

Abstract
Human genetic and pharmacological studies have demonstrated that voltage-gated sodium channels (VGSCs) are promising therapeutic targets for the treatment of pain. Spider venom contains many toxins that modulate the activity of VGSCs. To date, only 0.01% of such spider toxins has been explored, and thus there is a great potential for discovery of novel VGSC modulators as useful pharmacological tools or potential therapeutics. In the current study, we identified a novel peptide, µ-TRTX-Ca1a (Ca1a), in the venom of the tarantula Cyriopagopus albostriatus. This peptide consisted of 38 residues, including 6 cysteines, i.e. IFECSISCEIEKEGNGKKCKPKKCKGGWKCKFNICVKV. In HEK293T or ND7/23 cells expressing mammalian VGSCs, this peptide exhibited the strongest inhibitory activity on Nav1.7 (IC50 378 nM), followed by Nav1.6 (IC50 547 nM), Nav1.2 (IC50 728 nM), Nav1.3 (IC50 2.2 µM) and Nav1.4 (IC50 3.2 µM), and produced negligible inhibitory effect on Nav1.5, Nav1.8, and Nav1.9, even at high concentrations of up to 10 µM. Furthermore, this peptide did not significantly affect the activation and inactivation of Nav1.7. Using site-directed mutagenesis of Nav1.7 and Nav1.4, we revealed that its binding site was localized to the DIIS3-S4 linker region involving the D816 and E818 residues. In three different mouse models of pain, pretreatment with Cala (100, 200, 500 µg/kg) dose-dependently suppressed the nociceptive responses induced by formalin, acetic acid or heat. These results suggest that Ca1a is a novel neurotoxin against VGSCs and has a potential to be developed as a novel analgesic.
Keywords: µ-TRTX-Ca1a, tarantula spider, peptide, hNav1.7, electrophysiology, analgesic activity
Introduction
Chronic pain is pervasive. It has been estimated that 20% of adults worldwide suffer from pain, and 10% are newly diagnosed with chronic pain each year [1]. The prevalence of pain has generated a heavy global economic burden, and thus pain is a significant public health problem [2, 3]. However, the currently available drugs have limited efficacy and undesirable side effects. Accordingly, there is an unmet clinical need for novel analgesics to treat pain [4].
Voltage-gated sodium channels (VGSCs) play a vital role in the generation and propagation of action potentials. VGSCs consist of a pore-forming α subunit associated with one or more regulatory β subunits. The α subunit is organized into four homologous domains (DI–DIV), each containing six transmembrane segments (S1–S6). The S3–S4 segments form a voltage sensor domain, while the pore domain consists of the S5–S6 segments [5]. In mammals, nine distinct α subunits (Nav1.1–Nav1.9) have been cloned [6]. The Nav1.1–Nav1.3 subtypes are mainly expressed in the central nervous system, while the Nav1.6 subtype is found both centrally and peripherally. In contrast, the Nav1.7–Nav1.9 subtypes are predominantly located in the peripheral nervous system [7]. Nav1.4 is present in the skeletal muscle, while Nav1.5 is present in the cardiac muscle [8].
Nav1.1 has been implicated in mechanical pain [9], while Nav1.3, Nav1.7, Nav1.8, and Nav1.9 are strongly associated with chronic pain [10]. Nav1.6 is the predominant sodium channel distributed at the axon initial segment and mature nodes of Ranvier in myelinated fibers [11, 12]. Nav1.6 mediates the tetrodotoxin-sensitive (TTX-S) transient current, persistent current, and resurgent current [13]. Recent studies have revealed that Nav1.6 participates in chemically mediated pain pathways and that the knockdown of Nav1.6 relieves mechanical pain behavior in models of local inflammation and neuropathic pain [14–16]. These results suggest that Nav1.6 may be a valuable therapeutic target for the management of chronic pain. Additionally, compelling genetic studies have demonstrated that human Nav1.7 (hNav1.7) is an analgesic target. Loss-of-function mutations in the human SNC9A gene, encoding hNav1.7, result in congenital insensitivity to pain and anosmia [17–19], while human gain-of-function mutations lead to painful neuropathies, including erythromelalgia, small fiber neuropathy, and paroxysmal extreme pain disorder [20–22]. These studies indicate that inhibitors targeting hNav1.7 may be powerful analgesics. Because hNav1.7 is one of the most promising targets for alleviating chronic pain, peptide toxins targeting pain-related hNav1.7 continue to be developed.
Spider venom contains many toxins that modulate the activity of VGSCs in order to subdue prey and deter predators. To date, only 0.01% of such spider toxins has been explored, and thus there is potential for the discovery of novel VGSC modulators as useful pharmacological tools or potential therapeutics [23]. Disulfide-rich peptides account for the vast majority of the sodium channel toxins that have already been discovered [24, 25]. The µ-TRTX-Hs2a (HWTX-IV), µ-TRTX-Hhn2a (HNTX-III), and µ-TRTX-Hhn1b (HNTX-IV) peptides, isolated from tarantula spiders, have been shown to have excellent affinity and specificity for hNav1.7. HWTX-IV, isolated from the venom of Ornithoctonus huwena, is a 35-residue peptide that displays robust inhibitory activity against hNav1.7 (IC50 ~ 26 nM) [26], while HNTX-III, isolated from the venom of Ornithoctonus hainana, is a potent and specific antagonist of neuronal TTX-S VGSCs with high affinity for hNav1.7 (IC50 ~ 232 nM) [27]. HNTX-IV was also isolated from the venom of O. hainana and preferentially inhibits hNav1.7 (IC50 ~ 21 nM) [28]. All three of these toxins possess a classical inhibitor cystine knot motif with high stability and protease resistance. All three toxins interact with hNav1.7 by binding to the DIIS3-S4 linker and impede movement of the DII voltage sensor, leading to the inhibition of hNav1.7 activation by trapping the voltage sensor in a closed state [26–28].
Cyriopagopus albostriatus, a species belonging to the family Theraphosidae, is usually found in Myanmar, Thailand, and Cambodia. It is also known as the edible spider or the Thai zebra tarantula. The venom of C. albostriatus has not previously been investigated. Here we describe a novel peptide toxin µ-TRTX-Ca1a (Ca1a), isolated from the venom of C. albostriatus, showing potent inhibition of hNav1.7 with dose-dependent analgesic activity in three rodent models of pain.
Materials and methods
Venom collection and toxin purification
The crude venom of C. albostriatus was collected by electrostimulation as previously described [29]. Lyophilized crude venom was dissolved in ddH2O to a final concentration of 1 mg/mL and subjected to a first round of semipreparative reversed-phase high-performance liquid chromatography (RP-HPLC) purification (C18 column, 10 mm × 250 mm, 5 μm, Welch Materials Inc., Shanghai, China) with an acetonitrile gradient ranging from 10% to 55% at an increasing rate of 1% per minute (Waters e2695 Separations Module, Waters, CA, USA). The fraction containing Ca1a was collected, lyophilized, and subjected to a second round of RP-HPLC purification (C18 column, 4.6 mm × 250 mm, 5 μm, Welch Materials Inc.) with acetonitrile ranging from 20% to 40% at an increasing rate of 0.5% per minute (Waters 2795 Separations Module, Waters). The purified peptide was lyophilized and stored at −20 °C until further use.
cDNA library construction
Total RNA of C. albostriatus was extracted from the venom glands of the spider using a TRIzol Reagent Kit (Invitrogen, CA, USA) as previously described [30]. A cDNA library was constructed by using the SMART cDNA Library Construction Kit (Clontech, CA, USA) according to the instructions. PCR was performed using the M13 forward and reverse primers in the kit to rapidly screen recombinant clones. The clones from the original cDNA library were separated onto Luria-Bertani agar plates. The resulting colonies were randomly picked and sequenced with standard M13 reverse primers on an ABI 3730 automatic DNA sequencer (ABI, Waltham, MA, USA) according to the manufacturer’s instructions. The sequences of the expressed sequence tags were translated into amino acid sequences using the ExPASy program (https://web.expasy.org/translate/). The signal peptide was predicted with the SignalP 4.1 program online (http://www.cbs.dtu.dk/services/SignalP/).
cDNA sequence determination and mass spectrometric analysis
A portion of the amino acid sequence of Ca1a was determined by Edman degradation on an Applied Biosystems/PerkinElmer Life Science Procise 491-A protein sequencer (Applied Biosystems, CA, USA). The cDNA of this toxin was obtained by using BLAST to align the Edman degradation-determined amino acid sequence of Ca1a against the local cDNA library database of the spider C. albostriatus (unpublished data). The molecular mass of the peptide was determined using an AB SCIEX-TOF/TOF 5800 mass spectrometer (Applied Biosystems). The matrix used was α-cyano-4-hydroxy-cinnamic acid (saturated solution in 50% ACN:0.1% TFA, 1:1), and mass spectra were acquired in positive reflector mode. Mass spectra were recorded under the control of TOF/TOF Series Explorer Software (Applied Biosystems).
Plasmid construction and transient transfection
cDNAs encoding rat Nav1.2 and mouse Nav1.6 were subcloned into the vector pcDNA3.1. cDNAs encoding rat Nav1.3 and rat Nav1.4 were subcloned into the vectors pcDNA3.1 and pRGB4, respectively. cDNAs encoding hNav1.5 and hNav1.7 were subcloned into the vectors pcDNA3.1 and pcDNA3.1-mod, respectively. cDNAs encoding rNav1.8 and hNav1.9 were subcloned into the vectors pCMV-HA and pEGFP-N1, respectively. Mutations of rNav1.4 (N655D, Q657E, and N655D/Q657E) and hNav1.7 (D816N, E818Q, and D816N/E818Q) were generated using the Gene Tailor Site-Directed Mutagenesis System (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. Wild-type (WT) rNav1.2, rNav1.3, rNav1.4, mNav1.6, hNav1.5, and hNav1.7 and the abovementioned mutant plasmids were transiently transfected into HEK293T cells together with enhanced green fluorescent protein (eGFP), while rNav1.8 was transiently transfected into ND7/23 cells together with eGFP using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Additionally, β1- and β2-eGFP plasmids, encoding the sodium channel β1 and β2 subunits, were cotransfected with those encoding hNav1.7. hNav1.9 was transfected into ND7/23 cells according to a recent report from our laboratory [31]. HEK293T cells and ND7/23 cells were grown under standard tissue culture conditions (5% CO2, 37 °C) in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
Acute isolation of dorsal root ganglion neurons
Dorsal root ganglion (DRG) neurons were acutely dissociated from 18 to 20 g adult C57BL/6 mice according to the method reported previously [32]. Briefly, DRGs were transferred into ice-cold DMEM and minced with scissors. They were digested in oxygenated DMEM containing collagenase (1 mg/mL, type IA, Sigma) and trypsin (0.5 mg/mL, type III, Sigma) for 30 min with gentle agitation at 37 °C. Dissociated DRG cells were plated onto poly-D-lysine-coated glass coverslips in DMEM supplemented with 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 10% FBS. The DRG cells were incubated for 2–4 h before patch clamp experiments.
Acute isolation of dorsal unpaired median (DUM) neurons
According to a previous report [33], adult cockroaches (P. americana) were desheathed, and their abdominal ganglia were removed and incubated in insect physiological solution (in mM): 90 NaCl, 6 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 140 glucose at pH 6.6 containing 1 mg/mL trypsin for 5 min. After this, the ganglia were removed and stored in physiological solution for 1 h to recover. Thin silver needles were used to separate the large DUM cells situated in the dorsal midline of the ganglia. Cells that were bright under phase contrast microscopy were considered to be viable cells and were used for further experimental procedures.
Electrophysiological assays
Whole-cell patch clamp recordings were performed using an EPC 10 USB Patch Clamp Amplifier (HEKA Elektronik, Ludwigshafen, Germany). The recording pipettes were made from borosilicate glass capillary tubes (VWR micropipettes; VWR Co., West Chester, PA, USA) using a PC-10 puller (Narishige, Tokyo, Japan). The pipet resistance was controlled at 2.0–3.0 MΩ by adjusting the pulling temperature. The standard pipet solution for HEK293T cells contained (in mM): 140 CsCl, 10 NaCl, 1 EGTA, and 10 HEPES (pH 7.4). The bath solution for HEK293T cells contained (in mM): 140 NaCl, 2 CaCl2, 1 MgCl2, 5 KCl, 20 HEPES (pH 7.4), and 10 glucose. The external solution for mouse DRG neurons contained (in mM): 30 NaCl, 1 MgCl2, 1.8 CaCl2, 5 CsCl, 5 KCl, 25 D-glucose, 5 HEPES, and TEA-Cl (pH 7.4), and the micropipette internal solution for mouse DRG neurons contained 135 mM CsCl, 10 mM NaCl, and 5 mM HEPES (pH 7.4). The external solution for insect DUM neurons contained 80 mM NaCl, 30 mM TEA-Cl, 2 mM CaCl2, 4 mM KCl, 10 mM HEPES, 10 mM glucose, 50 mM choline-Cl, and 1 mM 4-AP (pH 6.8), and the micropipette internal solution for insect DUM neurons contained 140 mM CsF, 2 mM MgCl2, 10 mM EGTA, and 10 mM HEPES (pH 6.8). All the chemicals were purchased from Sigma Aldrich (St. Louis, MO, USA). All the experiments were conducted at room temperature (20–25 °C). Data were acquired by the PatchMaster software (HEKA Elektronik, Ludwigshafen, Germany). Membrane currents were filtered at 5 kHz and sampled at 20 kHz. Cells with GFP fluorescence were selected for whole-cell patch clamp recordings 24 h after transfection. HEK293T cells were maintained at −100 mV for all the parameters examined, and 80%–90% series resistance compensation was applied. Voltage-clamp recordings were acquired 5 min after establishing whole-cell configuration to allow adequate equilibration between the micropipette solution and the cell interior.
The Nav1.2–Nav1.7 channel current was elicited by 50 ms depolarization potential to −10 mV from the holding voltage of −100 mV. The depolarization potential for Nav1.8 was + 20 mV. The Nav1.9 current was elicited by 50 ms depolarization potential to −40 mV from the holding voltage of −120 mV in the presence of 1 µM TTX. To measure current–voltage (I–V) relationships, a range of potential from −100 mV to +100 mV in 5 mV increments were applied from the holding potential (−100 mV) for 50 ms at 5 s intervals. Peak values at each potential were plotted to form I–V curves. Steady-state inactivation was assessed with a 20-ms depolarizing test potential of −10 mV following a 500-ms prepulse at potentials that ranged from −130 to −10 mV with a 10-mV increment.
Animals
Healthy C57BL/6 mice (weight 18–20 g) and ICR mice (weight 18–22 g) were obtained from the Experimental Animal Center of SLac-kinda (Changsha, China), kept at 20–25 °C under a 12-h light/dark cycle, and fed with standard rodent chow and water ad libitum. All the experimental protocols were approved by the Animal Care and Use Committee (ACUC) at the Hunan Province Animal Management Office.
Abdominal writhing induced by acetic acid
Abdominal writhing responses were induced by intraperitoneal (i.p.) injection of 200 µL of 0.8% (v/v) acetic acid according to a previous report [34]. Briefly, mice were pretreated with Ca1a (100, 200, and 500 µg/kg) while control mice received an equal volume of saline (i.p.) for 15 min prior to acetic acid injection. After the challenge, mice were individually placed into open polyvinyl cages (30 × 40 × 30 cm3), and the abdominal writhing responses were counted for 30 min cumulatively.
Formalin-induced paw licking
Ca1a (100, 200, and 500 µg/kg) was administered 30 min prior to intraplantar formalin injection. A volume of 20 µL formalin (5%) solution was used as previously described [35] and injected into the plantar surface of right hind paw. The time spent licking the injected paw was counted every 5 min. Phase I was defined as 0–10 min, and phase II was defined as 15–40 min.
Hot plate
Mice preadministered with Ca1a (100, 200, and 500 µg/kg) were placed on a hot plate apparatus (model YLS-21A, Jinan, China) set at 55 ± 0.1 °C, and the time taken to observe a nociceptive response (hind paw licking or jumping) was recorded according to a previous report [36]. Latency time was determined using a stopwatch.
Data analysis
The data were analyzed with the Fit-Master (HEKA, Elektronik, Ludwigshafen, Germany), Igor Pro software (WaveMetrics, Lake Oswego, OR, USA), SigmaPlot 10 software (Sigma, St. Louis, MO, USA), and Prism 5 (GraphPad Software, San Diego, CA, USA). Concentration–response curves were fitted by the SigmaPlot sigmoidal equation as follows: y = a/(1 + exp(−(x − IC50)/b)), in which IC50 is the concentration of toxin at half-maximal efficacy, and a and b are the constants. The conductance–voltage (G–V) curves were obtained by calculating the conductance (G) at each voltage (V) using the equation G = I/(V − Vrev), with Vrev being the reversal potential determined for each cell individually. G–V curves were fitted using a Boltzmann equation: y = 1/(1 + exp[(Va − V)/κ]), in which Va, V, and κ represent the midpoint voltage of kinetics, test potential, and slope factor, respectively. Peak inward currents at the test pulse were normalized to the maximal inward current and fitted with Boltzmann functions: I/Imax = A + (1 − A)/{1 + exp[(V − Vh)/κ]}, where V represents the inactivating prepulse potential, Vh is the midpoint of the steady-state inactivation, A is the minimal channel availability, and κ is the slope factor.
Animals were randomly assigned to each treatment group. Data points are presented as the mean ± SEM, and n is presented as the number of separate experimental units. Statistical significance was assessed with GraphPad Prism using the paired Student’s t test or one-way analysis of variance with Tukey’s multiple comparison test. Statistical significance was accepted at P < 0.05.
Results
Purification and sequence analysis of Ca1a
Venom from C. albostriatus was collected by electrostimulation and then lyophilized for further use. The lyophilized crude venom was purified by RP-HPLC with solvent B (0.1% trifluoroacetic acid in acetonitrile) ranging from 10% to 55% over the course of 45 min. Comprehensive screening of each eluted fraction against hNav1.7, heterologously expressed in HEK293T cells, indicated that the fraction denoted with a red star possessed inhibitory activity (Fig. 1a). This fraction was further isolated using analytical RP-HPLC, and the purified toxin was named µ-TRTX-Ca1a (Ca1a) according to the standard nomenclature [37] (Fig. 1b). The molecular mass of this naturally occurring toxin was determined to be 4289.31 Da (M + H+) by matrix-assisted laser desorption/ionization time of flight mass spectrometry (Fig. 1c and 1c inset). A portion of the amino acid sequence of Ca1a was determined by N-terminal Edman degradation. The full-length amino acid sequence of the peptide was determined by combining Edman degradation and cDNA sequencing. Finally, the complete amino acid sequence of the toxin was determined to consist of 38 residues, including six cysteines: IFECSISCEIEKEGNGKKCKPKKCKGGWKCKFNICVKV (Fig. 1d). The theoretical molecular mass calculated from the sequence was 4294.21 Da, in good agreement with mass spectrometric data. BLAST results showed that the purified toxin shared 82% identity to a previously reported spider peptide toxin, HNTX-II, which was shown to be an insecticidal peptide [38] (Fig. 1e).
Fig. 1.

Purification and cDNA sequence of µ-TRTX-Ca1a. a RP-HPLC profile of the venom from the spider C. albostriatus. The red asterisk indicates the fraction containing Ca1a. b Further purification by analytical RP-HPLC of the fraction of interest. c MALDI-TOF MS analysis of purified Ca1a and inset showing an enlarged view of the peak. d The full cDNA sequence of Ca1a. The sequence of the mature peptide is highlighted in yellow. e Sequence alignment of Ca1a with related spider toxins
Effect of Ca1a on DRG VGSCs
The biological function of Ca1a was determined using adult mouse DRG neurons. Currents were elicited by a 50 ms depolarizing potential of −10 mV from a holding potential of −80 mV every 5 s. As shown in Fig. 2a, 0.5 µM Ca1a inhibited 50% DRG VGSC currents with an IC50 value of 565 nM (Fig. 2b). The I–V curve of DRG VGSCs showed that Ca1a did not alter the initial activation voltage or the reversal potential (Fig. 2c). In addition, the G–V curves of DRG VGSCs did not change in the presence of 0.5 µM Ca1a. The half-activation voltages of the G–V curve before and after toxin treatment were −21.9 mV and −22.0 mV, respectively (Fig. 2d).
Fig. 2.
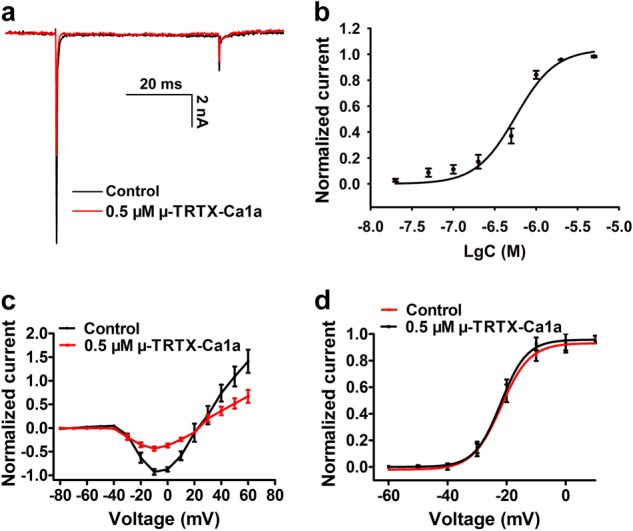
Effects of µ-TRTX-Ca1a on VGSCs in mice DRG neurons. a Inhibition of 0.5 µM Ca1a on DRG VGSC currents. b Dose-dependent inhibition of Ca1a on DRG VGSC currents. c Current–voltage (I–V) curve for the DRG Nav channel before (black) and after (red) application of 0.5 µM Ca1a (n = 8). d Conductance–voltage (G-V) curve for the Nav1.7 channel before (black) and after (red) application of 0.5 µM Ca1a (n = 8). Means ± SEM
Effect of Ca1a on mammalian VGSCs and insect DUM neurons
Ca1a exhibited high sequence similarity with HNTX-II, a peptide isolated from the Chinese bird spider (Haplopelma hainanum). HNTX-II exhibited higher insecticidal activity but lower lethality in mammals, and so we wondered whether Ca1a would also have an effect on insect VGSCs. Accordingly, we tested the activity of Ca1a on acutely isolated cockroach DUM neurons. Sodium currents were elicited by a 50 ms depolarization to −10 mV from a holding potential of −100 mV. Ca1a at 10 µM showed little inhibition on VGSCs in DUM neurons (Fig. 3a). We then tested its effect on mammalian VGSCs. Nav1.2−Nav1.7 channels were transiently expressed in HEK293T cells, while Nav1.8 and Nav1.9 channels were transiently expressed in ND723 cells. As shown in Fig. 3c–j, treatment with 1 µM Ca1a resulted in 86.5% inhibition of Nav1.7 current and decreased the Nav1.2 and Nav1.6 current amplitude by 63.7% and 68.1%, respectively. However, only 23.1% and 11.2% of the current was inhibited in cells expressing Nav1.3 and Nav1.4, respectively. No significant activity was observed against Nav1.5, Nav1.8, or Nav1.9 at concentrations up to 10 µM. Ca1a had the highest potency for hNav1.7 (IC50 378 nM), followed by Nav1.6 (IC50 547 nM) < Nav1.2 (IC50 728 nM) < Nav1.3 (IC50 2.2 µM) ~ Nav1.4 (IC50 3.2 µM) (Fig. 3b). Thus hNav1.7 was used to investigate the mechanism of Ca1a in VGSC activation and inactivation.
Fig. 3.

Activity of µ-TRTX-Ca1a for Nav1.2–Nav1.9 channels and DUM neurons. a Activity of Ca1a on whole-cell VGSC currents in DUM neurons at a dose of 10 µM. b Concentration–response curves of Ca1a at Nav1.2–Nav1.4, Nav1.6, and Nav1.7 assessed by whole-cell patch clamp experiments. Data are means ± SEM, n = 4–7. c–j Representative Nav1.2–Nav1.9 current traces before (black) and after addition of Ca1a (red). Ca1a at 1 µM potently inhibited Nav1.7 and moderately inhibited Nav1.2–1.4 and Nav1.6. Ca1a at 10 µM showed no obvious effect on Nav1.5, Nav1.8, and Nav1.9 currents
Effect of Ca1a on activation and inactivation of hNav1.7
The I–V curve, steady-state activation, and steady-state inactivation were analyzed in the presence and absence of Ca1a. Here 0.5 µM Ca1a inhibited 61.8% hNav1.7 current (Fig. 4a), and the toxin did not change the initial activation voltage or the reversal potential in the I–V curve (Fig. 4b). The half-activation voltages were −19.9 and −18.1 mV for hNav1.7 before and after treatment with 0.5 µM Ca1a, respectively (Fig. 4c), and the half-inactivation voltages were −67.3 and −69.6 mV for hNav1.7 before and after treatment with 0.5 µM Ca1a, respectively (Fig. 4d). These results indicate that Ca1a causes no obvious changes in the activation and inactivation of hNav1.7.
Fig. 4.
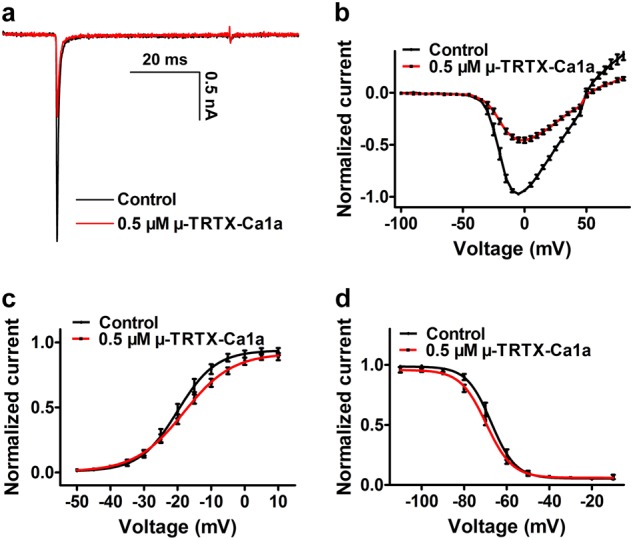
The inhibition of Ca1a on Nav1.7 expressed in HEK293T cells. a Inhibition of Nav1.7 currents by 0.5 µM Ca1a. b Current–voltage (I–V) curve for the Nav1.7 channel before (black) and after (red) application of 0.5 µM Ca1a (n = 10). c Conductance–voltage (G–V) curve for the Nav1.7 channel before (black) and after (red) application of 0.5 µM Ca1a (n = 10). d Steady-state inactivation of the Nav1.7 channel before (black) and after (red) treatment with 0.5 µM Ca1a (n = 10). Means ± SEM
Ca1a binds to the DIIS3-S4 linker of hNav1.7 expressed in HEK293T cells
Since Ca1a had the highest potency for Nav1.7 among TTX-S VGSCs, we compared the DIIS3-S4 linkers of these subtypes (Nav1.2–Nav1.7). The acidic residues D816 and E818 are present in the DIIS3-S4 linker of hNav1.7, while the corresponding residues in rNav1.4 are N655 and Q657 (Fig. 5a). We next investigated whether the two different residues in the DIIS3-S4 linker determined the sensitivity of Ca1a to hNav1.7. Mutations in the amino acid sequences of hNav1.7 (D816N, E816Q, and D816N/E816Q) and rNav1.4 (N655D, Q657E, and N655D/Q657E) were generated. The 1 µM Ca1a treatment resulted in the inhibition of rNav1.4 N655D, Q657E, and double-mutant N655D-Q657E current by 66.3%, 60.4%, and 78.0%, respectively (Fig. 5b), compared to 15.8% inhibition of WT rNav1.4. The IC50 values for Ca1a on N655D, Q657E, and N655D-Q657E were 666, 831, and 458 nM, respectively (Fig. 5c), which were close to the value for the WT hNav1.7 channel. These data indicate that N655 and Q657 in DIIS3-S4 of rNav1.4 confer channel resistance to Ca1a. This interpretation is supported by experiments indicating that 1 µM Ca1a inhibits hNav1.7 D816N, E818Q, and D816N-E818Q current by 3.1%, 37.9%, and 4.4%, respectively (Fig. 6a). The inhibitory activity of 1 µM Ca1a on these three mutants was significantly lower than that on WT hNav1.7. The IC50 value of E818Q was 1.218 µM, which was close to the value for the WT rNav1.4 channel (Fig. 6b). These results indicate that D816 and E818 in the DIIS3-S4 linker play a critical role in the action of Ca1a on Nav1.7 [26, 27].
Fig. 5.
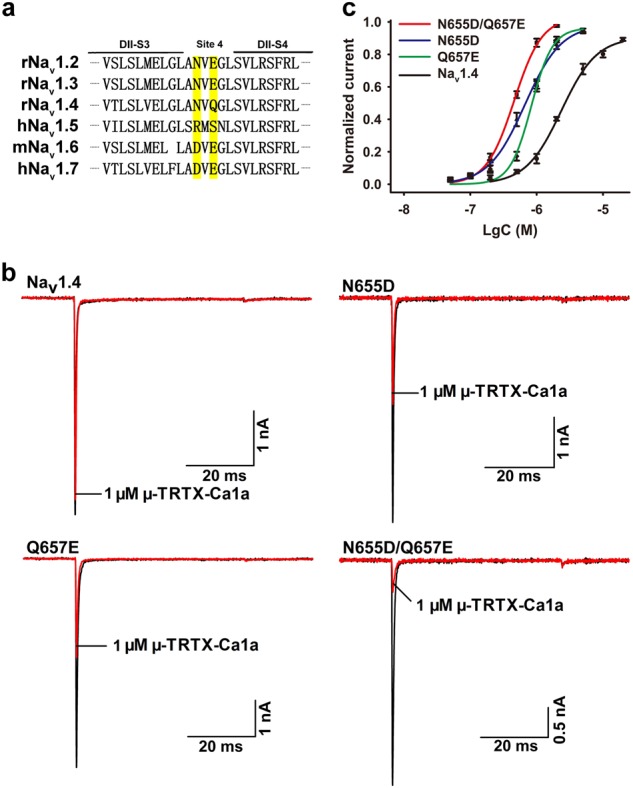
Effect of Ca1a on WT and mutant Nav1.4 expressed in HEK293T cells. a The sequence of the DIIS3-S4 linkers of Nav1.2–1.7. Amino acids of interest in the sequence are shaded in yellow. b Representative current traces for WT and mutant channels (N655D, Q657E, and N655D/Q657E) inhibited by 1 µM Ca1a. c Dose–response inhibition curves of Ca1a on WT and mutant Nav1.4 channels. Means ± SEM
Fig. 6.
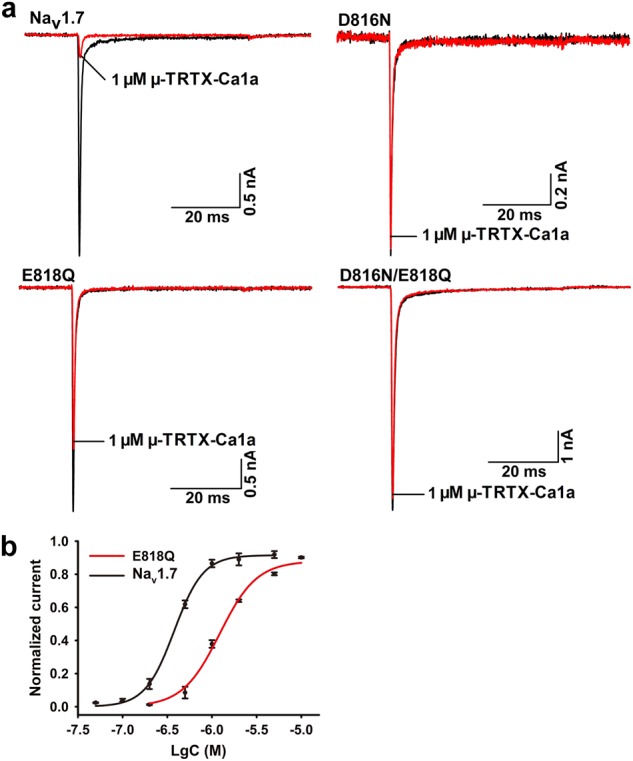
Effect of Ca1a on WT and mutant Nav1.7 expressed in HEK293T cells. a Representative current traces for WT and mutant channels (D816N, E818Q, and D816N-E818Q) inhibited by 1 µM Ca1a. b Dose–response inhibitory curves of Ca1a on WT and mutant Nav1.7 channels. Means ± SEM
Antinociceptive effects of Ca1a in mouse models of pain
The formalin-induced pain model includes two phases: phase I (0–10 min) and phase II (15–40 min). Pain is mediated by direct activation of nociceptive neurons in phase I, while in phase II, pain is attributed to the inflammatory response to tissue damage [39, 40]. Intraplantar injection of formalin caused nociceptive pain responses, including shaking and licking in the control group, and administration of Ca1a attenuated these pain behaviors (Fig. 7a). During phase I (0–10 min), the times spent in paw licking in the control group and in those injected with 100 µg/kg, 200 µg/kg, and 500 µg/kg Ca1a were 76.3, 70.1, 71.2, and 68.9 s, respectively. Morphine (100 µg/kg) reduced paw licking time to 57.2 s during phase I (Fig. 7b). During phase II (15–35 min), the time spent in paw licking was 174.9 s for the control group; after treatment with 100 µg/kg Ca1a, 200 µg/kg Ca1a, 500 µg/kg Ca1a, and 100 µg/kg morphine, the licking times were 91.8, 66.9, 27.0, and 125.1 s, respectively (Fig. 7c). These results showed that Ca1a attenuated the inflammatory pain behaviors caused by formalin during phase II in a dose-dependent manner, while no obvious analgesia was observed in phase I. In the hot plate test, Ca1a treatment significantly increased the pain threshold of mice in a dose-dependent manner (Fig. 7d). As shown in Fig. 7e, the latency of the control group was 9.6 s. Ca1a at doses of 100, 200, and 500 µg/kg increased the latency time to 11.0, 14.7, and 19.1 s, respectively. A dose of 2 mg/kg morphine was administered as a positive control, and it increased the latency to 12.4 s. In the acetic acid-induced writhing test, Ca1a at doses of 100, 200, and 500 µg/kg reduced the writhing count from 27 to 17, 11, and 6.5, respectively, while 100 µg/kg morphine caused a reduction to 11.3 (Fig. 7f).
Fig. 7.

Analgesic effect of Ca1a. a Time course of Ca1a on formalin-induced pain. Evaluation of the analgesic effect of Ca1a on phase I (b) or phase II (c). d Time course of Ca1a in a hot plate test in mice. e The analgesic effect of Ca1a at 30 min after injection was assessed. f The analgesic effect of Ca1a in the mouse abdominal writhing test. The data are shown as the means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle
Discussion
In the current study, we described the identification and characterization of a novel peptide toxin (µ-TRTX-Ca1a) isolated from the venom of the spider C. albostriatus. Ca1a contains 38 amino acids with three disulfide bonds and shows high affinity to hNav1.7 but weak inhibition of Nav1.2–1.4 and Nav1.6 and negligible activity against Nav1.5, Nav1.8, and Nav1.9. BLAST results show that Ca1a shares moderate sequence homology with some spider toxins such as HNTX-II, which was shown to possess insecticidal activity. However, 10 µM Ca1a did not show significant inhibition on VGSCs from Periplaneta americana cockroach DUM neurons. Therefore, Ca1a exhibits inhibitory activity only on mammalian VGSCs.
Many VGSC site 4 toxins have been identified in Chinese tarantula spiders, such as HWTX-IV from O. huwena venom and HNTX-III and HNTX-IV from the venom of O. hainana. Consistent with what has been reported regarding HNTX-III, HNTX-IV, and HWTX-IV, Ca1a did not significantly change activation and inactivation kinetics at low depolarization voltages, indicating that Ca1a was either a pore blocker or a gating modifier that inhibited the reopening of the toxin-bound channel. Moreover, site-directed mutagenesis analysis showed that Ca1a bound to the DIIS3-S4 linker of hNav1.7 by interacting with the D816 and E818 residues, which validated the hypothesis that the toxin is a gating modifier. Furthermore, as these two residues are also binding sites for HNTX-III, HNTX-IV, and HWTX-IV on Nav1.7, we were able to infer that Ca1a is also likely to be a site 4 toxin and therefore might trap the domain II voltage sensor of Nav1.7 in its resting conformation [26–28].
Morphine has potentially serious side effects, such as low blood pressure and respiratory depression, and long-term use can lead to tolerance and dependency with a high potential for addiction and abuse [41]. In recent years, studies have been undertaken with the aim of discovering novel analgesics with few side effects that target hNav1.7 [10, 42–45]. To date, several hNav1.7 inhibitors have been shown to have an analgesic effect in animal pain models [44, 46–48]. In the current study, the analgesic potential of Ca1a was measured in rodent models of formalin-, acetic acid-, and heat-induced pain. HWTX-IV (IC50 26 nM) and HNTX-IV (IC50 21 nM) have high affinity for hNav1.7 and showed similar analgesic efficiency in inflammatory pain and in acetic acid-induced pain. Similar to HNTX-IV and HWTX-IV, Ca1a (IC50 378 nM) exhibited analgesic effects in a dose-dependent manner against formalin-induced inflammatory pain. Ca1a at 100 µg/kg showed an analgesic efficiency similar to that of 100 µg/kg morphine against formalin-induced inflammatory pain, and Ca1a was also effective in reducing the abdominal writhing induced by i.p. injection of acetic acid. In this model, Ca1a at 200 µg/kg showed analgesia comparable to that of 100 µg/kg morphine. The robust analgesia that our data demonstrate against inflammatory pain indicates that Ca1a is an excellent candidate for drug development. Moreover, Ca1a increased the thermal stimulus threshold in the hot plate test, demonstrating that Ca1a can improve tolerance of harmful nociception. Notably, HWTX-IV and HNTX-IV were not tested against Nav1.6, but a triple-mutant of HWTX-IV (E1G, E4G, Y33W, m3-HWTX-IV) was, and it showed high affinity comparable to that of Nav1.7 [26, 49]. Therefore, Ca1a-induced analgesia likely depends on its activity against the Nav1.6 and Nav1.7 channels, and thus it could be possible to engineer peptides with increased affinity and isoform-specific selectivity [50–52]. Taken together, these data indicate that Ca1a is a promising candidate molecule for future clinical development as a novel therapeutic for the treatment of pain.
Acknowledgements
This work was supported by National Natural Science Foundation of China (81573320 and 31670783).
Author contributions
YXZ, MQR, and ZHL conceived and designed the experiments; YXZ, DZP, QFZ, BH, QCY, DFT, and MZC performed the experiments; YXZ, DZP, QFZ, and MQR analyzed the data; MQR and ZHL contributed reagents/materials/analysis tools. All the authors have read and approved the manuscript.
Competing interests
The authors declared no conflict of interests.
Contributor Information
Ming-qiang Rong, Phone: +86 731 8887 2556, Email: rongmq@hunnu.edu.cn.
Zhong-hua Liu, Email: liuzh@hunnu.edu.cn.
References
- 1.Goldberg DS, McGee SJ. Pain as a global public health priority. BMC Public Health. 2011;11:770. doi: 10.1186/1471-2458-11-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Institute of Medicine (US) Committee on Advancing Pain Research, Care and Education. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC (US): National Academies Press; 2011. [PubMed]
- 3.Holmes D. The pain drain. Nature. 2016;535:S2–3. doi: 10.1038/535S2a. [DOI] [PubMed] [Google Scholar]
- 4.Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14:162–73. doi: 10.1016/S1474-4422(14)70251-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/S0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 6.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411–25. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 7.Ogata N, Ohishi Y. Molecular diversity of structure and function of the voltage-gated Na+channels. Jpn J Pharmacol. 2002;88:365–77. doi: 10.1254/jjp.88.365. [DOI] [PubMed] [Google Scholar]
- 8.French RJ, Terlau H. Sodium channel toxins--receptor targeting and therapeutic potential. Curr Med Chem. 2004;11:3053–64. doi: 10.2174/0929867043363866. [DOI] [PubMed] [Google Scholar]
- 9.Osteen JD, Herzig V, Gilchrist J, Emrick JJ, Zhang C, Wang X, et al. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature. 2016;534:494–9. doi: 10.1038/nature17976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardoso FC, Dekan Z, Smith JJ, Deuis JR, Vetter I, Herzig V, et al. Modulatory features of the novel spider toxin mu-TRTX-Df1a isolated from the venom of the spider Davus fasciatus. Br J Pharmacol. 2017;174:2528–44. doi: 10.1111/bph.13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12:996–1002. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- 12.Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science. 2010;328:906–9. doi: 10.1126/science.1187958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, Yu FH, Sharp EM, Beacham D, Scheuer T, Catterall WA. Functional properties and differential neuromodulation of Na(v)1.6 channels. Mol Cell Neurosci. 2008;38:607–15. doi: 10.1016/j.mcn.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deuis JR, Zimmermann K, Romanovsky AA, Possani LD, Cabot PJ, Lewis RJ, et al. An animal model of oxaliplatin-induced cold allodynia reveals a crucial role for Nav1.6 in peripheral pain pathways. Pain. 2013;154:1749–57. doi: 10.1016/j.pain.2013.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie W, Strong JA, Ye L, Mao JX, Zhang JM. Knockdown of sodium channel NaV1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain. 2013;154:1170–80. doi: 10.1016/j.pain.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie W, Strong JA, Zhang JM. Local knockdown of the NaV1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience. 2015;291:317–30. doi: 10.1016/j.neuroscience.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmad S, Dahllund L, Eriksson AB, Hellgren D, Karlsson U, Lund PE, et al. A stop codon mutation in SCN9A causes lack of pain sensation. Hum Mol Genet. 2007;16:2114–21. doi: 10.1093/hmg/ddm160. [DOI] [PubMed] [Google Scholar]
- 18.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–8. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldberg YP, MacFarlane J, MacDonald ML, Thompson J, Dube MP, Mattice M, et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet. 2007;71:311–9. doi: 10.1111/j.1399-0004.2007.00790.x. [DOI] [PubMed] [Google Scholar]
- 20.McDonnell A, Schulman B, Ali Z, Dib-Hajj SD, Brock F, Cobain S, et al. Inherited erythromelalgia due to mutations in SCN9A: natural history, clinical phenotype and somatosensory profile. Brain. 2016;139:1052–65. doi: 10.1093/brain/aww007. [DOI] [PubMed] [Google Scholar]
- 21.Han C, Hoeijmakers JG, Ahn HS, Zhao P, Shah P, Lauria G, et al. Nav1.7-related small fiber neuropathy: impaired slow-inactivation and DRG neuron hyperexcitability. Neurology. 2012;78:1635–43. doi: 10.1212/WNL.0b013e3182574f12. [DOI] [PubMed] [Google Scholar]
- 22.Jarecki BW, Sheets PL, Jackson JO, 2nd, Cummins TR. Paroxysmal extreme pain disorder mutations within the D3/S4-S5 linker of Nav1.7 cause moderate destabilization of fast inactivation. J Physiol. 2008;586:4137–53. doi: 10.1113/jphysiol.2008.154906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klint JK, Senff S, Rupasinghe DB, Er SY, Herzig V, Nicholson GM, et al. Spider-venom peptides that target voltage-gated sodium channels: pharmacological tools and potential therapeutic leads. Toxicon. 2012;60:478–91. doi: 10.1016/j.toxicon.2012.04.337. [DOI] [PubMed] [Google Scholar]
- 24.King GF. The wonderful world of spiders: preface to the special Toxicon issue on spider venoms. Toxicon. 2004;43:471–5. doi: 10.1016/j.toxicon.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 25.King GF. Venoms as a platform for human drugs: translating toxins into therapeutics. Expert Opin Biol Ther. 2011;11:1469–84. doi: 10.1517/14712598.2011.621940. [DOI] [PubMed] [Google Scholar]
- 26.Xiao Y, Bingham JP, Zhu W, Moczydlowski E, Liang S, Cummins TR. Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain II voltage sensor in the closed configuration. J Biol Chem. 2008;283:27300–13. doi: 10.1074/jbc.M708447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z, Cai T, Zhu Q, Deng M, Li J, Zhou X, et al. Structure and function of hainantoxin-III, a selective antagonist of neuronal tetrodotoxin-sensitive voltage-gated sodium channels isolated from the Chinese bird spider Ornithoctonus hainana. J Biol Chem. 2013;288:20392–403. doi: 10.1074/jbc.M112.426627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai T, Luo J, Meng E, Ding J, Liang S, Wang S, et al. Mapping the interaction site for the tarantula toxin hainantoxin-IV (beta-TRTX-Hn2a) in the voltage sensor module of domain II of voltage-gated sodium channels. Peptides. 2015;68:148–56. doi: 10.1016/j.peptides.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Shu Q, Liang SP. Purification and characterization of huwentoxin-II, a neurotoxic peptide from the venom of the Chinese bird spider Selenocosmia huwena. J Pept Res. 1999;53:486–91. doi: 10.1034/j.1399-3011.1999.00039.x. [DOI] [PubMed] [Google Scholar]
- 30.Diao J, Lin Y, Tang J, Liang S. cDNA sequence analysis of seven peptide toxins from the spider Selenocosmia huwena. Toxicon. 2003;42:715–23. doi: 10.1016/j.toxicon.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Zhou X, Xiao Z, Xu Y, Zhang Y, Tang D, Wu X, et al. Electrophysiological and pharmacological analyses of Nav1.9 voltage-gated sodium channel by establishing a heterologous expression system. Front Pharmacol. 2017;8:852. doi: 10.3389/fphar.2017.00852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Y, Wu X, Zhang P, Duan Z, Zhou X, Chen M, et al. Peptide-rich venom from the spider Heteropoda venatoria potently inhibits insect voltage-gated sodium channels. Toxicon. 2017;125:44–9. doi: 10.1016/j.toxicon.2016.11.252. [DOI] [PubMed] [Google Scholar]
- 33.Wang M, Rong M, Xiao Y, Liang S. The effects of huwentoxin-I on the voltage-gated sodium channels of rat hippocampal and cockroach dorsal unpaired median neurons. Peptides. 2012;34:19–25. doi: 10.1016/j.peptides.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 34.Meng D, Wang L, Du J, Chen J, Chen C, Xu W, et al. The analgesic activities of Stauntonia brachyanthera and YM11 through regulating inflammatory mediators and directly controlling the sodium channel prompt. Sci Rep. 2017;7:7574. doi: 10.1038/s41598-017-07095-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Owoyele VB, Adediji JO, Soladoye AO. Anti-inflammatory activity of aqueous leaf extract of Chromolaena odorata. Inflammopharmacology. 2005;13:479–84. doi: 10.1163/156856005774649386. [DOI] [PubMed] [Google Scholar]
- 36.Zhu Y, Li Z, Liu H, He X, Zhang Y, Jin J, et al. Novel analgesic peptides from the tree frog of Hyla japonica. Biochimie. 2014;99:38–43. doi: 10.1016/j.biochi.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 37.King GF, Gentz MC, Escoubas P, Nicholson GM. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon. 2008;52:264–76. doi: 10.1016/j.toxicon.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 38.Pan JY, Yu ZQ. Isolation and characterization of Hainantoxin-II, a new neurotoxic peptide from the Chinese bird spider (Haplopelma hainanum) Dongwuxue Yanjiu. 2010;31:570–4. doi: 10.3724/SP.J.1141.2010.06570. [DOI] [PubMed] [Google Scholar]
- 39.Hong Y, Abbott FV. Peripheral opioid modulation of pain and inflammation in the formalin test. Eur J Pharmacol. 1995;277:21–8. doi: 10.1016/0014-2999(95)00045-M. [DOI] [PubMed] [Google Scholar]
- 40.Abbott FV, Franklin KB, Westbrook RF. The formalin test: scoring properties of the first and second phases of the pain response in rats. Pain. 1995;60:91–102. doi: 10.1016/0304-3959(94)00095-V. [DOI] [PubMed] [Google Scholar]
- 41.Beard EL., Jr The American Society of Health System Pharmacists. JONA’S Healthc law, Ethics Regul. 2001;3:78–9. doi: 10.1097/00128488-200109000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Deuis JR, Dekan Z, Wingerd JS, Smith JJ, Munasinghe NR, Bhola RF, et al. Pharmacological characterisation of the highly NaV1.7 selective spider venom peptide Pn3a. Sci Rep. 2017;7:40883. doi: 10.1038/srep40883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahuja S, Mukund S, Deng L, Khakh K, Chang E, Ho H, et al. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science. 2015;350:aac5464. doi: 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- 44.Yang S, Xiao Y, Kang D, Liu J, Li Y, Undheim EA, et al. Discovery of a selective NaV1.7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models. Proc Natl Acad Sci USA. 2013;110:17534–9. doi: 10.1073/pnas.1306285110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kornecook TJ, Yin R, Altmann S, Be X, Berry V, Ilch CP, et al. Pharmacologic characterization of AMG8379, a potent and selective small molecule sulfonamide antagonist of the voltage-gated sodium channel NaV1.7. J Pharmacol Exp Ther. 2017;362:146–60. doi: 10.1124/jpet.116.239590. [DOI] [PubMed] [Google Scholar]
- 46.London C, Hoyt SB, Parsons WH, Williams BS, Warren VA, Tschirret-Guth R, et al. Imidazopyridines: a novel class of hNav1.7 channel blockers. Bioorg Med Chem Lett. 2008;18:1696–701. doi: 10.1016/j.bmcl.2008.01.047. [DOI] [PubMed] [Google Scholar]
- 47.Bregman H, Berry L, Buchanan JL, Chen A, Du B, Feric E, et al. Identification of a potent, state-dependent inhibitor of Nav1.7 with oral efficacy in the formalin model of persistent pain. J Med Chem. 2011;54:4427–45. doi: 10.1021/jm200018k. [DOI] [PubMed] [Google Scholar]
- 48.Chowdhury S, Chafeev M, Liu S, Sun J, Raina V, Chui R, et al. Discovery of XEN907, a spirooxindole blocker of NaV1.7 for the treatment of pain. Bioorg Med Chem Lett. 2011;21:3676–81. doi: 10.1016/j.bmcl.2011.04.088. [DOI] [PubMed] [Google Scholar]
- 49.Revell JD, Lund PE, Linley JE, Metcalfe J, Burmeister N, Sridharan S, et al. Potency optimization of Huwentoxin-IV on hNav1.7: a neurotoxin TTX-S sodium-channel antagonist from the venom of the Chinese bird-eating spider Selenocosmia huwena. Peptides. 2013;44:40–6. doi: 10.1016/j.peptides.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 50.Murray JK, Ligutti J, Liu D, Zou A, Poppe L, Li H, et al. Engineering potent and selective analogues of GpTx-1, a tarantula venom peptide antagonist of the Na(V)1.7 sodium channel. J Med Chem. 2015;58:2299–314. doi: 10.1021/jm501765v. [DOI] [PubMed] [Google Scholar]
- 51.Shcherbatko A, Rossi A, Foletti D, Zhu G, Bogin O, Galindo Casas M, et al. Engineering highly potent and selective microproteins against Nav1.7 sodium channel for treatment of pain. J Biol Chem. 2016;291:13974–86. doi: 10.1074/jbc.M116.725978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moyer BD, Murray JK, Ligutti J, Andrews K, Favreau P, Jordan JB, et al. Pharmacological characterization of potent and selective NaV1.7 inhibitors engineered from Chilobrachys jingzhao tarantula venom peptide JzTx-V. PLoS ONE. 2018;13:e0196791. doi: 10.1371/journal.pone.0196791. [DOI] [PMC free article] [PubMed] [Google Scholar]