

Abstract
To characterize the physiological role of metallothioneins I and II (MT-I+II) in the brain, we have examined the chronological effects of a freeze injury to the cortex in normal and MT-I+II null mice. In normal mice, microglia/macrophage activation and astrocytosis were observed in the areas surrounding the lesion site, peaking at ∼1 and 3 d postlesion (dpl), respectively. At 20 dpl, the parenchyma had regenerated. Both brain macrophages and astrocytes surrounding the lesion increased the MT-I+II immunoreactivity, peaking at ∼3 dpl, and at 20 dpl it was similar to that of unlesioned mice. In situ hybridization analysis indicates that MT-I+II immunoreactivity reflects changes in the messenger levels. In MT-I+II null mice, microglia/macrophages infiltrated the lesion heavily, and at 20 dpl they were still present. Reactive astrocytosis was delayed and persisted at 20 dpl. In contrast to normal mice, at 20 dpl no wound healing had occurred. The rate of apoptosis, as determined by using terminal deoxynucleotidyl transferase-mediated dUTP–biotin nick end labeling, was drastically increased in neurons of ipsilateral cortex of the MT-I+II null mice. Our results demonstrate that MT-I+II are essential for a normal wound repair in the CNS, and that their deficiency impairs neuronal survival.
Keywords: brain inflammation, MT-I+II, superoxide dismutase, oxidative stress, zinc, brain macrophages, astrocytes, neurons, apoptosis, regeneration, degeneration
Metallothioneins (MTs) are a family of low molecular weight, heavy metal-binding, cysteine-rich proteins. In the mouse, there are four isoforms, MT-I to MT-IV (Palmiter et al., 1992; Quaife et al., 1994). In the CNS, MTs occur in the isoforms MT-I, MT-II, and MT-III. MT-I+II are expressed in virtually all tissues, and in the brain they are localized mainly in astrocytes, microglia, leptomeningeal cells, ependyma, and choroid plexus epithelium (Young et al., 1991; Masters et al., 1994b; Penkowa and Moos, 1995; Penkowa et al., 1997). MT-III is primarily confined to the brain, but the data regarding its cellular localization are conflicting, because in situ hybridization analysis suggests neurons as the main site of expression (Masters et al., 1994b), whereas immunocytochemistry studies suggest microglia/macrophages and astrocytes as the cells with the highest MT-III protein content (Hozumi et al., 1996; Yamada et al., 1996; Penkowa et al., 1999).
The actual physiological role(s) of the different MT isoforms in the brain still remains to be established. Those of MT-I+II could be related to their putative antioxidant functions as well as zinc and/or copper metabolism (Sato and Bremner, 1993; Kelly et al., 1996; Aschner et al., 1997; Hidalgo et al., 1997). Presumably MT-III functions will differ from those of their normal counterparts MT-I+II, as suggested from in vitro (Uchida et al., 1991; Erickson et al., 1994;Palmiter, 1995; Sewell et al., 1995) and in vivo studies (Quaife et al., 1998). The intracerebral expression of MT-I+II is clearly upregulated during pathological conditions induced by trauma (Penkowa and Moos, 1995), immobilization stress (Hidalgo et al., 1990), kainic acid-induced seizures (Dalton et al., 1995; Zheng et al., 1995), excitotoxic NMDA cortex damage (Hidalgo et al., 1997), and administration of 6-aminonicotinamide (Penkowa et al., 1997). Furthermore, MT-I+II expression is increased in the myelin-deficient jimpy mouse (Vela et al., 1997) and in several human adult neurodegenerative disorders such as Alzheimer’s disease (AD), Pick’s disease (Duguid et al., 1989), and amyotrophic lateral sclerosis (Sillevis Smitt et al., 1992), as well as in aging (Suzuki et al., 1992) and after brain ischemia (Neal et al., 1996).
MT-III was discovered unexpectedly as a factor decreased in AD (Uchida et al., 1991), and a number of animal models have shown that MT-III mRNA or protein levels are significantly altered during CNS damage (Hozumi et al., 1995, 1996; Yamada et al., 1996).
Taken together, these studies strongly suggest that MTs are important proteins in the brain for coping with the tissue damage elicited by a wide array of factors and diseases. However, what the protective roles are, if any, remains unknown. The MT-I+II (Masters et al., 1994a)-deficient mice used in this report represent a unique experimental approach for determining the putative importance of these MT isoforms in the CNS during traumatic conditions.
MATERIALS AND METHODS
Production of the MT-I+II- and MT-III-deficient mice.Homozygous MT-I+II knock-out (KO) mice were generated as previously described (Masters et al., 1994a). The KO mice were raised on the 129/Sv genetic background; therefore, mice from this strain were used as controls.
Experimental procedures. Normal and genetically MT-I+II null adult mice were lesioned under tribromethanol anesthesia. The skull over the right frontoparietal cortex was exposed, and a focal cryoinjury on the surface of the brain was produced with dry ice (−78°C) (Penkowa and Moos, 1995). The animals were housed in cages with free access to food and water. The handling of the animals were approved by the proper committees of animal research and ethics of Spain and Denmark.
We performed three experiments. In the first one, control and MT-I+II null mice were lesioned and killed 3 d postlesion (dpl) along with unlesioned animals. In a second experiment, the animals were killed after 1, 3, 10, and 20 dpl. In a third experiment designed for in situ hybridization studies, the animals were killed by decapitation without tissue fixation 6 and 24 hr after the lesion along with unlesioned mice. Three to six mice per group were used in each experiment.
Mice were deeply anesthetized with Brietal and perfused with Zamboni’s fixative for immunohistochemistry, histochemistry, and terminal deoxynucleotidyl transferase (TdT)-mediated dUTP–biotin nick end labeling (TUNEL). For Timm’s silver sulfide staining we added Na2S to the Zamboni’s fixative. For in situhybridization studies, mice were killed by cervical dislocation, and the brains were immediately frozen in liquid nitrogen and stored at −80°C.
Cellular countings were performed for lectin-, GFAP-, neuronal-specific enolase (NSE)-, MT-I+II-, Cu/Zn-superoxide dismutase (Cu/Zn-SOD)-, and TUNEL-positive cells for statistical evaluation of the results. To this end, positively stained cells were counted from a 0.4 mm2 area of unlesioned hemispheres (data not shown) as well as from the ipsilateral site of the lesioned mice. Representative counting areas are shown in Figures1, 2, and 4; they are in the border of the lesion, where gliosis is prominent.
Fig. 1.

Lectin staining of lesioned normal (A, C, E, H, K, M) and MT-I+II null (B, D, F, G, I, J, L, N) mice. Boxes in A andB depict the areas from which cells were counted (see Fig. 8). A, Lectin staining in normal mice at 1 dpl, showing microglia/macrophages surrounding the lesion site (arrows). B, Lectin staining in MT-I+II null mice at 1 dpl, showing a recruitment of microglia/macrophages similar to that of normal mice (arrows).C, At 3 dpl, the lesion of normal mice is confined to cortical layers and encircled by round microglia/macrophages.D, At 3 dpl, the lesion of MT-I+II null mice is also seen in cortical layers; however, the number of round microglia/macrophages is increased. Infiltrating microglia/macrophages are observed both around the lesion site (small arrows) and in deeper regions of the brain (curved arrows). These cells are by morphology divided into two populations, one being small and round cells adjacent to the lesion (D, small arrows) and the other population observed in deeper regions of the brain (D, curved arrows); these cells are large and amoeboid. E, Higher magnification of the framed area inC. F, Higher magnification of the large and amoeboid cells in D (curved arrowsand square in D). G, Higher magnification of the small and round cells in D(small arrows and square inD). H, At 10 dpl, normal mice display partly regenerated parenchyme. The size of the lesioned area has further diminished. Lectin staining product is seen in both round microglia/macrophages and small vessels (arrows).I, At 10 dpl, the lesion site is still extensive and heavily infiltrated by macrophages in MT-I+II null mice.J, Higher magnification of the macrophages inI. K, At 20 dpl, the brain parenchyma of normal mice has been repaired and is now similar to that of unlesioned mice. L, The lesion is still extensive at 20 dpl in MT-I+II null mice, and signs of wound repair are absent. The number of microglia/macrophages is continuously high, and regenerating vessels are few in number. M, Higher magnification ofK, showing that most of the lectin staining product is confined to vessels (arrows). The number of microglia/macrophages (arrowheads) is similar to that of unlesioned mice. N, Higher magnification ofL, showing that most of the lectin staining product is confined to round and amoeboid macrophages. Scale bars: A–D, H, I, 285 μm; E–G, J, M, N, 25 μm;K, 320 μm; L, 115 μm.
Fig. 2.
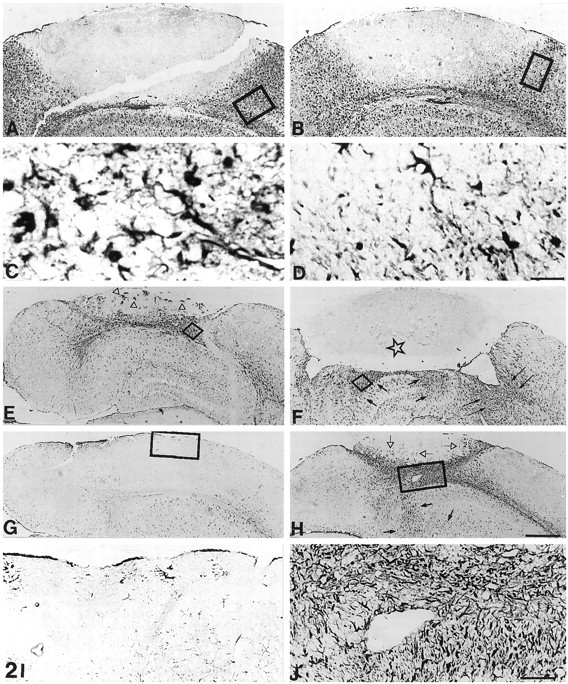
GFAP immunostaining of lesioned normal (A, C, E, G, I) and MT-I+II null (B, D, F, H, J) mice. A, At 3 dpl, numerous reactive astrocytes are surrounding the lesion in normal mice. B, In MT-I+II null mice at 3 dpl, reactive astrocytes are observed. However, the number of astrocytes is decreased compared with that of normal mice. C, Higher magnification ofA, showing reactive astrocytes. D, Higher magnification of B, showing less-reactive astrocytes than those of normal mice. E, At 10 dpl, the number of astrocytes surrounding the lesion is starting to decrease in normal mice, and simultaneously astrocytes are beginning to invade the injured area (arrowheads). F, The number of reactive astrocytes in lesioned MT-I+II null mice is increased at 10 dpl compared with that seen at 3 dpl. Several reactive astrocytes are seen at the border of the lesion (arrows); however, no astrocytes are invading the lesion (asterisk).G, At 20 dpl, the brain parenchyma is restored in normal lesioned mice, and the GFAP immunostaining is similar to that of unlesioned mice. H, At 20 dpl, the number of astrocytes surrounding the lesion is still high in MT-I+II null mice. Reactive astrocytes are seen as deep as in the basal nuclei (small arrows) as well as invading the lesioned area (arrowheads). The section showed is the neighboring section to that of Figure 1L. I, Higher magnification of the square in G, showing the same number of astrocytes as that of unlesioned mice.J, Higher magnification of the square inH. Scale bars: A, B, 200 μm; C, D, 20 μm; E–H, 280 μm; I, J, 57 μm.
Fig. 4.
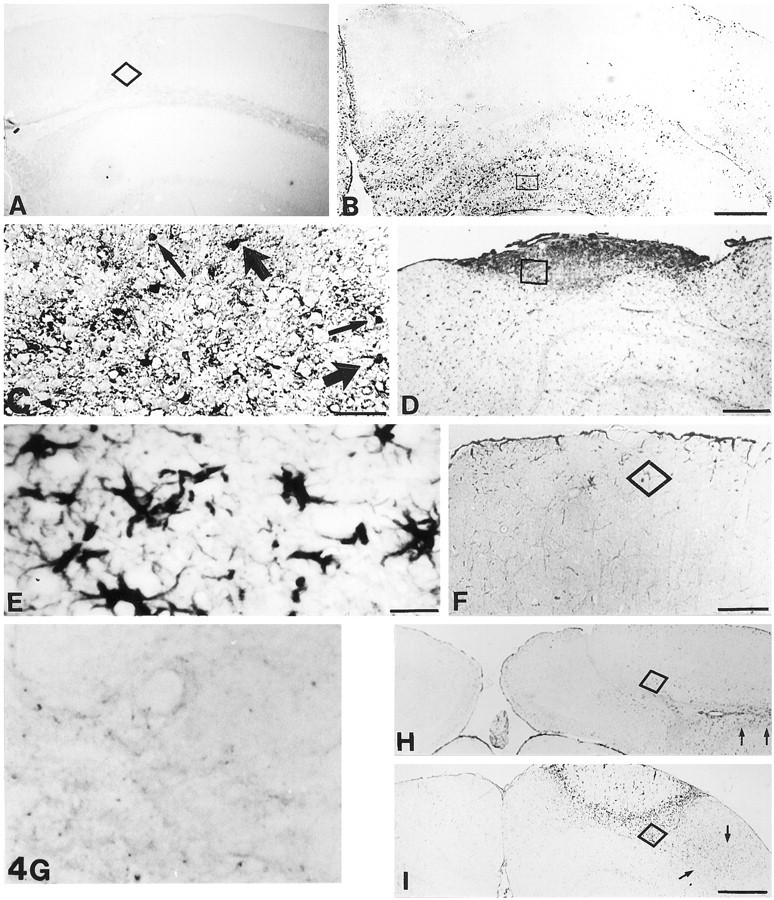
MT-I+II and Cu/Zn-SOD immunoreactivities in control and MT-I+II null mice. A, In unlesioned normal mice, MT-I+II expression is seen only in meninges, ependyma, and a few glial cells. B, In normal mice at 3 dpl, MT-I+II expression is significantly upregulated compared with that of unlesioned mice. MT-I+II is expressed in activated microglia/macrophages and reactive astrocytes surrounding the lesion.C, Higher magnification of the square inB, showing MT-I+II-positive cells. Both cells with macrophage phenotype (thin arrows) and astrocyte phenotype (thick arrows) are expressing MT-I+II.D, At 10 dpl in normal mice, MT-I+II expression is confined to the area containing activated microglia/macrophages and reactive astrocytes (seen in Figs. 1H, 2E, respectively). E, Higher magnification of the square in D, showing that stellate cells are the main source of MT-I+II expression at 10 dpl. F, At 20 dpl, the transient MT-I+II expression is decreased. G, As expected, MT-I+II immunoreactivity is absent in MT-I+II-deficient mice. H, Cu/Zn-SOD immunostaining in glia and neurons of normal mice at 20 dpl.I, At 20 dpl, MT-I+II null mice displayed increased SOD immunoreactivity compared with that of normal mice. The cells expressing Cu/Zn-SOD were microglia/macrophages, reactive astrocytes, and neurons situated around the continuously inflamed lesion site. Also, microglia/macrophages and reactive astrocytes situated more distantly to the lesion were expressing Cu/Zn-SOD (arrows). Scale bars: A, B, 200 μm;C, F, 57 μm; D, 114 μm; E, G, 20 μm; H, 285 μm; I, 450 μm.
Histochemistry. Biotinylated tomato lectin from theLycopersicon esculentum (Sigma, St. Louis, MO; code L9389), 1:500, was used as a marker for cells of the myelocytic and monocytic cell lineages, such as microglia/macrophages, as well as a marker for vessels. The lectin was developed using streptavidin–biotin–peroxidase complex (StreptABComplex/HRP; Dakopatts, Copenhagen, Denmark; code K377) prepared at the manufacturer’s recommended dilutions and performed for 30 min at room temperature. The reaction product was visualized using 0.015% H2O2 in 3,3′-diaminobenzidine/Tris-buffered saline (DAB/TBS), with DAB as a chromogen.
Immunohistochemistry. Sections were preincubated with pronase E (protease type XIV, Sigma, number P5147; 0.025 gm dissolved in 50 ml of TBS) for 10 min, pH 7.4, at 37°C followed by incubation in 10% goat serum in TBS (0.05 m Tris, pH 7.4, and 0.15m NaCl) with 0.01% Nonidet P-40 for 15 min at room temperature. Afterward, sections were incubated overnight with one of the following primary antibodies: polyclonal rabbit anti-bovine GFAP (1:250; Dakopatts, code Z 334) (as a marker for astrocytes), polyclonal rabbit anti-human NSE (1:1000; Dakopatts, code A589) (as a neuronal marker), monoclonal mouse anti-human neuro-filament (NF) (1:250; Dakopatts, code M762) (as a neuronal marker), polyclonal rabbit anti-rat liver MT-I+II (1:500; Gasull et al., 1993, 1994), and monoclonal mouse anti-human Cu/Zn-SOD (1:50; Sigma, code S2147). The NF and Cu/Zn-SOD antibodies were used after preincubation of the sections with blocking solutions from the HistoMouse-SP kit (Zymed, San Francisco, CA; code 95-9544) to quench endogenous IgG background staining by the secondary antibody used in mice tissue. The primary antibodies were detected using biotinylated monoclonal anti-rabbit IgG (Sigma, code B3275), 1:400, or biotinylated goat anti-mouse IgG (Sigma, code B8774), 1:200. These secondary antibodies were detected by StreptABComplex/HRP prepared at the manufacturer’s recommended dilutions. These secondary and tertiary steps in the immunoreaction were performed for 30 min at room temperature. The immunoreaction was visualized using 0.015% H2O2 in DAB/TBS with DAB as a chromogen.
Immunofluorescence. The presence of MT-I+II in microglia, macrophages, and astrocytes was verified by simultaneous staining for lectin and MT-I+II or GFAP and MT-I+II by using double-labeling immunofluorescence histochemistry. For this purpose we used Texas Red-labeled lectin (1:50; Sigma, number L-9139) simultaneously with monoclonal mouse anti-horse MT-I+II (1:40; Dakopatts, code M0638). The MT-I+II antibody was used after preincubation of the sections with blocking solutions from the HistoMouse-SP kit to quench endogenous IgG background staining by the secondary antibody in mice tissue. The MT-I+II were detected using goat anti-mouse IgG linked with amino-methylcoumarin (1:20) for 30 min at room temperature (Dakopatts, code W0477). Simultaneous detection of GFAP and MT-I+II was performed by using monoclonal rat anti-bovine GFAP (1:100; Zymed, code 13-0300) (as a marker for astrocytes). Anti-GFAP antibodies were detected using fluorescein-conjugated goat anti-rat IgG (H and L, 1:50) for 30 min at room temperature (Sigma, code F6258). Monoclonal mouse anti-horse MT-I+II were detected as mentioned above.
To evaluate the extent of nonspecific binding of the antisera in the immunohistochemical experiments, 1:100–1:1000 of normal rabbit or mouse serum or just the preincubation agent was substituted for the primary antibody step described above. Results were considered only if these controls were negative.
MT-I in situ hybridization. Brain MT-I mRNA levels were assayed by in situ hybridization in control and cryolesioned mice, which were killed 6 and 24 hr after the lesion. Serial coronal 30-μm sections were cut on a cryostat and mounted on poly-l-lysine-coated slides. For MT-I mRNA studies, we used the mouse cDNA kindly provided by Dr. R. D. Palmiter (University of Washington, Seattle, WA). MT-I and MT-II are isoforms regulated coordinately, and thus we assume that MT-I mRNA levels are representative of the MT-I+II isoforms (Masters et al., 1994b). The MT-I cDNA was labeled with [α-35S]UTP using an SP6/T7 transcription kit (Boehringer Mannheim, Mannheim, Germany). Preparation of sense and antisense probes and the in situ hybridization procedure were performed as previously described (Hernández et al., 1997a; Carrasco et al., 1998). Autoradiography was performed exposing the film (Hyperfilm-MP; Amersham, Arlington Heights, IL) to the slides for several days. All sections to be compared were prepared simultaneously and exposed to the same autoradiographic film.
Double immunohistochemistry–in situhybridization. To colocalize MT mRNA and MT protein, we simultaneously performed in situ hybridization for MT-I or MT-III mRNA and immunohistochemistry for MT-I+II and MT-III protein. The in situ hybridization procedure, except visualization, was performed before immunohistochemistry was carried out. The autoradiography of MT-I or MT-III mRNA (Hypercoat LM-1; Amersham, code RPN 40) was performed just previously to the visualization of MT-I+II and MT-III antibodies using DAB as chromogen.
Neo-Timm staining. Sections were physically (autometallographically) developed for 60 min at room temperature in solution containing silver lactate (Moos, 1993).
TUNEL technique. TUNEL staining was performed after tissue processing as mentioned above. Sections were deparaffinized and incubated with 20 μg/ml proteinase K (Sigma) for 5 min to strip off nuclear proteins. TUNEL was accomplished using the Apoptag Plusin situ apoptosis detection kit (Oncor, Gaithersburg, MD; code S7101-KIT). After immersion in equilibration buffer for 10 min, sections were incubated with TdT and dUTP-digoxigenin in a humidified chamber at 37°C for 1 hr and then incubated in the stop–wash buffer at 37°C for 30 min to stop the reaction. After washing in PBS buffer, the sections were incubated in anti-digoxigenin-peroxidase solution for 30 min. Afterward, DAB was used as chromogen, and the sections were counterstained with methyl green. Control sections were treated similarly but incubated in the absence of TdT enzyme, dUTP-digoxigenin, or anti-digoxigenin antibody. Furthermore, we performed control slides from Oncor (code S7115) for comparison. Besides detecting the staining reactivity for TUNEL, we also evaluated the morphological criteria of apoptosis, such as cell shrinkage and compactation of chromatin, to distinguish apoptosis from necrosis.
Double TUNEL–immunofluorescence histochemistry. To determine which cells were undergoing apoptosis, we performed double-labeling immunofluorescence histochemistry by using a fluorescein-linked apoptosis detection kit (TUNEL; Oncor, code S7110-KIT) simultaneously with one of the following antibodies: Texas Red-labeled lectin, polyclonal GFAP, and polyclonal NSE (as mentioned above). Anti-GFAP and anti-NSE were detected using swine anti-rabbit IgG linked with rhodamine (1:30) for 30 min at room temperature (Dakopatts, code R156).
Statistical analysis. Results were evaluated by two-way ANOVA, with strain and freeze lesion as main factors. When the interaction was significant, it was interpreted to be the consequence of a specific effect of the MT-I+II deficiency during the lesion. MT-I+II expression in MT-I+II null mice was evaluated by one-way ANOVA.
RESULTS
General
In unlesioned mice, gross inspection of the brain did not reveal significant differences between control and MT-I+II-deficient mice in the brain anatomy and histology of glial cells. In the lesioned mice, by gross examination of the brains a focal hemorrhage was seen in the right frontoparietal cortex at 1 and 3 dpl, whereas at 10 and 20 dpl only MT-I+II null mice displayed a visible hemorrhage. In toluidine blue-stained frontal sections, the freeze lesion was seen as a cortical area with no neuronal cells, but instead necrotic cells and debris were present. The tissue damage observed was higher in MT-I+II null mice.
Injury to the CNS produces a characteristic inflammatory response that results in migration of hematogenous cells into the damaged neural tissue and reactive gliosis (Ghirnikar et al., 1998). Activated microglia and bone marrow-derived monocytes transform to round phagocytes, generally referred to as microglia/macrophages (Stevens and Bähr, 1993; Perry et al., 1995). Astrocytes undergo hypertrophy and hyperplasia, generally referred to as reactive astrocytosis. These inflammatory cells of the brain are thought to be essential for maintaining neuronal survival and for tissue regeneration after CNS damage. These responses were thoroughly examined chronologically in the freeze lesion model used in this study, in line with previous results in the rat (Penkowa and Moos, 1995), providing a framework for characterizing the putative role of MTs in the CNS.
Microglia/macrophages
In normal mice, dramatic changes in the number and morphology of microglia/macrophages were observed surrounding the lesion in a highly temporal-specific manner (see Figs. 1, 8). Microglia/macrophages appeared to a great extent in the injured area at 1 dpl, and at 3 dpl the cells made a line of demarcation around the lesion (Fig.1A,C). Microglia/macrophages were round or amoeboid without ramifications (Fig. 1E). At 10 dpl, a majority of the recruited microglia/macrophages had disappeared, and instead, small capillaries appeared in the injured area (Fig. 1H). At 20 dpl, the tissue had regenerated, and the parenchyme appeared as that of unlesioned mice. Lectin staining product was seen in capillaries rather than round microglia/macrophages (Fig.1K,M).
Fig. 8.

Immunohistochemical cell countings in the brain cortex of 129/Sv and MT-I+II null mice (cells/0.4 mm2). We counted cells from the areas marked bysquares in Figures 1, 2, and 4. These areas are representative for all cell countings. Mice were either cryolesioned or left undisturbed and killed at the indicated times after the lesion. Cellular countings shown are mean ± SE (n = 3). Results were evaluated by two-way ANOVA. The effect of the cryolesion and the MT-I+II deficiency was significant (p at least < 0.01) in all cases. The main morphological features of these cells are shown in the previous figures in representative animals.
In MT-I+II null mice, an increase in microglia/macrophages was also observed around the lesion, but the temporal response was significantly different from that of normal mice (see Figs. 1, 8). At 1 dpl the number of microglia/macrophages in MT-I+II null mice was similar to that of normal mice (Fig. 1B). At 3 dpl the microglia/macrophages of MT-I+II null mice were significantly increased, and they appeared in two populations around the lesion (Fig.1D). One population, seen in the periphery of the lesioned area, was composed of large amoeboid cells (Fig.1D,F). The other population, seen adjacent to the necrotic area of the lesion, was composed of small and round cells (Fig. 1D,G). At 10 dpl, the parenchyma was heavily inflamed, and microglia/macrophages were still significantly increased in number in MT-I+II-deficient mice (Fig.1I,J). At 20 dpl, the freeze lesion still persisted, and the injured area was heavily infiltrated by round microglia/macrophages (Fig. 1L,N). Signs of tissue regeneration such as new vessel formation were almost absent in MT-I+II null mice.
Reactive astrocytes
After the freeze injury, a linear progression and afterward a decline in the number of reactive astrocytes were observed in normal mice, as verified from GFAP immunostaining (see Figs. 2, 8). Only a few reactive astrocytes appeared at 1 dpl, whereas numerous reactive astrocytes around the lesion were present at 3 dpl in normal mice (Fig.2A,C). At 10 dpl, reactive astrocytes were delineating the partly regenerated injury (Fig. 2E). At 20 dpl, the transient reactive astrocytosis had disappeared, and GFAP immunostainings were even lower than in unlesioned mice, probably because of alterations in the structure of cytoskeletal proteins as well as in the extracellular matrix caused by the lesion. Some astrocytes were seen around capillaries and as part of the glia limitans (Fig.2G,I).
In MT-I+II null mice, the expression of GFAP was delayed compared with that of normal mice (see Figs. 2, 8). Thus, at 1 dpl only a few reactive astrocytes were present, whereas at 3 dpl the number of reactive astrocytes had increased, but compared with that of normal mice at 3 dpl, the astrocytosis was reduced (see Figs.2A–D, 8). At 10 dpl, numerous reactive astrocytes appeared in MT-I+II null mice (Fig. 2F). Both around the lesion site and in deeper areas such as in the basal nuclei reactive astrocytosis was observed. In contrast to normal mice, in MT-I+II null mice the reactive astrogliosis continued at 20 dpl, with a thick layer of reactive astrocytes surrounding the lesion. In addition, several astrocytes were still identified in deeper areas of the injured hemisphere (Fig. 2H,J).
Neurons
As might be expected, the number of neurons was significantly decreased by the freeze lesion (see Figs. 3, 8). In normal mice, decreased numbers of NSE- and NF-expressing neurons were observed in neocortical layers surrounding the lesion zone at 1 and 3 dpl (Fig.3A,C). The neuronal morphology of these neurons was similar to that of unlesioned control mice, suggesting that they remained relatively unaffected. The number of NSE- and NF-expressing neurons was further decreased at 10 and 20 dpl (Fig. 3E,G).
Fig. 3.

NSE and NF immunostaining in lesioned normal (A, C, E, G) and MT-I+II null (B, D, F, H) mice. A, At 3 dpl, NSE-positive neurons (arrows) are seen in the parenchyma surrounding the lesion (triangles) of normal mice. B, At 3 dpl, the number of NSE-positive neurons around the lesion (triangles) of MT-I+II null mice is reduced compared with that of normal mice. C, NF immunostaining in normal mice at 3 dpl, which shows several apparently unaffected neurons in the parenchyma close to the injury (triangles).D, NF immunostaining in MT-I+II null mice at 3 dpl, which shows that neurons are present in the surroundings of the lesion (triangles), although almost no neurons contain NSE (seen in B). E, NSE immunostaining in normal mice at 20 dpl. Around the area that was lesioned, some NSE-containing neurons (thick arrows) are seen. Also, inside the regenerated parenchyme some small and shrunken NSE-positive cells are seen (small arrows). F, NSE immunostaining in MT-I+II null mice at 20 dpl. A few NSE-positive neurons (arrows) are seen surrounding the lesion, which is still present. G, NF immunostaining in normal mice at 20 dpl, showing several neurons in the parenchyme adjacent to the regenerated area. H, NF immunostaining in MT-I+II null mice at 20 dpl, showing neurons surrounding the lesion. Scale bars:A, B, 57 μm; C–F, 114 μm; G, H, 80 μm.
In MT-I+II null mice, the number of NSE-positive neurons in the cortex of unlesioned animals was decreased in comparison with normal mice (see Fig. 8). After the freeze lesion, the number of NSE- and NF-positive neurons dramatically decreased, significantly more in the MT-I+II null mice than in control mice (see Figs. 3, 8). Most of the neurons present in MT-I+II null mice at 3 dpl were either NSE-negative or showed a shrunken cytoplasm compared with those of normal mice at 3 dpl (Fig.3B,D). At 20 dpl, neurons surrounding the lesion in MT-I+II null mice either were still NSE-negative or had small, shrunken cell bodies with decreased levels of both NSE and NF (Fig.3F,H).
MT-I+II expression
In normal unlesioned mice, MT-I+II expression was confined to meninges, ependyma, and a few glial cells (Fig.4A). After the lesion, the expression of MT-I+II was increased in microglia/macrophages and reactive astrocytes of normal mice, peaking at ∼3 dpl (see Figs.4B–E, 8). At 20 dpl, the MT-I+II expression returned to normal (Fig. 4F).
As expected, in MT-I+II null mice no MT-I+II expression was detected (Fig. 4G). As verified from double immunofluorescence histochemistry, MT-I+II were detected in both microglia/macrophages and astrocytes (Fig. 5).
Fig. 5.

Double immunofluorescence for lectin, GFAP, and MT-I+II of lesioned normal mice at 10 dpl. A, Lectin staining showing microglia/macrophages from the borderline of the lesion. B, MT-I+II immunofluorescence in the same section as that seen in A. Small arrowsshow cells double-labeled with lectin and MT-I+II. Some microglia/macrophages are also seen as MT-I+II-negative (arrowheads). C, GFAP immunofluorescence from the borderline of the lesion in normal mice. D, MT-I+II in the same section as that seen in C.Small arrows show cells double-labeled with GFAP and MT-I+II. Some astrocytes are also seen as MT-I+II-negative (arrowheads). Scale bar, 20 μm.
Cu/Zn-SOD expression
In unlesioned normal and MT-I+II null mice, Cu/Zn-SOD expression was confined to glial cells and some neurons localized in the cortex. Cu/Zn-SOD expression followed the same pattern as that of gliosis. Thus, it was significantly increased by the lesion in a time-dependent manner, and the increase was higher and extended longer in MT-I+II-deficient mice (see Figs. 4H,I, 8). In normal mice, Cu/Zn-SOD immunoreactivity peaked at 3 dpl, and at 20 dpl it had returned to basal levels. Cu/Zn-SOD expression increased in microglia/macrophages, reactive astrocytes, and neurons, as verified by using double immunofluorescence histochemistry (data not shown). In MT-I+II-deficient mice, Cu/Zn-SOD expression increased significantly more than in normal mice and remained high even at 20 dpl (see Fig.8).
In situ hybridization of MT-I
A representative autoradiograph for MT-I and MT-III mRNA is shown in Figure 6. The quantifications performed in the border of the lesion of the ipsilateral and contralateral sites of the cortex of all the mice are also shown in Figure 6. It is clear that MT-I mRNA levels are significantly increased by the freeze lesion in a temporal manner, and that no effect is observed in the contralateral site. Thus, these results confirmed the immunocytochemical ones, suggesting that MT-I+II protein levels increase because of increased gene transcription.
Fig. 6.

Representative in situhybridization analysis of MT-I mRNA in normal unlesioned (Control) and lesioned (Lesion 24h) mice. The quantifications performed in all the animals are also shown (mean ± SE). MT-I mRNA levels in the ipsilateral and contralateral sites of normal mice were evaluated by one-way ANOVA, and only in the former was a significant effect of the lesion observed (p < 0.001).
Neo-Timm staining
Neo-Timm staining, which localizes reactive zinc, was only moderately increased after the freeze lesion in the ipsilateral cortex of the 129 mice, which returned to normal at 20 dpl. Timm staining was increased further in the MT-I+II null mice at all dpl studied (data not shown).
TUNEL labeling
In unlesioned normal and MT-I+II null mice, only a few cells dispersed in the cortex were labeled by TUNEL (see Fig. 8). In lesioned normal mice, a significant increase in TUNEL-positive cells was detected at 1 and 3 dpl (Figs.7A, 8). At 10 dpl, only a few apoptotic cells were detected, and at 20 dpl TUNEL labeling was limited to a few cells lying in the formerly lesioned area (Figs.7C, 8). The number of TUNEL-positive cells in normal mice at 20 dpl was similar to that of unlesioned mice.
Fig. 7.
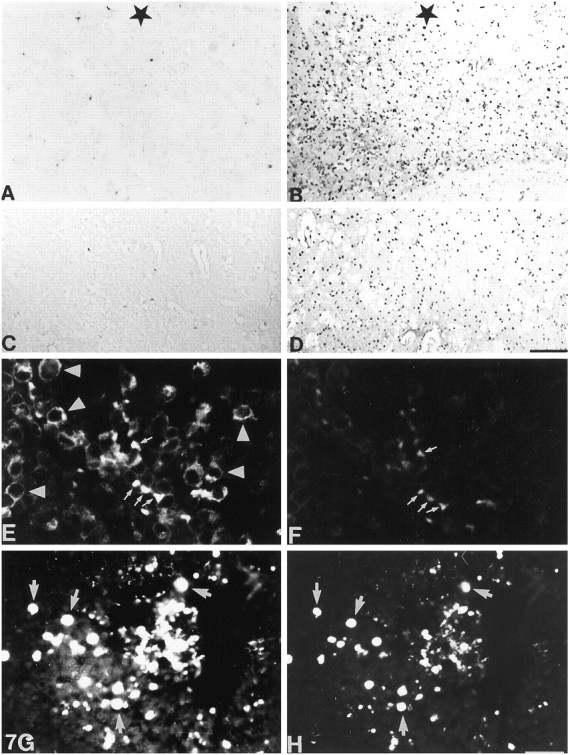
TUNEL staining in lesioned normal (A, C) and MT-I+II null (B, D) mice. Thestar indicates the lesion zone. A, In normal mice at 3 dpl, some cells are undergoing apoptosis.B, A dramatically high number of cells are undergoing apoptosis in MT-I+II null mice at 3 dpl. All around the lesion, cell nuclei are seen stained positively with TUNEL. C, In normal mice at 20 dpl, the tissue is regenerated, and almost no apoptotic cells are detected. D, A significantly high number of cells are still undergoing apoptosis in MT-I+II null mice at 20 dpl. E, At 3 dpl, apoptotic neurons (arrows) were present, and they appeared morphologically different from nonapoptotic neurons (arrowheads). Apoptotic neurons appeared round and shrunken, whereas nonapoptotic neurons showed normal cytoplasm staining. Most of the NSE-positive neurons were not undergoing postlesional apoptosis. F, Fluorescein-linked TUNEL staining in normal mice at 3 dpl, showing ongoing apoptosis in some of the neurons seen in E.G, H, Simultaneous immunofluorescence histochemistry for NSE (G) and TUNEL (H) in MT-I+II null mice at 3 dpl.G, NSE-positive neurons are seen around the lesion site. The morphology of these neurons is similar to that of apoptotic neurons observed in E. Indeed, most of the present NSE-positive neurons in G were undergoing apoptosis, as seen by fluorescein-linked TUNEL in H. Scale bars:A–D, 80 μm; E–H, 20 μm.
In MT-I+II null mice, a significant increase in TUNEL-labeled cells was also seen at 1 and 3 dpl, but the number of TUNEL-positive cells seen in these mice was drastically increased compared with that of normal lesioned mice (Figs. 7, 8). The MT-I+II null mice displayed plenty of TUNEL-labeled cells inside and surrounding the lesion site (Fig.7B). Also at 10 dpl, the number of TUNEL-positive cells in the lesioned area was drastically increased. At 20 dpl, MT-I+I null mice still displayed a significantly increased number of TUNEL-positive cells inside and surrounding the lesion (Fig. 7D). Double immunofluorescence histochemistry of NSE and fluorescein-linked TUNEL indicated that the apoptotic cells are neurons lying in the brain parenchyme ensheathing the lesion of both normal and MT-I+II null mice (Fig. 7E–H). The TUNEL-positive neurons displayed morphological criteria of apoptosis, whereas the TUNEL-negative neurons were showing a normal cytoplasmic NSE staining. In MT-I+II null mice, almost all neurons of the vicinity of the lesioned area were TUNEL-positive and were showing an apoptotic morphology. Double immunofluorescence histochemistry of fluorescein-linked TUNEL and lectin or GFAP or MTs, respectively, showed that neither activated microglia/macrophages nor reactive astrocytes were apoptotic, no matter whether they expressed MTs (data not shown).
DISCUSSION
As expected (Penkowa and Moos, 1995), in normal mice MT-I+II immunoreactivity increased transiently in microglia/macrophages and reactive astrocytes of the ipsilateral cortex after the freeze lesion. MT-I mRNA levels were also increased, clearly suggesting that the observed protein levels were the consequence of increased gene transcription. The temporal pattern of MT-I+II induction suggests that they form part of the plethora of mechanisms involved in protection against CNS inflammation. Consistent with such a role, MT-I+II levels have been observed to be increased in several human neurodegenerative disorders as well as in a number of experimental models (Hidalgo et al., 1997). Furthermore, in transgenic mice with interleukin 6 (IL-6) expression targeted to astrocytes, which show profound microgliosis and astrogliosis and upregulation of several inflammatory and other host response genes (Campbell et al., 1993), MT-I+II were also dramatically induced (Hernández et al., 1997b; Carrasco et al., 1998). These results suggest that MT-I+II upregulation during brain damage is mediated by cytokines such as IL-6. Indeed, it is believed that glial responses to brain damage are orchestrated by a number of cytokines and growth factors, including IL-6, IL-1, tumor necrosis factor-α, transforming growth factor-β, insulin-like growth factor I, and granulocyte- and/or macrophage colony-stimulating factor (Giulian et al., 1994; Perry et al., 1995). Results obtained in IL-6 null mice after the facial nerve transection (Klein et al., 1997) or experimental autoimmune encephalomyelitis (Eugster et al., 1998) demonstrate that IL-6 is a major cytokine controlling gliosis during CNS damage. Furthermore, this seems to be the case also in the cryolesion model (Penkowa et al., 1999). Importantly, MT-I+II upregulation was severely depressed by IL-6 deficiency.
Thus, the studies above suggest that MT-I+II appear to behave as acute-phase proteins in the brain, but their physiological roles remain uncertain. The MT-I+II null mice (Masters et al., 1994a) represent a unique experimental approach for determining the putative importance of these MT isoforms for coping with CNS injury. The results with the cryolesion model demonstrate that these proteins are of major importance.
A major finding of this study is that the activation of microglia/macrophages is dramatically extended in MT-I+II-deficient mice. As noted above, microglia/macrophages result from the activation of resident microglia as well as from the infiltration and activation of bone marrow-derived monocytes. The inspection of the temporal pattern of microglia/macrophages appearance suggests that both the activation process and the recruitment of blood monocytes are normal in the MT-I+II null mice. However, the normal functions of activated microglia/macrophages do not seem to proceed afterward, because no wound healing occurred even at 20 dpl, when complete healing was evident in normal mice. Thus, MT-I+II could be important for the normal functions of these cells in the brain. Strong support for this hypothesis is given by results obtained in the human monocyte-derived cell line THP-1 (Leibbrandt and Koropatnick, 1994; Leibbrandt et al., 1994; Koropatnick and Zalups, 1997), because the respiratory burst was significantly reduced by a number of experimental MT-I+II decreases, including antisense downregulation. Interestingly, monocytes of MT-I+II null mice also show an impaired respiratory burst in vitro(J. Koropatnick, personal communication). The present in vivo results with MT-I+II null mice suggest that microglia/macrophages are not functioning properly, which probably explains the lack of tissue regeneration and wound healing, as well as the delay in the astrocytosis. Both the sustained microgliosis and the heterogeneous populations of lectin-positive cells observed in the MT-I+II null mice, suggestive of the release of both undifferentiated and differentiated myelocytes and monocytes from the bone marrow, might be related to an unbalanced cytokine profile (our unpublished observations), which deserves further attention.
The present results also demonstrate that, in the absence of MT-I+II, neuronal apoptosis was dramatically increased in the cryolesion model. A role of MT-I+II in the control of apoptosis has received much support in recent in vitro and in vivo studies (Zheng et al., 1996a; Hamada et al., 1997; Hellquist, 1997; Houben et al., 1997;Kondo et al., 1997; Lie et al., 1998), but to our knowledge the present report is the first one to demonstrate a significant role of MT-I+II in apoptosis of brain cells. The actual mechanisms through which MT-I+II deficiency would be causing cells to engage in apoptosis are yet unknown, but several possibilities are likely. First, MT-I+II are major factors controlling zinc and copper metabolism (Palmiter and Findley, 1995; Dalton et al., 1996; Kelly and Palmiter, 1996; Kelly et al., 1996), and thus the absence of MT-I+II could likely increase the amount of free zinc, which in excessive amounts could affect neuronal survival on its own (Yokoyama et al., 1986; Koh et al., 1996). Indeed, the results obtained with Timm staining suggested that the amount of free zinc was increased in the MT-I+II null mice compared with controls in the lesioned area. However, further studies are needed considering the role of zinc transporters (Palmiter and Findley, 1995; Palmiter, 1998). Second, MT-I+II have also been suggested to be antioxidant proteins (Thornalley and Vasak, 1985; Sato and Bremner, 1993). MT-I+II null embryonic cells show enhanced sensitivity to oxidative stress caused by tert-butylhydroperoxide and the redox cycling toxin paraquat, as well as enhanced sensitivity to anticancer drugs such as cisplatin, melphalan, bleomycin, cytarabine, andN-methyl-N′-nitro-N-nitrosoguanidine (Kondo et al., 1995; Lazo et al., 1995; Woo and Lazo, 1997). In vivo, MT-I+II null mice are also more sensitive to the oxidative stress caused by paraquat (Sato et al., 1996), paracetamol (Rofe et al., 1998), and cisplatin (Lie et al., 1998). Also, hepatocytes in culture of MT-I+II null mice are more sensitive totert-butylhydroperoxide (Zheng et al., 1996b) and cisplatin (Lie et al., 1998). Thus, the absence of MT-I+II during traumatic injury to the CNS, such as after a cryolesion, which will undoubtedly have increased free radicals because of the cell disruption and the associated inflammatory response (Giulian et al., 1994; Perry et al., 1995), might render the CNS with less antioxidant defenses. This could lead to increased oxidative stress, which has been proposed as a mediator of apoptosis (Buttke and Sandstrom, 1994). We have observed that the heme oxygenase 1 (data not shown) and SOD levels were increased in the lesioned MT-I+II null mice, suggesting an increased oxidative stress (Maines, 1997). Third, the ongoing exacerbated inflammatory response in the MT-I+II null mice could also produce additional signals affecting the apoptosis pathway (Merrill and Jonakait, 1995; Thompson, 1995). Finally, neuronal apoptosis could also be attributable to the lack of proper glia function (see above).
In summary, the present study demonstrates that the MT-I+II isoforms appear to be essential for normal wound healing in the CNS after traumatic injury. Furthermore, they are also essential for neuronal survival. Thus, these functions provide some rationale for the importance of MT-I+II proteins, which are highly preserved and represented in nature but which, despite a tremendous number of studies, did not appear to have clear physiological functions (Palmiter, 1998).
Footnotes
This work was supported by The Novo Nordisk Fonden, Direktør Leo Nielsens Fond, and Warwara Larsen’s Fond (M.P. and T.M.) and by Comisión Interministerial de Ciencia y Tecnología Grant SAF96-0189, Programa Sectorial de Promoción General del Conocimiento Grant PM98-0170, and Fundación “La Caixa” Grant 97/102-00 (J.H.). J.C. is a fellow of Comissió Interdepartamental de Recerca i Innovació Tecnológica (Grant FI 96/2613). We acknowledge Dr. R. D. Palmiter for critically reading this manuscript and for the MT-I probe. Thanks are given to Hanne Hadberg, Pernille S. Thomsen, and Jordi Canto for excellent technical assistance and to Keld Stub and Birgit Risto for superb photographic assistance. The help of the Laboratori d’Análisi Bioquimica del Departament de Bioquimica i Biologia Molecular is acknowledged.
Correspondence should be addressed to Dr. Juan Hidalgo, Departamento de Biología Celular, de Fisiología y de Inmunología, Unidad de Fisiología Animal, Facultad de Ciencias, Universidad Autónoma de Barcelona, Bellaterra, Barcelona, Spain 08193.
REFERENCES
- 1.Aschner M, Cherian MG, Klaassen CD, Palmiter RD, Erickson JC, Bush AI. Metallothioneins in brain—the role in physiology and pathology. Toxicol Appl Pharmacol. 1997;142:229–242. doi: 10.1006/taap.1996.8054. [DOI] [PubMed] [Google Scholar]
- 2.Buttke T, Sandstrom P. Oxidative stress as a mediator of apoptosis. Immunol Today. 1994;15:7–10. doi: 10.1016/0167-5699(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 3.Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MBA, Mucke L. Neurologic disease in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci USA. 1993;90:10061–10065. doi: 10.1073/pnas.90.21.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carrasco J, Hernández J, González B, Campbell I, Hidalgo J. Localization of metallothionein-I and -III expression in the CNS of transgenic mice with astrocyte-targeted expression of interleukin 6. Exp Neurol. 1998;153:184–194. doi: 10.1006/exnr.1998.6861. [DOI] [PubMed] [Google Scholar]
- 5.Dalton T, Pazdernik TL, Wagner J, Samson F, Andrews GK. Temporalspatial patterns of expression of metallothionein-I and -III and other stress related genes in rat brain after kainic acid-induced seizures. Neurochem Int. 1995;27:59–71. doi: 10.1016/0197-0186(94)00168-t. [DOI] [PubMed] [Google Scholar]
- 6.Dalton T, Fu K, Palmiter RD, Andrews GK. Transgenic mice that overexpress metallothionein-I resist dietary zinc deficiency. J Nutr. 1996;126:825–833. doi: 10.1093/jn/126.4.825. [DOI] [PubMed] [Google Scholar]
- 7.Duguid JR, Bohmont CW, Liu NG, Tourtellotte WW. Changes in brain gene expression shared by scrapie and Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:7260–7264. doi: 10.1073/pnas.86.18.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erickson JC, Sewell AK, Jensen LT, Winge DR, Palmiter RD. Enhanced neurotrophic activity in Alzheimer’s disease cortex is not associated with down-regulation of metallothionein-III (GIF). Brain Res. 1994;649:297–304. doi: 10.1016/0006-8993(94)91076-6. [DOI] [PubMed] [Google Scholar]
- 9.Eugster H-P, Frei K, Kopf M, Lassmann H, Fontana A. IL-6 deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunol. 1998;28:2178–2187. doi: 10.1002/(SICI)1521-4141(199807)28:07<2178::AID-IMMU2178>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 10.Gasull T, Rebollo DV, Romero B, Hidalgo J. Development of a competitive double antibody radioimmunoassay for rat metallothionein. J Immunoassay. 1993;14:209–225. doi: 10.1080/15321819308019851. [DOI] [PubMed] [Google Scholar]
- 11.Gasull T, Giralt M, Hernandez J, Martinez P, Bremner I, Hidalgo J. Regulation of metallothionein concentrations in rat brain: effect of glucocorticoids, zinc, copper, and endotoxin. Am J Physiol. 1994;266:E760–E767. doi: 10.1152/ajpendo.1994.266.5.E760. [DOI] [PubMed] [Google Scholar]
- 12.Ghirnikar R, Lee Y, Eng L. Inflammation in traumatic brain injury: roles of cytokines and chemokines. Neurochem Res. 1998;23:329–340. doi: 10.1023/a:1022453332560. [DOI] [PubMed] [Google Scholar]
- 13.Giulian D, Li J, Li X, George J, Rutecki P. The impact of microglia-derived cytokines upon gliosis in the CNS. Dev Neurosci. 1994;16:128–136. doi: 10.1159/000112099. [DOI] [PubMed] [Google Scholar]
- 14.Hamada T, Tanimoto A, Sasaguri Y. Apoptosis induced by cadmium. Apoptosis. 1997;2:359–367. doi: 10.1023/a:1026401506914. [DOI] [PubMed] [Google Scholar]
- 15.Hellquist H. Apoptosis in epithelial hyperplastic laryngeal lesions. Acta Otolaryngol (Stockh) 1997;527:25–59. doi: 10.3109/00016489709124029. [DOI] [PubMed] [Google Scholar]
- 16.Hernández J, Carrasco J, Arbonés ML, Hidalgo J. IFN-γR−/− mice show an enhanced liver and brain metallothionein I+II response to endotoxin but not to immobilization stress. J Endotoxin Res. 1997a;4:363–370. [Google Scholar]
- 17.Hernández J, Molinero A, Campbell IL, Hidalgo J. Transgenic expression of interleukin 6 in the central nervous system regulates brain metallothionein-I and -III expression in mice. Mol Brain Res. 1997b;48:125–131. doi: 10.1016/s0169-328x(97)00087-9. [DOI] [PubMed] [Google Scholar]
- 18.Hidalgo J, Borras M, Garvey JS, Armario A. Liver, brain, and heart metallothionein induction by stress. J Neurochem. 1990;55:651–654. doi: 10.1111/j.1471-4159.1990.tb04182.x. [DOI] [PubMed] [Google Scholar]
- 19.Hidalgo J, Castellano B, Campbell IL. Regulation of brain metallothioneins. Curr Top Neurochem. 1997;1:1–26. [Google Scholar]
- 20.Houben R, Troppmair J, Hidalgo J, Rapp U. Differential gene expression in apoptotic 32Dcl3 cells: induction of metallothionein. Apoptosis. 1997;2:40–46. doi: 10.1023/a:1026483508306. [DOI] [PubMed] [Google Scholar]
- 21.Hozumi I, Inuzuka T, Hiraiwa M, Uchida Y, Anezaki T, Ishiguro H, Kobayashi H, Uda Y, Miyatake T, Tsuji S. Changes of growth inhibitory factor after stab wounds in rat brain. Brain Res. 1995;688:143–148. doi: 10.1016/0006-8993(95)00522-r. [DOI] [PubMed] [Google Scholar]
- 22.Hozumi I, Inuzuka T, Ishiguro H, Hiraiwa M, Uchida Y, Tsuji S. Immunoreactivity of growth inhibitory factor in normal rat brain and after stab wounds—an immunocytochemical study using confocal laser scan microscope. Brain Res. 1996;741:197–204. doi: 10.1016/s0006-8993(96)00912-2. [DOI] [PubMed] [Google Scholar]
- 23.Kelly EJ, Palmiter RD. A murine model of Menkes disease reveals a physiological function of metallothionein. Nat Genet. 1996;13:219–222. doi: 10.1038/ng0696-219. [DOI] [PubMed] [Google Scholar]
- 24.Kelly EJ, Quaife CJ, Froelick GJ, Palmiter RD. Metallothionein I and II protect against zinc deficiency and zinc toxicity in mice. J Nutr. 1996;126:1782–1790. doi: 10.1093/jn/126.7.1782. [DOI] [PubMed] [Google Scholar]
- 25.Klein M, Möller J, Jones L, Bluethmann H, Kreutzberg G, Raivich G. Impaired neuroglial activation in interleukin-6 deficient mice. Glia. 1997;19:227–233. doi: 10.1002/(sici)1098-1136(199703)19:3<227::aid-glia5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 26.Koh J, Suh S, Gwag B, He Y, Hsu C, Choi D. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- 27.Kondo Y, Woo ES, Michalska AE, Choo KH, Lazo JS. Metallothionein null cells have increased sensitivity to anticancer drugs. Cancer Res. 1995;55:2021–2023. [PubMed] [Google Scholar]
- 28.Kondo Y, Rusnak J, Hoyt D, Settineri C, Pitt B, Lazo J. Enhanced apoptosis in metallothionein null cells. Mol Pharmacol. 1997;52:195–201. doi: 10.1124/mol.52.2.195. [DOI] [PubMed] [Google Scholar]
- 29.Koropatnick J, Zalups R. Effect of non-toxic mercury, zinc or cadmium pretreatment on the capacity of human monocytes to undergo lipopolysaccharide-induced activation. Br J Pharmacol. 1997;120:797–806. doi: 10.1038/sj.bjp.0700975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lazo JS, Kondo Y, Dellapiazza D, Michalska AE, Choo KH, Pitt BR. Enhanced sensitivity to oxidative stress in cultured embryonic cells from transgenic mice deficient in metallothionein I and II genes. J Biol Chem. 1995;270:5506–5510. doi: 10.1074/jbc.270.10.5506. [DOI] [PubMed] [Google Scholar]
- 31.Leibbrandt ME, Koropatnick J. Activation of human monocytes with lipopolysaccharide induces metallothionein expression and is diminished by zinc. Toxicol Appl Pharmacol. 1994;124:72–81. doi: 10.1006/taap.1994.1010. [DOI] [PubMed] [Google Scholar]
- 32.Leibbrandt ME, Khokha R, Koropatnick J. Antisense down-regulation of metallothionein in a human monocytic cell line alters adherence, invasion, and the respiratory burst. Cell Growth Differ. 1994;5:17–25. [PubMed] [Google Scholar]
- 33.Lie J, Liu Y, Habeebu S, Klaassen C. Metallothionein (MT)-null mice are sensitive to cisplatin-induced hepatotoxicity. Toxicol Appl Pharmacol. 1998;149:24–31. doi: 10.1006/taap.1997.8325. [DOI] [PubMed] [Google Scholar]
- 34.Maines M. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 35.Masters BA, Kelly EJ, Quaife CJ, Brinster RL, Palmiter RD. Targeted disruption of metallothionein I and II genes increases sensitivity to cadmium. Proc Natl Acad Sci USA. 1994a;91:584–588. doi: 10.1073/pnas.91.2.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masters BA, Quaife CJ, Erickson JC, Kelly EJ, Froelick GJ, Zambrowicz BP, Brinster RL, Palmiter RD. Metallothionein III is expressed in neurons that sequester zinc in synaptic vesicles. J Neurosci. 1994b;14:5844–5857. doi: 10.1523/JNEUROSCI.14-10-05844.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merrill GM, Jonakait GM. Interactions of the nervous and immune systems in development, normal brain homeostasis, and disease. FASEB J. 1995;9:611–618. doi: 10.1096/fasebj.9.8.7768352. [DOI] [PubMed] [Google Scholar]
- 38.Moos T. Simultaneous application of Timm’s sulphide silver method and immunofluorescence histochemistry. J Neurosci Methods. 1993;48:149–156. doi: 10.1016/s0165-0270(05)80017-2. [DOI] [PubMed] [Google Scholar]
- 39.Neal JW, Singhrao SK, Jasani B, Newman GR. Immunocytochemically detectable metallothionein is expressed by astrocytes in the ischaemic human brain. Neuropathol Appl Neurobiol. 1996;22:243–247. [PubMed] [Google Scholar]
- 40.Palmiter R. The elusive function of metallothioneins. Proc Natl Acad Sci USA. 1998;95:8428–8430. doi: 10.1073/pnas.95.15.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palmiter RD. Constitutive expression of metallothionein-III (MT-III), but not MT-I, inhibits growth when cells become zinc deficient. Toxicol Appl Pharmacol. 1995;135:139–146. doi: 10.1006/taap.1995.1216. [DOI] [PubMed] [Google Scholar]
- 42.Palmiter R, Findley S. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 1995;14:639–649. doi: 10.1002/j.1460-2075.1995.tb07042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palmiter RD, Findley SD, Whitmore TE, Durnam DM. MT-III, a brain-specific member of the metallothionein gene family. Proc Natl Acad Sci USA. 1992;89:6333–6337. doi: 10.1073/pnas.89.14.6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Penkowa M, Moos T. Disruption of the blood-brain interface in neonatal rat neocortex induces a transient expression of metallothionein in reactive astrocytes. Glia. 1995;13:217–227. doi: 10.1002/glia.440130308. [DOI] [PubMed] [Google Scholar]
- 45.Penkowa M, Hidalgo J, Moos T. Increased astrocytic expression of metallothioneins I+II in brain stem of adult rats treated with 6-aminonicotinamide. Brain Res. 1997;774:256–259. doi: 10.1016/s0006-8993(97)81716-7. [DOI] [PubMed] [Google Scholar]
- 46.Penkowa M, Moos T, Carrasco J, Hadberg H, Molinero A, Bluethmann H, Hidalgo J. Strongly compromised inflammatory response to brain injury in interleukin-6 deficient mice. Glia. 1999;25:343–357. [PubMed] [Google Scholar]
- 47.Perry V, Bell M, Brown H, Matyszak M. Inflammation in the nervous system. Curr Opin Neurobiol. 1995;5:636–641. doi: 10.1016/0959-4388(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 48.Quaife CJ, Findley SD, Erickson JC, Froelick GJ, Kelly EJ, Zambrowicz BP, Palmiter RD. Induction of a new metallothionein isoform (MT-IV) occurs during differentiation of stratified squamous epithelia. Biochemistry. 1994;33:7250–7259. doi: 10.1021/bi00189a029. [DOI] [PubMed] [Google Scholar]
- 49.Quaife C, Kelly E, Masters B, Brinster R, Palmiter R. Ectopic expression of metallothionein-III causes pancreatic acinar cell necrosis in transgenic mice. Toxicol Appl Pharmacol. 1998;148:148–157. doi: 10.1006/taap.1997.8321. [DOI] [PubMed] [Google Scholar]
- 50.Rofe A, Barry E, Shelton T, Philcox J, Coyle P. Paracetamol hepatotoxicity inmetallothionein-null mice. Toxicology. 1998;125:131–140. doi: 10.1016/s0300-483x(97)00172-8. [DOI] [PubMed] [Google Scholar]
- 51.Sato M, Bremner I. Oxygen free radicals and metallothionein. Free Radic Biol Med. 1993;14:325–337. doi: 10.1016/0891-5849(93)90029-t. [DOI] [PubMed] [Google Scholar]
- 52.Sato M, Apostolova M, Hamaya M, Yamaki J, Choo K, Michalska A, Kodama N, Tohyama C. Susceptibility of metallothionein-null mice to paraquat. Environ Toxicol Pharmacol. 1996;1:221–225. doi: 10.1016/1382-6689(96)00011-7. [DOI] [PubMed] [Google Scholar]
- 53.Sewell AK, Jensen LT, Erickson JC, Palmiter RD, Winge DR. Bioactivity of metallothionein-3 correlates with its novel beta domain sequence rather than metal binding properties. Biochemistry. 1995;34:4740–4747. doi: 10.1021/bi00014a031. [DOI] [PubMed] [Google Scholar]
- 54.Sillevis Smitt PA, Blaauwgeers HG, Troost D, de Jong JM. Metallothionein immunoreactivity is increased in the spinal cord of patients with amyotrophic lateral sclerosis. Neurosci Lett. 1992;144:107–110. doi: 10.1016/0304-3940(92)90727-o. [DOI] [PubMed] [Google Scholar]
- 55.Stevens A, Bähr M. Origin of macrophages in central nervous tissue. J Neurol Sci. 1993;118:117–122. doi: 10.1016/0022-510x(93)90100-d. [DOI] [PubMed] [Google Scholar]
- 56.Suzuki K, Nakajima K, Kawaharada U, Uehara K, Hara F, Otaki N, Kimura M, Tamura Y. Metallothionein in the human brain. Acta Histochem Cytochem. 1992;25:617–622. [Google Scholar]
- 57.Thompson C. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 58.Thornalley PJ, Vasak M. Possible role for metallothionein in protection against radiation-induced oxidative stress. Kinetics and mechanism of its reaction with superoxide and hydroxyl radicals. Biochim Biophys Acta. 1985;827:36–44. doi: 10.1016/0167-4838(85)90098-6. [DOI] [PubMed] [Google Scholar]
- 59.Uchida Y, Takio K, Titani K, Ihara Y, Tomonaga M. The growth inhibitory factor that is deficient in the Alzheimer’s disease brain is a 68 amino acid metallothionein-like protein. Neuron. 1991;7:337–347. doi: 10.1016/0896-6273(91)90272-2. [DOI] [PubMed] [Google Scholar]
- 60.Vela JM, Hidalgo J, González B, Castellano B. Induction of metallothionein in astrocytes and microglia in the spinal cord from the myelin-deficient jimpy mouse. Brain Res. 1997;767:345–355. doi: 10.1016/s0006-8993(97)00628-8. [DOI] [PubMed] [Google Scholar]
- 61.Woo E, Lazo J. Nucleocytoplasmic functionality of metallothionein. Cancer Res. 1997;57:4236–4241. [PubMed] [Google Scholar]
- 62.Yamada M, Hayashi S, Hozumi I, Inuzuka T, Tsuji S, Takahashi H. Subcellular localization of growth inhibitory factor in rat brain: light and electron microscopic immunohistochemical studies. Brain Res. 1996;735:257–264. doi: 10.1016/0006-8993(96)00586-0. [DOI] [PubMed] [Google Scholar]
- 63.Yokoyama M, Koh J, Choi D. Brief exposure to zinc is toxic to cortical neurons. Neurosci Lett. 1986;71:351–335. doi: 10.1016/0304-3940(86)90646-4. [DOI] [PubMed] [Google Scholar]
- 64.Young JK, Garvey JS, Huang PC. Glial immunoreactivity for metallothionein in the rat brain. Glia. 1991;4:602–610. doi: 10.1002/glia.440040607. [DOI] [PubMed] [Google Scholar]
- 65.Zheng H, Berman NE, Klaassen CD. Chemical modulation of metallothionein I and III mRNA in mouse brain. Neurochem Int. 1995;27:43–58. doi: 10.1016/0197-0186(94)00167-s. [DOI] [PubMed] [Google Scholar]
- 66.Zheng H, Liu J, Choo KH, Michalska AE, Klaassen CD. Metallothionein-I and -II knock-out mice are sensitive to cadmium-induced liver mRNA expression of c-jun and p53. Toxicol Appl Pharmacol. 1996a;136:229–235. doi: 10.1006/taap.1996.0029. [DOI] [PubMed] [Google Scholar]
- 67.Zheng H, Liu J, Liu Y, Klaassen CD. Hepatocytes from metallothionein-I and II knock-out mice are sensitive to cadmium- and tert-butylhydroperoxide-induced cytotoxicity. Toxicol Lett. 1996b;87:139–145. doi: 10.1016/0378-4274(96)03770-8. [DOI] [PubMed] [Google Scholar]