

Abstract
Acquired docetaxel-resistance of prostate cancer (PCa) remains a clinical obstacle due to the lack of effective therapies. Acetyl-11-keto-β-boswellic acid (AKBA) is a pentacyclic triterpenic acid isolated from the fragrant gum resin of the Boswellia serrata tree, which has shown intriguing antitumor activity against human cell lines established from PCa, colon cancer, malignant glioma, and leukemia. In this study, we examined the effects of AKBA against docetaxel-resistant PCa in vitro and in vivo as well as its anticancer mechanisms. We showed that AKBA dose-dependently inhibited cell proliferation and induced cell apoptosis in docetaxel-resistant PC3/Doc cells; its IC50 value in anti-proliferation was ∼17 μM. Furthermore, AKBA dose-dependently suppressed the chemoresistant stem cell-like properties of PC3/Doc cells, evidenced by significant decrease in the ability of mammosphere formation and down-regulated expression of a number of stemness-associated genes. The activation of Akt and Stat3 signaling pathways was remarkably enhanced in PC3/Doc cells, which contributed to their chemoresistant stem-like phenotype. AKBA (10–30 μM) dose-dependently suppressed the activation of Akt and Stat3 signaling pathways in PC3/Doc cells. In contrast, overexpression of Akt and Stat3 significantly attenuated the inhibition of AKBA on PC3/Doc cell proliferation. In docetaxel-resistant PCa homograft mice, treatment with AKBA significantly suppresses the growth of homograft RM-1/Doc, equivalent to its human PC3/Doc, but did not decrease their body weight. In summary, we demonstrate that AKBA inhibits the growth inhibition of docetaxel-resistant PCa cells in vitro and in vivo via blocking Akt and Stat3 signaling, thus suppressing their cancer stem cell-like properties.
Keywords: acetyl-11-keto-β-boswellic acid, prostate cancer, docetaxel-resistance, chemoresistant stem cell-like properties, Akt, Stat3
Introduction
Prostate cancer (PCa) is the most common cancer diagnosis and the main cause of cancer-related deaths among males [1]. Hormone withdrawal therapy remains an effective treatment for advanced PCa. However, PCa proceeds from a hormone-dependent disease to invasive and metastatic hormone refractory prostate cancer (HRPC). Docetaxel is a taxane antimitotic chemotherapeutic agent for patients, and docetaxel-based regiments are currently used as the standard therapy for patients with HRPC [2, 3]. However, docetaxel only offers a modest survival advantage, largely because patients treated with this therapy inexorably experience disease progression and multidrug resistance (MDR) [4, 5]. In this context, acquired docetaxel resistance is commonly fatal due to the limited number of effective therapies. Molecular mechanisms that drive the development of docetaxel resistance in HRPC include an increase in the expression of α- and β-tubulin isoforms/mutations, activation of ATP-binding cassette drug transporters, and overexpression of antiapoptotic proteins such as BCL2, XIAP, and Survivin [4–6]. Unfortunately, targeting individual genes yields limited clinical impact in PCa [7, 8]. Therefore, the treatment of acquired docetaxel-resistant HRPC remains a critical clinical challenge, and alternative approaches for developing effective and less toxic agents are urgently required.
There is increasing evidence that has suggested that cancer stem cells (CSCs) or tumor-initiating cells (TICs), a subset of cancer cells with high self-renewal and stemness properties, are indispensable for tumorigenesis. CSCs exist in various solid tumors and are also considered the key contributor to chemoresistance and responsible for recurrence after conventional therapy [9–11]. In CSCs, various signaling pathways associated with stemness maintenance could abnormally activate different tumor types, mainly including Notch, Wnt, Hedgehog, Jak/Stat3, MAPK/ERK, PI3K/Akt, NF-κB, TGF-beta, and ROS, which provide ideal targets for the elimination of CSCs in clinical cancer treatment [10–12].
Recently, studies have shown that drug-resistant cancer cells have stem cell-like properties [13, 14]. In PCa, the occurrence of chemotherapy resistance also displays a close correlation with the existence of prostate cancer stem-like cells (PCSCs) and abnormal activation of stemness-associated signaling pathways [11, 15, 16]. It has been demonstrated that a subpopulation of cells that survive docetaxel exposure contributed to docetaxel resistance through both in vitro and in vivo studies [17]. These cells exhibited potent tumor-initiating capacity and lacked epithelial differentiation markers, which were depleted through inhibition of the survival molecules phosphor-Akt (Ser473) and Bcl2 [17]. Akt signaling was also reported to impart functional heterogeneity in CSCs during the acquisition of chemoresistance [16]. Inhibition of Akt abrogated CSC and the chemoresistant phenotype [18, 19]. Furthermore, the IL6/Stat3 signaling pathway is believed to contribute to PCSC activity and drug resistance [20, 21]. PCSCs from patients secrete high levels of IL6 that activate the downstream Jak2/Stat3 signaling. Anti-IL6 antibody or inhibitor of phosphor-Stat3 can inhibit clone formation of PCSCs [20, 21]. Therefore, agents that suppress the activity of PCSCs through blockade of the Akt and Stat3 signaling pathways could be used to treat docetaxel-resistant PCa.
Acetyl-11-keto-β-boswellic acid (AKBA), a pentacyclic triterpenic acid isolated from the fragrant gum resin of the Boswellia serrata tree, has been traditionally used in treatments for various inflammatory diseases [22, 23]. Recently, AKBA was also found to exhibit antitumor effects in human cell lines established from PCa, colon cancer, malignant glioma, and leukemia [24–26]. However, the potential therapeutic effect and the underlying molecular mechanism of AKBA on docetaxel-resistant PCa have yet to be elucidated. In the present study, we assessed the efficacy of AKBA in growth inhibition of docetaxel-resistant PCa in vitro and in vivo and proposed a novel mechanism by which AKBA exerts antitumor activity mediated via suppression of the tumor-initiating capacity of docetaxel-resistant cells.
Materials and methods
Cell culture and treatments
The human PCa cell line PC3 and murine PCa cell line RM-1 were obtained from The Cell Bank of Chinese Academy of Sciences (Shanghai, China) and maintained in RPMI 1640 medium (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (HyClone). The docetaxel-resistant cell lines PC3/Doc and RM-1/Doc were selected by growing the PC3 and RM-1 cells in increasing concentrations of docetaxel (Aventis Pharma Dagenham, Dagenham, UK), respectively. The procedure has been described in our previous reports [27, 28]. The resistant cells were maintained in medium with 1 nM docetaxel. All the cells were maintained in a humidified incubator with 5% CO2 at 37 °C.
AKBA was isolated as described previously [29] and dissolved in dimethyl sulfoxide (DMSO) at 20 mM as a stock solution stored at −20 °C. Dilution of AKBA was freshly performed before each experiment according to experimental requirements. The PI3K inhibitor LY294002 was purchased from Sigma-Aldrich (St. Louis, MO, USA). The Stat3 inhibitor S3I-201 was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The pan-caspase inhibitor Z-VAD-fmk was obtained from Enzo Life Sciences (Plymouth Meeting, PA, USA).
Cell viability and cell death assay
Cell viability was quantitatively tested by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT, Sigma-Aldrich) colorimetric assay on a plate reader (Bio-Rad, Hercules, CA, USA). Cell death was detected using an annexin V-FITC/PI apoptosis detection kit (Becton Dickinson, NJ, USA). Fluorescence was quantified by flow cytometry (Becton Dickinson).
Western blotting assay
After treatment as indicated, cell lysates were prepared with RIPA buffer for a western blotting assay as described previously [30]. Blots were incubated with primary antibodies against Poly (ADP-ribose) polymerase (PARP), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology, TX, USA), Akt, phospho-Akt (Ser473), Stat3, phospho-Stat3 (Tyr705), phosphor-Jak2 (Tyr1007/1008), IGF-1Rβ, Bcl-2 (Cell Signaling Technology, MA, USA), SOX2, OCT4, β-Actin, and MCL1 (Proteintech, Wuhan, China) overnight at 4 °C, followed by appropriate peroxidase-conjugated secondary antibodies. GAPDH or β-Actin served as a protein loading control. Immunocomplexes were visualized using chemiluminescence (Millipore, DT, Germany) by exposure to X-ray film.
Quantitative real-time PCR (qRT-PCR) analysis
Total RNA was obtained using an RNAiso plus kit (Takara Bio, Inc., Otsu, Japan). For qRT-PCR, cDNA was synthesized using a PrimeScript RT reagent Kit (Takara Bio, Inc.). qRT-PCR was performed using an Eppendorf qRT-PCR system. Changes in the mRNA levels of desired genes were normalized to the level of 18 s and calculated using the 2−ΔΔct method. The primer sequences are summarized in Supplementary Table 1.
Mammosphere formation assay
For the mammosphere formation assay, parental PC3 and docetaxel-selected resistant PC3/Doc cells were seeded on ultralow attachment 96-well plates (Corning, NY, USA). Cells were incubated with AKBA or DMSO vehicle in serum-free 1640 medium supplemented with 100 ng/mL EGF, 20 ng/mL bFGF, 1% N2, and 2% B27 (Invitrogen, CA, USA) for 10 days. The mammosphere growth was analyzed under a bright-field microscope (Nikon, Tokyo, Japan) at ×100 magnification.
Transient transfection assay
PC3/Doc cells were transiently transfected with pCMV6-Akt1-myr or pcDNA3.1-Stat3 using Lipofectamine 3000 (Invitrogen, CA, USA) according to the manufacturer’s instructions. Empty vectors pCMV6 or pcDNA3.1 served as controls. The plasmids have been described in our previous reports [27, 31]. After transfection for 24 h, the cells were treated with AKBA or vehicle for an additional 24 or 48 h. The effects of AKBA on the conditioned cells were determined by western blotting and cell viability assays as described above.
Antibody array analysis
For the detection of stemness-related protein expression, cell lysates were prepared, and antibody array analysis was performed using Human Stem Cell Array G1 (AAH-SC-G1, RayBiotech Inc., USA) according to the manufacturer’s protocol.
Murine homograft study
RM-1 cells from C57BL/6 mouse prostate tumors are androgen independent and commonly used to develop homograft animal models [32]. Here, RM-1 and RM-1/Doc cells were used for the establishment of docetaxel-sensitive and docetaxel-resistant PCa homografts in male C57BL/6 mice, respectively, as described previously [28, 30]. For the generation of homotransplantations, 6-week-old male C57BL/6 mice (Vital River Laboratories, Beijing, China) were inoculated subcutaneously in the backs with 1 × 105 RM-1 cells in 0.1 mL of physiological saline. After 1 week, treatment commenced by single daily intraperitoneal injections of 30 mg/kg AKBA for an additional 14 days. Simultaneously, treatment with 5 mg/kg docetaxel was used for medicine control. Tumor size and body weight were measured every 2 days. The tumor volume was calculated by using the formula 0.5 × L × W2 (L = length, W = width). At the end of the experiment, all mice were euthanized, and their tumors were resected for western blotting and immunohistochemistry. All animal experiments were approved by the Ethics Committee of the Second Hospital of Shandong University (Permit Number: KYLL-2016 (GJ)A-0012) and conducted accordingly.
Statistical analysis
The data are presented as means ± SD. Student’s unpaired t-test was used to determine the statistical significance of mean differences between treated and control groups. Multiple group comparisons were performed with one-way ANOVAs followed by Dunnett’s multiple comparison test. P < 0.05 was considered statistically significant (*), and P < 0.001 was highly significant (***).
Results
Docetaxel-resistant PC3/Doc cells are sensitive to AKBA
As an initial screening approach, a cell viability assay was carried out to identify the chemicals that potently inhibited cell proliferation in the established docetaxel-resistant PC3/Doc cells (Fig. 1a) [27, 28]. The chemical concentrations that induced cellular death at 50% (IC50) were calculated. As shown in Fig. 1b, PC3/Doc cells were sensitive to AKBA, with a relatively marginally decreased IC50 value at ~17 μM compared with ~21 μM in PC3 cells. AKBA at increasing concentrations of 5, 10, 20, 30 and 40 μM reduced cell viability in a dose-dependent manner, inhibiting PC3/Doc cell growth by about 6.3%, 25.8%, 49.2%, 75.7% and 93.3% while inhibiting PC3 cells by about 3.9%, 16.2%, 36.0%, 66.7% and 90.1%, respectively (Fig. 1b). The data demonstrated the potential of AKAB as a potent cytotoxic agent in resistant PC3/Doc cells.
Fig. 1.

Effect of AKBA on cell proliferation and apoptosis in docetaxel-resistant PCa cells. a The in vitro effect of docetaxel on established human docetaxel-resistant PC3/Doc cells and parental PC3 cells. ***P < 0.001. b Cell viability in response to AKBA was determined by the MTT assay after 48 h of treatment in PC3 and PC3/Doc cells. The results are the means ± SD of three independent experiments. *P < 0.05 versus AKBA-untreated control group. c The morphological changes after 48 h of treatment with 0, 10, 20, and 30 μM AKBA in PC3 and PC3/Doc cells. d AKBA-induced cell death was detected by annexin V/PI staining and flow cytometric analysis. e The expression of PARP in cells treated with AKBA at various concentrations was examined by western blotting. GAPDH served as the loading control. f Cell viability in the absence or presence of the pan-caspase inhibitor Z-VAD-fmk. PC3/Doc cells were exposed to 10 μM Z-VAD-fmk for 2 h prior to 20 μM AKBA or vehicle treatment. The results are the means ± SD of three independent experiments. **P < 0.01
Moreover, morphological analysis revealed that cell shrinkage was observed after AKBA treatment in PC3/Doc cells (Fig. 1c), thus suggesting that apoptosis was induced by AKBA. Flow cytometry was used to analyze the apoptotic cells exposed to AKBA in PC3/Doc cells. The results revealed that AKBA caused a significant increase in the fraction of apoptotic cells, with approximately 9.14%, 13.59%, and 37.73% of apoptotic cells after treatment with 10, 20, and 30 μM AKBA, respectively. By comparison, the apoptotic PC3 cell fractions in response to the same concentrations of AKBA were 7.42%, 11.65%, and 30.26%, respectively, showing a slight decrease under the same conditions (Fig. 1d). In addition, an increase in the cleavage of PARP, a hallmark of apoptosis, was initiated in PC3/Doc cells exposed to 10 μM AKBA and sustained with increasing concentrations (Fig. 1e). A pan-inhibitor of caspases, Z-VAD-fmk, was included to confirm the ability of AKBA to induce caspase-dependent cell apoptosis. The results in Fig. 1e provided evidence that Z-VAD-fmk markedly rescued cell death induced by AKBA in PC3/Doc cells (Fig. 1f). Together, the data indicated that chemoresistant PC3/Doc cells were sensitive to AKBA and that 10 and 20 μM AKBA were capable of inhibiting cell proliferation and efficiently triggering cell death via caspase-dependent apoptosis.
AKBA suppresses stem cell-like properties of docetaxel-resistant cells
Recent evidence has indicated that acquired docetaxel resistance is suppressed in PCa through depletion of TICs [17]. Since the ability to form mammospheres under low attachment growth conditions is a good index of the tumor-forming potential of stem cells, we substantiated mammosphere formation in drug-resistant PC3/Doc cells and parental PC3 cells. Consistent with previous studies, the number and size of mammospheres are much greater in drug-selected PC3/Doc cells than parental cells (Supplementary Figure S1a and 1b), which also corresponds to the view that chemotherapy reduces the bulk of tumors while sparing the CSC population [10, 11, 33, 34]. Interestingly, treatment with AKBA markedly reduced the mammosphere-forming potential in a dose-dependent manner. As shown in Fig. 2a, the size of the single sphere clearly declined. A rapid and significant decrease (33.3% ± 7.2%, P < 0.05) in the number of PC3/Doc-formed mammospheres was also noted when cells were exposed to 5 μM AKBA, reaching 91.6% ± 5.8% with 10 μM AKBA (Fig. 2b). Thus, our observation revealed that AKBA inhibited mammosphere formation in resistant PC3/Doc cells, suggesting its capacity to suppress stem cell-like properties in docetaxel-resistant PCa.
Fig. 2.

The inhibitory effect of AKBA on the stem cell-like properties of docetaxel-resistant cells. a AKBA repressed mammosphere formation of resistant cells. PC3/Doc cells were cultured in low-attachment sphere-forming medium with 5, 7.5, and 10 μM icaritin for 10 days. The mammospheres formed are imaged with ×10 objective. b The relative level of mammosphere formation compared with that in the AKBA-untreated control. The results are the means ± SD of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001. c Heatmap for mRNA levels of stemness maintenance-associated genes in PC3/Doc cells treated with different concentrations of AKBA as determined by qRT-PCR. green: underexpression, black: unchanged expression. d AKBA downregulated the expression of SOX2 and BCL2 in PC3/Doc cells as detected by western blotting assay. GAPDH served as the loading control. e Antibody array-based protein expression analysis by using a Human Stem Cell Array. After 48 h of treatment with 15 μM AKBA, cell lysates were prepared for antibody array detection
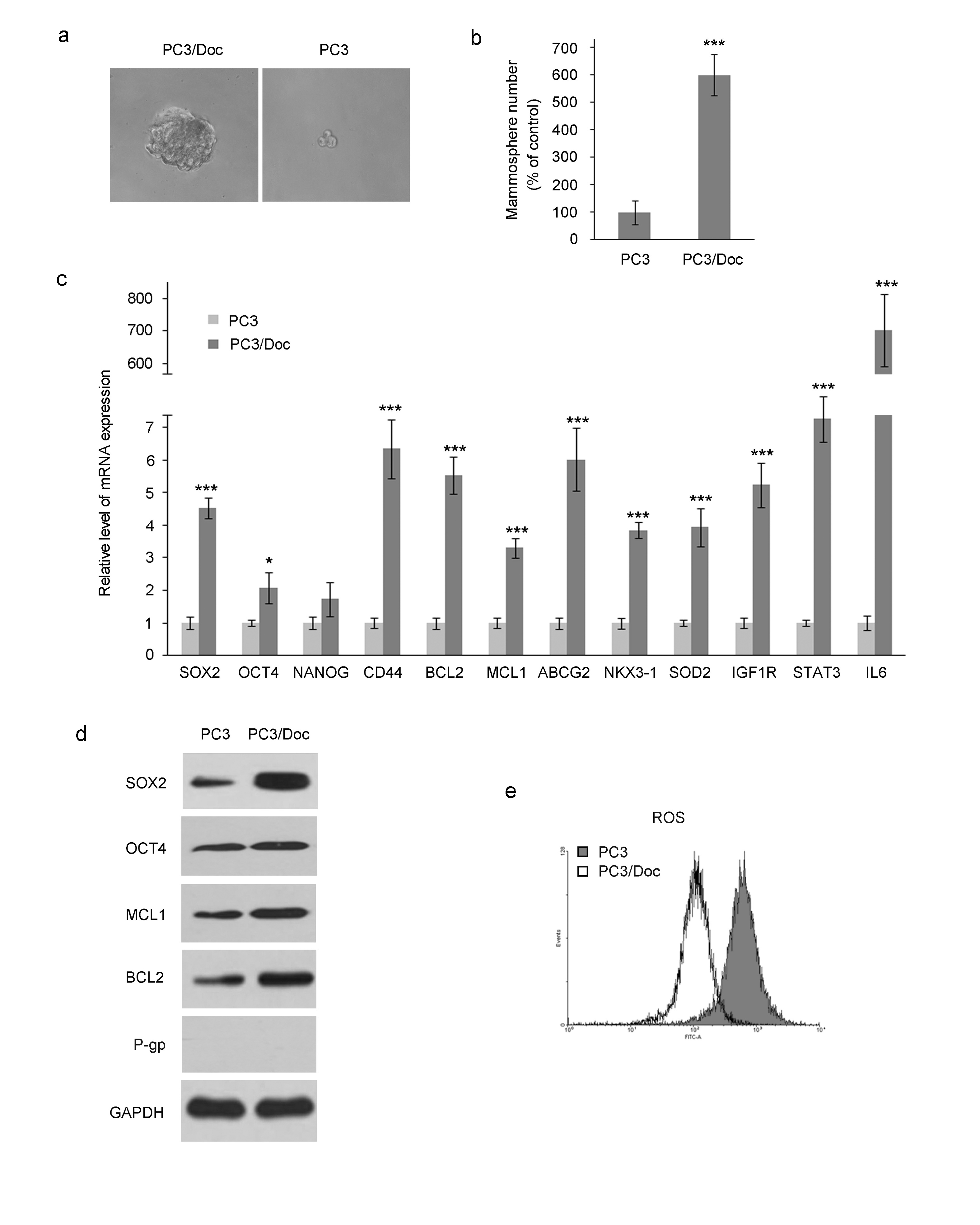
We then investigated the impact of AKBA on gene expression associated with cell survival as well as stemness in PC3/Doc cells. To make the study more targeted, we first analyzed expression changes in these genes in PC3/Doc versus PC3 cells by qRT-PCR and western blotting assays (Supplementary Figure S1c and 1d), which indicated that various genes clearly increased in resistant cells compared with PC3 cells. In particular, the mRNA levels of SOX2 and OCT4, two core stem cell markers [34], increased approximately 4.5- and 2-fold in resistant cells, respectively. High expression of CD44 (~6-fold increase) was observed in resistant cells [21]. We also detected some other genes related to stemness, such as ABCG2, NKX3-1, and MCL1, which displayed clearly enhanced expression. Additionally, Bcl2, an antiapoptosis gene that has been verified to have an important function in the survival of docetaxel-resistant PCa cells [17], was also profoundly upregulated in PC3/Doc cells. In addition, our previous data demonstrated that docetaxel-resistant PC3/Doc cells showed low levels of reactive oxygen species (ROS) and a significant increase in the antioxidant protein SOD2 [27], which have been suggested to be a chemoresistant stem-like phenotype (Supplementary Figure S1c and 1e) [33]. These results further confirmed the enhanced stem cell-like properties of our docetaxel-resistant cell models.
Based on the results above, qRT-PCR was performed to validate the changes in the response of these genes to AKBA in PC3/Doc cells. It was noticeable that AKBA treatment clearly resulted in a concentration-dependent decrease in the expression of SOX2, approximately 1.5-, 2.1-, and 3.2-fold at 10, 20, and 30 μM, respectively. AKBA had a mild effect on OCT4 expression, which remained unchanged upon low concentration treatment and decreased in a moderate manner in response to a high concentration of AKBA. In addition, the expression of BCL2, MCL1, and NKX3-1 was also downregulated pronouncedly. We next detected the protein levels of SOX2 and BCL2 in response to AKBA in PC3/Doc cells. In line with the mRNA level, treatment with increased concentrations of AKBA (10–30 μM) resulted in downregulation of the expression of these proteins to different degrees. The inhibitory effect of AKBA on stemness was further confirmed by using an antibody-based human stem cell array (Fig. 2e and Supplementary Figure S2). The results revealed that the expression of SOX2, E-Cadherin, and VEGFR2 was obviously reduced, whereas that of OCT4, NANOG, CD38, and NESTIN decreased moderately. Therefore, these data indicated that AKBA significantly reduced the expression levels of multiple cell survival- and stemness-associated genes, which were overexpressed in docetaxel-resistant PC3/Doc cells compared with sensitive PC3 cells.
Blockade of STAT3 and Akt signaling by AKBA contributes to inhibition of stem-like properties and cell growth
We further explored the potential mechanisms of AKBA action in resistance cells. As shown in Supplementary Figure S1c and Fig. 3a, the IGF-1R/Akt and Jak/Stat3 signaling pathways were overactivated in PC3/Doc cells compared to PC3 cells, as supported by the dramatically upregulated expression levels of IGF-1R, phosphor-Akt, IL6, and phosphor-Stat3 [27, 28]. Elevated levels of IGF-1R/Akt downstream genes such as SOX2 and BCL2 [18] and IL6/Jak/Stat3 downstream genes such as MCL1, OCT4, and CyclinD1 [35] were also observed from the above results. Since enhancement of IGF-1R/Akt and Jak/Stat3 signaling has been shown to be associated with enrichment of stem-like features during the acquisition of drug resistance [19–21], we next investigated their roles in the AKBA-mediated inhibitory effects on PC3/Doc cells. As shown in Fig. 3b, treatment of PC3/Doc cells with AKBA led to significant downregulation of IGF-1R and phosphor-Akt, as well as phosphor-Jak2 and phosphor-Stat3. Indeed, activated Akt and Stat3 signaling contributed to the stem-like properties in PC3/Doc cells, as evidenced by the decreased mammosphere formation after treatment with a specific PI3K inhibitor (LY294002) or Stat3 inhibitor (S3I-201) (Fig. 3c, d). Thus, the observations suggested the involvement of attenuated Akt and Stat3 signaling in AKBA-mediated inhibition of stem cell-like properties.
Fig. 3.

Akt and Stat3 signaling are involved in AKBA-mediated suppression of mammosphere formation and cell proliferation. a Elevated expression of IGF-1R, phosphor-Akt (Ser473), phosphor-Stat3 (Tyr705), and total Stat3 in PC3/Doc versus PC3 cells. b Effect of AKBA on the expression of IGF-1R, phosphor-Akt (Ser473), phosphor-Stat3 (Tyr705), and phosphor-Jak2 (Tyr1007/1008) in PC3/Doc cells was estimated by western blotting analysis. GAPDH served as an internal control. c, d Akt and Stat3 were critical for mammosphere formation of PC3/Doc cells. PC3/Doc cells treated with 20 μM LY294002 (c) or 100 μM S3I-201 (d) were used for mammosphere formation analysis. Western blotting was performed with the indicated antibodies. **P < 0.01 compared to the control group. e–h Impact of Stat3 or Akt overexpression on cell survival in AKBA-treated PC3/Doc cells. The overexpression efficiency was examined using western blotting (e, g). Cell viability was tested by the MTT assay (f, h). The results are the means ± SD of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the AKBA-untreated control group
To determine whether Akt and Stat3 played an important role in AKBA-mediated proliferation inhibition in PC3/Doc cells, we modulated the levels of Akt and Stat3 in PC3/Doc cells by overexpression. Western blotting analysis showed that Akt and Stat3 expression plasmids indeed enhanced the expression and phosphorylation levels of Stat3 and Akt, respectively (Fig. 3e, g). Overexpression of either Stat3 or Akt resulted in a partial rescue of the consequences of cell viability inhibition induced by AKBA (Fig. 3f, h). These data suggested a possible mechanism of AKBA suppressing cell growth as well as stemness of resistant PC3/Doc cells mediated through blockade of Akt and Stat3 signaling.
AKBA exhibits potent antitumor activity in docetaxel-resistant PCa homograft mice
On the basis of the above results, it is of great value to further elucidate the antitumor efficiency of AKBA in vivo. First, we examined the cytotoxic activity of AKBA in docetaxel-resistant RM-1/Doc cells and parental RM-1 cells, demonstrating that RM-1/Doc cells were sensitive to AKBA (Supplementary Figure S3). Thus, the RM-1/Doc cell line is equivalent to its human counterparts and was used for the establishment of docetaxel-resistant PCa homografts in C57BL/6 mice. As shown in Fig. 4a, after 14 days of treatment, the initial and final body weights of tumor-bearing mice remained almost unchanged with the administration of AKBA compared with the administration of the vehicle control in both the docetaxel-sensitive RM-1 and docetaxel-resistant RM-1/Doc homograft groups. However, administration of docetaxel caused a significant decrease in body weight in the two groups. Moreover, compared to vehicle control administration, AKBA treatment exerted a similar inhibitory effect on the average tumor volume in the RM-1 and RM-1/Doc homograft groups (P < 0.05, Fig. 4b, c). Correspondingly, tumor weights were also decreased significantly in the AKBA-treated RM-1/Doc and RM-1 groups in comparison with the respective vehicle control group (1.10 ± 0.22 g versus 1.83 ± 0.17 g, P < 0.05 in RM-1/Doc group; 1.31 ± 0.17 g versus 2.01 ± 0.16 g, P < 0.05 in RM-1 group, Fig. 4d). The inhibition rates of tumor weights by AKBA were 39.83% and 35.18% in the RM-1/Doc and RM-1 groups, respectively. For docetaxel treatment, the average volume and weight of the RM-1/Doc tumor had no significant change versus control (P > 0.05), while a remarkable volume and weight reduction in RM-1 group was observed (P < 0.001, Fig. 4a–c). These results indicated that docetaxel was effective for PCa but to a lesser extent for drug-resistant PCa, whereas the inhibitory effect of AKBA on chemoresistant tumor growth was clearly evidenced.
Fig. 4.

AKBA exhibits potent antitumor activity in docetaxel-resistant homograft mice. a The body weights were measured every 2 days after the indicated treatment. b, c The volume of tumors from different groups was recorded every 2 days. Data are represented as the means ± SEM. In a–c, *P < 0.05 and ***P < 0.001 compared with the vehicle control in RM-1/Doc or RM-1 group. d Effect of AKBA on tumor weight was detected at the time of sacrifice. Data are shown as the means ± SEM. e Representative tumors from the six groups are shown. f, g Inhibitory effect of AKBA on tumor cell proliferation was also confirmed with the decreased Ki67-positive cells in RM-1/Doc homografts. Representative images of H&E and Ki67 staining of the indicated treated groups (magnification, 100×) (f). Ki67-positive rates in the two groups (g). Data are shown as the means ± SEM. **P < 0.01 compared with the control. h Western blotting analysis of phosphor-Akt and phosphor-Stat3 expression in RM-1/Doc homografts treated with vehicle or AKBA
Given the antitumor efficacy of AKBA in resistant PCa, we determined RM-1/Doc tumor cell proliferation using Ki67 immunohistochemistry. As shown in Fig. 4f, g, AKBA treatment resulted in a statistically significant decrease in positively stained Ki67 cells (19.54% ± 4.973% versus 49.84% ± 4.219% in the control group), supporting the observations that AKBA potently inhibited resistant cell proliferation. Western blotting analysis revealed that the expression levels of phosphor-Akt and phosphor-Stat3 in RM-1/Doc tumors were high and that AKBA treatment pronouncedly reduced the expression of phospho-Akt and phospho-Stat3 (Fig. 4h), indicating the suppression of Akt and Stat3 signaling by AKBA in vivo.
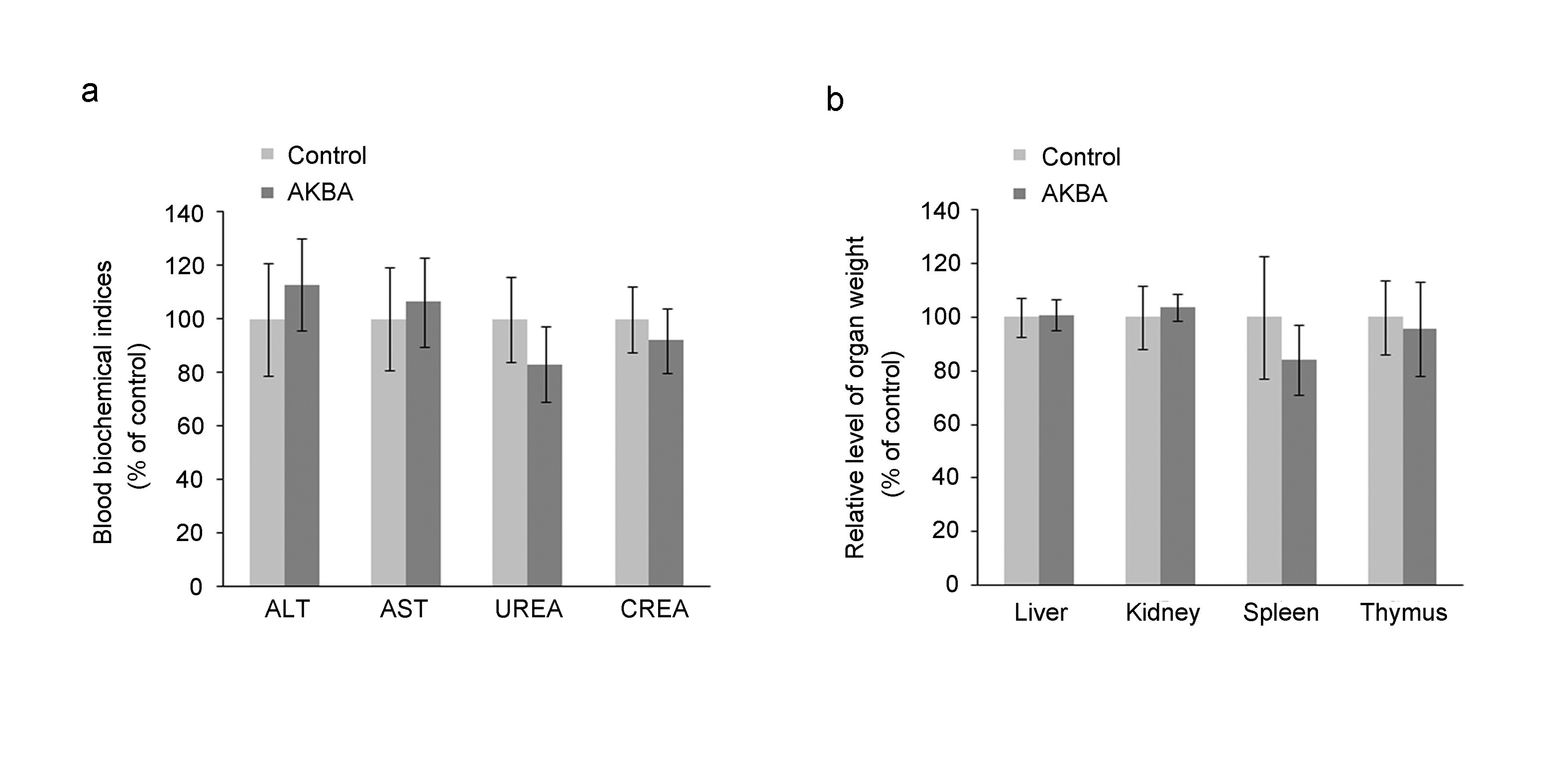
Additionally, we analyzed the toxicity of AKBA in RM-1/Doc tumor-bearing mice. Glutamic-pyruvic transaminase (ALT), glutamate–oxaloacetate transaminase (AST), urea (UREA), and serum creatinine (CREA) levels in the blood were examined to evaluate liver and kidney toxicity. The results revealed that there were no significant differences in any of the blood biochemical indices between the AKBA and control groups (Supplementary Figure S4a). We also observed that AKBA did not significantly affect the weights of the liver, kidney, spleen, and thymus (Supplementary Figure S4b). Together, our data suggest that AKBA might be a potential candidate for therapy of docetaxel-resistant PCa.
Discussion
AKBA, a member of the family of natural pentacyclic triterpenic acids, has recently gained considerable attention because of its potential antitumor activity in many types of cancer. The present study was designed to uncover the cellular/molecular basis by which AKBA exerted its antitumor action in acquired docetaxel-resistant PCa. In this study, we first identified the potent growth inhibition effect of AKBA in PC3/Doc cells, a docetaxel-resistant PCa cell model selected by treating PC3 cells with increasing concentrations of docetaxel and characterized by notable stem-like phenotype, in agreement with previous studies [17, 18, 33]. In this study, we also demonstrated that the mechanism by which AKBA exerted its antitumor activity was suppressing chemoresistant stem cell-like properties in PC3/Doc cells, which was associated with the blockade of Akt and Stat3 signaling. Moreover, the discovery was further supported by the in vivo study results.
The relapse of cancer after chemotherapy remains a major obstacle to successful cancer treatment. As a subpopulation of cancer cells, CSCs are able to elude conventional treatments and eventually regenerate a more aggressive tumor. Presently, accumulating evidence suggests the emergence and enrichment of aggressive drug-resistant cells with stem cell properties after chemotherapy. Previous studies demonstrated that despite the rare escape of cancer cells from drug-induced cell death, some cells escaped and generated stable colonies with stem cell-like phenotypes [4, 5]. Many widely used antitumor agents, such as doxorubicin, paclitaxel, docetaxel, cisplatin, and camptothecin, have been reported to have this capacity [4]. The drug-selected cells contained low levels of ROS and displayed reactivation of antioxidant enzymes and stem cell markers such as SOX2, OCT4, and Nanog [4, 5]. Likewise, the population of PCa cells with resistance to docetaxel was identified to exhibit an undifferentiated phenotype and high tumor-initiating capacity in both cell lines and patient samples [5]. In this study, we generated an in vitro docetaxel-resistant model PC3/Doc using the well-established human PCa cell line PC3 [6, 27]. In agreement with previous results [5, 17, 18], PC3/Doc cells were characterized by potent CSC-like properties, as validated by their high levels of cell viability, mammosphere formation and stem cell marker expression compared with those in the sensitive parental PC3 cells. Since the therapeutic strategies for docetaxel-resistant HRPC are limited, our study sought to discover an effective and safe treatment based on the stem cell-like activities of docetaxel-resistant cells.
Naturally occurring chemicals provide rich resources and powerful tools to obtain essential bioactive agents. In our program searching for effective antitumor drugs, AKBA drew our attention because of its remarkable inhibitory effect on docetaxel-resistant PC3/Doc cells and C57BL/6 homografts. It has been demonstrated that AKBA has the ability to effectively promote cell apoptosis by reducing the expression of P-glycoprotein (P-gp), which is highly expressed in drug-resistant cells [36, 37]. However, our study and others have revealed that docetaxel-resistant PCa cells have no P-gp expression (Supplementary Figure 1d) [7, 38], whereas multiple mechanisms, especially enrichment of CSCs, contribute to the acquisition of docetaxel resistance. In the present study, AKBA significantly suppressed the malignant growth of docetaxel-resistant PCa in vitro and in vivo and notably exhibited potent inhibitory activity on stem cell-like properties, as evidenced by the reduction in mammosphere formation and stem cell marker expressions. These findings suggest that AKBA can be developed into a potent agent to treat docetaxel-resistant PCa by targeting CSC-like properties.
Constitutive Stat3 activation has been reported to play an essential role in prostate stem and progenitor cells [20, 21]. In PCa, stem-like cells secrete high levels of IL-6, which binds to gp80 first and then recruits gp130. After gp130 dimerization, Jak2 becomes activated and leads to tyrosine phosphorylation of cytoplasmic transcription factors, primarily Stat3, in prostate cancer [20, 21, 35]. Stat3 translocates from the cytoplasm to the nucleus and regulates the downstream transcription of genes such as OCT4, BCL2, and CyclinD1 [12, 35]. Blockade of activated Stat3 signaling suppresses clonogenic recovery of stem-like cells in patients with high grade disease [21]. Increasing evidence has demonstrated that the activation of Akt signaling is involved in the characteristics of stem-like cells and the development of drug resistance in cancer, including PCa [17, 18, 39, 40]. The addition of activated forms of Akt to prostate cancer cells promotes drug resistance [40]. SOX2, one of the core stem cell markers, has been shown to be regulated by Akt in stem cells through the inhibition of proteasomal degradation [39, 41]. Furthermore, upregulated IGF-1R expression contributes to resistance to cytotoxic treatments, and IGF-1R-mediated Akt signaling activation assists in the maintenance of chemoresistant stem cell-like properties [18]. IGF-1R/Akt inhibition resulted in a drastic decrease in the levels of stem cell markers (SOX2, OCT4, and Nanog) and mammosphere formation compared to those in their parental cells [18]. Our data and the results from other studies have shown the remarkable enhancement in IL-6/Stat3 and IGF-1R/Akt in resistant PC3/Doc cells compared to PC3 cells [27, 28]. In this study, intriguingly, the abolishment of both Stat3 and Akt signaling was pronounced after AKBA treatment, as indicated by a decrease in the expression levels of IGF-1R, phosphor-Akt, phosphor-Stat3, and phosphor-Jak2 as well as their downstream genes. Stat3 and Akt signaling are critical for stem cell-like properties and drug resistance, thus suggesting that the blockade of Stat3 and Akt by AKBA was at least in part essential for the suppression of stem cell-like activity and cell survival in docetaxel-resistant cells. In addition, AKBA was found to exert its antitumor action by inhibiting the NF-κB and Wnt/β-catenin signaling pathways [24, 42], which are also associated with stem cell properties [12, 43]. Therefore, it remains to be further clarified whether these signaling pathways are involved in the response to resistant cell growth inhibition induced by AKBA.
In conclusion, this study suggested a mode of action for the exertion of antitumor activity by AKBA, namely, by inhibiting chemoresistant stem cell-like properties through blockade of Akt and Stat3 signaling in docetaxel-resistant PCa. Since acquired resistance to docetaxel precedes fatality in PCa and strategies that target docetaxel-resistant cells remain limited, our study provides a strong rationale for further development of AKAB for the treatment of docetaxel-resistant PCa.
Electronic supplementary material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81603140), Shandong Provincial Natural Science Foundation, China (Doctoral Foundation; Grant No. ZR2016HB69), Jinan Science and Technology Development Program (201704087), and Shandong Medicine and Health Science Technology (2017WS628, 2017WS202).
Author contributions
HXL, RMW, and YQL designed the research. YQL, QQX, YXG, and ZML performed the experiments. YQL, HXL, and HQY contributed new reagents and analytic tools. SKW, FK, and QW analyzed the data. YQL Liu wrote the paper.
Competing interests
The authors declare no competing interests.
Contributor Information
Rong-mei Wang, Email: rongmeiwang@hotmail.com.
Hong-xiang Lou, Email: louhongxiang@sdu.edu.cn.
Electronic supplementary material
The online version of this article (10.1038/s41401-018-0157-9) contains supplementary material, which is available to authorized users.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 3.McKeage K. Docetaxel: a review of its use for the first-line treatment of advanced castration-resistant prostate cancer. Drugs. 2012;72:1559–77. doi: 10.2165/11209660-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Mahon KL, Henshall SM, Sutherland RL, Horvath LG. Pathways of chemotherapy resistance in castration-resistant prostate cancer. Endocr Relat Cancer. 2011;18:R103–23. doi: 10.1530/ERC-10-0343. [DOI] [PubMed] [Google Scholar]
- 5.Seruga B, Ocana A, Tannock IF. Drug resistance in metastatic castration resistant prostate cancer. Nat Rev Clin Oncol. 2011;8:12–23. doi: 10.1038/nrclinonc.2010.136. [DOI] [PubMed] [Google Scholar]
- 6.Ranganathan S, Benetatos CA, Colarusso PJ, Dexter DW, Hudes GR. Altered beta-tubulin isotype expression in paclitaxel resistant human prostate carcinoma cells. Br J Cancer. 1998;77:562–6. doi: 10.1038/bjc.1998.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeda M, Mizokami A, Mamiya K, Li YQ, Zhang J, Keller ET, et al. The establishment of two paclitaxel-resistant prostate cancer cell lines and the mechanisms of paclitaxel resistance with two cell lines. Prostate. 2007;67:955–67. doi: 10.1002/pros.20581. [DOI] [PubMed] [Google Scholar]
- 8.Zalcberg J, Hu XF, Slater A, Parisot J, El-Osta S, Kantharidis P, et al. MRP1 not MDR1 gene expression is the predominant mechanism of acquired multidrug resistance in two prostate carcinoma cell lines. Prostate Cancer Prostatic Dis. 2000;3:66–75. doi: 10.1038/sj.pcan.4500394. [DOI] [PubMed] [Google Scholar]
- 9.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–68. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 10.Mimeault M, Hauke R, Mehta PP, Batra SK. Recent advances in cancer stem/progenitor cell research: therapeutic implications for overcoming resistance to the most aggressive cancers. J Cell Mol Med. 2007;11:981–1011. doi: 10.1111/j.1582-4934.2007.00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ni J, Cozzi P, Hao J, Duan W, Graham P, Kearsley J, et al. Cancer stem cells in prostate cancer chemoresistance. Curr Cancer Drug Targets. 2014;14:225–40. doi: 10.2174/1568009614666140328152459. [DOI] [PubMed] [Google Scholar]
- 12.Sell S. Cancer and stem cell signaling: a guide to preventive and therapeutic strategies for cancer stem cells. Stem Cell Rev. 2007;3:1–6. doi: 10.1007/s12015-007-0015-5. [DOI] [PubMed] [Google Scholar]
- 13.Barr MP, Gray SG, Hoffmann AC, Hilger RA, Thomale J, O’Flaherty JD, et al. Generation and characterisation of cisplatin-resistant non-small cell lung cancer cell lines displaying a stem-like signature. PLoS ONE. 2013;8:e54193. doi: 10.1371/journal.pone.0054193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu LL, Chen XH, Zhang G, Liu ZC, Wu N, Wang H, et al. CCL21 facilitates chemoresistance and cancer stem cell-like properties of colorectal cancer cells through AKT/GSK-3. Oxid Med Cell Longev. 2016;2016:5874127. doi: 10.1155/2016/5874127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rybak AP, Bristow RG, Kapoor A. Prostate cancer stem cells: deciphering the origins and pathways involved in prostate tumorigenesis and aggression. Oncotarget. 2015;6:1900–19. doi: 10.18632/oncotarget.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maitland NJ, Collins AT. Prostate cancer stem cells: a new target for therapy. J Clin Oncol. 2008;26:2862–70. doi: 10.1200/JCO.2007.15.1472. [DOI] [PubMed] [Google Scholar]
- 17.Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012;22:373–88. doi: 10.1016/j.ccr.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh RK, Dhadve A, Sakpal A, De A, Ray P. An active IGF-1R-AKT signaling imparts functional heterogeneity in ovarian CSC population. Sci Rep. 2016;6:36612. doi: 10.1038/srep36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh RK, Gaikwad SM, Jinager A, Chaudhury S, Maheshwari A, Ray P, et al. IGF-1R inhibition potentiates cytotoxic effects of chemotherapeutic agents in early stages of chemoresistant ovarian cancer cells. Cancer Lett. 2014;354:254–62. doi: 10.1016/j.canlet.2014.08.023. [DOI] [PubMed] [Google Scholar]
- 20.Qu Y, Oyan AM, Liu R, Hua Y, Zhang J, Hovland R, et al. Generation of prostate tumor-initiating cells is associated with elevation of reactive oxygen species and IL-6/STAT3 signaling. Cancer Res. 2013;73:7090–100. doi: 10.1158/0008-5472.CAN-13-1560. [DOI] [PubMed] [Google Scholar]
- 21.Kroon P, Berry PA, Stower MJ, Rodrigues G, Mann VM, Simms M, et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013;73:5288–98. doi: 10.1158/0008-5472.CAN-13-0874. [DOI] [PubMed] [Google Scholar]
- 22.Abdel-Tawab M, Werz O, Schubert-Zsilavecz M. Boswellia serrata: an overall assessment of in vitro, preclinical, pharmacokinetic and clinical data. Clin Pharmacokinet. 2011;50:349–69. doi: 10.2165/11586800-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 23.Syrovets T, Buchele B, Krauss C, Laumonnier Y, Simmet T. Acetyl-boswellic acids inhibit lipopolysaccharidemediated TNF-α induction in monocytes by direct interaction with IκB kinases. J Immunol. 2005;174:498–506. doi: 10.4049/jimmunol.174.1.498. [DOI] [PubMed] [Google Scholar]
- 24.Syrovets T, Gschwend JE, Buchele B, Laumonnier Y, Zugmaier W, et al. Inhibition of IkappaB kinase activity by acetyl-boswellic acids promotes apoptosis in androgen-independent PC-3 prostate cancer cells in vitro and in vivo. J Biol Chem. 2005;280:6170–80. doi: 10.1074/jbc.M409477200. [DOI] [PubMed] [Google Scholar]
- 25.Liu JJ, Nilsson A, Oredsson S, Badmaev V, Zhao WZ, Duan RD. Boswellic acids trigger apoptosis via a pathway dependent on caspase-8 activation but independent on Fas/Fas ligand interaction in colon cancer HT-29 cells. Carcinogenesis. 2002;23:2087–93. doi: 10.1093/carcin/23.12.2087. [DOI] [PubMed] [Google Scholar]
- 26.Hostanska K, Daum G, Saller R. Cytostatic and apoptosis-inducing activity of boswellic acids toward malignant cell lines in vitro. Anticancer Res. 2002;22:2853–62. [PubMed] [Google Scholar]
- 27.Zhang D, Cui Y, Niu L, Xu X, Tian K, Young CY, et al. Regulation of SOD2 and β-arrestin1 by interleukin-6 contributes to the increase of IGF-1R expression in docetaxel resistant prostate cancer cells. Eur J Cell Biol. 2014;93:289–98. doi: 10.1016/j.ejcb.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Xu Q, Liu X, Zhu S, Hu X, Niu H, Zhang X, et al. Hyper-acetylation contributes to the sensitivity of chemo-resistant prostatecancer cells to histone deacetylase inhibitor Trichostatin A. J Cell Mol Med. 2018;22:1909–22. doi: 10.1111/jcmm.13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan HQ, Kong F, Wang XL, Young CY, Hu XY, Lou HX. Inhibitory effect of acetyl-11-keto-beta-boswellic acid on androgen receptor by interference of Sp1 binding activity in prostate cancer cells. Biochem Pharmacol. 2008;75:2112–21. doi: 10.1016/j.bcp.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 30.Liu YQ, Hu XY, Lu T, Cheng YN, Young CY, Yuan HQ, et al. Retigeric acid B exhibits antitumor activity through suppression of nuclear factor-κB signaling in prostate cancer cells in vitro and in vivo. PLoS ONE. 2012;7:e38000. doi: 10.1371/journal.pone.0038000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang H, Sun J, Xu Q, Liu Y, Wei J, Young CY, et al. Marchantin M: a novel inhibitor of proteasome induces autophagic cell death in prostate cancer cells. Cell Death Dis. 2013;4:e761. doi: 10.1038/cddis.2013.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson TC, Southgate J, Kitchener G, Land H. Multistage carcinogenesis induced by ras and myc oncogenes in a reconstituted organ. Cell. 1989;56:917–30. doi: 10.1016/0092-8674(89)90625-9. [DOI] [PubMed] [Google Scholar]
- 33.Achuthan S, Santhoshkumar TR, Prabhakar J, Nair SA, Pillai MR. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J Biol Chem. 2011;286:37813–29. doi: 10.1074/jbc.M110.200675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brambrink T, Foreman R, Welstead GG, Lengner CJ, Wernig M, Suh H, et al. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell. 2008;2:151–9. doi: 10.1016/j.stem.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao H, Guo Y, Li S, Han R, Ying J, Zhu H, et al. A novel anti-cancer agent Icaritin suppresses hepatocellular carcinomainitiation and malignant growth through the IL-6/Jak2/Stat3 pathway. Oncotarget. 2015;31:31927–43. doi: 10.18632/oncotarget.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruefli AA, Bernhard D, Tainton KM, Kofler R, Smyth MJ, Johnstone RW. Suberoylanilide hydroxamic acid (SAHA) overcomes multidrug resistance and induces cell death in P-glycoprotein-expressing cells. Int J Cancer. 2002;99:292–8. doi: 10.1002/ijc.10327. [DOI] [PubMed] [Google Scholar]
- 37.Xue X, Chen F, Liu A, Sun D, Wu J, Kong F, et al. Reversal of the multidrug resistance of human ileocecal adenocarcinoma cells by acetyl-11-keto-β-boswellic acid via downregulation of P-glycoprotein signals. Biosci Trends. 2016;10:392–9. doi: 10.5582/bst.2016.01115. [DOI] [PubMed] [Google Scholar]
- 38.O’Neill AJ, Prencipe M, Dowling C, Fan Y, Mulrane L, Gallagher WM, et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol Cancer. 2011;10:126. doi: 10.1186/1476-4598-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh S, Trevino J, Bora-Singhal N, Coppola D, Haura E, Altiok S, et al. EGFR/Src/Akt signaling modulates Sox2 expression and self-renewal of stem-like side-population cells in non-small cell lung cancer. Mol Cancer. 2012;11:73. doi: 10.1186/1476-4598-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul. 2006;46:249–79. doi: 10.1016/j.advenzreg.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Jeong CH, Cho YY, Kim MO, Kim SH, Cho EJ, Lee SY, et al. Phosphorylation of Sox2 cooperates in reprogramming to pluripotent stem cells. Stem Cells. 2010;28:2141–50. doi: 10.1002/stem.540. [DOI] [PubMed] [Google Scholar]
- 42.Liu HP, Gao ZH, Cui SX, Wang Y, Li BY, Lou HX, et al. Chemoprevention of intestinal adenomatous polyposis by acetyl-11-keto-beta-boswellic acid in APC(Min/+) mice. Int J Cancer. 2013;132:2667–81. doi: 10.1002/ijc.27929. [DOI] [PubMed] [Google Scholar]
- 43.Civenni G, Longoni N, Costales P, Dallavalle C, García Inclán C, Albino D, et al. EC-70124, a novel glycosylated indolocarbazole multi-kinase inhibitor, reverts tumorigenic and stem cell properties in prostate cancer by inhibiting STAT3 and NF-κB. Mol Cancer Ther. 2016;15:806–18. doi: 10.1158/1535-7163.MCT-15-0791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.