

Abstract
Pharmacological and biochemical approaches were used to elucidate the involvement of growth factor signaling pathways mediating estrogen neuroprotection in primary cortical neurons after glutamate excitotoxicity. We addressed the activation of mitogen-activated protein kinase (MAPK) signaling pathways, which are activated by growth factors such as nerve growth factor (NGF). Inhibition of MAPK signaling with the MAPK kinase inhibitor PD98059 blocks both NGF and estrogen neuroprotection in these neurons. These results correlate with a rapid and sustained increase in MAPK activity within 30 min of estrogen exposure. The involvement of signaling molecules upstream from MAPK was also examined to determine whether activation of MAPK by estrogen is mediated by tyrosine kinase activity. Estrogen produces a rapid, transient activation of src-family tyrosine kinases and tyrosine phosphorylation of p21ras-guanine nucleotide activating protein. Effects of estrogen on neuroprotection, as well as rapid activation of tyrosine kinase and MAPK activity, are blocked by the anti-estrogen ICI 182,780. This provides evidence that activation of the MAPK pathway by estrogen participates in mediating neuroprotection via an estrogen receptor. These results describe a novel mechanism by which cytoplasmic actions of the estrogen receptor may activate the MAPK pathway, thus broadening the understanding of effects of estrogen in neurons.
Keywords: estrogen, MAPK, neuroprotection, growth factors, excitotoxicity, src, GAP
Although estrogen is generally thought to act as an activator of transcription via nuclear receptors, including estrogen receptors ERα and ERβ, increasing evidence suggests that estrogen may also cause rapid activation of signal transduction pathways. Intracellular calcium is immediately increased with estrogen treatment in granulosa cells (Morley et al., 1992) and modulates neuronal calcium channels in striatal neurons (Mermelstein et al., 1996). Estrogen potentiates kainate currents in the hippocampus via cAMP-dependent phosphorylation (Wong and Moss, 1992; Gu and Moss, 1996), increases cAMP accumulation in breast cancer cells (Aronica et al., 1994), and rapidly activates mitogen-activated protein kinase (MAPK) in neuroblastoma (Watters et al., 1997) and non-neuronal cells (Migliaccio et al., 1996).
The MAPKs are a family of serine–threonine kinases activated by phosphorylation in response to a variety of mitogenic signals. Activation of MAPK transduces extracellular signals from multiple membrane receptors to intracellular targets, including transcription factors, cytoskeletal proteins, and other enzymes. Analysis of components of MAPK signaling activated by growth factors such as nerve growth factor (NGF) has elucidated a pathway in which a tyrosine kinase receptor initiates sequential phosphorylation and stimulation of adapter molecules to activate a guanine nucleotide exchange factor and a family of small guanine nucleotide binding proteins, including p21ras (Seger and Krebs, 1995). In breast cancer cells, MAPK activation by estrogen has been attributed to the initial activation of the nonreceptor tyrosine kinase src (Migliaccio et al., 1996). Transient transfection of ERα into COS-7 cells was sufficient for estrogen-mediated activation of src and MAPK, providing direct evidence of the early involvement of ER in the MAPK signal transduction cascade. Recent evidence has further indicated that activation of the MAPK pathway by another ovarian steroid, progesterone, is also mediated via cross talk of the progesterone receptor with the ER (Migliaccio et al., 1998).
Estrogen enhances neuronal survival resulting from oxidative stress, excitotoxic insults, and β-amyloid (Behl et al., 1995; Goodman et al., 1996; Green et al., 1996; Singer et al., 1996, 1998). Our laboratory has reported that a 24 hr pretreatment with 10–15 nm estrogen before an excitotoxic glutamate exposure significantly reduces cell death in primary cortical neurons (Singer et al., 1996). The regulation of neurotrophins (Gibbs et al., 1994; Singh et al., 1995) and neurotrophin receptors (Sohrabji et al., 1994a,b;McMillan et al., 1996) by estrogen, as well as the known role of growth factors in cell survival and recovery from injury (Hefti, 1986; Kromer, 1987; Cheng et al., 1994; Lindholm, 1994), has led to the hypothesis that estrogen neuroprotection may be mediated, in part, via signaling pathways similar to those used by growth factors. In the experiments reported here, we have examined MAPK and src tyrosine kinase activity in cultured cortical neurons treated with estrogen to ascertain whether growth factor-related signaling pathways may participate in mediating protective effects of estrogen in neurons after glutamate toxicity.
MATERIALS AND METHODS
Cell culture. Primary cortical neurons were prepared from rat embryos at 17–19 d gestation in dissociation buffer containing 0.14 m NaCl, 5.4 mm KCl, 24 mm HEPES, 4.2 mm NaHCO3, 2.5 mm NaH2 PO4, 14 mm glucose, and 0.01 gm/l phenol red. Cerebral neocorticies were removed, taking care to avoid the olfactory bulbs and hippocampus. The tissue was minced, treated with 0.25 mg/ml trypsin, and dissociated by trituration in 130 U/ml DNase and trypsin inhibitor. Cells were plated at 0.9 × 106 cells/ml on poly-d-lysine-coated 24-well plates for toxicity experiments or 100 mm cultures dishes for immunoblots in Neurobasal media containing the serum and estrogen-free B27 supplement (Life Technologies, Gaithersburg, MD), 0.5 mm glutamine, and 50 μg/ml gentamicin. Cultures were maintained at 37°C in a humidified 6% CO2 atmosphere, and all experiments were performed after 12 d in culture.
Glutamate toxicity. Forty-eight hours before glutamate exposure, cultures were placed in phenol red-free Neurobasal media containing B27 supplement, 0.5 mm glutamine, and 50 μg/ml gentamicin. Estrogen or other drug exposures took place as described at the indicated times. These compounds included a 0.01% ethanol vehicle, 2-amino-5-phosphopentanoic acid (AP-5), CNQX, estrogen in the form of 17β-estradiol (Sigma, St. Louis, MO), MK-801 (Research Biochemicals, Natick, MA), NGF (Promega, Madison, WI), ICI 182,780 (ICI) (a gift from Zeneca Pharmaceuticals, Cheshire, England), PD98059, dephostatin, and 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d] pyrimidine (PP1) (Calbiochem, La Jolla, CA). Glutamate exposure was performed for 5 min at 37°C in buffer containing 2 mm KCl, 1 mm MgSO4, 2.5 mmCaCl2, 1 mm NaH2PO4, 4.2 mm NaHCO3, 12.5 mm HEPES, 10 mm glucose, 0.1 mNaCl, and 0.1 mml-glutamic acid. Cultures were then washed and returned to fresh phenol red-free media.
Evaluation of cell death. Except where alternatively described, glutamate toxicity was evaluated by measuring lactate dehydrogenase (LDH) activity released in the media 24 hr after glutamate exposure using the CytoTox96 nonradioactive assay (Promega) and quantitated by measuring wavelength absorbance at 490 nm. Data are normalized to the amount of LDH released from vehicle-treated cells receiving glutamate (100%) and are corrected for baseline LDH release from vehicle-treated cells exposed to buffer only. Where noted, LDH release was also corrected from vehicle-treated cells exposed to drugs. This procedure is necessary to ensure that the reported effects of estrogen are not attributable to the presence of the 0.01% ethanol vehicle solution and to take into account the effects, if any, of the ethanol vehicle on glutamate receptors and buffer or drug treatment on cell viability. Statistical analysis was performed by one-way ANOVA, followed by Fisher’s least significant difference test post hoc.
MAPK phosphorylation. An antibody recognizing the dual threonine and tyrosine phosphorylation sequence from MAPK necessary for activation of the enzyme (Anti-Active MAPK; Promega) was used to evaluate extracellular signal-regulated kinase (ERK1/ERK2) MAPK phosphorylation. Twenty micrograms of total protein from aliquots of whole-cell lysates obtained as described below for MAPK activity or prepared in immunoprecipitation buffer were separated under denaturing and reducing conditions by SDS-PAGE on 10–20% gradient polyacrylamide Tris–glycine gels (Novex Corporation, San Diego, CA). After transfer to Immobilin-P (Millipore, Bedford, MA), the membrane was blocked with 5% milk in 0.2% Tween 20 in PBS (TPBS) and incubated with Anti-Active MAPK at a 1:20,000 dilution in TPBS as recommended by the manufacturer. Membranes were then incubated in horseradish peroxidase (HRP)-tagged sheep anti-rabbit IgG diluted in TPBS, and results were visualized by chemiluminescence (Renaissance; NEN Life Sciences, Boston, MA). To examine total ERK2 present in the samples, the same membranes were then stripped of antibody in 62.5 mm Tris, pH 6.8, 2% SDS, and 100 mmβ-mercaptoethanol at 50°C, washed in TPBS, and incubated with an anti-ERK2 antibody (C-14) at 0.25 μg/ml (Santa Cruz Biotechnology, Santa Cruz, CA), followed by chemiluminescence. Optical density analysis with a computer imaging system (MCID, St. Catharines, Ontario, Canada) was used to quantitate immunoreactivity in terms of fold phospho-ERK2 induction relative to total ERK2 present in the sample.
MAPK activity. MAPK activity was measured as described previously (Seger et al., 1992). Cells were treated with estrogen or other drugs as indicated, washed in PBS containing 137 mm NaCl, 2.7 mm KCl, 4.3 mmNa2HPO4, and 1.5 mmKH2PO4, and scraped in 50 mmβ-glycerolphosphate, 1.5 mm EGTA, 0.1 mmsodium orthovanadate, 1 mm DTT, 1 mmbenzamidine, 0.2 mm PMSF, 10 μg/ml aprotinin, 2 μg/ml pepstatin, 10 μg/ml leupeptin, and 1% Triton X-100. After centrifugation at 15,000 rpm, whole-cell lysates were added to equilibrated DE-52 columns, washed, and MAPK eluted in 0.2m NaCl. Extracts were then incubated with 25 mmβ-glycerolphosphate, 1.25 mm EGTA, 0.15 mmsodium orthovanadate, 1 mm DTT, 10 mmMgCl2, 2 μm PKI, 10 μmcalmidizolium, 1 mg/ml bovine serum albumin (BSA), 0.1 mmATP, and [γ-32P]ATP in the presence or absence of 2 mg/ml myelin basic protein (MBP) substrate for 15 min at 30°C. Reactions were terminated by spotting on P81 phosphocellulose, followed by exhaustive washes in 150 mm phosphoric acid and scintillation counting. Protein concentrations were determined using a modified Bradford assay (Bio-Rad, Hercules, CA).
Phosphotyrosine and p21ras-GTPase activating protein immunoblotting. Primary cortical neuron cultures were treated with estrogen, ICI, or PP1 as indicated. Cultures were then washed in PBS (137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, and 1.5 mm KH2PO4), and whole-cell lysates were prepared by centrifugation at 15,000 rpm in immunoprecipitation buffer containing 25 mm HEPES, pH 7.5, 10% glycerol, 5 mm EGTA, 5 mm EDTA, 100 mm NaCl, 100 mm sodium pyrophosphate, 50 mm NaF, 0.1 mm sodium orthovanadate, 1% Triton X-100, 1 mmbenzamidine, 1 mm PMSF, 10 μg/ml aprotinin, 1 μg/ml pepstatin, and 10 μg/ml leupeptin. Protein concentrations were determined using bicinchoninic acid (BCA) (Pierce, Rockford, IL) and 20 μg of protein separated under denaturing and reducing conditions by SDS-PAGE on 10–20 or 4–20% gradient polyacrylamide Tris–glycine gels (Novex Corporation). After transfer to Immobilin-P (Millipore), membranes were blocked in 5% nonfat milk in TPBS (0.2% Tween 20, 137 mm NaCl, 2.7 mm KCl, 4.3 mmNa2HPO4, and 1.5 mmKH2PO4) and incubated with mouse monoclonal anti-p21ras-GTPase activating protein (p21ras-GAP) (B4F8; Santa Cruz Biotechnology) or mouse monoclonal anti-phosphotyrosine (PY20; Santa Cruz Biotechnology) overnight at 4°C. Membranes were then incubated in HRP-conjugated mouse IgG, and results were visualized by chemiluminescence (Renaissance; NEN Life Sciences).
Protein tyrosine kinase assay. Whole-cell lysates were prepared from primary cortical neurons treated with estrogen or other drugs in 25 mm HEPES, pH 7.5, 10% glycerol, 5 mm EGTA, 5 mm EDTA, 100 mm NaCl, 100 mm sodium pyrophosphate, 50 mm NaF, 0.1 mm sodium orthovanadate, 1% Triton X-100, 1 mmbenzamidine, 1 mm PMSF, 10 μg/ml aprotinin, 1 μg/ml pepstatin, and 10 μg/ml leupeptin. Protein tyrosine kinase (PTK) activity was measured using a peptide substrate with the SignaTECT assay system (Promega). Extracts were diluted in 8 mm imidazole hydrochloride, pH 7.3, 8 mmβ-glycerolphosphate, and 0.1 mg/ml BSA and incubated for 20 min at 30°C in assay mix containing 8 mm imidazole2hydrochloride, 8 mm β-glycerolphosphate, 0.2 mm EGTA, 20 mm MgCl2, 1 mm MnCl2, 0.1 mg/ml BSA, 1 mm DTT, 0.125 mm sodium orthovanadate, 0.2 mm ATP, 0.2 μCi/μl [γ-32P]ATP, and 0.3 mmbiotinylated peptide substrate. Reactions were terminated by addition of 7.5 m guanidine hydrochloride, spotted onto SAM2 biotin capture membrane squares, washed in 2m NaCl and 2 m NaCl in 1% H3PO4, and counted by scintillation. Protein concentrations were determined using a BCA assay.
RESULTS
Initial experiments were performed to characterize glutamate excitotoxicity in these primary neuronal cultures under the dissociation and culture conditions described. In all experiments, cell death was evaluated by measuring LDH release into the media from dead or dying cells 24 hr after glutamate exposure. Although other methods of evaluating cell death are available, LDH release is a reliable biochemical indicator of cell death amenable to the large number of cultures used in these studies. LDH release has also been extensively used to quantitate cell death in neuronal cells (Choi, 1987; Behl et al., 1994; Singer et al., 1996). As reported previously, concentrations of glutamate ranging from 0.1 to 5 mm cause cell death in 55% of the cells in this culture preparation (Singer et al., 1996). The presence of glutamate-insensitive neurons or non-neuronal cells may account for the inability of glutamate to induce death in all cells (Choi, 1992). Subsequent experiments were performed using 0.1 mm glutamate, representing the lowest dose at which the maximal amount of cell death is achieved in these cultures.
Other laboratories have demonstrated that cell death mediated by an acute toxic glutamate exposure in primary cortical neurons, as performed in these experiments, is dependent on the NMDA glutamate receptor and the presence of extracellular calcium (Choi, 1985; Choi et al., 1988). To examine the involvement of NMDA receptors in mediating excitotoxicity in this preparation, cultures were treated with the non-NMDA receptor CNQX (10 μm), the competitive NMDA receptor antagonist AP-5 (200 μm), and the noncompetitive NMDA receptor antagonist MK-801 (10 μm) in the presence of 0.1 mm glutamate for 5 min. Glutamate excitotoxicity is partially inhibited by the presence of CNQX or AP-5 and completely blocked in the presence of MK-801 (Fig.1). Excitotoxins acting through non-NMDA receptors, such as kainate, do not produce cell death in these cultures after an acute 5 min exposure (data not shown). The results demonstrate that the acute glutamate excitotoxicity induced in these cultures corresponds to that reported in the literature (Choi et al., 1988; Choi, 1992).
Fig. 1.

Effects of glutamate receptor antagonists on glutamate toxicity in primary cortical neurons. Primary cortical neurons were exposed to 0.1 mm glutamate alone for 5 min at 37°C or in the presence of the glutamate receptor antagonists CNQX (10 μm), AP-5 (200 μm), or MK-801 (10 μm). Cultures were then washed and returned to fresh media, and LDH release was measured 24 hr later. Data are normalized to the amount of LDH present in cultures treated with glutamate alone (100%) and corrected for baseline LDH release in cultures exposed to buffer only. Results are from three to four separate platings;n = 3 per plating; mean ± SE. *p < 0.05, statistical significance between cultures exposed to antagonists and those treated with glutamate alone.
Previous experiments had determined an optimal dose of estrogen (10 nm) required to achieve maximal neuroprotection when the hormone is added 24 hr before glutamate exposure in primary cortical neurons. This effect was not seen with other steroid hormones (Singer et al., 1996). It was of interest, however, to determine whether shorter treatment times necessary to mediate nongenomic effects of estrogen could also promote cell survival. In addition, because the expression (Gibbs et al., 1994; Singh et al., 1995) and presumably function of growth factors can be altered in the presence of estrogen, it was also of interest to determine whether a representative growth factor, NGF, could elicit neuroprotection in these cultures. A 24 hr pretreatment of 10 nm estrogen before a 5 min 0.1 mm glutamate exposure decreases LDH release by 20% when compared with vehicle-treated cells exposed to glutamate (Fig.2, filled bars). This same magnitude of neuroprotection is seen with 20 ng/ml NGF pretreatment for 24 hr. A 5 min pretreatment with either 10 nm estrogen or 20 ng/ml NGF also results in a 20% decrease in LDH release (Fig. 2,open bars). These results indicate that at either time point, 20% of the cells in this culture preparation treated with estrogen or NGF survive an acute toxic glutamate exposure.
Fig. 2.

Neuroprotective effects of 10 nmestrogen (E2) and 20 ng/ml NGF after a 24 hr (filled bars) or 5 min (open bars) pretreatment before a 5 min 0.1 mm glutamate exposure. Data are representative from four separate platings; n = 3–4 per plating; mean ± SE. Cell death was evaluated by measuring LDH release into the media 24 hr later and normalized as described in Materials and Methods. **p < 0.01, statistical significance between vehicle (V) and NGF at both time points, and vehicle and estrogen at both time points.
In previous studies, pretreatment of these cultures with glucocorticoids, progesterone, or cholesterol failed to elicit the neuroprotective effects seen with estrogen. Immunoreactivity for the ERα is also present in these cultures (Singer et al., 1996). To determine the requirement for ER in mediating the neuroprotective effects of estrogen, the cultures were treated with ER antagonists. It had been reported previously that treatment with the anti-estrogen tamoxifen can block neuroprotection elicited by estrogen in these cultures (Singer et al., 1996). Tamoxifen, however, may act as a partial agonist in some systems, so it was of interest to determine the effects of ICI, an ER antagonist lacking agonist activity. In Figure3, primary cortical neurons were treated with 10 nm estrogen in the presence of 1 μmICI 24 hr before a toxic glutamate exposure. The dose of ICI used in these experiments does not affect neuronal viability in the absence of glutamate, and in the presence of estrogen, ICI completely blocks any protective effect observed with estrogen. ICI alone does not to alter glutamate toxicity.
Fig. 3.

ICI 182,780 blocks estrogen neuroprotection. Rat primary cortical neurons were treated with vehicle (V), 10 nm estrogen (E2), estrogen plus 1 μm ICI 182,780 (E2+ICI), or ICI alone for 24 hr before a 5 min 0.1 mm glutamate exposure. Data are normalized as described in Materials and Methods and are representative from five separate platings; n = 3–4 per plating; mean ± SE. *p < 0.05, statistical significance between estrogen alone, ICI alone, or estrogen plus ICI; **p < 0.01, statistical significance between vehicle and estrogen.
The finding that shorter treatment times of 5 min are sufficient to mediate neuroprotection by estrogen and NGF led to the possibility that the mechanism of estrogen neuroprotection may involve rapid activation of signaling pathways. To determine whether activation of growth factor signaling through MAPK is a component of estrogen neuroprotection, rat primary cortical neurons were treated with the MAPK kinase (MEK) inhibitor PD98059 (Fig. 4). Neuroprotection elicited by either estrogen or NGF is blocked by the addition of 50 μm PD98059. Addition of PD98059 alone does not significantly affect cell viability in the presence or absence of glutamate. Subsequent experiments demonstrate that 50 μm PD98059 is sufficient to block MAPK activation and phosphorylation by MEK in these cultures (see Fig. 7A).
Fig. 4.

PD98059 blocks estrogen and NGF-induced neuroprotection. Rat primary cortical neurons were treated with vehicle (V), 10 nm estrogen (E2), and 20 ng/ml NGF, all alone or plus 50 μm PD98059 24 hr before a 5 min 0.1 mmglutamate exposure. Data are representative from three separate platings; n = 3–4 per platings; mean ± SE. Cell death was evaluated by measuring LDH release into the media 24 hr later and normalized to LDH release from vehicle- or drug-treated cultures as described in Materials and Methods. *p< 0.05, statistical significance between vehicle and NGF, NGF alone, or NGF plus PD98059; **p < 0.01, statistical significance between estrogen and PD98059; ***p < 0.001, statistical significance between vehicle and estrogen.
Fig. 7.
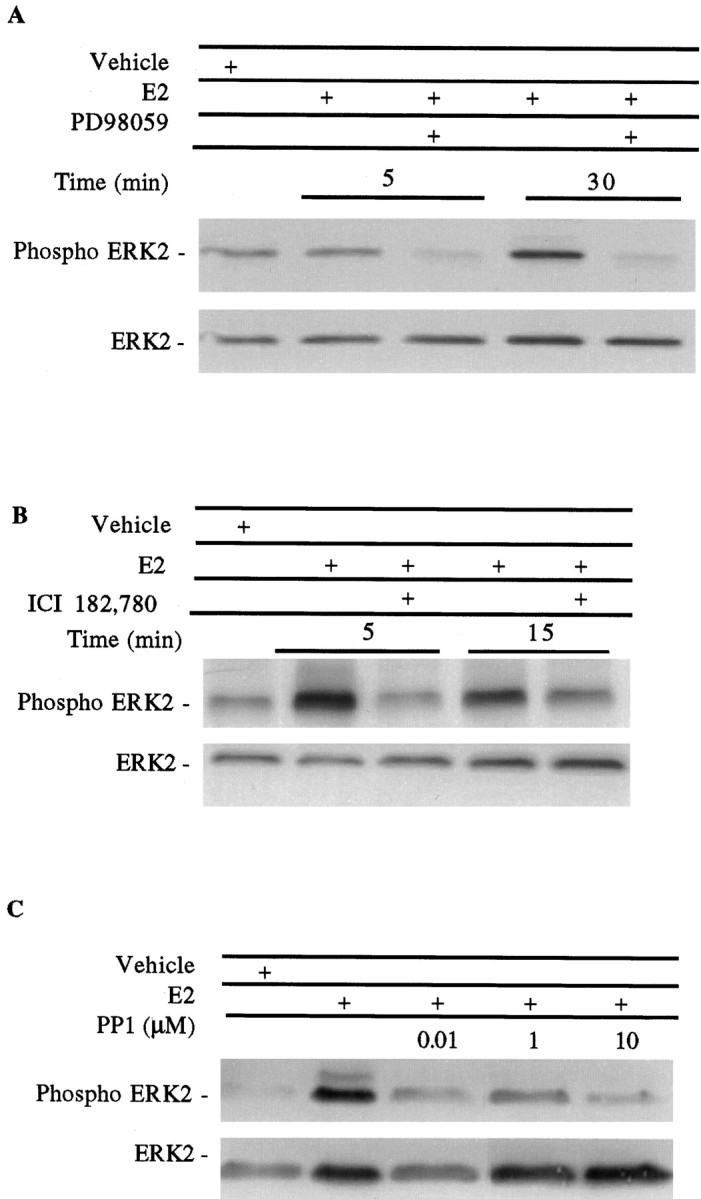
MAPK phosphorylation in primary cortical neurons treated at the times indicated with 10 nm estrogen (E2) with or without the addition of 50 μmPD98059 (A), 1 μm ICI 182,178 (B), or increasing concentrations of PP1 (C) for 30 min. Twenty micrograms of total protein from whole-cell lysates were separated by SDS-PAGE and incubated with the Anti-Active MAPK antibody. The single bands shown in A and B represent p42 ERK2 MAPK. The membrane was then stripped and reprobed for ERK2 as a control. In C, p44 ERK1 phosphorylation can be seen with estrogen treatment.
The role of tyrosine phosphorylation and tyrosine kinase activation, signaling components presumably upstream from MAPK in these cultures, were examined pharmacologically after glutamate toxicity (Table1). To examine the role of tyrosine phosphorylation in mediating neuroprotection, primary cortical neurons were pretreated for 24 hr in the presence of 10 nm estrogen with dephostatin, a membrane-permeable protein tyrosine phosphatase inhibitor (Braunton et al., 1998). Dephostatin augmented estrogen neuroprotection against glutamate toxicity as measured by a 40% decrease in LDH release from cells receiving estrogen. The role of nonreceptor src-family PTKs was also examined in primary cortical neurons exposed to glutamate using PP1, an inhibitor of src tyrosine kinases (Hanke et al., 1996). A 24 hr pretreatment with PP1 in the presence of estrogen completely blocks neuroprotection, indicating that tyrosine kinase activation through a src-family kinase may be another component mediating estrogen neuroprotection and MAPK activity. Dephostatin or PP1 alone did not significantly affect LDH release in the presence or absence of glutamate (data not shown).
Table 1.
Role of tyrosine phosphorylation and tyrosine kinase activation after glutamate toxicity
| % LDH Release | % LDH Release | |
|---|---|---|
| Vehicle | 99.9 ± 6.2 | 100.2 ± 13.6 |
| Estrogen | 77.6 ± 6.2* | 71.6 ± 4.3* |
| Dephostatin | 36.7 ± 13.5 | ND |
| PP1 | ND | 113.9 ± 6.7 |
Primary cortical neurons were treated for 24 hr with vehicle or 10 nm estrogen in the presence of 10 μmdephostatin, a protein tyrosine phosphatase inhibitor, or 10 μm PP1, a src-family tyrosine kinase inhibitor. Cultures were then exposed to 0.1 mm glutamate for 5 minutes as described, and LDH release into the media was measured to evaluate cell death after 24 hr. Data were normalized to the amount of LDH release from cells treated with vehicle in the presence of glutamate (100%) from representative experiments; n = 3; mean ± SE. ND, Not determined.
* p < 0.05, significant differences between estrogen and all drug treatments.
To determine whether estrogen activates MAPK in these cultures,in vitro phosphorylation assays were performed to assess MAPK activation in the presence of estrogen. Treatment with 10 nm estrogen results in a rapid twofold increase in MAPK activity, as measured by phosphorylation of MBP, that is sustained with 30 min of estrogen exposure before returning to basal levels (Figs.5,6). No MAPK activation was observed in response to the ethanol vehicle. As a comparison, NGF, a growth factor known to activate MAPK, rapidly phosphorylates MBP within 5 min of treatment (data not shown). Although other enzymes capable of phosphorylating MBP may be found in the 0.2 m NaCl eluate used in this assay, treatment with 50 μm PD98059 completely blocks MAPK activity (data not shown) and phosphorylation (Fig. 7A) in estrogen-treated cultures, indicating that this activity can be attributed to MAPK.
Fig. 5.

Activation of MAPK by estrogen. Rat primary cortical neurons were treated with 10 nm estrogen at the indicated time points, and whole-cell lysates were prepared to evaluate MBP phosphorylation in duplicate with or without the addition of MBP. Data points represent picomoles of 32P incorporated on phosphocellulose filters in 15 min at 30°C normalized to the amount of protein present in each sample; n = 6–7; mean ± SE; three to four separate platings.
Fig. 6.

Phosphorylation of ERK MAPK by estrogen. Rat primary cortical neurons were treated with 10 nm estrogen at the indicated times points. Aliquots of whole-cell lysates obtained for activity assays were also separated by SDS-PAGE, transferred to membranes, and incubated with the Anti-Active MAPK antibody, which recognizes threonine and tyrosine phosphorylation of the active enzyme.A, Representative immunoblots are shown demonstrating ERK2 phosphorylation by estrogen in the top panel with total ERK2 shown from the same blot in the bottom panel.B, Fold induction of MAPK phosphorylation after optical density analysis of phosphorylated and total ERK2 levels.
Dual phosphorylation of ERK MAPK on the threonine and tyrosine residues necessary for activation was also evaluated using the Anti-Active MAPK antibody, which has been developed to correlate ERK1/ERK2 MAPK activation with its phosphorylation state (White et al., 1996). Aliquots of whole-cell lysates used for activity assays were separated by SDS-PAGE and incubated with the Anti-Active MAPK antibody. A time course of ERK2 MAPK phosphorylation by estrogen is shown (Fig.6A, top). Addition of 5% serum for 15 min resulted in increased phosphorylation of p44 ERK1 and p42 ERK2 (data not shown) and, in comparison, demonstrated that the band shown represents ERK2 phosphorylation by estrogen. The same blots were stripped and reprobed for total ERK2 as a control for protein loading (Fig. 6A, bottom). Optical density analysis of phospho-ERK2 immunoreactivity relative to total ERK2 immunoreactivity correlates with the twofold induction seen using the substrate-based enzymatic assay (Fig. 6B). The Anti-Active MAPK antibody was then used in subsequent experiments as a measure of MAPK activity.
To examine the possibility that the activation of MAPK by estrogen is caused by the actions of MEK, the MEK inhibitor PD98059 was added to the cultures. In the presence of 10 nm estrogen, MAPK phosphorylation again increases twofold from vehicle-treated cells within 30 min of estrogen treatment as examined by optical density analysis (data not shown). Addition of 50 μm PD98059 blocks this ERK2 phosphorylation (Fig. 7A). Total ERK2 immunoreactivity is also shown as a control in the bottom panel. The ability of PD98059 to block unstimulated ERK2 phosphorylation at the 5 min time point may be attributed to effects of PD98059 on basal ERK2 phosphorylation.
Data from other laboratories has indicated that an ER may mediate MAPK activity in breast cancer cells (Migliaccio et al., 1996) and can be found complexed with heat shock protein 90 (hsp90), MEK, and raf in cortical explants (Singh et al., 1998), leading us to pharmacologically examine the role of an ER in MAPK activation in neurons. Primary cortical neurons were treated at the times indicated in Figure7B with 10 nm estrogen in the presence of 1 μm ICI. In the experiment shown, ERK2 phosphorylation peaks within 5–15 min of estrogen treatment and is blocked with the addition of ICI. ICI alone did not have any effect on ERK2 phosphorylation in these cultures (data not shown). Total ERK2 from the same immunoblot is shown in the bottom panel.
Pharmacological inhibition of src-family tyrosine kinases (Table 1) has thus far implicated tyrosine kinase activity as a component in the mechanism of estrogen neuroprotection. It was of interest to determine the involvement of src-family tyrosine kinases in MAPK activation by estrogen. This was done by evaluating the phosphorylation and activation of MAPK in the presence of the src-family tyrosine kinase inhibitor PP1. After 30 min of estrogen treatment, MAPK phosphorylation increases twofold (Fig. 7C). In the presence of increasing doses of PP1, MAPK phosphorylation is decreased, thus indicating that MAPK is no longer active after estrogen treatment.
p21ras-GAP is phosphorylated after activation of many growth factor receptors and associates with GTP-bound p21ras to catalyze its intrinsic GTPase activity (Tocque et al., 1997). p21ras-GAP also contains an src homology 3 (SH3) and two SH2 phosphotyrosine-containing sequences phosphorylated by a variety of effectors, including src tyrosine kinases (Brott et al., 1991). Figure 8shows phosphotyrosine and p21ras-GAP immunoreactivity at 120 kDa after treatment with 10 nmestrogen in the presence of the src-family inhibitor PP1. Phosphotyrosine immunoreactivity is seen within 2.5–5 min of estrogen treatment and is not present with addition of 10 μm PP1, suggesting that tyrosine phosphorylation of a 120 kDa protein, which may be p21ras-GAP, requires src-family tyrosine kinase activity.
Fig. 8.

Primary cortical neurons were treated with 10 nm estrogen (E2) with or without PP1 (10 μm) for 30 min. Twenty micrograms of total protein from whole-cell lysates were separated by electrophoresis, transferred to membranes, and immunoblotted with anti-p21ras-GAP shown at 120 kDa. The same blot was then stripped of antibody and incubated with anti-phosphotyrosine (p-Tyr). The phosphotyrosine-containing protein at 120 kDa comigrates p21ras-GAP at 120 kDa. Results shown are from a representative experiment.
Activation of PTK activity by estrogen in primary cortical neuron cultures was examined using a high-affinity peptide substrate for src-family tyrosine kinases. In the presence of estrogen, PTK activity increases twofold within 1 min of estrogen treatment (Fig.9A). Previous reports have indicated that activation of src in breast cancer cells occurs in the presence of a ligand-activated estrogen receptor complex (Migliaccio et al., 1996). The role of estrogen receptor in mediating src-family PTK activity in the presence of estrogen is shown in Figure 9Bin which addition of ICI blocks PTK activity induced by estrogen, suggesting that an ICI-sensitive ER is necessary to mediate the effects of estrogen on PTK activity. The addition of the src-family kinase inhibitor PP1 blocks PTK activity measured in this assay, thus indicating that the PTK activity measured in this assay is attributable to src-family tyrosine kinases.
Fig. 9.
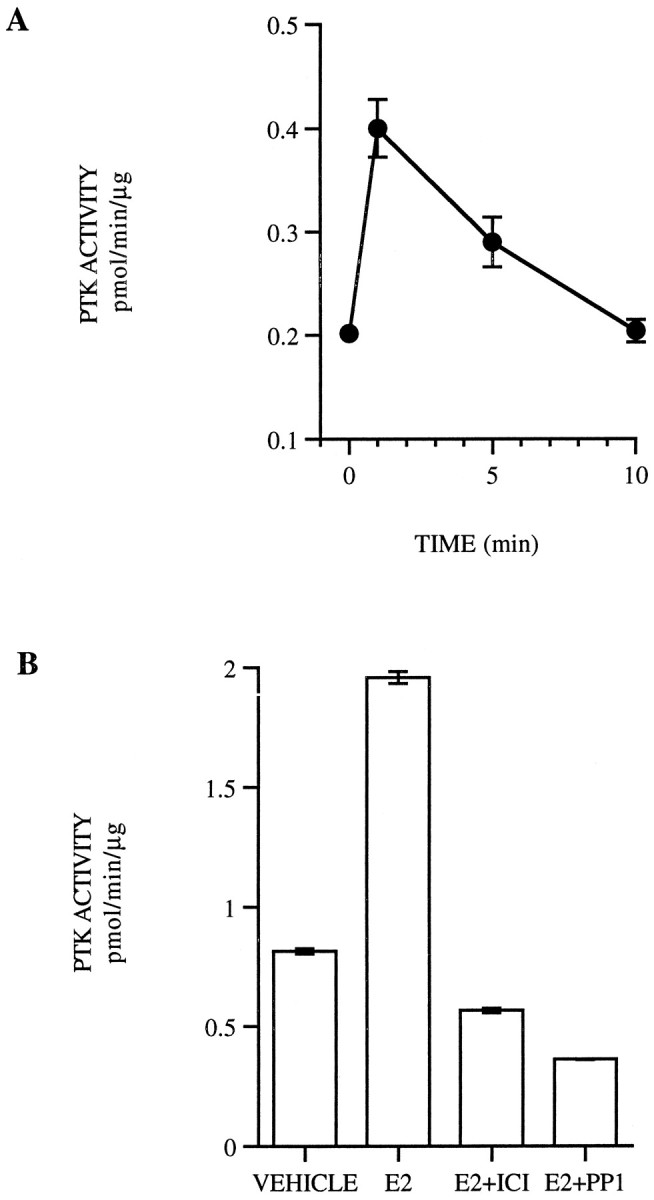
A, PTK activity was evaluated at the indicated time points in primary cortical neurons treated with 10 nm estrogen (E2) with or without peptide substrate in the SignaTECT assay system, followed by scintillation counting. The results are reported as picomoles of ATP added to the substrate during the assay time and normalized to the amount of protein in each sample. The resulting values represent the mean ± SE from duplicate samples representative from three experiments.B, Primary cortical neurons were also treated with vehicle, 10 nm estrogen with or without 1 μmICI 182,780 (ICI), or 10 μm PP1 for 1 min. PTK activity was then evaluated with or without peptide substrate as described and the results described above. The resulting values represent the mean ± SE from duplicate samples representative from three experiments.
DISCUSSION
Trophic effects of estrogen affecting neuronal survival have been well documented, but the mechanisms underlying these effects are not fully understood. Recent reports from other laboratories demonstrating estrogen neuroprotection against oxidative stress, excitotoxicity, and β-amyloid toxicity (Behl et al., 1995; Goodman et al., 1996; Green et al., 1996; Gridley et al., 1997) have emphasized possible antioxidant properties of the steroid as a mechanism of action. The glutamate toxicity experiments presented here were performed using substantial levels of antioxidants contained in the B27 media supplement. The contribution of the antioxidant properties of estrogen to neuroprotection after glutamate toxicity in these experiments may therefore be masked by the use of B27. The fact that estrogen can elicit neuroprotection in the presence of antioxidants, however, indicates that growth factor signaling pathways are another component of estrogen protection that should be assessed.
Results for our laboratory have demonstrated that a 24 hr or 5 min pretreatment with 10 nm estrogen protects primary cortical neurons from glutamate toxicity and that this neuroprotection is mediated by activation of a tamoxifen and ICI-sensitive ER. The specificity of these effects had been reported previously, showing that treatment with other steroid hormones, including progesterone, dihydrotestosterone, dexamethasone and cholesterol, failed to positively affect neuronal survival in these cultures (Singer et al., 1996). The wide variety of protective actions of estrogen and the range of doses (2 nm-10 μm) and time points presented in the literature (2–48 hr) creates difficulties in comparing results. To our knowledge, the results presented here are the first to demonstrate neuroprotection with 5 min of estrogen treatment. In addition, the 10 nm dose of estrogen used in these experiments is the same as that used by others (Migliaccio et al., 1996) in which ER-expressing cells also exhibited rapid effects on signaling pathways.
The regulation of neurotrophin expression by estrogen (Gibbs et al., 1994; Singh et al., 1995) and the role of various growth factors in mediating cell survival (Kromer, 1987; Cheng et al., 1994;Lindholm, 1994) has led to the examination of growth factor signaling pathways as a possible mechanism in estrogen neuroprotection. NGF was used as a representative growth factor that activates MAPK signaling pathways and significantly increases cell viability after glutamate toxicity in the neuronal cultures used in these experiments. Coaddition of NGF and estrogen does not promote greater neuroprotection than either compound used alone (C. A. Singer, unpublished observations), suggesting that growth factors, such as NGF and estrogen, may be acting on the same signaling pathway and that estrogen may not increase the secretion of NGF in these cultures to enhance cell viability. It is also likely that the effects of NGF and estrogen reported here occur in a small percentage of neurons expressing neurotrophin receptor or estrogen receptors, which may account for the modest effects of both compounds on neuroprotection. The role of growth factor signaling pathways in mediating neuroprotection was examined using the MEK inhibitor PD98059 (50 μm), which blocks neuroprotection elicited by estrogen and NGF. Doses of PD98059 from 10–100 μm are sufficient to completely block MAPK activation by MEK and do not affect activities of other protein kinases, including raf, cAMP-dependent protein kinase, protein kinase C, phosphatidylinositol 3-kinase, c-jun N-terminal kinase, or p38 MAPKs (Dudley et al., 1995; Pang et al., 1995). The present observation that activation of ERK MAPK may lead to neuronal survival is consistent with a previous suggestion by Xia et al. (1995) in which cell death induced by NGF withdrawal in PC12 cells was observed to activate JNK and p38 MAPK and inhibit ERK MAPK. In such a model, activation of ERK MAPK and inhibition of JNK and p38 MAPK would be predicted to promote neuronal survival.
The mechanism of MAPK activation by estrogen is not well understood. Results from several laboratories, however, now indicate this activity may represent a novel mechanism for steroid hormone actions in the cell. In breast cancer cells, a ligand-induced estrogen receptor complex capable of activating the MAPK pathway within 2 min of estrogen treatment has been reported, indicating that the estrogen receptor may function in the cytosol to modulate signaling pathways and that the initial signal is likely to come from activation of src (Migliaccio et al., 1996). Previous work in primary cortical neurons demonstrated that the estrogen receptor antagonist tamoxifen blocks neuroprotection elicited by estrogen (Singer et al., 1996). Further experiments have now indicated that ICI, a more potent antagonist, also inhibits neuroprotection and that addition of ICI inhibits MAPK activation by estrogen, indicating that an ICI-sensitive ER is involved in MAPK activation within 30 min of estrogen exposure. Activation of growth factor signaling pathways, such as the MAPK cascade, by estrogen appears then to involve cytosolic actions of an estrogen receptor. To the best of our knowledge, the expression of ER is not affected by the use of the phenol red-free culture conditions described here, and the results obtained in the absence of phenol red are essentially the same as published previously with media containing phenol red (Singer et al., 1996).
Estrogen treatment of rat primary cortical neurons causes a rapid, yet sustained, phosphorylation and activation of ERK2 MAPK that appears to involve upstream components of the signaling pathway. PD98059 inhibits MAPK activation by estrogen, indicating that MEK phosphorylates MAPK in the presence of estrogen. Inhibition of tyrosine phosphatase activity also enhances neuroprotection, thus making it likely that upstream tyrosine phosphorylation is a prelude to estrogen-mediated activation of MAPK in neurons. Stimulation of tyrosine kinase activity attracts adapter proteins and guanine nucleotide exchange factors, such as grb2 and sos, leading to subsequent activation of p21ras (Egan et al., 1993). The GTPase activating protein GAP stimulates p21ras GTPase activity after activation, leading to p21ras inactivation. In primary cortical neurons, estrogen appears to rapidly increase src-family kinase-mediated tyrosine phosphorylation of a 120 kDa protein that may be p21ras-GAP. These results differ from previous observations in breast cancer cells demonstrating no change in p21ras-GAP but increases in GAP-associated p190 phosphorylation (Migliaccio et al., 1996).
The activity of estrogen on src tyrosine kinases and MAPK activity described here pharmacologically appears to require an ER, consistent with results in breast cancer cells (Migliaccio et al., 1996). The nature of the involvement of the ER with this signaling pathway remains unclear, but recent results suggest that ER may exist in a cytosolic complex containing hsp90, raf, and MEK (Singh et al., 1998). Sequence analysis has also suggested that ER interaction with src tyrosine kinases or other phosphotyrosine-containing proteins may be facilitated by a previously unrecognized SH2 domain (Arnold and Notides, 1995).
The data presented here do not address the specific involvement of ERα or ERβ in direct activation MAPK or whether this activation occurs via an intermediate transcriptional mechanism. It is likely that activation of the ERK MAPK signaling pathway by estrogen may ultimately be mediating the genomic activity of ER or other transcription factors. MAPK phosphorylation of the ER can regulate transcriptional activity (Kato et al., 1995), and src-mediated phosphorylation of the ER, presumably via MAPK, has been demonstrated to modulate ER dimerization necessary for DNA binding (Arnold and Notides, 1995; Arnold et al., 1995). It is not known whether dimerization of ER is necessary to mediate the effects of estrogen on MAPK activity or whether estrogen may activate MAPK while rapidly entering the nucleus to activate genomic responses through the ER. In addition, although our results thus far are in agreement with those reported in breast cancer cells, it is not clear whether estrogen may differentially affect MAPK activity in nonproliferating neuronal cultures. The time course of tyrosine kinase activation by estrogen within 1 min of treatment further suggests that ER may be acting directly on a src tyrosine kinase or on a closely associated molecule. In addition, the time course of ERK2 activation by estrogen indicates that ER is not acting on ERK2 through an effect on transcription, which would require hours rather than minutes of estrogen treatment. Based on this interpretation, these data suggest that ER is acting directly on one or more components of this pathway.
The results from these studies clearly demonstrate that activation of MAPK by estrogen in neurons is mediated via phosphorylation from a src tyrosine kinase. Stimulation of this signal transduction pathway in the presence of estrogen confers the neuroprotective effects of estrogen demonstrated after glutamate toxicity, although it is unclear whether further transcriptional activity mediated by the ER or MAPK is required to mediate neuroprotection. Although estrogen may act as an antioxidant to increase cell viability, these results indicate that alterations in signal transduction pathways by estrogen represent another mechanism of action in a complex chain of events that occur to bring about neuronal death and survival. Although some of the effects of estrogen on src tyrosine kinase and MAPK activity reported here have been reported in breast cancer cells, it is imperative to relate effects of estrogen reported in cell lines to results in primary neurons to determine that these effects can occur in nontransformed cells. The neuroprotective effects of estrogen reported here support clinical observations suggesting that estrogen replacement therapy after menopause reduces the risk of developing Alzheimer’s disease (Henderson et al., 1994;Kawas et al., 1997). Among the implications of these findings are that understanding the mechanisms of estrogen neuroprotection will allow for the development of more effective estrogenic agents useful for the treatment of neurodegenerative disorders, in addition to well documented beneficial actions in preventing cardiovascular and osteoporetic diseases.
Footnotes
This work was supported by National Institutes of Health Grants NS07332 (C.A.S.) and NS20311 (D.M.D), a University of Washington Presidential Fellowship (C.A.S.), and University of Washington Alzheimer’s Disease Research Center Grant AG05136 (D.M.D.). We thank Dr. Robert Shapiro for helpful scientific discussions and critical review of this manuscript, Zeneca Pharmaceuticals for the use of ICI 182,780, and Rachael Crickman for excellent technical assistance.
Correspondence should be addressed to Daniel M. Dorsa, University of Washington, Psychiatry and Behavioral Sciences, Box 356560, Seattle, WA 98195-6560.
REFERENCES
- 1.Arnold SF, Notides AC. An antiestrogen: a phosphotyrosyl peptide that blocks dimerization of the human estrogen receptor. Proc Natl Acad Sci USA. 1995;92:7475–7479. doi: 10.1073/pnas.92.16.7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnold SF, Obourn JO, Jaffe HJ, Notides AC. Phosphorylation of the human estrogen receptor on tyrosine 537 in vivo and by src family tyrosine kinases in vitro. Mol Endocrinol. 1995;9:24–33. doi: 10.1210/mend.9.1.7539106. [DOI] [PubMed] [Google Scholar]
- 3.Aronica SM, Kraus WL, Katzenellenbogen BS. Estrogen action via the cAMP signaling pathway: stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc Natl Acad Sci USA. 1994;91:8517–8521. doi: 10.1073/pnas.91.18.8517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 5.Behl C, Widmann M, Trapp T, Holsboer F. 17β-estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Commun. 1995;216:473–482. doi: 10.1006/bbrc.1995.2647. [DOI] [PubMed] [Google Scholar]
- 6.Braunton JL, Wong V, Wang W, Salter MW, Roder J, Liu M, Wang YT. Reduction of tyrosine kinase activity and protein tyrosine dephosphorylation by anoxic stimulation in vitro. Neuroscience. 1998;82:161–170. doi: 10.1016/s0306-4522(97)00286-8. [DOI] [PubMed] [Google Scholar]
- 7.Brott BK, Decker S, Shafer J, Gibbs JB, Jove R. GTPase activating protein interactions with viral and cellular Src kinases. Proc Natl Acad Sci USA. 1991;88:755–759. doi: 10.1073/pnas.88.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng B, Goodman Y, Begley JG, Mattson MP. Neurotrophin-4/5 protects hippocampal and cortical neurons against energy deprivation- and excitatory amino acid-induced injury. Brain Res. 1994;650:331–335. doi: 10.1016/0006-8993(94)91801-5. [DOI] [PubMed] [Google Scholar]
- 9.Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58:293–297. doi: 10.1016/0304-3940(85)90069-2. [DOI] [PubMed] [Google Scholar]
- 10.Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 12.Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell cultures: attenuation by NMDA antagonists. J Neurosci. 1988;7:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egan SE, Giddings BW, Brooks MW, Buday L, Sizeland AM, Weinberg R. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature. 1993;363:45–51. doi: 10.1038/363045a0. [DOI] [PubMed] [Google Scholar]
- 15.Gibbs RB, Wu D, Hersh LB, Pfaff DW. Effects of estrogen replacement on the relative levels of choline acetyltransferase, trkA, and nerve growth factor messenger RNAs in the basal forebrain and hippocampal formation of adult rats. Exp Neurol. 1994;129:70–80. doi: 10.1006/exnr.1994.1148. [DOI] [PubMed] [Google Scholar]
- 16.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid β-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 17.Green PS, Gridley KE, Simpkins JW. Estradiol protects against β-amyloid (25–35) induced toxicity in SK-N-SH human neuroblastoma cells. Neurosci Lett. 1996;218:165–168. doi: 10.1016/s0304-3940(96)13148-7. [DOI] [PubMed] [Google Scholar]
- 18.Gridley KE, Green PS, Simpkins JW. Low concentrations of estradiol reduce β-amyloid toxicity, lipid peroxidation and glucose utilization in human SK-N-Sh neuroblastoma cells. Brain Res. 1997;778:158–165. doi: 10.1016/s0006-8993(97)01056-1. [DOI] [PubMed] [Google Scholar]
- 19.Gu Q, Moss RL. 17β-estradiol potentiates kainate-induced currents via activation of the cAMP cascade. J Neurosci. 1996;16:3620–3629. doi: 10.1523/JNEUROSCI.16-11-03620.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 21.Hefti F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J Neurosci. 1986;6:2155–2162. doi: 10.1523/JNEUROSCI.06-08-02155.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henderson VW, Paganini Hill A, Emanuel CK, Dunn ME, Buckwalter JG. Estrogen replacement therapy in older women. Comparisons between Alzheimer’s disease cases and nondemented control subjects. Arch Neurol. 1994;51:896–900. doi: 10.1001/archneur.1994.00540210068014. [DOI] [PubMed] [Google Scholar]
- 23.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 24.Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Lingle DD, Metter E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer’s disease: the Baltimore longitudinal study of aging. Neurology. 1997;48:1517–1521. doi: 10.1212/wnl.48.6.1517. [DOI] [PubMed] [Google Scholar]
- 25.Kromer LF. Nerve growth factor treatment after brain injury prevents neuronal death. Science. 1987;235:216–218. doi: 10.1126/science.3798108. [DOI] [PubMed] [Google Scholar]
- 26.Lindholm D. Role of neurotrophins in preventing glutamate induced neuronal cell death. J Neurol. 1994;241:S16–S18. doi: 10.1007/BF00939234. [DOI] [PubMed] [Google Scholar]
- 27.McMillan PJ, Singer CA, Dorsa DM. Effects of ovariectomy and estrogen replacement on trkA and choline acetyltransferase mRNA expression in the rat basal forebrain. J Neurosci. 1996;16:1860–1865. doi: 10.1523/JNEUROSCI.16-05-01860.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mermelstein PJ, Becker JB, Surmeier DJ. Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. J Neurosci. 1996;16:595–604. doi: 10.1523/JNEUROSCI.16-02-00595.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Migliaccio A, DiDomenico M, Castoria G, deFalco A, Bontempo P, Nola E, Auricchio F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996;15:1292–1300. [PMC free article] [PubMed] [Google Scholar]
- 30.Migliaccio A, Piccolo D, Castoria G, DiDomenico M, Bilancio A, Lombardi M, Gong W, Beato M, Auricchio F. Activation of the src/p21 ras/erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J. 1998;17:2008–2018. doi: 10.1093/emboj/17.7.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morley P, Whitfield JF, Vanderhyden BC, Tsang BK, Schwartz J-L. A new, nongenomic estrogen action: the rapid release of intracellular calcium. Endocrinology. 1992;131:1305–1312. doi: 10.1210/endo.131.3.1505465. [DOI] [PubMed] [Google Scholar]
- 32.Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of MAP kinase kinase blocks differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 33.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 34.Seger R, Ahn NG, Posada J, Munar ES, Jensen AM, Cooper JA, Cobb MH, Krebs EG. Purification and characterization of mitogen-activated protein kinase activators from epidermal growth factor-stimulated A431 cells. J Biol Chem. 1992;267:25628–25631. [PubMed] [Google Scholar]
- 35.Singer CA, Rogers KL, Strickland TM, Dorsa DM. Estrogen protects primary cortical neurons from glutamate neurotoxicity. Neurosci Lett. 1996;212:13–16. doi: 10.1016/0304-3940(96)12760-9. [DOI] [PubMed] [Google Scholar]
- 36.Singer CA, Rogers KL, Dorsa DM. Modulation of Bcl-2 expression: a potential component of estrogen neuroprotection in NT2 neurons. NeuroReport. 1998;9:2565–2568. doi: 10.1097/00001756-199808030-00025. [DOI] [PubMed] [Google Scholar]
- 37.Singh M, Meyer EM, Simpkins JW. The effect of ovariectomy and estradiol replacement on brain-derived neurotrophic factor messenger ribonucleic acid expression in cortical and hippocampal brain regions of female Sprague Dawley rats. Endocrinology. 1995;136:2320–2324. doi: 10.1210/endo.136.5.7720680. [DOI] [PubMed] [Google Scholar]
- 38.Singh M, Sétáló G, Jr, Guan X, Warren M, Toran-Allerand CD. Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J Neurosci. 1999;19:1179–1188. doi: 10.1523/JNEUROSCI.19-04-01179.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sohrabji F, Greene LA, Miranda RC, Toran-Allerand CD. Reciprocal regulation of estrogen and NGF receptors by their ligands in PC12 cells. J Neurobiol. 1994a;25:974–988. doi: 10.1002/neu.480250807. [DOI] [PubMed] [Google Scholar]
- 40.Sohrabji F, Miranda RC, Toran-Allerand CD. Estrogen differentially regulates estrogen and nerve growth factor receptor mRNAs in adult sensory neurons. J Neurosci. 1994b;14:459–471. doi: 10.1523/JNEUROSCI.14-02-00459.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tocque B, Delumeau I, Parker F, Maurier F, Multon M-C, Schweighoffer F. Ras-GTPase activating protein (GAP): a putative effector for Ras. Cell Signalling. 1997;9:153–158. doi: 10.1016/s0898-6568(96)00135-0. [DOI] [PubMed] [Google Scholar]
- 42.Watters JJ, Campbell JS, Cunningham MJ, Krebs EG, Dorsa DM. Rapid membrane effects of steroids on neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology. 1997;138:4030–4033. doi: 10.1210/endo.138.9.5489. [DOI] [PubMed] [Google Scholar]
- 43.White MA, Vale T, Camonis JH, Schaefer E, Wigler MH. A role of ral guanine nucleotide dissociation stimulator in mediating ras-induced transformation. J Biol Chem. 1996;271:16439–16442. doi: 10.1074/jbc.271.28.16439. [DOI] [PubMed] [Google Scholar]
- 44.Wong M, Moss RL. Long-term and short-term electrophysiological effects of estrogen on the synaptic properties of hippocampal CA1 neurons. J Neurosci. 1992;12:3217–3225. doi: 10.1523/JNEUROSCI.12-08-03217.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]