

Abstract
The data we report in this study concern the types, location, numbers, forms, and composition of microscopic huntingtin aggregates in brain tissues from humans with different grades of Huntington’s disease (HD). We have developed a fusion protein antibody against the first 256 amino acids that preferentially recognizes aggregated huntingtin and labels many more aggregates in neuronal nuclei, perikarya, and processes in human brain than have been described previously. Using this antibody and human brain tissue ranging from presymptomatic to grade 4, we have compared the numbers and locations of nuclear and neuropil aggregates with the known patterns of neuronal death in HD. We show that neuropil aggregates are much more common than nuclear aggregates and can be present in large numbers before the onset of clinical symptoms. There are also many more aggregates in cortex than in striatum, where they are actually uncommon. Although the striatum is the most affected region in HD, only 1–4% of striatal neurons in all grades of HD have nuclear aggregates. Neuropil aggregates, which we have identified by electron microscopy to occur in dendrites and dendritic spines, could play a role in the known dendritic pathology that occurs in HD. Aggregates increase in size in advanced grades, suggesting that they may persist in neurons that are more likely to survive. Ubiquitination is apparent in only a subset of aggregates, suggesting that ubiquitin-mediated proteolysis of aggregates may be late or variable.
Keywords: Huntington’s disease, huntingtin, neuropil aggregates, nuclear inclusions, ubiquitin, neuropathology
Huntington’s disease (HD) is a progressive autosomal dominant neurodegenerative disease characterized by movement disorders, psychiatric manifestations, and dementia (Harper, 1996). The neuropathology of HD consists of selective neuronal loss occurring most prominently in the striatum and deep layers of the cerebral cortex (Hersch and Ferrante, 1997). Based on the severity of striatal neuropathology, HD cases have been classified as grades 0–4 (Vonsattel et al., 1985). Striatal degeneration also occurs in a gradient, starting dorsomedially and extending ventrolaterally (Vonsattel et al., 1985). In grade 1, 50% of caudate nucleus neurons are lost, yet there is preservation of much of the putamen and ventral striatum. In grade 4, there are almost no neurons left in the dorsal striatum, whereas neurons in the ventral striatum are still relatively spared (Vonsattel et al., 1985). Grade 4 is associated with end stage disease in patients who are bedridden and nearly vegetative (Myers et al., 1988).
The molecular basis of HD is the expansion of a CAG repeat encoding a polyglutamine tract in the N terminus of the HD protein huntingtin (Huntington’s Disease Collaborative Research Group, 1993). The CAG expansion is translated within the mutant HD gene and expressed in brain (DiFiglia et al., 1995; Gutekunst et al., 1995; Sharp et al., 1995; Trottier et al., 1995). Normally, huntingtin is a cytoplasmic protein expressed at high levels in the striatal neurons vulnerable to degeneration in HD and at low or undetectable levels in the neurons resistant to degeneration (Ferrante et al., 1997). The normal function of huntingtin is unknown; however, a role in intracellular transport has been proposed (DiFiglia et al., 1995; Gutekunst et al., 1995).
In HD brain, N-terminal fragments of mutant huntingtin were reported to accumulate and form inclusions in the nucleus (DiFiglia et al., 1997;Becher et al., 1998). Abnormal nuclear accumulations have also been observed in other glutamine repeat disorders (Paulson et al., 1997;Skinner et al., 1997; Becher et al., 1998; Holmberg et al., 1998) and in various animal and cell models (Davies et al., 1997; Ordway et al., 1997; Skinner et al., 1997; Cooper et al., 1998; Martindale et al., 1998). Thus, aggregation of mutant proteins in the nucleus might be a common cause of neuronal death in these diseases (Ross, 1997; Davies et al., 1998). If nuclear inclusions are pathogenic in HD, they should be readily identified in the regions and in specific neuronal types that are known to degenerate in HD. We have developed a fusion protein antibody that selectively recognizes aggregated huntingtin N-terminal fragments and labels many more aggregates in neurons and their processes in human brain than have been described previously. Using this antibody and human brain tissue of different pathological grades, we have compared the numbers and locations of nuclear and neuropil aggregates with the known patterns and gradients of neuronal death in HD. We observed that the distribution of nuclear aggregates does not correspond to the neuropathology of HD. Our results suggest a potential role for neuropil aggregates in dendritic pathology and indicate that aggregate ubiquitination may be late or variable.
MATERIALS AND METHODS
Antibodies and Western blot analysis. To generate an antibody specific to N-terminal fragments of huntingtin and without cross-reactivity with other proteins containing polyglutamine repeats, we used RT-PCR to obtain a truncated human cDNA that encodes the first 256 amino acids with an in-frame deletion of the polyglutamine stretch. The sense oligonucleotide primer used was 5′-TCGAGGTCGACCATGGCTACGTTAGAGAAATTAATGAAGGCTTTT-GAGAGTTTAAAAAGTTTTCAACAGCCGCCA, and the antisense primer used was 5′-GAAGGCCTTTAACAAAACCTTAATTTC. The resulting huntingtin cDNA had two CAGs and an in-frame deletion of the polyproline stretches (see Fig. 1). This cDNA was inserted into the pGEX vector to generate a glutathioneS-transferase (GST)-fusion protein in bacterial strain BL21. The purified GST-fusion protein was used as immunogen to produce a rabbit polyclonal antiserum (Covance, Denver, PA). The antiserum (EM48) was affinity-purified by incubation with a nitrocellulose strip containing transferred GST-huntingtin. Antibodies bound to the strip were then eluted with 0.2 mm Tris-glycine, pH 2.8, and neutralized by 1 m Tris-HCl, pH 8. Characterization of EM48 involved the use of brain tissues and huntingtin-transfected cells [Li and Li (1998); and also see below]. Protein samples were solubilized in SDS sample buffer and resolved by 8 or 10% SDS-PAGE. Blots were incubated with EM48 (1:500), and immunoreactive bands were visualized by chemiluminescence (Amersham, Arlington Heights, IL). EM48 immunoreactivity could be eliminated by overnight preabsorption of the antibody with 20 μg/ml GST-huntingtin but not GST alone. A rat monoclonal antibody (mHD549) to the internal region of huntingtin (amino acids 549–679) (Gutekunst et al., 1995) and rabbit polyclonal antibody to ubiquitin (Dako, Carpinteria, CA) were also used in the study.
Fig. 1.

Antibody EM48 and its reaction with N-terminal fragments of huntingtin. A, Schematic structure showing a truncated human huntingtin cDNA generated by PCR. Its corresponding region in wild-type huntingtin cDNA is shown above. The truncated protein contains only two glutamines in the glutamine repeat region and deletes the polyproline stretches. The numbers in parentheses represent the number of glutamines (Q) or prolines in normal human huntingtin.B, Western blots showing that EM48 recognizes native huntingtin (350 kDa band) in human brain cortex. C, EM48 immunofluorescent staining of 293 cells transfected with a series of cDNA constructs encoding different N-terminal fragments of human huntingtin with 120 glutamine repeats. The numbers in parentheses are N-terminal amino acid residues not including glutamine repeats. Note that only the huntingtin fragment (1–311) forms aggregates.
Huntingtin constructs and transfection. A partial huntingtin cDNA containing 150 CAG repeats was isolated from a λ phage DNA that contains exon 1 of the human HD gene [provided by Dr. Gillian Bates (University of College of London); see Mangiarini et al. (1996)]. Because of the instability of the CAG repeat in bacteria, we obtained a series of cDNAs encoding N-terminal huntingtin fragments with 23–150 glutamine (Li and Li, 1998). The sizes of the CAG repeats in these constructs were confirmed by sequencing or Southern blotting, as described previously (Li and Li, 1998). The full-length human huntingtin cDNA with 120 CAG repeats obtained in the previous study (Li and Li, 1998) was digested with KpnI, Ecor V, and BglI to generate cDNAs encoding N-terminal fragments of huntingtin (amino acids 1–586, 1–528, and 1–311). Because of the varied sizes of the polyglutamine in these truncated proteins, the indicated amino acids do not include the glutamine repeat. These N-terminal huntingtin fragments were expressed using the pCIS vector, which provides a stop codon. Subconfluent 293 cells were transfected with the same amount of huntingtin cDNAs (1 μg/well of a two-well chamber slide or 7 μg/10 cm dish) using lipofectAMINE (Life Technologies, BRL, Gaithersburg, MD). The cells were used for immunofluorescence 24 hr after transfection.
Immunofluorescent labeling of cultured cells. Transfected cells in chamber slides (Nalge Nunc, Naperville, IL) were fixed in 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.4% Triton X-100 in PBS for 30 min, blocked with 5% normal goat serum (NGS) in PBS for 1 hr, and incubated with primary antibodies in 2% NGS in PBS overnight. After several washes, the cells were incubated with secondary antibodies conjugated with either FITC or rhodamine (Jackson ImmunoResearch Lab, West Grove, PA). Huntingtin aggregates were readily recognized as large spherical structures (0.5–2 μm) labeled by EM48. On average, 200–300 cells transfected with huntingtin were randomly selected per experimental sample to count the spherical aggregates in the cells. A fluorescence microscope (Zeiss) and video system (Optronics DEI-470) were used to capture images. The captured images were stored and processed using Adobe Photoshop software.
Human brain tissues. Brain tissue from 12 HD patients, four controls without any evidence of neurological disease, and seven neuropathological controls, including Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, schizophrenia, and stroke were studied (see Table 1). The HD brains included neuropathological grades 1–4 (Vonsattel et al., 1985). Two of the HD cases had juvenile onset disease and advanced neuropathological severity. Some of the HD patients were followed, during life, at the Emory Huntington’s Clinic, and their clinical status at the time of death was well characterized. The other cases were contributed from the tissue archives at the Bedford VA Medical Center. For some of the early grade cases, which are extraordinarily rare, only a few sections were available. Thus, we were only able to examine a few brain regions in some of the cases. In every case, however, regions of cerebral cortex and striatum were examined. In as many cases as possible, tissue from the globus pallidus, additional regions of cerebral cortex, hippocampus, substantia nigra, and cerebellum were examined. CAG repeat length analysis could not be performed for the majority of the HD cases for which only fixed tissue was available. The number of CAG repeats in the HD allele was identified as 48 in case HD1 and 89 in HD11. Each HD brain originated from patients who had been clinically diagnosed on the basis of known family history and typical symptoms of HD. The presence of HD was confirmed by neuropathology, and the extent of neurodegeneration in the striatum was assessed using the grading system of Vonsattel (Vonsattel et al., 1985). The average age at death was 44 for the HD patients and 55 for the normal controls. All of the postmortem intervals before fixation were 26 hr or less and averaged 11 hr. Immunocytochemical studies were performed in representative coronal planes sampling the entire brain and including rostral, central, and caudal portions of the striatum. In two grade 1 cases, sections were available only from a plane rostral to the crossing of the anterior commissure.
Table 1.
Case information
| Case | Age (years) | Sex | PMI (hr) | Pathology |
|---|---|---|---|---|
| HD1 | 29 | F | 6 | HD grade 1 |
| HD2 | 82 | F | 17.4 | HD grade 1 |
| HD3 | 40 | M | 12 | HD grade 1 |
| HD4 | 46 | F | 20 | HD grade 2 |
| HD5 | 74 | M | 12.5 | HD grade 2 |
| HD6 | 86 | M | 2 | HD grade 3 |
| HD7 | NA | M | 7 | HD grade 4 |
| HD8 | 47 | M | 3.5 | HD grade 4 |
| HD9 | 78 | F | 10 | HD grade 3–4 |
| HD10 | 42 | F | 13 | HD grade 3–4 |
| HD11 | 17 | M | 5 | JHD grade 4 |
| HD12 | 22 | M | 3 | JHD grade 4 |
| CT1 | 68 | F | 3 | Control |
| CT2 | 31 | M | 26 | Control |
| CT3 | 60 | M | 20 | Control |
| CT4 | 64 | M | 11 | Control |
| ND1 | NA | F | 4 | AD |
| ND2 | 67 | M | 16 | AD |
| ND3 | 78 | M | 14 | AD |
| ND4 | 71 | M | 5 | PD |
| ND5 | 77 | M | 10 | MS |
| ND6 | 69 | M | 14 | Multiple infarcts |
| ND7 | 53 | M | 7 | Schizophrenia |
PMI, Postmortem interval time; HD, Huntington’s disease; AD, Alzheimer’s disease; PD, Parkinson’s disease; MS, multiple sclerosis; ND, not determined.
Case description: huntingtin aggregates in a presymptomatic patient. Brain tissue from presymptomatic individuals at risk for HD is extraordinarily rare because such young and healthy individuals only die accidentally. One such individual that we (S.M.H. and R.J.) have followed very closely in the Emory Huntington’s Disease Clinic died in a car accident. This individual was a 32-year-old woman who had been followed since undergoing predictive genetic testing in 1994. The CAG repeat lengths of her two IT15 alleles were 20 and 48, the latter being diagnostic of the HD mutation. Detailed neuropsychological testing was performed using the Wechsler Adult Intelligence Test-revised, Wechsler Memory Scale-revised, Wide-Range Achievement test-revised, controlled oral word association, and the California Verbal Learning Test, at a time of significant depression. Results indicated average intelligence and performance with impairments in mathematics and in the more challenging memory tasks. Her only past neuropsychiatric symptom was intermittent depression for which she at times received medications. She also was routinely tardy for work and appointments, had difficulties in her interpersonal relations, changed jobs several times, and was involved in multiple past motor vehicle accidents. In the months leading up to her death, she was not clinically depressed, was not taking antidepressant medications, and was performing well at a stable job. Her neurological examination, using the Huntington’s disease rating scale (Group, 1996), was completely normal, with no evidence of motor dysfunction. Her scores on the functional capacity scales of the United Huntington’s Disease Rating Scale did not indicate any decrements in her day-to-day functioning. Despite past hints of dysfunction, these examinations indicate that she was mentally and neurologically normal at the time of her death. As a result of the motor vehicle accident, she suffered multiple fractures, organ injuries, and a severe closed head injury. She was maintained in intensive care on a respirator for ∼2 d, during which she continued to deteriorate. After meeting clinical criteria for brain death, supportive care was discontinued. At autopsy, her brain was swollen, and contused areas of the cerebral cortex were edematous and bloody. Brainstem herniation was present and was considered to be the cause of brain death. Areas of contusion included the anterior portions of the temporal lobes, the frontal poles, and the posterior cerebellum, especially on the left. Many areas of the brain were well preserved and had no evident edema or hemorrhage and were examined by immunocytochemistry. These preserved regions included the most posterior areas of frontal cortex, the parietal lobes, the posterior portions of the temporal lobes, the hippocampus, most of the inferior cerebellar cortex, and deep forebrain structures including the basal ganglia, substantia nigra, and thalamus.
Immunocytochemistry in human brain. At autopsy, entire hemispheres were placed in cold (4°C) 2% paraformaldehyde-lysine-periodate solution for 8–15 hr, then removed, sliced, and returned to fresh fixative for a total of 24–36 hr of fixation. Tissue slices were then rinsed in 0.1 m sodium phosphate buffer (PB), pH 7.3, and placed in cold 20% glycerol/2% DMSO solution or 30% sucrose/30% ethylene glycol. Tissue blocks were then dissected from the basal ganglia, cerebral cortex, hippocampus, and cerebellum, and serial sections were cut at 50 μm using a freezing microtome or vibratome (Technical Products International). The cut sections were stored in cryoprotectant at −20°C for subsequent immunocytochemistry performed as described elsewhere (Gutekunst et al., 1995). Briefly, free-floating sections were incubated with primary antibodies in TBS containing NGS, 0.1% Triton X-100, and biotin (50 μg/ml). After rinses in TBS, the sections were incubated in biotinylated goat anti-rabbit antibody (Vector ABC Elite, Burlingame, CA) in TBS containing NGS. After several rinses in TBS, the sections were incubated in avidin–biotin complex (Vector ABC Elite). Immunoreactivity was visualized by incubation in 0.05% 3–3′-diaminobenzidine tetrahydrochloride (DAB) (Sigma, St. Louis, MO) and 0.01% hydrogen peroxide in 0.05 m Tris, pH 7. Controls included the omission of primary antibody and the preabsorption of the antibody with 20 μg/ml of the homologous fusion protein overnight. When used, counterstaining was with cresyl violet or thionine. We performed double immunoperoxidase labeling with rabbit polyclonal EM48 and rat monoclonal mHD549. Immunolabeling of adjacent sections by mHD549 or EM48 alone was included for comparison with the double labeling.
Electron microscopic immunocytochemistry in human brain.Immunoperoxidase staining was performed using vibratome sections as described above except that Triton X-100 was omitted from the blocking steps and from primary antibody solutions. After DAB visualization, sections were further fixed in 1% glutaraldehyde in 0.1 mPB, osmicated (1% OsO4 in 0.1 m cacodylate buffer), and stained overnight in 2% aqueous uranyl acetate. For more precise resolution, we used a pre-embedding immunogold method. In this method, sections were incubated overnight in Fab fragments of goat anti-rabbit secondary antibodies (1:50) conjugated to 1.4 nm gold particles (Nanoprobes, Stony Brook, NY) in PBS with 2% NGS. After rinsing in PBS and 0.1 m PB, sections were fixed in 2% glutaraldehyde in 0.1 m PB for 1 hr. After several washes in PB, sections were silver-intensified using the IntenSEM kit (Amersham International, Buckinghamshire, UK), post-fixed for 10 min in 0.5% OsO4 in PB, and processed for electron microscopy as described below.
All sections used for electron microscopy were dehydrated in ascending concentrations of ethanol and propylene oxide and embedded in Eponate12 (Ted Pella, Redding, CA). Ultrathin sections (90 nm) were cut using a Leica Ultracut S ultramicrotome. Thin sections were counterstained with 5% aqueous uranyl acetate for 5 min followed by lead citrate for 5 min and examined using a Hitachi H-7500 electron microscope. To determine the subcellular localization of the aggregates, several blocks were serially sectioned, and two to three sections were mounted on formvar-coated single-slot grids.
Quantification of aggregates in the brain. Examination of the HD brain tissue by light microscopy revealed differences in the density of EM48 immunoreactive aggregates between various grades of HD. To better analyze these differences we compared the densities and sizes of aggregates in grade 1, grade 2, and grade 4 cases. Counts and measurements were performed using single coronal sections from each case containing the striatum and insular cortex at a level just rostral to the crossing of the anterior commissure. In insular cortex, counts were performed in layers III and V/VI. Densities of aggregates were obtained as follows in three grade 1, two grade 4, and two juvenile cases. Immunoreacted sections (50 μm) were visualized on a Nikon Labophot-2 microscope using a 40× lens equipped with a 1 mm2 ocular grid. For each case, a random starting point in each layer was selected. From the starting point, counts were made using 10 240 μm2 frames obtained systematically according to a preset zigzag pattern within layer III or layers V/VI. This was repeated three times in each section and in each layer for a total of 30 frames in each. Within each frame, aggregates were counted and categorized as intranuclear or neuropil on the basis of their subcellular localization. In the striatum, a similar counting scheme was used; however, frames composed of >50% white matter or ventricle were excluded. A total of 30 frames each were counted from two grade 1, two grade 2, and two grade 4 cases.
The sizes of aggregates in layers V/VI of insular cortex were compared between two grade 1 and two grade 4 adult onset cases. EM48-immunoreacted sections were visualized using a Leitz microscope at 40×. The individual performing the counts was blinded to the identity and grade of the cases. For each case, five 40× images were selected using a similar systematic sampling scheme as described above and captured using an MCID image analysis system (Imaging Research, St. Catherines, Ontario, Canada). Images were then transferred to NIH Image (Rasband), and the areas of the first 10 aggregates touching a horizontal reference line were obtained.
We also compared densities of EM48 versus ubiquitin-positive aggregates in one grade 1 case and one grade 4 case. Counts were performed as described above in adjacent sections from layers V/VI of insular cortex. To determine whether aggregate size correlated with ubiquitination, we compared the size of EM48 aggregates and ubiquitinated aggregates in layers V/VI of a grade 1 case. Sizes were measured as described above using video microscopy and NIH Image.
RESULTS
Production of a fusion protein antibody (EM48) to the N-terminal region of huntingtin
We used PCR to generate a truncated huntingtin cDNA that encodes the first 256 amino acids of human huntingtin with a deletion of the polyglutamine and polyproline stretches (Fig.1A). This truncated cDNA was expressed as a GST-fusion protein that served as the immunogen to generate rabbit polyclonal antibody EM48. On Western blots EM48 reacted with native huntingtin in human brain. However, it reacted very weakly with huntingtin in rat brain (Fig. 1B), suggesting that it is specific to primate huntingtin.
We have shown previously that EM48 reacts with N-terminal huntingtin fragments (amino acids 1–212) with a normal or expanded glutamine repeat and that expanded polyglutamine caused N-terminal fragments of huntingtin (<212 amino acids) to form aggregates in transfected 293 cells (Li and Li, 1998). To further examine the specificity of EM48 and the relationship between the size of huntingtin and the formation of huntingtin aggregates, we performed an in vitro assay by transfecting HEK 293 cells with various N-terminal huntingtin fragments (amino acids 1–67, 1–212, 1–311, 1-528, and 1-586) and full length huntingtin, which contain >120 glutamine repeats (150Q or 120Q). The expressed proteins display various sizes on blots that parallel the length of the truncated proteins (data not shown). Immunofluorescent staining of transfected cells revealed that EM48 labeled spherical aggregates that were formed by an N-terminal huntingtin fragment containing the first 311 amino acids and an expanded glutamine repeat (Fig. 1C). The percentage of transfected cells that have aggregates appears to be dependent on the size of huntingtin protein expressed. We transfected equal amounts of each DNA construct, and the DNA sizes of the constructs did not differ enough to affect transfection efficiency. We found that 35, 45, and 17% of transfected cells with 150Q(1–67), 120Q(1–212), and 120Q(1–311) contained the aggregates, respectively. Because the N-terminal region was extended to 528 amino acids [120Q(1–528)], only 2% of transfected cells contained these spherical aggregates. The next longer fragment 120Q(1–586) and full length mutant huntingtin [120Q(1–3121)] with 120 glutamine repeats did not show any spherical aggregates; all of the expressed huntingtin was diffusely distributed throughout the cytoplasm. These results substantiate the view that huntingtin aggregates are composed of N-terminal fragments of huntingtin that contain fewer than the first 528 amino acids and an expanded glutamine repeat.
EM48 selectively labels huntingtin aggregates in HD brain
We used EM48 to examine postmortem brains from patients with HD and other neurological disorders (Table1). In sections from HD cases, EM48 predominantly and intensely labeled puncta of varying size and location (Fig. 2A). Diffuse cytoplasmic staining was very faint, suggesting that EM48 selectively recognized an aggregated form of huntingtin. Most EM48 immunoreactive puncta were larger than organelles and will be referred to as aggregates. Normal and disease control brains exhibited faint cytoplasmic staining. Interestingly, some plaques in the hippocampus and cortex of brain sections from the Alzheimer’s disease cases (data not shown) were immunoreactive, although not nearly as intensely as the aggregates in HD brain. All EM48 immunoreactivity was abolished when the primary antibody was omitted or preadsorbed with excess antigen.
Fig. 2.

Neuropil aggregates are not labeled by an antibody to the internal region of huntingtin. Adjacent sections through cerebral cortex from a grade 1 HD case were immunostained with (A) EM48 alone, (B) mHD549 alone, or (C) EM48 and mHD549 combined. mHD549 staining is found in the perikarya and proximal dendrites of the pyramidal neurons (A, B) but is not found in aggregates. In contrast, EM48 intensely labels the aggregates (arrowheads). Scale bar, 30 μm.
To investigate whether the EM48 aggregates contained full-length or N-terminal fragments of huntingtin, we performed double labeling with EM48 and mHD549, a rat monoclonal antibody that reacts with an internal region of human huntingtin (amino acids 549–679) (Gutekunst et al., 1995). A series of adjacent brain sections of the insular cortex, caudate, and putamen from a grade 1 HD case were examined. Sections stained with mHD549 alone (Fig. 2B) exhibited intense but diffuse staining in neuronal perikarya and proximal dendrites as described previously (Gutekunst et al., 1995). Fine punctate labeling was sometimes observed; however, mHD549 immunoreactive puncta were comparable in size to mitochondria (Ferrante et al., 1997) and smaller than those labeled with EM48. Thus, the aggregates recognized by EM48 were not labeled with mHD549. Sections stained with both EM48 and mHD549 showed complementary labeling of cytoplasm and aggregates (Fig.2C). This indicates that EM48 selectively recognizes aggregated huntingtin. Because EM48 was raised against the first 256 amino acids of huntingtin and mHD549 recognizes amino acids 549–679 of huntingtin, these data are consistent with the in vitro data above and with earlier studies (DiFiglia et al., 1997) indicating that the aggregates are formed by N-terminal fragments of mutant huntingtin (<549 amino acids).
Distribution and types of aggregates
EM48 labeled many more aggregates than did other previously reported antibodies (DiFiglia et al., 1997; Becher et al., 1998), providing a more complete and quite different picture of their morphology and distribution. These aggregates were heterogeneously distributed in different regions of the HD brain. They were primarily observed in gray matter and were infrequent in white matter. Aggregates were especially frequent in layers V and VI of cerebral cortex. However, differences between cortical areas within individual HD brains were observed. For example, in case HD1, insular and cingulate cortex had significantly higher densities of aggregates than prefrontal, temporal association and premotor cortex. In other regions, EM48-immunoreactive aggregates were at lower densities than in the cortex. These other regions include the caudate, putamen, substantia nigra, hypothalamic nuclei, thalamus, and brainstem nuclei such as nucleus cuneatus. In the striatum, aggregates were widely scattered, without any groupings suggestive of patch or matrix compartmentation. In the substantia nigra, most aggregates were in the pars compacta neuropil and very few were in pars reticulata. Aggregates were rarely seen in the globus pallidus, a major target of striatal axons. Aggregates were also rare in the hippocampus and cerebellum in which a few were visible in the molecular and granule cell layers, respectively. The distribution of cortical and striatal aggregates was studied much more intensely because these regions are particularly vulnerable in HD (see below).
In gray matter, EM48-immunoreactive aggregates were visible in three compartments: nuclear, perikaryal, and neuropil (Fig.3). Aggregates located in the neuropil (defined by not being present in nuclei or perikarya) were by far the most common. Their frequency and complexity have not been previously appreciated, primarily because earlier studies using other N-terminal huntingtin antibodies identified only a small fraction of aggregates.
Fig. 3.

Types of EM48-immunoreactive aggregates. Light micrographs showing EM48-labeled aggregates of different shapes and cellular localization in HD cortex. Aggregates were found in the neuropil (A–C) and in neuronal nuclei (D, long arrows) and perikarya (D, small arrows). In the neuropil, small spherical or fusiform aggregates were either scattered (A, arrowheads) or arranged in linear arrays (C) reminiscent of neuronal process. EM48 immunoreactivity is also found in long tubular (A, long arrow) or serpentine elements (B) reminiscent of short dendritic segments and branch points, some of which appeared to give rise to immunoreactive dendritic spines (A, B, small arrows). Scale bar (shown inD for A–D), 10 μm.
We have also confirmed our findings using an antibody against the first 17 amino acids of human huntingtin (provided by Dr. Peter Detloff, University of Alabama at Birmingham). The major axis of most neuropil aggregates ranged from 1.35 to 21.38 μm. Although most neuropil aggregates were round to oval (Fig. 3A–C), more tubular forms that could be hundreds of micrometers long and appeared to fill neuronal processes were also common (Fig. 3A). An anti-transglutaminase antibody that is selective for vascular endothelium did not label these tubular aggregates, indicating that they are not vascular structures. In the cerebral cortex, these tubular aggregates often had the size and orientation of apical dendrites. These tubular forms were not as intensely stained as the more punctate forms, suggesting that the protein they contain may not be as condensed. Curved multiform aggregates reminiscent of short dendritic segments and branch points were also seen (Fig. 3C), some of which appeared to give rise to aggregate-containing dendritic spines. Linear arrays of more intensely stained neuropil aggregates were also observed (Fig. 3B). In cortex, they were oriented like apical dendrites, whereas in striatum they tended to be curvilinear. These arrays suggest that the tubular and multiform aggregates might condense into a series of separate punctate aggregates. Because larger punctate aggregates are seen in later grade cases (see below), they may continue to grow in size. These different types of neuropil aggregates constituted the overwhelming majority of the aggregates in most of the brain regions of early grade HD cases, including cortical and subcortical regions. The dystrophic neurites that have been described previously, which averaged 5 μm in diameter (DiFiglia et al., 1997), are most likely a subset of the punctate neuropil aggregates we have observed. Because most neuropil aggregates are small and do not obviously alter the morphology of the processes that contain them, “dystrophic neurites” is not the best term for them. We have thus adopted the term “neuropil aggregates” so as not to suggest that the processes containing them are necessarily abnormal.
As described previously with other antibodies (DiFiglia et al., 1997;Becher et al., 1998), nuclear aggregates were round to fusiform in shape, usually one but occasionally two in number per nucleus, and had long axes ranging from ∼3 to 5 μm (Fig. 3D). Neurons containing nuclear aggregates also commonly contained smaller immunolabeled puncta in their perikarya (Fig. 3D). Labeled perikaryal puncta were round to oval and generally smaller than nuclear or neuropil aggregates, having diameters ranging from 0.3 to 1.5 μm. We were unable to determine whether these were small aggregates or labeled organelles by electron microscopy.
Ultrastructure of neuropil aggregates
Light microscopy suggested that at least some of the neuropil aggregates were contained within dendrites. To confirm this impression and to examine their subcellular structure, we performed immunoelectron microscopy on insular cortical layers V/VI of a grade 1 HD case. This region was chosen because it is especially enriched in smaller neuropil aggregates (<3 μm). Because these aggregates could only be identified by EM48 labeling, we used DAB and immunogold immunocytochemistry and examined serial ultrathin sections to help reconstruct the cellular elements containing the aggregates. Both DAB and immunogold yielded similar results, so we will describe only the immunogold results because labeling is more precise spatially (Fig.4). It is important to note that many of the processes containing aggregates were not identifiable, either because of the poor ultrastructural preservation of the postmortem tissue or a lack of distinctive morphology. Elements containing the largest aggregates were particularly difficult to identify, presumably because of mechanical distortion. However, all identifiable profiles (n = 30) containing aggregates were either dendrites or dendritic spines, identified on the basis of their size, shape, and postsynaptic association with axon terminals (Fig. 4). Often, the aggregates completely filled the cross-section of the process being examined. Very few immunogold particles were found outside the aggregates, confirming our impression that EM48 reacted more strongly with the aggregated than with soluble huntingtin and suggesting that there may be low levels of N-terminal fragments that are not within aggregates. Aggregates were not membrane bound. Likewise, no extracellular aggregates were identified that might have been released from dead and phagocytosed neurons. In addition, the neuropil aggregates did not contain any apparent membrane-bound structures such as vesicles or vacuoles. Neither were neuropil aggregates obviously surrounded by particular organelles such as mitochondria or vesicular organelles. No gold particles were seen in control sections for which the primary antibody was omitted. EM examination also showed that the neuropil aggregates were made up of granular and filamentous material that resembled the filaments seen in intranuclear aggregates in humans and in several transgenic models (Davies et al., 1997;DiFiglia et al., 1997; Ordway et al., 1997; Paulson et al., 1997). The width of the filaments was ∼10 nm, which is similar to those observed in huntingtin aggregates in exon-1 HD transgenic mice (Davies et al., 1997) and in vitro (Scherzinger et al., 1997). The filaments were often ordered and aligned along the axis of the dendrites (Fig.4C). These results demonstrate that neuropil aggregates are intracellular, that many exist within dendrites, and that their structure is similar to those of nuclear aggregates or huntingtin proteins aggregated in vitro.
Fig. 4.
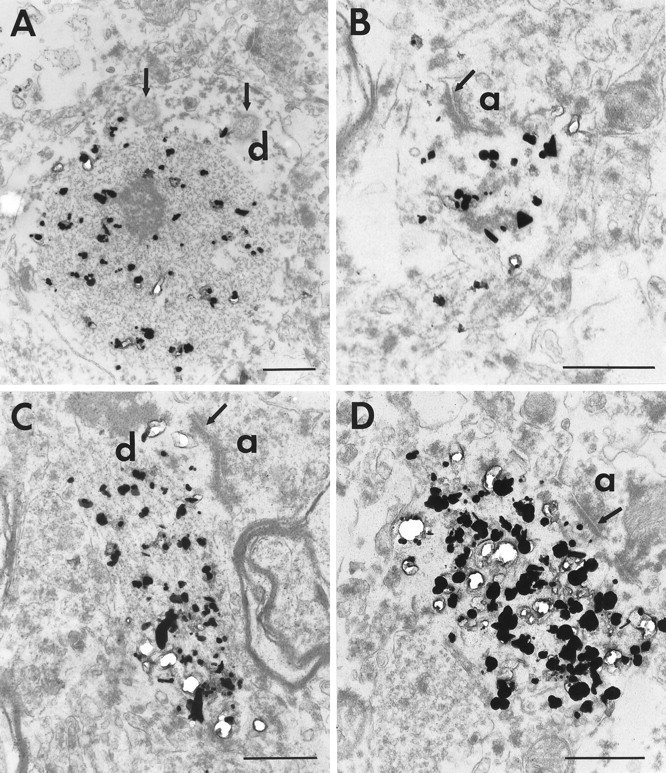
Neuropil aggregates in dendritic profiles. Electron micrographs of EM48 immunogold-labeled aggregates in insular cortex from an adult HD brain of grade 1. Immunogold particles are associated with aggregates made of filamentous material within dendritic processes. A is an example of a large caliber dendrite (d) containing an immunolabeled aggregate. Mitochondria (arrows) are seen in the cytoplasm adjacent to the aggregate. B shows a labeled aggregate in a dendritic spine receiving synaptic contact (arrow) from an axon terminal (a). In C and D, labeled aggregates are shown in longitudinal sections through two dendrites. In C, the filaments constituting the aggregates align with the orientation of the dendrite. Near the aggregates, the dendrites are receiving synaptic contact (arrow) from an axon terminal (a). Synaptic vesicles can be seen in the axon terminals (B, C). Scale bars, 500 nm.
Case description: huntingtin aggregates in a presymptomatic patient
A remarkably high density of neuropil aggregates but almost no nuclear aggregates were found in the areas of cerebral cortex that were examined (Fig. 5A). Aggregates were especially frequent in insular, cingulate, and dorsolateral prefrontal cortex but were less common in calcarine and superior parietal cortex and in hippocampus. The striatum showed degenerative changes dorsally, consistent with a grade 1 classification (the grading system applies to symptomatic cases, however). All types of striatal aggregates, however, were very rare, even in the dorsal striatum where degeneration had already started (Fig.5B). This unique case indicates that neuropil but not nuclear aggregates may be very common in the brain before the development of neurological symptoms. At the same time, nuclear aggregates were exceedingly rare, despite the overall burden of aggregation, at least in cerebral cortex. Most importantly, despite significant neuronal loss in a striatum still containing many normal areas, striatal aggregates seem too rare to correlate with the active degenerative process occurring there.
Fig. 5.

Huntingtin aggregates in human postmortem cerebral cortex and striatum from a presymptomatic case. Light micrographs are from the insular cortex (A) and dorsal striatum (B). Large numbers of EM48-immunoreactive aggregates of a wide variety of shapes and sizes are visible in cortex. All of these aggregates are in the neuropil. In contrast, striatal aggregates are exceedingly uncommon. Scale bar, 70 μm.
The relationship of aggregate and neuropathology
In the presymptomatic case described above, the cerebral cortex was found to contain an enormous number of aggregates at a stage during which cortical degeneration has not been described. At the same time, aggregates were quite rare in the striatum, despite the presence of significant neuronal loss dorsally. We studied this more closely by examining the density of aggregates in both regions more quantitatively (Fig. 6). In the striatum, aggregates were surprisingly uncommon in every HD brain. In grade 1, the deep layers of cortex had nearly 10-fold more aggregates (all types) per microscopic field than the striatum (24 aggregates per 240 μm2 field, compared with 2.6 aggregates per 240 μm2 field). The average 240 μm2 fields from which these counts were made in both the striatum and the deep layers of cortex contained 42 neurons. Thus, unbalanced neuronal loss was not a factor in this comparison. These results demonstrate that aggregates are much more common in cerebral cortex than in striatum at a time when cortical cell loss has not been identified, yet striatal cell loss is significant but not extensive enough to explain the low number of aggregates.
Fig. 6.

Neuropil and nuclear aggregates in cortex and striatum. Micrographs of coronal sections through the top layer (layer III, top panel) and the bottom layers (layers V/VI, middle panel) of insular cortex from adult HD brain of grade 1 (A, D, G), grade 4 (B, E, H), or juvenile HD brain (C, F). In the cortex of the grade 1 HD brain, neuropil aggregates are more frequent in layers V/VI than in layer III. More nuclear aggregates (arrowheads) are present in grade 4 and juvenile HD brain. Layer III from grade 4 adult HD brain has more aggregates than from grade 1. In the striatum, EM48-immunoreactive aggregates are present in grade 1 (G) and grade 4 (H), but at a much lower density than in cortex. Aggregates were absent in controls (I). Arrowheads indicate intranuclear aggregates. Scale bar, 50 μm.
If nuclear aggregates do cause neuronal death, they are likely to be present at a high density in more preserved areas of the striatum containing the largest numbers of dysfunctional and dying neurons. However, when areas of the striatum containing the highest aggregate densities were selected, the highest percentage of striatal cells containing nuclear aggregates was 10%. With systematic random sampling of 240 μm2 fields throughout the dorsal striatum, only 1–4% of neurons had nuclear aggregates. Within this range, higher-grade cases had the lowest numbers of aggregates, presumably because of the extensive loss of neurons. Thus it appears that aggregates are not distributed similarly to the known patterns of cell loss in HD.
We also examined the locations and relative numbers of neuropil and nuclear aggregates in cortex and striatum according to neuropathological grade. In insular cortex of grade 1 cases, the majority of aggregates were found in layers V and VI (Fig. 6). Aggregates were also present in layer III but at a much lower density (4.05 aggregates in layer III vs 24.04 aggregates in layers V/VI per 240 μm2). In all layers of cerebral cortex, virtually all (98.2%) EM48-immunoreactive aggregates were located in the neuropil. In grade 4 HD cases, there was a reduction in aggregate density in the deep layers of cortex, probably related to known neuronal loss, but an increased density was found in layer III. Although the majority of these aggregates were still located in the neuropil, 22% of them were found in neuronal nuclei. In two advanced juvenile HD cases, up to 32% of aggregates in insular cortex were nuclear. Similar results were found in cingulate cortex (data not shown). The opposite trend occurred in the striatum. Although all types of aggregates were infrequent, almost equal proportions of neuropil aggregates (51%) and nuclear aggregates (49%) were present in grade 1 striatum. In grade 2 and 4 cases, a higher proportion of aggregates were in the neuropil (up to 84%) than in nuclei (Fig.7), which might be the result of severe neuronal loss in advanced HD.
Fig. 7.

Quantitative analysis of neuropil aggregates in HD brains. This graph shows the percentages of neuropil and nuclear aggregates in layers III and V/VI of insular cortex and caudate nucleus from grade 1, grade 4, and juvenile HD cases. The proportion of aggregates that are in the neuropil decreases with disease grade in cortex but increases with disease grade in striatum.
If aggregates do not correlate with neuropathology, they could represent storage of huntingtin fragments occurring in neurons resistant to degeneration. If so, they might be expected to increase in size as the disease progresses. To help examine why we have identified so many more neuropil aggregates than previous studies and to determine whether there is any correlation between aggregate size and grade, we categorized the size and localization of the neuropil aggregates in HD of various grades. Because previous studies have shown that neuropil aggregates (dystrophic neurites) range from 3 to >10 μm (DiFiglia et al., 1997), we counted aggregates of various sizes (0.15–15 μm) in the lower layers of the insular cortex (Fig.8). In grades 1 and 4 of adult onset HD brains, we found that 82 and 34% of neuropil aggregates were smaller than 3 μm, respectively, indicating that EM48 recognizes many small aggregates not previously identified. In contrast, large neuropil aggregates (>3 mm) were more frequent in grade 4 (46%) than in grade 1 (18%), suggesting that the size of the aggregates increased with the duration of the disease.
Fig. 8.

The frequency of aggregates of various sizes in grade 1 and grade 4 cases. This graph shows the relative frequencies of aggregates based on their size using increments in their area of 15 nm2. Small aggregates are more frequent in earlier grade cases.
Most EM48-immunoreactive aggregates were not ubiquitinated
Abnormal aggregates in HD, as well as in other glutamine repeat disorders and their animal models, are commonly ubiquitin immunoreactive. This has been interpreted to indicate that resistance to proteolysis likely contributes to aggregation. EM48, however, appears to label many more aggregates than in previous studies in which similar numbers of aggregates were labeled with huntingtin and ubiquitin antibodies. We thus compared ubiquitin and EM48 labeling of aggregates in layers V and VI of grades 1 and 4 HD insular cortex (Fig.9). Some nuclear and neuropil aggregates but not the smaller perikaryal aggregates were ubiquitin immunoreactive (Fig. 9). The density of ubiquitin-immunoreactive aggregates was 31% of the density of EM48-immunoreactive aggregates in grade 1 (Fig.9A,C). In grade 4 (Fig. 9B,D), however, the density of ubiquitin-immunoreactive aggregates was 75% of the density of EM48-immunoreactive aggregates. Therefore, an increasing proportion of aggregates is ubiquitinated with a longer duration of disease. To examine whether ubiquitin-immunoreactive aggregates might make up a particular subset of aggregates, we compared their location and size to those labeled with EM48. Consistent with the low frequency of nuclear aggregates in early grades, all of the ubiquitinated aggregates in grade 1 were in the neuropil. In grade 4, each antibody labeled equal proportions of nuclear versus neuropil aggregates. In grade 1, EM48 and ubiquitin-immunoreactive aggregates had similar size and distribution, although a subset of EM48-positive aggregates were ubiquitinated.
Fig. 9.
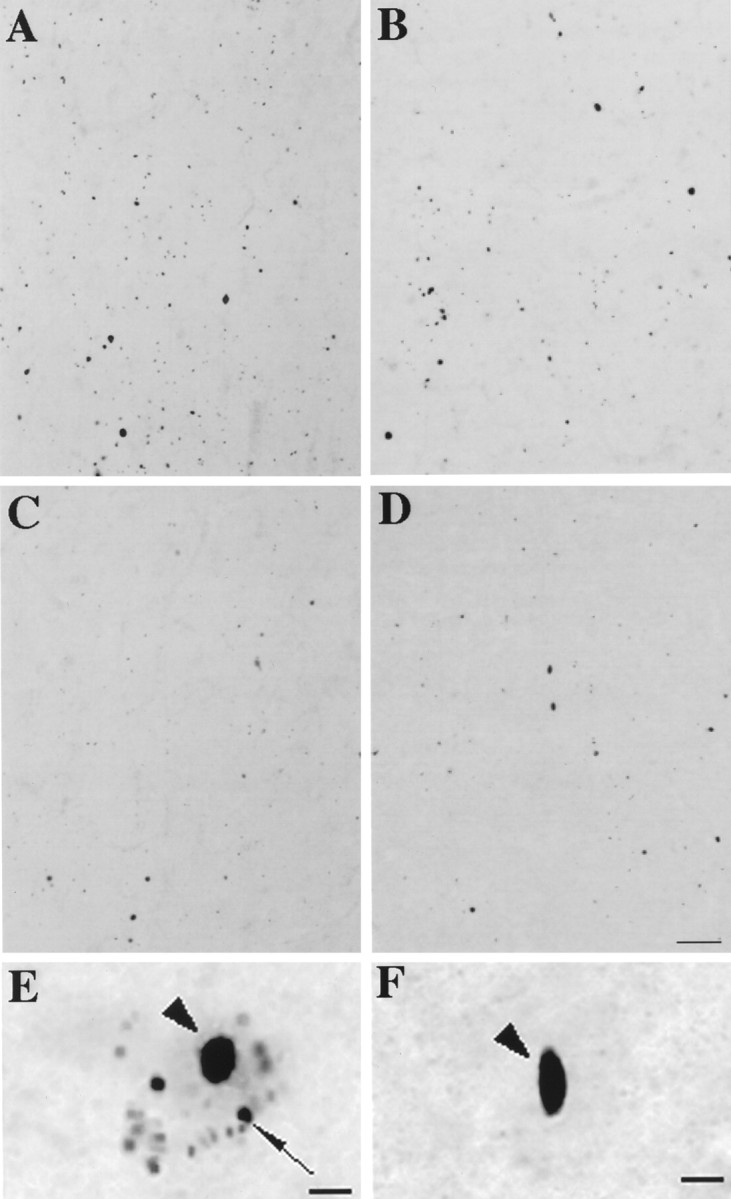
Ubiquitination of EM48 aggregates. Adjacent sections through layers V/VI of HD cerebral cortex immunolabeled with EM48 (A, B) and ubiquitin (C, D). In grade 1, the density of EM48-immunostained aggregates is three times that of ubiquitin-labeled aggregates (A, C). In grade 4, however, the densities are more similar (B, D). At higher magnification, it is evident that small perikaryal aggregates are stained by (E) EM48 but not by (F) anti-ubiquitin antibody. Scale bars:A–D, 70 μm; E, F, 10 μm.
DISCUSSION
The data we report in this study concern the types, location, numbers, forms, and composition of microscopic huntingtin aggregates in brain tissues from humans with different grades of HD and how well these aggregates correspond to the known patterns of neuronal loss that occur in the disease.
EM48 selectively labels huntingtin aggregates
We have made an antibody against the N-terminal 256 amino acids of huntingtin. EM48 appears to be much more sensitive for recognizing the huntingtin aggregates recently reported in HD brain (DiFiglia et al., 1997; Becher et al., 1998). In immunocytochemistry, EM48 also appears to be less sensitive to full-length huntingtin than it is to aggregates containing portions of its N terminus. The intense cytoplasmic staining that antibodies against internal epitopes give is only faintly apparent with EM48. This may suggest that modifications and interactions that the N terminus of huntingtin undergoes as a result of its fragmentation and aggregation makes it more recognizable to EM48.
Antibodies against internal regions of human huntingtin (amino acids 549–679) do not reveal huntingtin aggregates (DiFiglia et al., 1995;Gutekunst et al., 1995; Sharp et al., 1995; Trottier et al., 1995;DiFiglia et al., 1997; Becher et al., 1998). Similarly, when we expressed various N-terminal fragments of mutant huntingtin in transfected cells, we observed that aggregates were formed by fragments smaller than the first 538 residues and containing a polyglutamine expansion. This is consistent with other studies (Cooper et al., 1998;Hackam et al., 1998; Li and Li, 1998; Martindale et al., 1998) and confirms the selectivity of EM48 for aggregated N-terminal fragments of huntingtin. These findings support the hypothesis that proteolytically cleaved N-terminal fragments containing expanded polyglutamine lead to the formation of aggregates perhaps by acting as a polar zipper (Perutz et al., 1994) or through cross-linking by transglutaminases (Kahlem et al., 1996).
Nuclear aggregates and the pathogenesis of HD
Nuclear aggregates have been reported in neurons of HD patients (DiFiglia et al., 1997; Becher et al., 1998; Gourfinkel-An et al., 1998) and in patients with other glutamine repeat disorders including dentatorubral and pallidoluysian atrophy (Becher et al., 1998), spinocerebellar ataxia type 1 (SCA1) (Skinner et al., 1997), SCA3 (Paulson et al., 1997), and SCA7 (Holmberg et al., 1998). Nuclear aggregates have also been reported in transgenic mice expressing exon 1 of the HD gene (Davies et al., 1997) as well as in transgenic models of SCA1 and SCA3 (Paulson et al., 1997; Skinner et al., 1997). It has also been shown that the aggregation of truncated huntingtin in vitro is dependent on the presence of the polyglutamine expansion (Cooper et al., 1998; Li and Li, 1998;Martindale et al., 1998). All of these data led to the hypothesis that nuclear aggregates of mutant proteins are the common cause of neurodegeneration in these disorders and that the molecular context of the neurons in which they are expressed accounts for the differences between these diseases (Ross, 1997; Davies et al., 1998). In HD, however, it has not yet been shown that huntingtin aggregation or even the presence of nuclear huntingtin is injurious to neurons and not a secondary phenomenon. Our data suggest that nuclear aggregates in the striatum are not present in the known temporal and spatial patterns of neuronal loss in HD. More importantly, they seem to be too rarely found in the neurons most vulnerable in HD. Thus, our data suggest that nuclear aggregates, as visualized microscopically, may not play a causative role in HD. Alternatively, nuclear aggregates could form quickly and kill striatal neurons quickly, leaving few behind to be visualized. It also remains open whether nuclear huntingtin or even whether submicroscopic aggregates of huntingtin in the nucleus might be toxic. An additional possibility we have proposed previously (Ordway et al., 1997) is that huntingtin aggregation in the nucleus could represent benign sequestration of a protein fragment resistant to proteolysis. Intriguing support for this comes from our finding that many of the nuclear aggregates in the striatum are present in neurons that resist neurodegeneration in HD (Kuemmerle et al., 1998).
Our finding that EM48 does not label significant numbers of nuclear aggregates in the striatum is unlikely to be explained by the existence of many unrecognized aggregates or their loss in a degenerating striatum. First, EM48 recognizes many more aggregates than have been reported previously. Second, we have replicated our principal findings with EM48 using an antibody against the first 17 amino acids of huntingtin. Third, it is difficult to find nuclear aggregates in early grade human striatum by examining neuronal nuclei by electron microscopy. Finally, there are too many regions of well preserved striatum in our early grade cases for cell loss to explain not finding more aggregates.
Other emerging data also suggest that microscopic nuclear aggregates may not be pathogenic. It has been very difficult to show neuronal death in an animal model of HD in which there are nuclear aggregates (Davies et al., 1997). Transfected cells with cytoplasmic but not nuclear aggregates experience heightened cell death (Hackam et al., 1998). A transgenic model using a polyglutamine expansion to cause an unrelated cytoplasmic protein to translocate to the nucleus and aggregate did not show any neuronal death (Ordway et al., 1997). A recent cell culture study has suggested that nuclear but not aggregated huntingtin is toxic (Saudou et al., 1998). Finally, transgenic models of SCA1 have recently shown that nuclear but not aggregated ataxin-1 is responsible for the neurodegenerative phenotype (Klement et al., 1998). The presence of nuclear aggregates may thus be insufficient in cell or animal models to recreate the specific phenotype of HD.
Potential role of neuropil aggregates
One of the striking findings in this study was the previously unappreciated number and complexity of neuropil aggregates in HD. Aggregates in the neuropil were identified previously and termed dystrophic neurites (DiFiglia et al., 1997). We prefer the term neuropil aggregates, however, because they are present in processes that cannot be considered dystrophic. We were able to positively identify many neuropil aggregates in dendrites but not in axons, and we saw comparably few in white matter. Their presence in axon terminals, however, could not be excluded. Interestingly, degenerative and proliferative dendritic alterations have been described in the cortex and striatum in HD (Graveland et al., 1985; Ferrante et al., 1991;Sotrel et al., 1993), including changes in sizes and numbers of dendrites and dendritic spines. It is conceivable that the intradendritic formation of aggregates might underlie these changes and lead to functional alterations in the affected neurons. Many of the dendritic aggregates we observed seem large enough to disturb dendritic transport mechanically. Disturbance of intracellular transport at a molecular level is also possible because huntingtin has been shown to interact with proteins likely to play a role in cytoskeleton-based transport (Li et al., 1995; Kalchman et al., 1997; Wanker et al., 1997;Li et al., 1998). We have not been able to localize HAP1 in neuropil aggregates (Gutekunst et al., 1998), so perhaps the mechanisms that normally transport huntingtin are unable to clear the fragments that aggregate. Although striatal aggregates are uncommon, a connection between the large numbers of cerebral cortical neuropil aggregates and striatal pathology could exist by virtue of the potential importance of glutamatergic corticostriatal projections.
We observed large numbers of cerebral cortical neuropil aggregates in a presymptomatic individual in whom both nuclear and striatal aggregates were very rare. This finding suggests that neuropil aggregates might be present long before detectable symptoms or neuropathology. This is supported by cortical cell loss not being detected in grades 0 and 1 cases (Cudkowicz and Kowall, 1990; Hedreen et al., 1991; Sotrel et al., 1993; Rajkowska et al., 1998) and by volumetric magnetic resonance imaging that has not revealed significant cortical neuropathology until there are significant clinical symptoms (Aylward et al., 1998). Because cerebral cortex is affected less and later in HD, it might suggest that neuropil aggregates are relatively benign. However, a slow degenerative process from gradually accumulating toxicity would be consistent with the course of HD.
Aggregate ubiquitination
It has been suggested from an ataxin-1 model that misfolding of polyglutamine expansion containing proteins could prevent proteosomal proteolysis and lead to aggregate formation (Cummings et al., 1998). Evidence that this might be a general mechanism in polyglutamine disorders is that ubiquitination has been found to be a common feature of aggregates. We found a minority of huntingtin aggregates to be ubiquitinated in early grades, suggesting that in HD ubiquitination might occur after aggregates have been present for some time. Alternatively, only a subset of aggregates might become ubiquitinated, or ubiquitination may be transient. Another explanation might be the relative insensitivity of the ubiquitin antibodies; however, our small numbers are consistent with the findings of other investigators. Because aggregates increase in size with a longer duration of disease and a higher proportion become ubiquitinated, many aggregates and the neurons containing the aggregates may be relatively long-lived.
Footnotes
This work was supported by National Institutes of Health Grants NS36232 (X.-J.L.) and NS35255 (S.M.H., C.-A.G., R.J.F.), The Hereditary Disease Foundation and The Wills Foundation (X.-J.L.), the Huntington’s Disease Society of America (R.J.F., X.-J.L.,S.M.H.), the Veteran’s Administration (R.J.F.), and the Emory Huntington’s Disease Society of America Center of Excellence for Family Services (S.M.H., R.J.). S.K. is a medical student from Technische Universitaet Muenchen Medical School, Munich, Germany.
Correspondence should be addressed to Dr. Xiao-Jiang Li, Department of Genetics, Emory University School of Medicine, 1462 Clifton Road N.E., Atlanta GA 30322.
REFERENCES
- 1.Aylward EH, Anderson NB, Bylsma FW, Wagster MV, Barta PE, Sherr M, Feeney J, Davis A, Rosenblatt A, Pearlson GD, Ross CA. Frontal lobe volume in patients with Huntington’s disease. Neurology. 1998;50:252–258. doi: 10.1212/wnl.50.1.252. [DOI] [PubMed] [Google Scholar]
- 2.Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, Ross CA. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis. 1998;4:387–397. doi: 10.1006/nbdi.1998.0168. [DOI] [PubMed] [Google Scholar]
- 3.Cooper J, Schilling G, Peters M, Herring W, Sharp A, Kaminsky Z, Masone J, Khan F, Delanoy M, Borchelt D, Dawson V, Dawson T, Ross C. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum Mol Genet. 1998;7:783–790. doi: 10.1093/hmg/7.5.783. [DOI] [PubMed] [Google Scholar]
- 4.Cudkowicz M, Kowall NW. Degeneration of pyramidal projection neurons in Huntington’s disease cortex. Ann Neurol. 1990;27:200–204. doi: 10.1002/ana.410270217. [DOI] [PubMed] [Google Scholar]
- 5.Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 6.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 7.Davies SW, Beardsall K, Turmaine M, DiFiglia M, Aronin N, Bates GP. Are neuronal intranuclear inclusions the common neuropathology of triplet-repeat disorders with polyglutamine-repeat expansions? Lancet. 1998;351:131–133. doi: 10.1016/S0140-6736(97)08360-8. [DOI] [PubMed] [Google Scholar]
- 8.DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, Boyce FM, Aronin N. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 9.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 10.Ferrante RJ, Kowall NW, Richardson EP., Jr Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;11:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrante R, Gutekunst C-A, Persichetti F, McNeil S, Kowall N, Gusella J, MacDonald M, Beal M, Hersch S. Heterogeneous topographic and cellular distribution of huntingtin expression in the normal human neostriatum. J Neurosci. 1997;17:3052–3063. doi: 10.1523/JNEUROSCI.17-09-03052.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gourfinkel-An I, Cancel G, Trottier Y, Devys D, Tora L, Lutz Y, Imbert G, Saudou F, Stevanin G, Agid Y, Brice A, Mandel JL, Hirsch EC. Differential distribution of the normal and mutated forms of huntingtin in the human brain. Ann Neurol. 1997;42:712–719. doi: 10.1002/ana.410420507. [DOI] [PubMed] [Google Scholar]
- 13.Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- 14.Group HS. Unified Huntington’s disease rating scale: reliability and consistency. Huntington Study Group. Mov Disord. 1996;11:136–142. doi: 10.1002/mds.870110204. [DOI] [PubMed] [Google Scholar]
- 15.Gutekunst CA, Levey AI, Heilman CJ, Whaley WL, Yi H, Nash NR, Rees HD, Madden JJ, Hersch SM. Identification and localization of huntingtin in brain and human lymphoblastoid cell lines with anti-fusion protein antibodies. Proc Natl Acad Sci USA. 1995;92:8710–8714. doi: 10.1073/pnas.92.19.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutekunst C-A, Li S-H, Ferrante R, Li X-J, Hersch S. The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): comparison with huntingtin in rat and human. J Neurosci. 1998;18:7674–7686. doi: 10.1523/JNEUROSCI.18-19-07674.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hackam A, Singaraja R, Wellington C, Metzler M, McCutcheon K, Zhang T, Kalchman M, Hayden M. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harper P. Huntington’s disease. W. B. Saunders; London: 1996. [Google Scholar]
- 19.Hedreen JC, Peyser CE, Folstein SE, Ross CA. Neuronal loss in layers V and VI of cerebral cortex in Huntington’s disease. Neurosci Lett. 1991;133:257–261. doi: 10.1016/0304-3940(91)90583-f. [DOI] [PubMed] [Google Scholar]
- 20.Hersch S, Ferrante R. Neuropathology and pathophysiology of Huntington’s disease. movement disorders. Neurologic principles and practice (Watts RL, Koller WC, eds), pp 503–526. McGraw-Hill; New York: 1997. [Google Scholar]
- 21.Holmberg M, Duyckaerts C, Durr A, Cancel G, Gourfinkel-An I, Damier P, Faucheux B, Trottier Y, Hirsch EC, Agid Y, Brice A. Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum Mol Genet. 1998;7:913–918. doi: 10.1093/hmg/7.5.913. [DOI] [PubMed] [Google Scholar]
- 22.Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 23.Kahlem P, Terre C, Green H, Djian P. Peptides containing glutamine repeats as substrates for transglutaminase-catalyzed cross-linking: relevance to diseases of the nervous system. Proc Natl Acad Sci USA. 1996;93:14580–14585. doi: 10.1073/pnas.93.25.14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalchman MA, Koide HB, McCutcheon K, Graham RK, Nichol K, Nishiyama K, Kazemi-Esfarjani P, Lynn FC, Wellington C, Metzler M, Goldberg YP, Kanazawa I, Gietz RD, Hayden MR. HIP1, a human homologue of S. cerevisiae Sla2p: interacts with membrane-associated huntingtin in the brain. Nat Genet. 1997;16:44–53. doi: 10.1038/ng0597-44. [DOI] [PubMed] [Google Scholar]
- 25.Klement I, Skinner P, Kaytor M, Yi H, Hersch S, Clark H, Zoghbi H, Orr H. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 26.Kuemmerle S, Klein A, Gutekunst C-A, Li X-J, Li S-H, Hersch S, Ferrante R. Cellular distribution of huntingtin aggregation in spared and vulnerable neurons in Huntington’s disease. Soc Neurosci Abstr. 1998;24:973. [Google Scholar]
- 27.Li S-H, Li X-J. Aggregation of N-terminus of huntingtin is dependent on the length of its glutamine repeat. Hum Mol Genet. 1998;7:777–782. doi: 10.1093/hmg/7.5.777. [DOI] [PubMed] [Google Scholar]
- 28.Li S-H, Gutekunst C, Hersch S, Li X-J. Interaction of huntingtin associated protein with dynactin p150Glued. J Neurosci. 1998;18:1261–1269. doi: 10.1523/JNEUROSCI.18-04-01261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li XJ, Li SH, Sharp AH, Nucifora FC, Jr, Schilling G, Lanahan A, Worley P, Snyder SH, Ross CA. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378:398–402. doi: 10.1038/378398a0. [DOI] [PubMed] [Google Scholar]
- 30.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 31.Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 32.Myers RH, Vonsattel JP, Stevens TJ, Cupples LA, Richardson EP, Martin JB, Bird ED. Clinical and neuropathologic assessment of severity in Huntington’s disease. Neurology. 1988;38:341–347. doi: 10.1212/wnl.38.3.341. [DOI] [PubMed] [Google Scholar]
- 33.Ordway JM, Tallaksen-Greene S, Gutekunst CA, Bernstein EM, Cearley JA, Wiener HW, Dure LS, Lindsey R, Hersch SM, Jope RS, Albin RL, Detloff PJ. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell. 1997;91:753–763. doi: 10.1016/s0092-8674(00)80464-x. [DOI] [PubMed] [Google Scholar]
- 34.Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 35.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci USA. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajkowska G, Selemon LD, Goldman-Rakic PS. Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Arch Gen Psychiatry. 1998;55:215–224. doi: 10.1001/archpsyc.55.3.215. [DOI] [PubMed] [Google Scholar]
- 37.Ross CA. Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron. 1997;19:1147–1150. doi: 10.1016/s0896-6273(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 38.Saudou F, Finkbeiner S, Devys D, Greenberg M. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 39.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 40.Sharp A, Loev S, Schilling G, Li S-H, Li X-J, Bao J, Wagster M, Kotzuk J, Steiner J, Lo A, Hedreen J, Sisodia S, Snyder S, Dawson T, Ryugo D, Ross C. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron. 1995;14:1065–1074. doi: 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- 41.Skinner PJ, Koshy BT, Cummings CJ, Klement IA, Helin K, Servadio A, Zoghbi HY, Orr HT. Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature. 1997;389:971–974. doi: 10.1038/40153. [DOI] [PubMed] [Google Scholar]
- 42.Sotrel A, Williams RS, Kaufmann WE, Myers RH. Evidence for neuronal degeneration and dendritic plasticity in cortical pyramidal neurons of Huntington’s disease: a quantitative Golgi study. Neurology. 1993;43:2088–2096. doi: 10.1212/wnl.43.10.2088. [DOI] [PubMed] [Google Scholar]
- 43.Trottier Y, Devys D, Imbert G, Saudou F, An I, Lutz Y, Weber C, Agid Y, Hirsch EC, Mandel JL. Cellular localization of the Huntington’s disease protein and discrimination of the normal and mutated form. Nat Genet. 1995;10:104–110. doi: 10.1038/ng0595-104. [DOI] [PubMed] [Google Scholar]
- 44.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Wanker EE, Rovira C, Scherzinger E, Hasenbank R, Walter S, Tait D, Colicelli J, Lehrach H. HIP-I: a huntingtin interacting protein isolated by the yeast two-hybrid system. Hum Mol Genet. 1997;6:487–495. doi: 10.1093/hmg/6.3.487. [DOI] [PubMed] [Google Scholar]