.
The possibility that METH may produce neurotoxicity by affecting intracellular DA levels has been suggested previously (Liu and Edwards, 1997; Wrona et al., 1997; Uhl, 1998). Cubells and associates (1994)have reported that METH promotes intraneuronal DA-dependent formation of oxygen radicals in vitro, suggesting that interaction of METH with intracellular DA pools might represent a key mechanism of its neurotoxicity. METH, like amphetamine, is known to displace DA from presynaptic storage vesicles and to simultaneously inhibit its intracellular degradation by MAO (Seiden et al., 1993; Sulzer et al., 1995; Jones et al., 1998b; Fumagalli et al., 1998). The resulting elevation of cytosolic DA may facilitate its own oxidation. Here, we show that interruption of catecholamine synthesis by αMPT, which in the short term affects primarily newly synthesized cytoplasmic pool of DA (Fairbrother et al., 1990; Seiden et al., 1993), significantly decreased DA levels in VMAT2 +/− compared with wild-type mice, suggesting higher basal levels of cytoplasmic DA in heterozygous mice.
It is reasonable to propose in a situation of altered vesicular transport that both the perpetually higher basal levels of cytoplasmic oxidizable DA and the additional portion of DA redistributed from vesicular stores to cytoplasmic compartment in response to METH may together result in a higher oxidation rate, which exceeds the scavenging ability of cellular protective systems. Several studies support this interpretation. Reserpine-stimulated elevations in striatal cytoplasmic DA levels produce an increase in cytoplasmic levels of highly reactive 5-S-cysteinyldopamine, a product of DA oxidation (Fornstedt and Carlsson, 1989), and potentiate METH-induced dopaminergic degeneration (Wagner et al., 1983). Conversely, αMPT, which preferentially depletes the cytoplasmic pool of DA, attenuates METH-induced neurotoxicity (Wagner et al., 1983;Schmidt et al., 1985). Low vesicular storage of DA in DAT knock-out mice (Gainetdinov et al., 1998b; Jones et al., 1998a) might be responsible for the lack of METH neurotoxicity in these animals. In this case, the ability of METH to displace enough DA from vesicles to attain toxic cytoplasmic concentrations may be prevented (Fumagalli et al., 1998). These considerations imply an involvement of oxidative mechanisms in METH toxicity; however, in the present study, increased toxicity in VMAT2 +/− mice did not correspond to the degree of elevation of the indices of hydroxyl radical formation, at least at the time points examined. Instead, these indices were correlated to differences in extracellular DA dynamics. Thus, it is possible that the technique used in this study may be limited by the detection of hydroxyl free radicals originating solely from extracellular DA oxidation. Alternatively, the possibility that enhanced METH toxicity in VMAT2+/− mice might involve the reactive species undetectable by the in vivo salycilate microdialysis method cannot be ruled out.
Although the most likely explanation for the increased vulnerability to METH in the VMAT2+/− mice is intraneuronal redistribution of DA from the vesicular to the cytoplasmic compartment, other possibilities (such as alterations in MAO function or regulation of DA synthesis in mutant mice) cannot be completely excluded. Further studies will be required to examine other possibilities.
Several reports have suggested a direct correlation between METH-induced hyperthermia and the severity of dopaminergic neurotoxicity in mice (Bowyer et al., 1992, 1994; Albers and Sonsalla, 1995). In contrast, other studies indicate that hyperthermia cannot solely account for the neuron-damaging effect elicited by METH (Ricaurte et al., 1983; Wagner et al., 1983; Fumagalli et al., 1998), suggesting that factors other than thermal responses contribute to its neurotoxic effects. In our experiments, both genotypes displayed approximately the same degree of hyperthermia after METH injection, indicating that the increased toxicity observed in VMAT2 +/− mice is unlikely to be caused by a greater increase in body temperature.
In conclusion, we have demonstrated that genetic disruption of vesicular transport results in a potentiation of METH-induced dopaminergic neurotoxicity. Moreover, our data point to cytoplasmic DA, not released DA, as potentially a more important contributor of this effect. Together, our results demonstrate that compromised vesicular transport may represent an important mechanism involved in the susceptibility to METH-induced neurotoxicity. The addictive nature and recent escalation in abuse of METH and other related drugs stress the importance of understanding the mechanisms leading to their neurotoxicity. In addition, these observations may have important implications in the pathogenesis of neurodegenerative diseases, because cytoplasmic oxidation of DA is suggested to play a role in Parkinson’s disease (Liu and Edwards, 1997). Therefore, our findings, aside from providing insight into the factors that account for METH-induced neurotoxicity, might contribute to a better understanding of the basic mechanisms involved in DA-related neurodegeneration.
Footnotes
This work was supported in part by National Institutes of Health Grants ES-09248 (G.W.M.), NS-19576, and MH-40159, and unrestricted gifts from Bristol-Myers Squibb and Zeneca Pharmaceuticals (M.G.C.). M.G.C. is an Investigator of the Howard Hughes Medical Institute. R.R.G. is a visiting scientist from the Institute of Pharmacology, Russian Academy of Medical Sciences, Baltiyskaya 8, 125315 Moscow, Russia. We thank S. T. Suter, J. A. Holt, and S. N. Penland for excellent technical assistance.
Correspondence should be addresssed to Dr. Marc G. Caron, Howard Hughes Medical Institute, Box 3287, Duke University Medical Center, Durham, North Carolina 27710.
Dr. Fumagalli’s present address: Center of Neuropharmacology, Institute of Pharmacological Sciences, University of Milan, Via Balzaretti 9, 20133 Milano, Italy.
Dr. Valenzano’s present address: Pharmacopeia, Inc., 3000 Eastpark Boulevard, Cranbury, New Jersey 08512.
Dr. Miller’s present address: Division of Pharmacology and Toxicology, College of Pharmacy, University of Texas, Austin, Texas 78712-1074.
REFERENCES
- 1.Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective agents. J Pharmacol Exp Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- 2.Axt KJ, Commins DL, Vosmer G, Seiden LS. α-Methyl-p-tyrosine pretreatment partially prevents methamphetamine-induced endogenous neurotoxin formation. Brain Res. 1990;515:269–276. doi: 10.1016/0006-8993(90)90606-c. [DOI] [PubMed] [Google Scholar]
- 3.Bannon MJ, Bunney EB, Roth RH. Mesocortical dopamine neurons: rapid neurotransmitter turnover compared to other brain catecholamine system. Brain Res. 1981;218:376–382. [PubMed] [Google Scholar]
- 4.Bowyer JF, Tank AW, Newport GD, Slikker W, Jr, Ali SF, Holson RR. The influence of environmental temperature on the transient effects of methamphetamine on dopamine levels and dopamine release in striatum. J Pharmacol Exp Ther. 1992;260:817–824. [PubMed] [Google Scholar]
- 5.Bowyer JF, Davied DL, Schmued L, Broening HW, Newport GD, Slikker W, Jr, Holson RR. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- 6.Brodie BB, Pletscher A, Shore PA. Evidence that serotonin has a role in brain function. Science. 1955;122:968–972. doi: 10.1126/science.122.3177.968. [DOI] [PubMed] [Google Scholar]
- 7.Cadet JL, Sheng P, Ali S, Rothman R, Carlson E, Epstein CJ. Attenuation of methamphetamine-induced neurotoxicity in copper/zinc superoxide dismutase transgenic mice. J Neurochem. 1994;62:380–383. doi: 10.1046/j.1471-4159.1994.62010380.x. [DOI] [PubMed] [Google Scholar]
- 8.Callaway CW, Kuczensky R, Segal DS. Reserpine enhances amphetamine stereotypies without increasing amphetamine-induced changes in striatal dialysate dopamine. Brain Res. 1989;505:83–90. doi: 10.1016/0006-8993(89)90118-2. [DOI] [PubMed] [Google Scholar]
- 9.Carlsson A. Monoamines of the central nervous system: a historical perspective. In: Meltzer HY, editor. Psychopharmacology: the third generation of progress. Raven; New York: 1987. pp. 37–48. [Google Scholar]
- 10.Chiueh CC, Murphy DL, Miyake H, Lang K, Tulsi PK, Huang SJ. Hydroxyl free radical (·OH) formation reflected by salicylate hydroxylation and neuromelanin. In vivo markers for oxidant injury of nigral neurons. Ann NY Acad Sci. 1993;679:370–375. doi: 10.1111/j.1749-6632.1993.tb18324.x. [DOI] [PubMed] [Google Scholar]
- 11.Colzi A, D’Agostini F, Cesura AM, Borroni E, Da Prada M. Monoamine oxidase-A inhibitors and dopamine metabolism in rat caudatus: evidence that an increased cytosolic level of dopamine displaces reversible monoamine oxidase-A inhibition in vivo. J Pharmacol Exp Ther. 1993;265:103–111. [PubMed] [Google Scholar]
- 12.Coudray C, Talla M, Martin S, Fatome M, Favier A. High-performance liquid-chromatography electrochemical determination of salycilate hydroxylation products as an in vivo marker of oxidative stress. Anal Biochem. 1995;227:101–111. doi: 10.1006/abio.1995.1258. [DOI] [PubMed] [Google Scholar]
- 13.Cubells JF, Rayport S, Rajindron G, Sulzer D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular stress. J Neurosci. 1994;14:2260–2771. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Vito MJ, Wagner GC. Methamphetamine-induced neuronal damage: a possible role for free radicals. Neuropharmacology. 1989;28:1145–1150. doi: 10.1016/0028-3908(89)90130-5. [DOI] [PubMed] [Google Scholar]
- 15.Fairbrother IS, Arbuthnott GW, Kelly JS, Butcher SP. In vivo mechanisms underlying dopamine release from rat nigrostriatal terminals. I. Studies using veratrine and ouabain. J Neurochem. 1990;54:1835–1843. doi: 10.1111/j.1471-4159.1990.tb04880.x. [DOI] [PubMed] [Google Scholar]
- 16.Fleckenstein AE, Wilkins D, Gibb JW, Hanson GR. Interaction between hyperthermia and oxygen radical formation in the 5-hydroxytryptaminergic response to a single methamphetamine administration. J Pharmacol Exp Ther. 1997;283:281–285. [PubMed] [Google Scholar]
- 17.Fon EA, Pothos EN, Sun BC, Killeen N, Sulzer D, Edwards RH. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- 18.Fornstedt B, Carlsson A. A marked rise in 5-S-cysteinyl-dopamine levels in guinea pig striatum following reserpine treatment. J Neural Transm. 1989;76:155–161. doi: 10.1007/BF01578755. [DOI] [PubMed] [Google Scholar]
- 19.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; San Diego: 1996. [Google Scholar]
- 20.Fumagalli F, Gainetdinov RR, Valenzano KJ, Caron MG. Role of dopamine transporter in methamphetamine-induced neurotoxicity: evidence from mice lacking the transporter. J Neurosci. 1998;18:4861–4869. doi: 10.1523/JNEUROSCI.18-13-04861.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gainetdinov RR, Fumagalli F, Jones SR, Caron MG. Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J Neurochem. 1997;69:1322–1325. doi: 10.1046/j.1471-4159.1997.69031322.x. [DOI] [PubMed] [Google Scholar]
- 22.Gainetdinov RR, Fumagalli F, Wang YM, Jones SR, Levey AI, Miller GW, Caron MG. Increased MPTP neurotoxicity in vesicular monoamine transporter knockout mice. J Neurochem. 1998a;70:1973–1978. doi: 10.1046/j.1471-4159.1998.70051973.x. [DOI] [PubMed] [Google Scholar]
- 23.Gainetdinov RR, Jones SR, Fumagalli F, Wightman RM, Caron MG. Re-evaluation of the role of the dopamine transporter in dopamine system homeostasis. Brain Res Rev. 1998b;26:148–153. doi: 10.1016/s0165-0173(97)00063-5. [DOI] [PubMed] [Google Scholar]
- 24.Gibb JW, Kogan FJ. Influence of dopamine synthesis on methamphetamine-induced changes in striatal and adrenal tyrosine hydroxylase. Naunyn Schmiedebergs Arch Pharmacol. 1979;310:185–187. doi: 10.1007/BF00500283. [DOI] [PubMed] [Google Scholar]
- 25.Hotchkiss AJ, Gibb JW. Long-term effects of multiple doses of methamphetamine on tryptophane hydroxylase activity in rat brain. J Pharmacol Exp Ther. 1980;214:257–262. [PubMed] [Google Scholar]
- 26.Javoy F, Glowinsky G. Dynamic characteristics of the “functional” compartment of dopamine in dopaminergic terminals of the rat striatum. J Neurochem. 1971;18:1305–1311. doi: 10.1111/j.1471-4159.1971.tb00230.x. [DOI] [PubMed] [Google Scholar]
- 27.Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci USA. 1998a;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998b;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lesch KP, Heils A, Riederer P. The role of neurotransporters in excitotoxicity, neuronal cell death, and other neurodegenerative processes. J Mol Med. 1996;74:365–378. doi: 10.1007/BF00210631. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Edwards RH. The role of vesicular transport proteins in synaptic transmission and neural degeneration. Annu Rev Neurosci. 1997;20:125–156. doi: 10.1146/annurev.neuro.20.1.125. [DOI] [PubMed] [Google Scholar]
- 31.Marek GJ, Vosmer G, Seiden LS. Dopamine uptake inhibitors block long-term neurotoxic effects of methamphetamine upon dopaminergic neurons. Brain Res. 1990a;513:274–279. doi: 10.1016/0006-8993(90)90467-p. [DOI] [PubMed] [Google Scholar]
- 32.Marek GJ, Vosmer G, Seiden LS. The effects of monoamine uptake inhibitors and methamphetamine on neostriatal 6-hydroxydopamine (6-OHDA) formation, short-term monoamine depletions and locomotor activity in the rat. Brain Res. 1990b;516:1–7. doi: 10.1016/0006-8993(90)90889-j. [DOI] [PubMed] [Google Scholar]
- 33.Mignot E, Laude D. Study of dopamine turnover by monitoring the decline of dopamine metabolites in rat csf after α-methyl-p-tyrosine. J Neurochem. 1985;45:1527–1533. doi: 10.1111/j.1471-4159.1985.tb07223.x. [DOI] [PubMed] [Google Scholar]
- 34.Miller GW, Staley JK, Heilman CJ, Perez JT, Mash DC, Rye DB, Levey AI. Immunochemical analysis of dopamine transporter protein in Parkinson’s disease. Ann Neurol. 1997;41:530–539. doi: 10.1002/ana.410410417. [DOI] [PubMed] [Google Scholar]
- 35.Obata T, Chiueh CC. In vivo trapping of hydroxyl free radicals in the striatum utilizing intracranial microdialysis perfusion of salicylate: effects of MPTP, MPDP+ and MPP+. J Neural Transm. 1992;89:139–145. doi: 10.1007/BF01245361. [DOI] [PubMed] [Google Scholar]
- 36.O’Dell SJ, Weihmuller FB, Marshall JF. Multiple methamphetamine injections induce marked increases in extracellular striatal dopamine which correlate with subsequent neurotoxicity. Brain Res. 1991;564:256–260. doi: 10.1016/0006-8993(91)91461-9. [DOI] [PubMed] [Google Scholar]
- 37.O’Dell SJ, Weihmuller FB, Marshall JF. Methamphetamine-induced DA overflow and injury to striatal DA terminals: attenuation by DA D1 or D2 antagonists. J Neurochem. 1993;60:1792–1799. doi: 10.1111/j.1471-4159.1993.tb13405.x. [DOI] [PubMed] [Google Scholar]
- 38.Pifl C, Drobny H, Reither H, Hornykiewicz O, Singer EA. Mechanism of the dopamine-releasing actions of amphetamines and cocaine: plasmalemmal dopamine transporter and vesicular monoamine transporter. Mol Pharmacol. 1995;47:368–373. [PubMed] [Google Scholar]
- 39.Pu C, Vorhees CV. Developmental dissociation of methamphetamine-induced depletion of dopaminergic terminals and astrocyte reaction in rat striatum. Dev Brain Res. 1993;72:325–328. doi: 10.1016/0165-3806(93)90201-k. [DOI] [PubMed] [Google Scholar]
- 40.Reinhard JF, Jr, Daniels AJ, Viveros OH. Potentiation by reserpine and tetrabenazine of brain catecholamine depletions by MPTP in the mouse: evidence for subcellular sequestration as basis for cellular resistance to the toxicant. Neurosci Lett. 1988;90:349–353. doi: 10.1016/0304-3940(88)90214-5. [DOI] [PubMed] [Google Scholar]
- 41.Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- 42.Ricaurte GA, Fuller RW, Perry KW, Seiden LS, Schuster CR. Fluoxetine increases long-lasting neostriatal dopamine depletion after administration of d-methamphetamine and d-amphetamine. Neuropharmacology. 1983;22:1165–1169. doi: 10.1016/0028-3908(83)90075-8. [DOI] [PubMed] [Google Scholar]
- 43.Rudnick G, Clark J. From synapse to vesicle: the reuptake and storage of biogenic amine neurotransmitters. Biochim Biophys Acta. 1993;1144:249–263. doi: 10.1016/0005-2728(93)90109-s. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt CJ. l-DOPA potentiates the neurotoxicity of some amphetamine analogues. Ann NY Acad Sci. 1992;648:343–344. doi: 10.1111/j.1749-6632.1992.tb24576.x. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt CJ, Gibb JW. Role of the dopamine uptake carrier in the neurochemical response to methamphetamine: effects of amfonelic acid. Eur J Pharmacol. 1985;109:73–80. doi: 10.1016/0014-2999(85)90541-2. [DOI] [PubMed] [Google Scholar]
- 46.Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 1985;233:539–544. [PubMed] [Google Scholar]
- 47.Schuldiner S, Shirvan A, Linial M. Vesicular neurotransmitter transporters: from bacteria to humans. Physiol Rev. 1995;75:369–392. doi: 10.1152/physrev.1995.75.2.369. [DOI] [PubMed] [Google Scholar]
- 48.Seiden LS, Ricaurte G. Neurotoxicity of methamphetamine and related drugs. In: Meltzer HY, editor. Psychopharmacology: the third generation of progress. Raven; New York: 1987. pp. 359–365. [Google Scholar]
- 49.Seiden LS, Sabol KE. Neurotoxicity of methamphetamine-related drugs and cocaine. In: Chang LW, Dyer RS, editors. Handbook of neurotoxicology. Marcel Dekker; New York: 1995. pp. 825–843. [Google Scholar]
- 50.Seiden LS, Vosmer G. Formation of 6-hydroxydopamine in caudate nucleus of the rat brain after a single large dose of methylamphetamine. Pharmacol Biochem Behav. 1984;21:29–31. doi: 10.1016/0091-3057(84)90125-4. [DOI] [PubMed] [Google Scholar]
- 51.Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;32:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- 52.Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takahashi N, Miner LL, Sora I, Ujike H, Revay RS, Kostic V, Jackson-Lewis V, Przedborski S, Uhl GR. VMAT2 knockout mice: heterozygotes display reduced amphetamine-conditioned reward, enhanced amphetamine locomotion, and enhanced MPTP toxicity. Proc Natl Acad Sci USA. 1997;94:9938–9943. doi: 10.1073/pnas.94.18.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uhl GR. Hypothesis: the role of dopaminergic transporters in selective vulnerability of cells in Parkinson’s disease. Ann Neurol. 1998;43:555–560. doi: 10.1002/ana.410430503. [DOI] [PubMed] [Google Scholar]
- 55.Varoqui H, Erickson JD. Vesicular neurotransmitters transporters. Mol Neurobiol. 1997;15:165–192. doi: 10.1007/BF02740633. [DOI] [PubMed] [Google Scholar]
- 56.Wagner GC, Ricaurte G, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- 57.Wagner GC, Lucot JB, Schuster CR, Seiden LS. α-methyltyrosine attenuates and reserpine increases methamphetamine-induced neuronal changes. Brain Res. 1983;270:285–288. doi: 10.1016/0006-8993(83)90602-9. [DOI] [PubMed] [Google Scholar]
- 58.Wagner GC, Carelli RM, Jarvis MF. Pretreatment with ascorbic acid attenuates the neurotoxic effects of methamphetamine in rats. Res Commun Chem Pathol Pharmacol. 1985;47:221–228. [PubMed] [Google Scholar]
- 59.Wang YM, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, Miller GW, Wightman RM, Caron MG. Knockout of the vesicular monoamine transporter-2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron. 1997;19:1285–1296. doi: 10.1016/s0896-6273(00)80419-5. [DOI] [PubMed] [Google Scholar]
- 60.Weihmuller FB, O’Dell SJ, Marshall JF. l-DOPA pretreatment potentiates striatal dopamine overflow and produces dopamine terminal injury after a single dose of methamphetamine. Brain Res. 1993;623:303–307. doi: 10.1016/0006-8993(93)91442-u. [DOI] [PubMed] [Google Scholar]
- 61.Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med. 1996a;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- 62.Wilson JM, Levey AI, Rajput A, Ang L, Guttman M, Shannak K, Niznik H, Hornykiewicz O, Pifl C, Kish S. Differential changes in neurochemical markers of striatal dopamine nerve terminals in idiopathic Parkinson’s disease. Neurology. 1996b;47:718–726. doi: 10.1212/wnl.47.3.718. [DOI] [PubMed] [Google Scholar]
- 63.Wrona MZ, Yang Z, Zhang F, Dryhurst G. Potential new insights into the molecular mechanisms of methamphetamine-induced neurodegeneration. Natl Inst Drug Abuse Res Monogr. 1997;173:146–174. [PubMed] [Google Scholar]
- 64.Zetterstrom T, Sharp T, Collin AK, Ungerstedt U. In vivo measurement of extracellular dopamine and DOPAC in rat striatum after various dopamine-releasing drugs: implications for the origin of extracellular DOPAC. Eur J Pharmacol. 1988;148:327–334. doi: 10.1016/0014-2999(88)90110-0. [DOI] [PubMed] [Google Scholar]