

Abstract
Synaptobrevin is a key constituent of the synaptic vesicle membrane. The neuronal-synaptobrevin(n-syb) gene in Drosophilais essential for nerve-evoked synaptic currents, but miniature excitatory synaptic currents (mESCs) remain even in the complete absence of this gene. To further characterize the defect in these mutants, we have examined conditions that stimulate secretion. Despite the inability of an action potential to trigger fusion, high K+ saline could increase the frequency of mESCs 4- to 17-fold in a Ca2+-dependent manner, and the rate of fusion approached 25% of that seen in wild-type synapses under the same conditions. Similarly, the mESC frequency in n-sybnull mutants could be increased by a Ca2+ ionophore, A23187, and by black widow spider venom. Thus, the ability of the vesicles to fuse in response to sustained increases in cytosolic Ca2+ persisted in the absence of this protein. Tetanic stimulation could also increase the frequency of mESCs, particularly toward the end of a train and after the train of stimuli. In contrast, these mutants did not respond to an elevation of cAMP induced by an activator of adenylyl cyclase, forskolin, or a membrane-permeable analog of cAMP, dibutyryl cAMP, which in wild-type synapses causes a marked increase in the mESC frequency even in the absence of external Ca2+. These results are discussed in the context of models that invoke a special role for n-syb in coupling fusion to the transient, local changes in Ca2+ and an as yet unidentified target of cAMP.
Keywords: neuronal-synaptobrevin, miniature synaptic current, high potassium stimulation, tetanic stimulation, cAMP, forskolin, neuromuscular junction, Drosophila
The specialization of the presynaptic nerve terminal for fast exocytosis has been widely investigated. It is postulated that molecules on the synaptic vesicle membrane specifically bind to the receptors on the presynaptic membrane and form a docked state. After arrival of an action potential at the presynaptic nerve terminal and the resulting entry of Ca2+ through voltage-gated Ca2+channels, the local concentration of Ca2+ rises abruptly to a submillimolar level (Simon and Llínas, 1985;Roberts et al., 1990; Adler et al., 1991). This high concentration of Ca2+ is detected by a low-affinity Ca2+ sensor on the vesicular membrane that initiates rapid vesicle fusion resulting in synchronous transmitter release. On the other hand, spontaneous fusions of synaptic vesicles at the resting state generate miniature synaptic potentials. The frequency of these events changes when the cytosolic Ca2+ concentration is altered in the micromolar range (Delaney and Tank, 1994). Therefore, requirements for these two types of transmitter release, namely, action potential-triggered synaptic currents and miniature excitatory synaptic currents (mESCs), may be different.
Synaptobrevin is a major protein of synaptic vesicle membranes and the target of several clostridial neurotoxins (Schiavo et al., 1992). It is thought to be a crucial determinant of transmitter release and may contribute to membrane fusion or assist in tethering vesicles to appropriate target membranes (Söllner et al., 1993a,b).Neuronal synaptobrevin (n-syb) is the predominant synaptobrevin gene in Drosophila (DiAntonio et al., 1993). At the fly neuromuscular junction, cleavage of this protein by expression of a tetanus toxin transgene removed nerve-evoked synaptic transmission, but mESCs remained (Sweeney et al., 1995).Deitcher et al. (1998) confirmed these findings with a genetic deletion of the n-syb locus. These findings raised questions concerning the nature of the residual release of transmitter. Do the vesicles lacking n-syb protein retain the ability to be regulated by Ca2+? Can the absence of a response to an action potential be attributed to a lack of function of the voltage-gated Ca2+ channels? In the present study, we have stimulated the fusion of synaptic vesicles by several treatments that produce sustained changes in cytosolic Ca2+ and found that the loss of this major vesicular protein produces a specific deficit in the ability of the vesicles to respond to fast, transient increases in cytosolic Ca2+.
At many synapses, cAMP can also facilitate synaptic transmission and is presumed to play a key role in synaptic plasticity (Dixon and Atwood, 1989; Salin et al., 1996; Chen and Regehr, 1997). At theDrosophila neuromuscular junction, cAMP has been shown to facilitate synaptic transmission (Zhong and Wu, 1991) and also to increase the frequency of spontaneous transmitter release (Zhang et al., 1999a,b). How does cAMP interact with the two types of transmitter release? We report here that n-syb mutants completely lack the response to agents that increase cAMP. It, therefore, appears that n-syb will be essential for facilitation of transmitter release by cAMP and that cAMP cannot influence the tonic n-syb-independent pathway.
MATERIALS AND METHODS
Fly stocks. A detailed genetic and molecular description of the mutants used in this study has been presented inDeitcher et al. (1998). Briefly, the allelen-sybΔF33B contains a deletion encompassing most of the coding region and represents a null allele. We also used n-sybF33-R, which is homozygous lethal and closely resembles a null allele but could produce some n-syb protein. As a control, the parental strain, line 34, was used in the majority of experiments, although Canton S was also used in some early experiments. Each stock was maintained in a y, w background and balanced with a TM6, y+ Ubxchromosome. Homozygous mutant embryos were identified by yellow mouth hooks. Mutant embryos were used at 21–24 hr after egg laying. Because by this time line 34 or Canton S larvae were already hatched, those newly hatched larvae were used for controls.
Electrophysiology. The dissecting and procedures for recording synaptic currents were described elsewhere (Kidokoro and Nishikawa, 1994; Nishikawa and Kidokoro, 1995). Dissection was done in Ca2+-free, Mg2+ saline. The ventral ganglion was kept intact. After dissection, the preparation was treated for 1–3 min with 1 mg/ml collagenase (type IV; Sigma, St. Louis, MO) in 0.1 mm Ca2+ saline. To measure the frequency of spontaneous synaptic currents in high K+ saline, we kept the preparation in Ca2+-free solution for 3 min before changing to the solution containing a desired Ca2+concentration.
Mainly longitudinal muscles 6, 7, and 4 were voltage-clamped at −60 mV, and currents were filtered at 5 kHz. In the majority of experiments, the mESC frequency was visually counted for 5 min on a CRT screen. To avoid missing events during fly-back time of CRT sweeps, we simultaneously used a paper recorder (Nihon-Kohden, Tokyo, Japan), but when the frequency was high, a computer was used to count synaptic currents for 1 min. Spontaneous synaptic currents with a slow time course were mixed with fast ones because of electrical coupling of muscle cells with those in neighbor segments (Kidokoro and Nishikawa, 1994; Gho, 1994; Ueda and Kidokoro, 1996). In this study only synaptic currents with a fast time course were counted. Each synaptic current was observed on the CRT screen, and mESCs with slow time courses (>2 msec in the rise time) were rejected from counting.
In both n-syb mutant embryos and line 34 larvae, stretching nerve terminals in higher concentrations of external Ca2+ (≥0.5 mm) caused high frequencies of synaptic currents even in the presence of tetrodotoxin (TTX). In this study we tried to avoid stretching preparations during dissection and did not record from cells that were distorted because of the contraction of neighboring muscle cells. Even with these precautions we did observe cells with very high frequencies of mESCs inn-syb mutant embryos as well as controls. However, these anomalously high frequency fibers were excluded from the data.
All electrophysiology experiments were performed at room temperature (22–26°C)
Solutions. The ionic composition of solutions used in the experiments are as follows (in mm): normal external saline: NaCl, 140; KCl, 2; MgCl2, 5.5, CaCl2, 0.5; and HEPES-NaOH, 5, pH 7.1; Ca2+-free external solution: NaCl, 140; KCl, 2; MgCl2, 6; and HEPES-NaOH, 5, pH 7.1; and high K+ external saline: NaCl, 122; KCl, 20; MgCl2, 5.5; CaCl2, 0.5, and HEPES-NaOH, 5, pH 7.1. High K+ external salines with various concentrations of Ca2+ were prepared by replacing MgCl2 with CaCl2. The ionic composition of the internal solution was (in mm); CsCl, 158; ATP, 2; EGTA, 5; and HEPES-NaOH, 10, pH 7.1.
Biochemicals and toxins. TTX and Ca2+ionophore A23187 were purchased from Sigma. A23187 was dissolved in DMSO at 5 mm and used at 10 μm in the external solutions with various Ca2+ concentrations. A synthetic toxin, PLTX-II, was purchased from Peptide Institute (Osaka, Japan).
Black widow spider venom (BWSV) was a gift from Drs. Joy A. Umbach and Cameron B. Gundersen at University of California at Los Angeles. A gland was dissolved in 100 μl of Na/Tris-saline (150 mm NaCl and 20 mm Tris-HCl, pH 7.5) and stored at −70°C. Shortly before use, the stock solution was thawed and diluted into the external solution (1/60) with 0.5 or 2 mmCa2+ for n-syb mutant embryos and 0.2 or 0.5 mm Ca2+ for line 34 larvae. With perfusion, ∼0.7 ml of the toxin-containing solution was introduced into the bath with a volume of 1.2–1.5 ml. Thus, there was another approximately threefold dilution of the toxin in this process. The final concentration was ∼0.056 gland/ml.
Forskolin was dissolved in 100% ethanol at the concentration of 10 mm. Forskolin-containing Ca2+-free solution (0.5 ml) of two times a desired concentration was added to the bath solution of 0.5 ml and stirred so that the final concentration of forskolin became the desired concentration. In this experimental condition, when the highest concentration (500 μm) of forskolin was used, the ethanol concentration became as high as 5%, but 5% ethanol dissolved in Ca2+-free external solution by itself did not have any effect on the mESC frequency. Dibutyryl cAMP is water soluble and used at 1 mm. For application of dibutyryl cAMP, the volume of bath solution was reduced to 0.15 ml before infusion of the drug containing solution with a volume of 1.1 ml. Consequently, there was a slight dilution of dibutyryl cAMP to a final concentration of 0.86 mm. The perfusion rate was 0.73 ml/min, and the pump was stopped after infusion of dibutyryl cAMP. Forskolin and dibutyryl cAMP were purchased from Wako Chemicals (Osaka, Japan).
RESULTS
Movements in n-syb null embryos suggest that some neuromuscular communication persists
Our previous studies (Deitcher et al., 1998) have indicated that spontaneous mESCs are present in n-syb null mutants, but that action potentials in the nerve could not evoke a response in the muscle. Nerves were stimulated at 0.3 Hz in 2 mmCa2+ saline and in the presence of 2 mm4-AP to prolong Ca2+ entry into the terminal, and recordings from the postsynaptic muscle fiber were examined for a response in the 10 msec interval after the stimulus. In control lines, such a protocol consistently would have produced large, multiquantal responses to each action potential. Yet in the n-sybmutants, every stimulus failed. These results were in accord with similar low-frequency stimulation experiments in tetanus toxin-expressing transformants (Broadie et al., 1995; Sweeney et al., 1995); yet, observations of the behavior of the embryos suggested to us that some nerve-evoked contractions might be occurring in the muscles. In their gross morphological features, n-sybnull mutant embryos resembled those of control larvae (line 34) of a similar age. At 21–24 hr after egg laying, the mutant embryos had aerated tracheae but did not move spontaneously within the vitelline membrane; yet, when the membrane was torn by a sharp needle, they invariably displayed wiggly movements. For several minutes, segmental muscles at various positions contracted sporadically until the embryos eventually became placid. When embryos were prepared for electrophysiological recordings by dissection in the presence of 1 or 2 mm Ca2+ in the external saline, spontaneous and uncoordinated contractions of the muscles were again observed. If these contractions and those observed after devitellinization were entirely myogenic, they should be insensitive to TTX. We observed, however, the 3 μm TTX caused most of the movements to disappear, suggesting a neural origin. These observations and a more general interest in the nature of the n-syb-independent pathway to transmitter release led us to examine the neuromuscular junctions more closely in these mutants. As described below, we have found that sustained Ca2+ influx into the nerve terminal, as a result of K+ depolarization or tetanic stimuli, can indeed stimulate the release of transmitter.
The frequency of mESCs in n-syb null mutants can be modulated
In all cells examined inn-sybΔF33B andn-sybF33-R embryos, spontaneous synaptic currents were observed at low frequencies in normal saline (∼1/min) (Deitcher et al., 1998). The frequency was higher in high K+ saline in the presence of external Ca2+. This observation suggests that Ca2+ ions do enter the terminal in a voltage-dependent manner in mutant embryos and are detected by the Ca2+ sensor for an acceleration of spontaneous release of transmitter. These spontaneous synaptic currents were mESCs because they remained even in the presence of 3 μm TTX. Because the mESC frequency was low in normal saline, it was difficult to collect enough events for a detailed analysis. Therefore, we used high K+ external solution to facilitate the occurrence of mESCs and examined the effect of external Ca2+ on their frequency.
The mESCs were studied in the presence of 3 μm TTX and 20 mm KCl in the external solution, and the concentration of external Ca2+ was varied between 0 and 2 mm. Spontaneous synaptic currents were observed in low frequencies even in the absence of external Ca2+ in mutant embryos. The mean frequency was 1.6 ± 0.7/min (mean ± SD, n = 10; data will be expressed in this format throughout the text unless otherwise specified) inn-sybΔF33B embryos and 1.5 ± 1.5/min (n = 14) inn-sybF33-R embryos. These frequencies were slightly smaller than that in line 34 larvae (3.3 ± 2.8/min;n = 10). This observation at zero external Ca2+ is in accord with that reported for tetanus toxin-expressing transformants (Broadie et al., 1995)
As the external Ca2+ concentration was increased, the frequency was elevated. With 2 mmCa2+ in the external solution, the mean frequency was 13.5 ± 13.0/min (n = 16) inn-sybΔF33B embryos and 10.5 ± 11.8/min (n = 15) inn-sybF33-R embryos, whereas it was 68.2 ± 85.9/min (n = 8) in line 34 larvae. In Figure 1, the mean frequency was plotted against the external Ca2+ concentration. The curves for mutant embryos (n-sybΔF33B embryos,dotted line with squares andn-sybF33-R embryos, broken line with triangles) were always below that for line 34 larvae (solid line with circles). This could be either caused by less Ca2+ entering the presynaptic terminal or a less efficient vesicle fusion machinery for spontaneous release in n-syb mutant embryos. We favor the latter possibility based on observations with a Ca2+ ionophore and black widow spider venom as described below.
Fig. 1.

The relation between the frequency of mESCs and external Ca2+ concentration in the 20 mmK+ solution in the presence of 3 μmTTX. The frequency was plotted against external Ca2+concentration. Open circles are for line 34 larvae,squares are forn-sybΔF33B, andtriangles are forn-sybF33-R. Error bars indicate SEM. Neighboring data points were connected by straight lines.
The increase in mESC frequency in the high K+solution was blocked by Cd2+ or PLTX-II inn-syb mutant embryos
To test whether the increase in the mESC frequency with external Ca2+ in high K+ saline is caused by activation of voltage-gated Ca2+ channels, we examined the effect of 1 mm Cd2+. When 1 mm Cd2+ was included in the external solutions together with 2 mm Ca2+, the mean frequency was reduced to the level seen in 0 Ca2+ (Fig. 2). Because Cd2+ is known to be an effective blocker of voltage-gated Ca2+ channels, this result suggests that the effect of external Ca2+ on the mESC frequency was caused by Ca2+ influx through voltage-gated Ca2+ channels.
Fig. 2.

Effects of Ca2+ channel blockers on the mESC frequency inn-sybΔF33B. The frequency of mESCs were counted in the 20 mm K+ solution in the presence of 3 μm TTX. Conditions for each experiment are written below the abscissa. Error bars indicate SEM.Two asterisks on each column indicate statistical difference at p = 0.01 from control (blank column).
Furthermore, a spider toxin, PLTX-II, is known to inhibit Ca2+ currents in Drosophila neurons (Leung et al., 1989) and block neuromuscular transmission at theDrosophila neuromuscular junction (Branton et al., 1987). Therefore, it has been postulated that PLTX-II blocks voltage-gated Ca2+ channels at the presynaptic terminals and thereby eliminates nerve-evoked synaptic currents. We tested the effect of PLTX-II on the increase of mESC frequency in high K+ saline in n-syb mutant embryos. As shown in Figure 2 (horizontally striped column), in n-sybΔF33B embryos the increase of mESC frequency in the high K+ solution in the presence of 2 mm Ca2+ was completely blocked by 50 nm PLTX-II.
Similar effects were also found in line 34 larvae in the presence of 1 mm Ca2+. Cd2+ (0.5 mm) reduced the mESC frequency from 18.4 ± 4.6/min (n = 9) to 3.8 ± 1.7/min (n = 8). The latter frequency was similar to the level observed in 0 mm Ca2+ (3.3 ± 0.9/min;n = 10). PLTX-II (50 nm) also reduced the increase of mESC frequency in 0.5 mmCa2+ [7.7 ± 2.2/min (n = 13) to 1.7 ± 0.2/min (n = 5)]. These results suggest that Cd2+- and PLTX-II-blockable Ca2+ channels are operating in n-sybmutant embryos.
Ca2+ ionophore, A23187, also increased the mESC frequency in the presence of external Ca2+ inn-sybΔF33B embryos
The lower frequency of mESCs in n-syb mutant embryos relative to line 34 larvae at a given concentration of external Ca2+ could either be caused by less Ca2+ entry or reduced efficiency of the fusion mechanism. Syntaxin is known to reduce currents through N-type Ca2+ channels by shifting the steady-state inactivation curve to the left along the voltage axis (Bezprozvanny et al., 1995). Therefore, it is plausible that neuronal synaptobrevin, which binds to syntaxin together withN-ethylmaleimide-sensitive factor and solubleN-ethylmaleimide-sensitive factor attached protein (Söllner et al., 1993a,b), might indirectly affect Ca2+ channels and that the lack of neuronal synaptobrevin in n-syb mutant embryos might reduce Ca2+ influx associated with an action potential. To distinguish these two alternatives, it is necessary to increase the cytoplasmic Ca2+ concentration in the presynaptic terminal by other routes than voltage-gated Ca2+channels.
We used a Ca2+ ionophore, A23187, to increase Ca2+ concentrations in the nerve terminal independently from voltage-gated Ca2+ channels. Inn-sybΔF33B embryos treated with A23187, the frequency of mESCs increased in the presence of 0.5 mmCa2+ compared with that in 0 mmCa2+ (p < 0.01) (Fig.3, circles). With 0.2 mm Ca2+, the effect was intermediate. A23187 itself did not increase the mESC frequency independently of Ca2+ entry: in 0 Ca2+ the frequency was 1.3 ± 0.9/min (n = 10) with the ionophore and 1.1 ± 0.9 (n = 5) without.
Fig. 3.

Effects of Ca2+ ionophore A23187. The number of mESCs per minute is plotted against the external Ca2+ concentration. Squares are for line 34 larvae, and open circles are forn-sybΔF33B embryos. Error bars indicate SEM, and numbers are the number of cells examined. Anasterisk indicates statistical difference between line 34 and n-sybΔF33B atp = 0.05.
The extent of the increase inn-sybΔF33B embryos was less than that in line 34 (Fig. 3, squares). Because we have no reason to think the ionophore behavior of A23187 would be affected by the mutation, we suggest that the vesicle fusion machinery inn-syb mutant embryos is less effective after elevation of cytosolic Ca2+ than that in line 34 larvae, as when high K+ was used to elevate intracellular Ca2+, the rate of release inn-sybΔF33B was ∼20% of wild-type control.
BWSV also increased the mESC frequency inn-sybΔF33B embryos
BWSV increases the internal Ca2+ concentration and causes massive release of transmitter (Meldolesi et al., 1986). Considering the results described above with high K+saline and Ca2+ ionophore, we would expect that BWSV also increases the mESC frequency in n-syb mutant embryos, although the mechanism of action of BWSV on insect synapses is not well characterized. Contrary to this expectation, Broadie et al. (1995)reported that BWSV did not increase the mESC frequency in aDrosophila transformant in which neuronal synaptobrevin was cleaved by tetanus toxin. To resolve this discrepancy we tested BWSV inn-sybΔF33B embryos. BWSV was added in the bath by perfusion while synaptic current recordings were made. All solutions used in these experiments contained 3 μm TTX to avoid an indirect effect of BWSV through the ventral ganglion. After ∼0.7 ml of the solution containing BWSV was introduced in the bath, perfusion was stopped while the mESC frequency was continuously monitored. The effect became suddenly evident 3–5 min after addition of the toxin to the bath and slowly declined within ∼10 min, even in the presence of the toxin (Fig. 4), but the extent of increase, measured at the peak, was less inn-syb mutant embryos (circles) compared with line 34 larvae (squares). As shown in Table1, BWSV significantly increased the mESC frequency in n-sybΔF33B embryos with 0.5 and 2 mm Ca2+ in the external solution. The effect was more prominent in 2 mm than in 0.5 mm external Ca2+. While in line 34 larvae the effect was vigorous in 0.5 mmCa2+, and a significant increase in the mESC frequency was observed even in 0.2 mmCa2+.
Fig. 4.

Effects of BWSV on the frequency of mESCs. BWSV was applied during the period shown by a horizontal dark line at the bottom. Squares are for line 34 larvae (4 preparations), and open circlesare for n-sybΔF33B embryos (3 preparations). Error bars indicate SEM. The frequency of mESCs was measured in the external solution containing 0.5 mmCa2+ and 3 μm TTX.
Table 1.
Effects of black widow spider venom on the mESC frequency
| External Ca2+ concentration (mm) | n-sybΔ F33B | Line 34 | ||
|---|---|---|---|---|
| BWSV (−) | BWSV (+) | BWSV (−) | BWSV (+) | |
| 0.2 | 2.3 ± 0.6 n = 3 | 58 ± 21 n = 3 | ||
| 0.5 | 0.7 ± 0.2 n = 4 | 162 ± 59 n = 3 | 2.0 ± 0.7 n = 5 | 553 ± 225 n = 4 |
| 2.0 | 0.8 ± 0.1 n = 6 | 381 ± 38 n = 3 | ||
Numbers are the average number of mESCs per minute ± the SEM and the number of cells examined (n). BWSV (−) indicates measurements in the absence of black widow spider venom, and BWSV (+) shows those in the presence of the venom.
Thus, BWSV did increase the mESC frequency in n-syb mutant embryos, and the extent of the effect was dependent on the external Ca2+ concentration. BWSV was, however, less effective in n-syb mutant embryos than in line 34 larvae: the peak rate of release was ∼ that of the control. This result is in accord with reports on the effect of BWSV on tetanus toxin-treated preparations (Janicki and Habermann, 1983; Dreyer et al., 1987; Gansel et al., 1987). However, it is not clear at this moment why the effect of BWSV was not observed in the tetanus toxin-expressing transformants by Broadie et al. (1995).
Tetanic stimulation induced release of transmitter inn-syb mutant embryos
So far we have shown that the mESCs in n-sybmutant embryos respond to an elevation of internal Ca2+. However, nerve stimulation at the low frequency does not evoke synaptic currents in the mutant embryos (Deitcher et al., 1998). Is it possible that nerve stimulation does not elevate internal Ca2+ in mutants? To test this possibility, we stimulated the nerve repetitively at 10 Hz and examined the frequency of mESCs during and after the tetanus.
Spontaneous synaptic currents inn-sybΔF33B andn-sybF33-R embryos were rare in the 2 mm Ca2+ solution with 2 mmK+. However, with tetanic stimulation (10–20 pulses at 10 Hz), release of individual quanta of transmitter was observed toward the end of the tetanus and for ∼1 sec thereafter (Fig.5Aa,Ba). When the frequency histogram was constructed along the time axis and was aligned to the tetanus, this trend became evident (Fig.5Ab,Bb). Some of these fusions occurred with the right latency for evoked release, namely between 4 and 10 msec after onset of the individual stimuli within the tetanic train (Fig.5Aa,Ba, arrows). This timing suggested that they were indeed coupled to the preceding action potential.
Fig. 5.
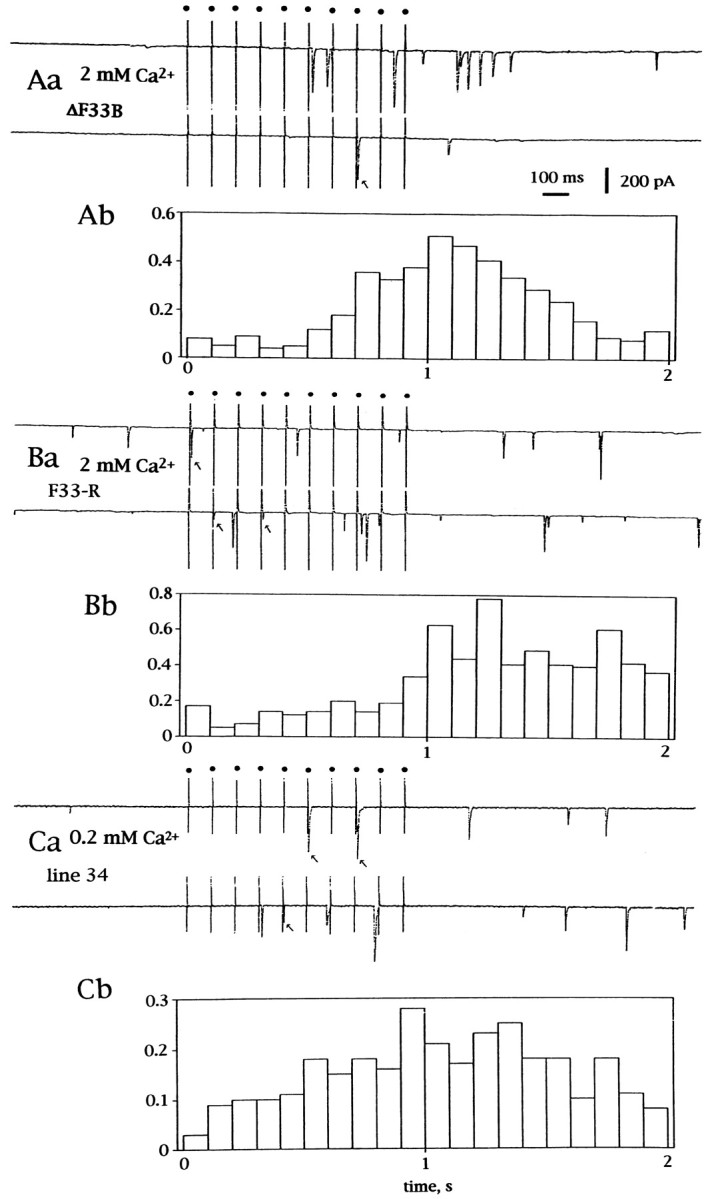
Release of transmitter during and after tetanic stimulation. Ten pulses at 10 Hz were delivered while the muscle cell was voltage-clamped at −60 mV. Aa, Current traces on a paper recorder during and after stimulation recorded in ann-sybΔF33B embryo. The external solution contained 2 mm Ca2+ and 4 mm Mg2+. Each stimulus was given at adot indicated at the top. Downward deflections indicate inward currents. Ab, The number of events per 100 msec of tetanic stimulation is plotted on the ordinate. A total of 76 sets of 10 tetanic stimuli were delivered. Timing is aligned to the top traces. Ba, Current traces on a paper recorder during and after tetanic stimulation recorded in an n-sybF33-R.Bb, Number of events per 100 msec of tetanic stimulation. A total of 59 sets of 10 stimuli were delivered. The external solution contained 2 mm Ca2+and 4 mm Mg2+. Ca, Current traces on a paper recorder during and after stimulation recorded in a line 34 larva. The external solution contained 0.2 mm Ca2+ and 5.8 mmMg2+. Arrows indicate evoked synchronized synaptic currents. Cb, Number of events per 100 msec of tetanic stimulation. A total of 119 sets of 10 tetanic stimuli were delivered.
In line 34 larvae with low concentrations of external Ca2+ (0.2 mm), individual stimuli did not evoke synaptic currents in the majority of cases, but tetanic stimulation did evoke release of transmitter as shown in Figure5Ca. The frequency histogram shown in Figure 5Cbwas similar to that in n-syb mutant embryos described above except that the synaptic currents that were evoked were synchronized to stimuli more frequently than those in n-syb mutant embryos at 2 mm Ca2+ (Fig.5Ab,Bb, arrows).
Tetanic stimulation in the absence of Ca2+ in the external solution did not evoke synaptic currents in n-sybmutant embryos, indicating that Ca2+ influx induced by tetanic stimulation is causing the observed release of transmitter. This observation again suggests that voltage-gated Ca2+ channels are operating in n-sybmutant nerve terminals. In accord with this observation, tetanus toxin treatment of the squid giant synapse markedly reduced synaptic transmission but did not affect Ca2+ influx (Hunt et al., 1994).
Because, in addition to mESCs after tetanic stimulation, some of the quanta appeared to have the appropriate latency for having been evoked by the immediately preceding stimulus within each tetanus, we looked at the timing of the mESCs in greater detail (Fig.6A). To distinguish a genuine coupling of stimulus and release from coincidental occurrence of asynchronous events during the 4–10 msec poststimulus interval, we constructed a latency histogram (Fig. 6Ab) to describe the interval between the onset of the stimulating pulse and each of the observed synaptic currents. In these histograms it became clear that the earlier bins had more entries than later ones. Thus, stimuli did evoke synaptic currents, although the latency was fluctuating, and the synchronous release was extremely rare even in the presence of 2 mm Ca2+. We defined synchronized transmission as synaptic currents that appeared within 10 msec after the onset of stimuli and calculated the percentage of individual stimuli that successfully evoked synchronized transmission. It was 1.5 ± 1.6% (n = 7) forn-sybΔF33B embryos in 2 mmCa2+, which was smaller than that of line 34 larvae in 0.2 mm Ca2+, 13.4 ± 9% (n = 5). Interestingly, the percentage of synchronized transmission for n-sybF33-R embryos at 2 mm Ca2+ was 9.1 ± 4.7% (n = 8), which is significantly larger than that ofn-sybΔF33B embryos in 2 mmCa2+ (p < 0.002, student’s two-tailed t test). This result suggests thatn-sybF33-R is not a complete null allele, and some n-syb protein being synthesized in these embryos might be making this difference.
Fig. 6.

Loosely synchronous release of transmitter in ann-sybΔF33B embryo in 2 mm Ca2+ (Aa,Ab), in an n-sybF33-R in 2 mm Ca2+ (Ba,Bb), and synchronous release in a line 34 larva in 0.2 mm Ca2+ (Ca,Cb). Aa, Sample traces during tetanic stimulation that had synaptic currents were selected.Ab, The latency histogram of synaptic currents occurred after 1841 stimuli during tetani. On the ordinate the cumulative number of events belonging to a bin is plotted. The abscissa is aligned to the above sample traces. Upward arrows indicate the stimulus onset. Ba, Selected sample traces during tetanic stimulation. Bb, The latency histogram was constructed out of synaptic currents occurred after 792 stimuli during tetanic stimulation. Ca, Sample traces that occurred during tetanic stimulation. Cb, The latency histogram was constructed out of synaptic currents occurred after 1094 stimuli during tetani.
In accord with this interpretation, the latency histogram had relatively more entries in the first bin (between 4 and 10 msec) in ann-sybF33-R embryo (Fig.6Bb). To document this observation in populations of mutant embryos, we calculated the percentage of events that belonged to the first bin among all events that occurred within 54 msec after stimuli. It was 17.8 ± 6.5% (n = 6) forn-sybΔF33B embryos, whereas that forn-sybF33-R embryos was 31.3 ± 10.0% (n = 6). These values are statistically different (p < 0.02). This observation suggests that a role of n-syb protein in synaptic transmission is to facilitate fast vesicle fusion after arrival of an action potential in the presynaptic terminal.
cAMP failed to elevate the frequency of mESCs inn-syb mutants
At the Drosophila neuromuscular junction, metabotropic glutamate receptors cause a tonic increase in the frequency of mESCs by increasing cAMP levels (Zhang et al., 1999a,b). This action appears to be caused by a stimulation of the Ca2+/calmodulin-dependent adenylate cyclase and is responsible for the phenomena of synaptic facilitation and post-tetanic potentiation in this preparation. The means by which cAMP stimulates vesicle fusion is not as yet known, but it occurs in the absence of external Ca2+ and therefore cannot entirely consist of the modulation of ion channels in the terminal membrane (Siegelbaum et al., 1982). We have investigated the interaction of cAMP with the two pathways defined by the n-syb mutations in the experiments above. If cAMP were acting by raising cytosolic Ca2+ levels, we would predict that agents that increase mESC frequency (forskolin and dibutyryl cAMP) would continue to be effective in the mutants: stimulation of the cAMP pathway would resemble the tonic elevation of intracellular Ca2+produced by high K+ or ionophores. On the other hand, if cAMP acts on a component of the exocytotic apparatus that is unique to the phasic pathway, it would be ineffective in then-syb mutations that lack this pathway.
Experiments with the adenylate cyclase activator forskolin and with dibutyryl cAMP were conducted in Ca2+-free saline to separate the intracellular actions of these agents from any potential actions on Ca2+ influx. When applied to a control preparation (line 34), forskolin increased the mESC frequency at relatively high concentrations (a partial dose–response curve is shown in Fig. 7). Forskolin (at a final concentration of 500 μm) increased the mESC frequency 2–7/min (3.8 ± 2.3/min; n = 4) to 6–295/min (133.6 ± 137.1/min; n = 4) (Fig.8Aa). Although the time course was variable, the response was observed in all four cases examined. This result is in accord with the previous report on wild-type larvae (Zhang et al., 1999a,b). In contrast, inn-sybΔF33B embryos, the mESC frequency was 5.6 ± 1.9/min (n = 5) before application of the drug and was 1.9 ± 1.4/min (n = 5) in 500 μm forskolin for >10 min (Fig. 8Ab). In all five cases examined, no response was observed. Similarly, inn-sybF33-R embryos, the mESC frequency was 4.3 ± 1.4/min (n = 5) before and 1.0 ± 0.5/min (n = 5) in 500 μm forskolin (Fig.8Ac). Thus, the response to forskolin was completely absent in both n-syb mutant embryos.
Fig. 7.

Effects of forskolin on the mESC frequency in line 34 larvae. The concentration-dependent increase of mESC frequency is shown in the ordinate as the difference of mESC events per minute between the average of frequency during a 5 min interval shortly after application of forskolin and that of a 5 min interval between 25 and 30 min after drug application. Data from three cells for 50 μm, from three cells for 100 μm, from six cells for 200 μm, from three cells for 300 μm, from six cells for 400 μm, and from four cells for 500 μm were averaged. Error bars indicate SEM.
Fig. 8.

Effects of forskolin (A) and dibutyryl cAMP (B) on the mESC frequency in the absence of external Ca2+. Forskolin (500 μm) or dibutyryl cAMP (0.86 mm) was applied during a horizontal bar below the abscissa. Aa andBa are from line 34 (data from 4 cells forAa and 7 cells for Ba were averaged). Error bars indicate SEM. Ab and Bb are from n-sybΔF33B(data from 5 cells for Ab and from 6 cells forBb were averaged). Ac andBc are fromn-sybF33-R (data from 5 cells for Ac and from 4 cells for Bcwere averaged).
As an independent means of elevating cAMP levels in the terminals, we used a membrane-permeable analog, dibutyryl cAMP, at 0.86 mm. When the responses from seven control larvae (line 34) were averaged, dibutyryl cAMP transiently increased the frequency of mESCs between 8 and 22 min of application, although the time course and amplitude of the response was somewhat variable for cell to cell. The averaged frequency at the peak 10 min after application of the drug was 17.1 ± 30.2/min (n = 7), while that before application was 5.8 ± 4.5/min (n = 7, three consecutive points, 1 min per point, were averaged) (Fig.8Ba). In contrast, inn-sybΔF33B embryos no response was observed in all six cells examined (Fig. 8Bb). Similarly, in n-sybF33-R no response was observed in all six cells examined (Fig. 8Bc). Thus, the response to dibutyryl cAMP was completely absent inn-syb mutant embryos. The response to stimulation of the cAMP pathway appears to depend absolutely to the presence of n-syb.
DISCUSSION
v-SNARE proteins such as synaptobrevin are thought to be essential in membrane trafficking. In this study, we have observed that vesicles retain the ability to fuse both spontaneously and in response to elevated levels of cytosolic Ca2+. These fusions could occur in a pathway that is independent of a v-SNARE protein or, alternatively, the fusions may involve another v-SNARE: the homologous but largely non-n-syb gene, an additional homolog that has not yet been discovered, or perhaps vesicular syntaxin acting as a v-SNARE. The purpose of this study was to probe this n-syb-independent pathway to learn its capacities and limitations. Although this pathway could not respond to the Ca2+ transient elicited by an individual action potential, we found that it was still capable of regulation by Ca2+. In contrast, the n-syb-independent pathway was unresponsive to cAMP.
n-syb null mutants retain Ca2+-dependent release
In n-sybΔF33B andn-sybF33-R embryos, we found that nerve stimulation at 0.3 Hz did not evoke synaptic currents under any circumstances we tried. To determine whether the n-syb-independent pathway could respond to changes in cytosolic Ca2+other than action potential-triggered changes, we studied the effects of depolarizing the terminal with high K+ salines. We demonstrated that the mESC frequency was increased in an external Ca2+-dependent manner, even inn-sybΔF33B, which is unequivocally a null allele (Deitcher et al., 1998). Thus Ca2+ ions entered the terminal as the membrane was depolarized, and this influx was blocked by the Ca2+ channel blockers Cd2+ and PLTX-II. These observations are in accord with a previous report in synaptosomes, in which an increase in internal Ca2+ was induced by K+stimulation despite the presence of tetanus toxin (McMahon et al., 1992).
Although the rate of release in the n-syb mutant did not match that of wild-type, this difference cannot be attributed to an impairment of Ca2+ channel function because the same diminution of release was observed when the Ca2+channels were bypassed with the ionophore A23187 or with black widow spider venom. We therefore ascribe the downward shift in the curve relating Ca2+ to mESC frequency to a decrease in the efficiency of transmitter release.
The decrease in mESC frequency in the n-syb mutants did not correlate with the complete absence of a response to an individual action potential, even when the Ca2+ influx in response to the action potential had been augmented by high Ca2+ concentrations or 4-AP (Deitcher et al., 1998). This discrepancy raised the possibility that there was a change in the kinetics of the Ca2+ channel that prevented its gating by an action potential. This possibility also appears to be excluded by studies of tetanic stimulation: the long-lasting increase in mESC frequency that was observed during and after the tetani confirmed that action potentials could raise cytosolic Ca2+. The latency of these events was not compatible with a decrease in the probability of channel opening; such a decrease would have produced normal latency responses, as was seen in wild-type synapses in low Ca2+.
n-syb null mutants do not respond to an elevation of cAMP
In the absence of external Ca2+, forskolin evoked marked transmitter release in line 34 larvae. Dibutyryl cAMP had a similar, although less robust, effect. In the n-syb null allele, these responses were entirely absent. cAMP probably does not act in these synapses by releasing Ca2+ from intracellular stores: such elevations would, like those evoked by high K+ or ionophores, be expected to act in the absence of n-syb. Instead, cAMP would appear directly to facilitate the fusion of synaptic vesicles. This action could be accomplished by increasing the number of fusion-competent docked vesicles, by altering the fusion apparatus to increase the probability of release, or by altering a Ca2+ sensor so that it was capable of responding to resting Ca2+ levels. These observations are consistent with studies in rat cerebellar slices in which forskolin increases the mESC frequency without raising internal Ca2+ (Chen and Regehr, 1997).
Unlike Ca2+-dependent stimulation of release, which was only partially inhibited by loss of n-syb, cAMP-dependent stimulation was completely removed. The n-syb product is therefore essential to the processes of synaptic facilitation and post-tetanic potentiation that involve cAMP (Zhang et al., 1999a,b;Weisskopf et al., 1994). Although very little is known about the intracellular action of cAMP at synapses, the n-sybphenotype suggests that a phosphorylation target of protein kinase A could be either n-syb itself or a protein upstream of n-syb in the exocytotic apparatus.
Plausible models for the n-syb phenotype
What then is the critical difference between the n-syb-mediated and the n-syb-independent pathways for Ca2+-dependent release? What allows only the former to respond to an individual action potential? One indication may come from the time course of the response to tetanic stimulation. The latency histogram suggested a slow step in the coupling of Ca2+ entry to fusion. Two attractive models present themselves at present.
The first model invokes a difference in the spatial relationship of the active Ca2+ channels to the fusion-competent vesicles. Specifically, in the absence of n-syb, the vesicles would no longer be anchored in the immediate vicinity of the channels. Thus, in the null mutants, the brief and focal rise in Ca2+concentration would dissipate because of diffusion and buffering before reaching the Ca2+ sensor. A similar spatial separation has been invoked to explain the Ca2+sensitivity and time course of release in chromaffin cells (Chow et al., 1996). Moreover, such an explanation would be consonant with known biochemical properties of mammalian synaptic proteins. Syntaxin binds to synaptic isoforms of Ca2+ channels, and vesicle-associated membrane protein–synaptobrevin binds to syntaxin (Söllner et al., 1993a,b). Thus, the absence of n-syb could prevent the association of the vesicle with the channel while leaving the vesicle free to sense Ca2+ and fuse with the membrane when sustained Ca2+ influx allowed the ions to reach the sensor. A variant of this hypothesis would invoke two classes of Ca2+ channel in the terminal. The class that normally dominates release can be inhibited by syntaxin (Bezprozvanny et al., 1995). n-syb protein, by binding to syntaxin, could dissociate it from the channel and thereby allow it to trigger fusion when opened by an action potential. In the absence of n-syb, fusion would have to rely on more distant channels that are not inhibited by syntaxin.
The second model invokes two distinct Ca2+ sensors. In this model, the n-syb-mediated pathway is coupled to a low-affinity Ca2+ sensor and leads to rapid fusion. The n-syb-independent pathway is coupled to a high-affinity Ca2+ sensor, but the rate at which Ca2+ binding leads to vesicle fusion is slower than in the n-syb-dependent pathway. Evidence from several systems has already pointed to the existence of two classes of Ca2+ sensor. After tetanic stimulation, evoked release is enhanced because of residual Ca2+ binding to high-affinity sites (micromolar range) that, perhaps because of slow kinetics, are rarely occupied by Ca2+ entering during a single action potential. These high-affinity sites are distinct from the low-affinity sites that are occupied only when a high concentration of cytosolic Ca2+ is achieved after the arrival of an action potential (Yamada and Zucker, 1992; Kamiya and Zucker, 1994; van der Kloot, 1994).
Occupation of high-affinity sites may be sufficient to cause spontaneous fusion of synaptic vesicles. Although the frequency of mESCs has not yet been correlated directly to presynaptic Ca2+, conditions that are likely to raise Ca2+ to the 1–3 μm range appear to promote spontaneous fusions (Miledi and Thies, 1971; Erulkar and Rahamimoff, 1978; Kamiya and Zucker, 1994). Therefore, the low-affinity sites need not be occupied for spontaneous release of transmitter to occur, albeit at a relatively slow rate. To accomplish the rapid release of vesicles evoked by an action potential, the low-affinity sites must be occupied as well, which occurs only in the microenvironment of the open Ca2+ channel.
We would add to this description that the high- and low-affinity sites appear to trigger distinct fusion mechanisms: only the slow, high-affinity site can operate in the absence of n-syb. The Ca2+ sensor for the n-syb-independent pathway would largely go unoccupied and would not trigger release after an individual action potential. This pathway would be triggered by sustained Ca2+ increases caused by tetanic stimulation, high K+, or Ca2+ ionophores, but does not appear to be modulated by cAMP.
Exocytosis in secretory cells has been divided into constitutive and regulated pathways (Kelly, 1988). The genetic ablation of the evoked ESC and persistence of mESCs might have reflected the ability of the constitutive pathway to mediate a portion of the spontaneous release. The present experiments, however, indicate that this spontaneous release arises from a Ca2+-regulated pathway that is genetically distinguishable from the evoked pathway.
Footnotes
This research was supported by grant-in-aids from the Ministry of Education, Science, Sports, and Culture of Japan to Y.K. and M.Y., and by a Silvio Conti Center for Neuroscience Award from the National Institute of Mental Health to T.L.S. D.L.D. was a National Institute of Neurological Disorders and Stroke postdoctoral fellow. We thank Professor Richard Tsien for insightful discussions.
Correspondence should be addressed to Dr. Yoshi Kidokoro, Gunma University School of Medicine, 3-39-22 Showa-machi, Maebashi, 371–8511, Japan.
REFERENCES
- 1.Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- 3.Branton WD, Kolton L, Jan YN, Jan LY. Neurotoxins from Plectreurys spider venom are potent presynaptic blockers in Drosophila. J Neurosci. 1987;7:4195–4200. doi: 10.1523/JNEUROSCI.07-12-04195.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broadie K, Prokop A, Bellen HJ, O’Kane CJ, Schulze KL, Sweeney ST. Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron. 1995;15:663–673. doi: 10.1016/0896-6273(95)90154-x. [DOI] [PubMed] [Google Scholar]
- 5.Chen C, Regehr WG. The mechanism of cAMP-mediated enhancement at the cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chow RH, Klingauf J, Heinemann C, Zucker RS, Neher E. Mechanisms determining the time course of secretion in neuroendcrine cells. Neuron. 1996;16:369–376. doi: 10.1016/s0896-6273(00)80054-9. [DOI] [PubMed] [Google Scholar]
- 7.Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL. Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene, neuronal-synaptobrevin. J Neurosci. 1998;18:2028–2039. doi: 10.1523/JNEUROSCI.18-06-02028.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaney KR, Tank DW. A quantitative measurement of the dependence of short-term synaptic enhancement on presynaptic residual calcium. J Neurosci. 1994;14:5885–5902. doi: 10.1523/JNEUROSCI.14-10-05885.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiAntonio A, Burgess RW, Chin AC, Deitcher DL, Sheller RH, Schwarz TL. Identification and characterization of Drosophila genes for synaptic vesicle proteins. J Neurosci. 1993;13:4924–4935. doi: 10.1523/JNEUROSCI.13-11-04924.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon D, Atwood HL. Adenylate cyclase system is essential for long term facilitation at the crayfish neuromuscular junction. J Neurosci. 1989;9:4246–4252. doi: 10.1523/JNEUROSCI.09-12-04246.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dreyer F, Rosenberg F, Becker C, Bigalke H, Penner R. Differential effects of various secretagogues on quantal transmitter release from mouse motor nerve terminals treated with botulinum A and tetanus toxin. Naunyn Schmiedebergs Arch Pharmacol. 1987;335:1–7. doi: 10.1007/BF00165027. [DOI] [PubMed] [Google Scholar]
- 12.Erulkar SD, Rahamimoff R. The role of calcium ions in tetanic and post-tetanic increase of miniature endplate potential frequency. J Physiol (Lond) 1978;278:501–511. doi: 10.1113/jphysiol.1978.sp012320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gansel M, Penner R, Dreyer F. Distinct sites of action of clostridial neurotoxins revealed by double-poisoning of mouse motor nerve terminal. Pflügers Arch. 1987;409:533–539. doi: 10.1007/BF00583812. [DOI] [PubMed] [Google Scholar]
- 14.Gho M. Voltage-clamp analysis of gap-junction between embryonic muscles in Drosophila. J Physiol (Lond) 1994;481:371–383. doi: 10.1113/jphysiol.1994.sp020446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunt JM, Bommert K, Charlton MP, Kistner A, Habermann E, Augustine GJ, Betz H. A post-docking role for synaptobrevin in synaptic vesicle fusion. Neuron. 1994;12:1269–1279. doi: 10.1016/0896-6273(94)90443-x. [DOI] [PubMed] [Google Scholar]
- 16.Janicki PK, Habermann E. Tetanus and botulinum toxins inhibit, and black widow spider venom stimulates the release of methionine-enkephalin-like material in vitro. J Neurochem. 1983;41:395–402. doi: 10.1111/j.1471-4159.1983.tb04755.x. [DOI] [PubMed] [Google Scholar]
- 17.Kamiya H, Zucker RS. Residual Ca2+ and short-term synaptic plasticity. Nature. 1994;371:603–606. doi: 10.1038/371603a0. [DOI] [PubMed] [Google Scholar]
- 18.Kelly RB. The cell biology of the nerve terminal. Neuron. 1988;1:431–438. doi: 10.1016/0896-6273(88)90174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kidokoro Y, Nishikawa K. Miniature endplate currents at the newly formed neuromuscular junction in Drosophila embryos and larvae. Neurosci Res. 1994;19:143–154. doi: 10.1016/0168-0102(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 20.Leung H-T, Branton WD, Phillips HS, Jan L, Byerly WL. Spider toxins selectively block calcium currents in Drosophila. Neuron. 1989;3:767–772. doi: 10.1016/0896-6273(89)90245-6. [DOI] [PubMed] [Google Scholar]
- 21.McMahon HT, Foran P, Dolly JO, Verhage M, Wiegant VM, Nicholls DG. Tetanus toxin and botulinum type A and B inhibit glutamate, γ-aminobutyric acid, aspartate, and met-enkephalin release from synaptosomes. J Biol Chem. 1992;267:21338–21343. [PubMed] [Google Scholar]
- 22.Meldolesi J, Scheer H, Madeddu L, Wanke E. Mechanism of action of α-latrotoxin: the presynaptic stimulatory toxin of the black widow spider venom. Trends Pharmacol. 1986;7:151–155. [Google Scholar]
- 23.Miledi R, Thies R. Tetanic and post-tetanic rise in frequency of miniature endplate potentials in low-calcium solutions. J Physiol (Lond) 1971;212:246–257. doi: 10.1113/jphysiol.1971.sp009320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishikawa K, Kidokoro Y. Junctional and extrajunctional glutamate receptor channels in Drosophila embryos and larvae. J Neurosci. 1995;15:7905–7915. doi: 10.1523/JNEUROSCI.15-12-07905.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts WM, Jacobs RA, Hudspeth AJ. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J Neurosci. 1990;10:3664–3684. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- 27.Schiavo G, Benfenati F, Poulain B, Rosetto O, de Laureto P, DasGupta B, Montecucco C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature. 1992;359:832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- 28.Siegelbaum SA, Camardo JS, Kandel ER. Serotonin and cyclic AMP close single K+ channels in Aplysia sensory neurones. Nature. 1982;299:413–417. doi: 10.1038/299413a0. [DOI] [PubMed] [Google Scholar]
- 29.Simon SM, Llínas RR. Compartmentalization of the submembrane calcium activity during calcium influx and its significance in transmitter release. Biophys J. 1985;48:485–498. doi: 10.1016/S0006-3495(85)83804-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Söllner T, Whiteheart SW, Brunner M, Erdjumer-Bromage H, Geromonos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993a;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 31.Söllner T, Benett MK, Whiteheart SW, Sheller RH, Rothman JE. A protein assembly/disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993b;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 32.Sweeney ST, Broadie K, Keane J, Niemann H, O’Kane CJ. Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron. 1995;14:341–351. doi: 10.1016/0896-6273(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 33.Ueda A, Kidokoro Y. Longitudinal body wall muscles are electrically coupled across the segmental boundary in the third instar larva of Drosophila melanogaster. Invertebr Neurosci. 1996;1:315–322. doi: 10.1007/BF02211911. [DOI] [PubMed] [Google Scholar]
- 34.Van der Kloot W. Facilitation of transmission at the frog neuromuscular junction at 0°C is not maximal at time zero. J Neurosci. 1994;14:5722–5724. doi: 10.1523/JNEUROSCI.14-09-05722.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- 36.Yamada WM, Zucker RS. Time course of transmitter release calculated from simulations of a calcium diffusion model. Biophys J. 1992;61:671–682. doi: 10.1016/S0006-3495(92)81872-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang D, Kuromi H, Kidokoro Y (1999a) Synaptic transmission at the Drosophila neuromuscular junction: effects of metabotropic glutamate receptor activation. Jpn J Physiol, in press. [DOI] [PubMed]
- 38.Zhang D, Kuromi H, Kidokoro Y (1999b) Activation of metabotropic glutamate receptors enhances synaptic transmission at the larval Drosophila neuromuscular synapse. Neuropharmacology, in press. [DOI] [PubMed]
- 39.Zhong Y, Wu C-F. Altered synaptic plasticity in Drosophila memory mutants with a defective cyclic AMP cascade. Science. 1991;251:198–201. doi: 10.1126/science.1670967. [DOI] [PubMed] [Google Scholar]