

Abstract
Arthrobacter globiformis Uricase (AgUricase) is a homotetrameric uricase with the potential for therapeutic use in treating hyperuricemia-related diseases. To achieve sufficient therapeutic effects, it is essential for this enzyme to have high thermostability and long half-life in physiological condition. To improve the thermostability of this enzyme, we introduced a series of cysteine pair mutations into the AgUricase subunits based on its structural model and studied the thermostability of the mutant enzymes with introduced disulfide bridges. Two intersubunit cysteine pair mutations, K12C–E286C and S296C–S296C, were found to markedly increase the melting temperatures of the corresponding mutant enzymes compared with WT AgUricase. The crystal structure of the K12C–E286C mutant at 1.99 Å resolution confirmed the formation of a distinct disulfide bond between the two subunits in the dimer. Structural analysis and biochemical data revealed that the C-terminal loop of AgUricase was flexible, and its interaction with neighboring subunits was required for the stability of the enzyme. We introduced an additional intersubunit K244C–C302 disulfide bond based on the crystal structure of the K12C–E286C mutant and confirmed that this additional disulfide bond further stabilized the flexible C-terminal loop and improved the thermostability of the enzyme. Disulfide cross-linking also protected AgUricase from protease digestion. Our studies suggest that the introduction of disulfide bonds into proteins is a potential strategy for enhancing the thermostability of multimeric proteins for medical applications.
Keywords: uricase, thermostability, disulfide cross-linking, crystal structure
Introduction
Uricase or urate oxidase (EC 1.7.3.3) catalyzes the oxidation of uric acid to 5-hydroxyisourate and hydrogen peroxide (H2O2) [1]. Subsequent hydrolysis and decarboxylation lead to the formation of allantoin, which is easily excreted by the kidney in most prokaryotic and eukaryotic organisms [1]. However, all five genera of hominoids (human, chimpanzee, gorilla, orangutan, and gibbon) have lost uricase activity during hominoid evolution [2]. Thus, elevated levels of uric acid in the blood (hyperuricemia) can result in urate crystal deposition, which is involved in gout, chronic kidney disease, tumor lysis syndrome (TLS), hypertension and various cardiovascular diseases [1, 3].
Arthrobacter globiformis Uricase (AgUricase) is a homotetrameric enzyme composed of four identical 34 kDa subunits with high expression levels in Escherichia coli and excellent activity at neutral pH; thus, it has the potential for therapeutic applications [4]. To achieve sufficient therapeutic efficacy, it is essential for this enzyme to have high thermostability and a long half-life in physiological conditions [5]. It was reported that the introduction of disulfide bonds into Bacillus sp. TB-90 uricase (Bt Uricase) via the R298C mutation increased the transition temperature by 15 °C, at which the enzyme retained 50% of its initial activity [6]. Compared with single-residue substitution, the rational design of cysteine mutations in adjacent subunits using a structural model is a more efficient method to generate disulfide bonds to stabilize proteins [7–9]. The wild-type (WT) AgUricase crystal structure has been solved (PDB entry 2YZB) and shows four identical chains that enclose a closed tunnel, and each chain consists of two tandem tunneling-fold motifs. In this study, we introduced a series of disulfide bonds between AgUricase subunits based on the WT crystal structure and identified intersubunit disulfide bonds formed in AgUricase mutants with mutations K12C–E286C and K244C using SDS–PAGE and thermal shift assays (TSA). We solved the crystal structure of the K12C–E286C mutant, which confirmed that two disulfide bonds formed between the cysteine pair K12C–E286C at the interface of two subunits within a dimer. Furthermore, our data suggested that these cysteine pair mutants retained full WT enzyme activity and also had significantly improved thermostability and heat resistance, thus providing promising therapeutic options for the treatment of hyperuricemia-related diseases.
Materials and methods
Materials
Uric acid, ampicillin, isopropyl β-d-l-thiogalactopyranoside (IPTG), and all media supplements were purchased from Sigma-Aldrich (St. Louis, MO). All of the other chemicals were local products of analytical grade.
Molecular cloning and site-directed mutagenesis
The complementary DNA (cDNA) of AgUricase (UniProtKB entry sequence D0VWQ1) was synthesized by Synbio Tech. The cDNA of AgUricase was cloned into the pET21 vector. An 8X His tag was introduced into the N terminus of AgUricase for affinity purification. The site-specific mutagenesis of AgUricase was conducted by overlap extension using polymerase chain reaction [10]. The primer sequences are listed in Supplementary Table S1. The mutations were verified by DNA sequencing.
Protein expression and purification
The pET21–8X His-AgUricase plasmid was transformed into BL21 (DE3) cells. The cells were grown in Lysogeny broth medium with appropriate antibiotics in a flask at 37 °C with shaking at 225 r/min, and protein expression was induced at an OD600 = 1.0 with the addition of 0.1 mM IPTG at 16 °C overnight. The cells were collected by centrifugation at 4000 r/min at 4 °C for 20 min. The cell pellets were resuspended in 3 mL lysis buffer (Buffer A: 50 mM Tris, pH 8.0, 400 mM NaCl, 25 mM imidazole and phenylmethylsulfonyl fluoride) and lysed by sonication. After ultracentrifugation, the supernatant was loaded into a gravity column containing 5 mL nickel resin (GE Health) pre-equilibrated with lysis buffer. After binding at 4 °C for 2 h, the column was washed with 10 column volume (CV) lysis buffer. The target protein was eluted with 5 CV elution buffer (50 mM Tris, pH 8.0, 400 mM NaCl, and 500 mM imidazole). The protein was concentrated to ~5 mg/mL and further purified with a Superdex 200 Hiload 26/60 gel filtration column (GE Healthcare) equilibrated with SEC buffer (20 mM HEPES, pH 7.5, 100 mM NaCl). The AgUricase fractions were pooled and concentrated for the subsequent experiments. The concentration of AgUricase was quantified using a Qubit protein assay kit and a Qubit 2.0 fluorometer (Invitrogen).
Thermal shift assay
The thermostability of the uricases was evaluated by a TSA as described previously [11]. In brief, 1000 × SYPRO Orange (Sigma-Aldrich, USA) was diluted with phosphate-buffered saline and mixed with the protein. All reactions were performed in triplicate in 384-well plates with a final volume of 10 µL per well. The thermal melting curve was determined using a LightCycler 480 II Real-Time PCR System (Roche Diagnostics, Rotkreuz, Switzerland) with a ramp rate of 1 °C and a temperature range from 30 °C to 100 °C. The melting temperatures (Tm) were calculated by fitting the sigmoidal melting curve to the Boltzmann equation using GraphPad Prism 7.0 with R2 values > 0.99.
Uricase activity assay
Uricase activity was assayed by monitoring the depletion of uric acid, which was detected by a decrease in absorbance at 292 nm in the presence of the enzyme [4, 6]. The uricase was diluted to 1 μg/mL and combined with different concentrations of UA in 0.1 M phosphate buffer (PB), pH 7.4. Absorbance measurements at 292 nm were recorded every 30 s for a period of 10 min. The extinction coefficient for uric acid was assumed to be 1.22 × 104 M−1· cm−1. The rate of uric acid degradation (μmol/min) was calculated. The enzyme kinetics parameters were calculated by fitting the data to the Michaelis–Menten equation using the GraphPad Prism 7.0 program.
Heat resistance assay
The AgUricases were incubated for 30 min at various temperatures, and then the temperature was reduced to 25 °C, after which the residual activity was assayed using the methods described above. The data were fit according to a two-state mechanism using the following equations to determine the temperature at which the enzyme retained half of its activity (Tm) and the van’t Hoff enthalpy (ΔH) value: [12]
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
where Y is the experimentally measured enzymatic activity at temperature T, YN, and YU are the enzymatic activity of AgUricase in the native state and the unfolded state, respectively, fN and fU are the fractions of protein in the native state and the unfolded state, respectively, K is the apparent equilibrium constant, ΔG is the free energy change, Tm is the temperature at which the enzyme retains half of its activity, ΔH is the enthalpy change, R is the gas constant and T is the absolute temperature [12]. The software GraphPad Prism 7.0 was used for the data fitting.
Proteinase K resistance assay
AgUricases (80 μg) were treated with 20 μg Proteinase K at room temperature, and the residual activity of AgUricase at various times was assayed using the methods described above.
Protein crystallization, structure determination, and refinement
The K12C–E286C AgUricase crystals were obtained with the sitting drop method in a buffer containing 12% (w/v) polyethylene glycol 3350 and 0.1 M sodium malonate (pH 5.0). The crystals were transferred to a reservoir solution with 20% glycerol, which served as a cryoprotectant, before being flash-frozen in liquid nitrogen. The diffraction data for the K12C–E286C AgUricase crystals were collected at beamline BL19U1 at the Shanghai Synchrotron Radiation Facility (Shanghai, China). The X-ray diffraction data were integrated and scaled using the HKL3000 program [13]. The structure of K12C–E286C AgUricase was initially solved by molecular replacement using Phenix.phaser [14] with the wild-type AgUricase structure (PDB entry: 2YZB) as the starting model. The model was manually adjusted in COOT [15] and refined with Phenix.refine [16] and CCP4 [17]. The data and refinement statistics are listed in Supplementary Table S3.
Results
Cysteine mutations K12C–E286C and S296C introduced disulfide bonds between the AgUricase subunits
AgUricase is a homotetramer composed of four identical subunits. High thermostability of this enzyme is a highly desirable property that is correlated with its tolerance to heat denaturation, which involves a two-step process consisting of the dissociation of the tetramer into dimers and the dissociation of the dimers into monomers, followed by the unfolding of the monomers [18–21]. Based on the crystal structure, we designed a series of cysteine mutations to introduce cysteine pairs at the dimer interface that would form disulfide bonds that would crosslink the subunits to prevent the disassociation and unfolding of the multimeric enzyme (Fig. 1 and Supplementary Table S2). The formation of intersubunit disulfide bonds was verified by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE)SDS–PAGE with and without the reducing reagent dithiothreitol (DTT) [21] (Fig. 2a and Supplementary Fig. S1). Wild-type (WT) AgUricase showed a single band at 35 kDa in both the presence and absence of DTT, which corresponded to the AgUricase monomer (Fig. 2a and Supplementary Fig. S1). Many of the cysteine pair mutants showed a weak band at 70 kDa during SDS–PAGE analysis that was similar to that of WT AgUricase, indicating that the cysteine pairs did not form proper disulfide bonds between the two subunits (Supplementary Fig. S1). However, the F125C/L156C and K29/E267C mutants did not show any distinct band corresponding to this protein on the SDS–PAGE gel, suggesting that there was no soluble expression of these mutants (Supplementary Fig. S1). Interestingly, K12C–E286C and S296C–S296C showed dimer bands at 70 kDa in the absence of DTT, suggesting that proper disulfide bonds between the AgUricase subunits were successfully introduced through the K12C–E286C and S296C mutations (Fig. 2a). WT AgUricase also showed a weak dimer band at 70 kDa in the absence of DTT, which is probably owing to intersubunit cross-linking via the C-terminal residue C302 since the C termini of the subunits in an AgUricase dimer are close to each other, as shown in the crystal structure. To avoid the nonspecific cross-linking of C302 with S296C, we introduced the additional mutation C302S into the S296C mutant. Thus, the S296C mutant in this study actually contained two mutations (S296C/C302S).
Fig. 1.

Introduction of disulfide bonds to stabilize AgUricase based on its crystal structure. a The introduction of disulfide bonds based on the structural model of WT AgUricase (PDB code 2YZB). Chain A, green; chain B, cyan; chain C, magenta; chain D, orange. b Cartoon model showing the disulfide bond design. Monomer–monomer, dimer–dimer, and intra-subunit disulfide bonds were generated by introducing cysteine pair mutations based on the crystal structure of WT AgUricase
Fig. 2.

Cysteine mutations K12C–E286C and S296C introduced disulfide bonds between the AgUricase subunits and significantly improved the thermostability of AgUricase. a K12C–E286C and S296C–S296C AgUricase mutants formed disulfide bonds between subunits. Disulfide bridge formation was verified by SDS–PAGE with and without DTT. b Thermal shift assays (TSA) of AgUricases. The thermal stabilities of wild-type (blue), K12C–E286C (red), and S296C–C302S (green) mutant AgUricase were evaluated by TSA using the method described in the methods section
Intersubunit disulfide bonds improved the thermostability of AgUricase
A TSA experiment was conducted to further investigate the effect of the introduced intersubunit disulfide bonds on the thermostability of AgUricase. The TSA curves of WT AgUricase showed two distinct transition peaks. Double-peaked transitions are often interpreted as indicating the existence of multiple structural domains or subunits within a molecule that unfold independently [22]. The first peak corresponds to the disassociation of the tetramer into dimers, and the second peak represents the disassociation of the dimer into monomers [4, 22]. The K12C–E286C mutations delayed the first transition, whereas the S296C–S296C mutations sped it up, suggesting that K12C–E286C prevented tetramer disassociation, whereas S296C did not (Fig. 2b). This observation is consistent with the results showing that the K12C–E286C mutant formed both monomer–monomer and dimer–dimer disulfide cross-links, whereas S296C formed only monomer–monomer disulfide cross-links. As shown in Fig. 2b, the second peak of the transition of the K12C–E286C and S296C–S296C mutants shifted to 83.3 °C and 80 °C, respectively, compared with that of the WT enzyme, which occurred at 72 °C, indicating that both the K12C–E286C and S296C mutations strengthened the interaction between the two subunits within the dimers and delayed the dissociation of the dimer in the enzyme into monomers. Other cysteine pair mutations we introduced did not improve the thermostability of the enzyme based on TSA analysis, except H123C–S158C, for which the TSA profile showed that the second peak was shifted from 72 °C to 74.5 °C, whereas the first peak disappeared, indicating that the H123C/S158C mutations slightly strengthened the monomer–monomer interaction but weakened the dimer–dimer interaction (Supplementary Fig. S2). Therefore, we focused our study on the K12C–E286C and S296C–S296C mutations.
K12C–E286C and S296C–S296C mutations significantly improved the heat resistance of AgUricase
We tested the effect of disulfide cross-linking on the heat resistance of AgUricase. The activities of WT and mutant AgUricase were assayed at various temperatures, and the residual activity data were fit to the equations (1–6) [23]. The Tm value is used to define the temperature at which half of the total enzyme is denatured [24]. The K12C–E286C and S296C–S296C mutants showed markedly increased heat resistance (Fig. 3). The Tm values of the K12C–E286C and S296C–S296C mutants were 14 °C and 9 °C higher, respectively, than those of the wild-type enzyme (61 °C vs 75 °C and 61 °C vs 70 °C) (Fig. 3). These increases in thermal resistance were likely due to the formation of intersubunit disulfide bridges [25–27].
Fig. 3.
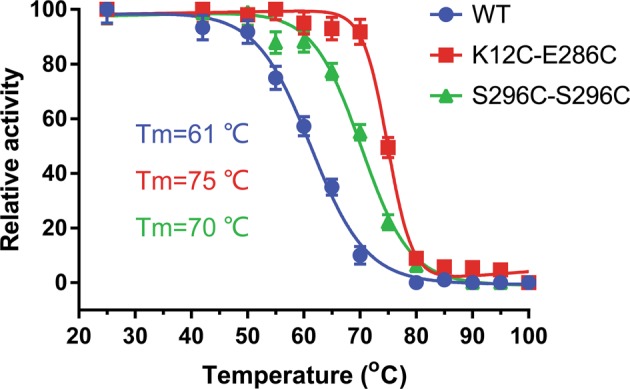
K12C–E286C and S296C–S296C mutations significantly improved the heat resistance of AgUricase. The relative activity of wild-type (circle), K12C–E286C (square), and S296C–C302S (triangle) mutant AgUricase were measured after 30 min of incubation at various temperatures. The temperatures at which the enzymes retained half of their activity (Tm) were determined by fitting the curve to the equations (1–6) using GraphPad Prism 7 with R2 values > 0.99. The data are presented as the mean ± standard error of the means (SEM) of at least three independent experiments
Disulfide cross-linking did not affect the catalytic efficiency of AgUricase
We tested the effect of disulfide cross-linking on the enzyme kinetics of AgUricase. The K12C–E286C mutant showed a kcat of 5.8 ± 0.4 s−1 and a Km value of 104.5 ± 10.2 μM, and the S296C/C302S mutant exhibited a kcat of 5.89 ± 0.3 s−1 and a Km value of 126.8 ± 17 μM; the kcat/Km values were (5.55 ± 0.3) × 104 s−1· M−1 and (4.64 ± 0.2) × 104 s−1· M−1, respectively. All these kinetic parameters of the mutants were close to those of the wild-type enzyme (kcat = 6.64 ± 0.3 s−1, Km = 140.3 ± 18.3 μM, and kcat/Km = (4.73 ± 0.2) × 104 s−1· M−1) (Fig. 4). These results suggested that disulfide cross-linking did not greatly affect the catalytic efficiency of AgUricase.
Fig. 4.
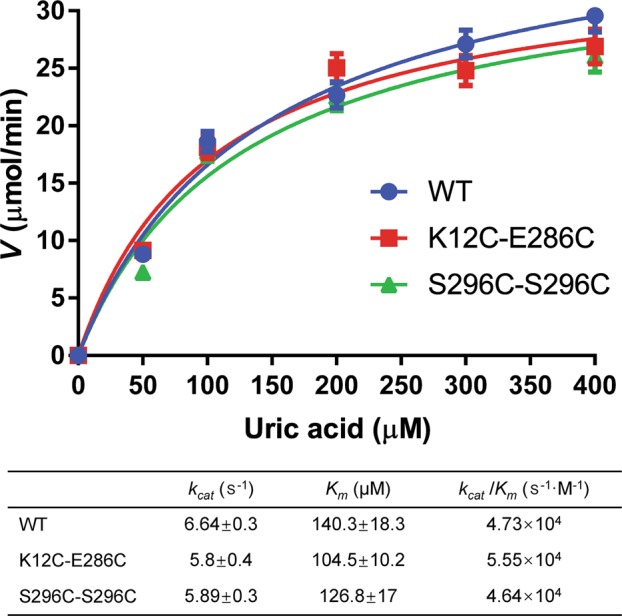
Enzyme kinetics of AgUricases. The activities of wild-type (circle), K12C–E286C (square), and S296C–C302S (triangle) mutant AgUricase were measured by substrate (UA) depletion assays. The rate (specific activity) of UA oxidation was calculated based on the linear decrease in absorbance at 292 nm. The data were fit to the Michaelis–Menten equation by GraphPad Prism 7 to determine the kcat and Km values. The data are presented as the mean ± standard error of the mean (SEM) of at least three independent experiments
Disulfide cross-linking hindered the disassociation of tetramer and dimer
To confirm the formation of disulfide bond cross-linking and further investigate its effects on the thermostability of AgUricase, we solved the crystal structure of the K12C–E286C mutant at 1.99 Å resolution (Fig. 5, PDB entry 6OE8). The crystal structure of K12C–E286C clearly showed four disulfide bonds between C12 and C286 in neighboring subunits (Fig. 5a). K12C in subunit A formed a disulfide bond with E286C in subunit D. K12C in subunit D also formed a disulfide bond with E286C in subunit A. Similar disulfide bonds were formed between the introduced cysteine residues in subunit B and subunit C. The Cα–Cα distance between residue 12 and residue 286 remained 5.4 Å, which is the same as that found in the wild-type enzyme. The parameters of these disulfide bonds are listed in Supplementary Table S4.
Fig. 5.

Structure of the K12C–E286C AgUricase mutant. The structure of the K12C–E286C AgUricase mutant was generated with the PyMOL program (PDB entry: 6OE8). a Side and top views of the schematic model of the AgUricase tetramer: chain A (TV green), chain B (cyan), chain C (purple), and chain D (light blue). Disulfide bonds are shown in yellow and indicated by black arrows. b Side and top views of the 2│Fo│−│Fc│density map of the disulfide bonds between Cys12 of subunit B and Cys286 of subunit C. Disulfide bonds are shown in yellow
To investigate the effect of disulfide bond formation on subunit assembly, the wild-type and K12C–E286C mutant enzymes were analyzed using the PISA program [6], and the results are shown in Table 1. ΔGdiss indicates the free energy of tetramer or dimer dissociation in kcal/mol. Positive values of ΔGdiss indicate that an external driving force must be applied to result in the dissociation of the assembly. The ΔGdiss values of the K12C–E286C-cross-linked dimers (A–D and B–C) increased from 33.4 and 31.5 kcal/mol to 59.1 and 49.8 kcal/mol, respectively, compared with those of the relevant WT dimers, indicating that both the A–D and B–C dimers in the K12C/E286C mutant enzyme are more difficult to dissociate than those in the wild-type enzyme.
Table 1.
Assembly analysis of AgUricases
| Subunits | ΔGdiss (kcal/mol) | ASA, Å2 | BSA, Å2 | |
|---|---|---|---|---|
| Wild-type | Tetramer | 77.6 | 39484 | 25175 |
| A–D dimer | 33.4 | 25324 | 7045 | |
| B–C dimer | 31.5 | 25114 | 7176 | |
| K12C–E286C | Tetramer | 77.7 | 38389 | 30069 |
| A–D dimer | 59.1 | 25869 | 8126 | |
| B–C dimer | 49.8 | 25988 | 8475 |
ASA accessible surface area, BSA buried surface area, ΔGdiss free energy of disassociation
We also analyzed the buried surface area of the wild-type and K12C–E286C AgUricase with the PISA program [6] (Table 1). The solvent-accessible areas of the WT and K12C–E286C AgUricases were 39484 Å2 and 38389 Å2, respectively, which were calculated based on the crystal structures. Disulfide cross-linking decreases the solvent-accessible area by 2.8%, which may contribute to the improved stability of the mutant enzyme. The total buried interface areas for the wild-type and K12C–E286C enzymes were 25175 and 30069 Å2, respectively. The buried surface areas of the monomer–monomer interfaces of the wild-type and K12C–E286C AgUricases were 7045 Å2 (subunit A–D, WT), 7176 Å2, (subunit B–C, WT), 8126 Å2 (subunit A–D, K12C–E286C mutant), and 8475 Å2 (subunit B–C, K12C–E286C mutant). The K12C–E286C mutant showed an increase in the total buried interface area of 19% and in the buried surface areas of subunit A–D and subunit B–C of 15% and 18%, respectively. The increases in the buried interface areas contributed to the stabilization of the subunit assembly, leading to higher thermostability.
The overall main-chain trace of the K12C–E286C mutant was in good agreement with that of the wild-type enzyme (PDB entry 2YZB) (Supplementary Fig. S3). However, the residue ranges 11–17, 258–263, and 281–297 in the mutant structure showed different conformations compared to those in the WT enzyme owing to the formation of the disulfide bonds that stabilized these flexible loop regions. The dynamic movement of these flexible loop regions may affect the thermostability of AgUricase. Disulfide cross-linking between these flexible loop regions can fix their conformations, thereby enhancing the stability of the loop regions and the whole protein. The K12C–E286C mutation did not affect the catalytic site of the enzyme; therefore, disulfide cross-linking had no effect on the enzymatic activity of AgUricase (Fig. 4).
The C-terminal loop affected the stability of AgUricase
Our structure showed a clear density map for the C-terminal residues 298 through 302 (Fig. 6a), which were missing in the structure of wild-type AgUricase (PDB: 2YZB) (Fig. 6b). We compared the mean B-factors of wild-type and K12C–E286C AgUricase. The mean B-factors of the residues in the C-terminal loop of wild-type AgUricase were relatively high, indicating that the C-terminal loop is highly flexible. The mean B-factors of the residues in the C-terminal loop of K12C–E286C AgUricase decreased dramatically, indicating that disulfide cross-linking between C12 and C286 stabilized the flexible C-terminal loop (Fig. 6c). To investigate the effect of this loop on the stability of AgUricase, we analyzed the thermostability of the full-length enzyme, the C-terminal-truncated WT enzyme and the K12C–E286C mutant enzyme. TSA data showed that the Tm1 values of the wild-type and K12C–E286C AgUricase decreased to 11.5 °C (50.6–39.1 °C) and 7.9 °C (52.3–44.4 °C), respectively. The Tm2 values of wild-type (72 °C) and K12C–E286C AgUricase (83.3 °C) did not change when the C-terminal residues 298–302 were removed (Fig. 6d). Our structure showed that the C-loops in subunits A and D and B and C were close; for example, the distance between S296 in chain A and S296 in chain D was 4.2 Å, and the distance between C302 in chain A and K244 in chain D was 6.7 Å. Therefore, the C-loops of subunits A and D interacted with each other and contributed to the interface of the A–D dimer and the stability of the intact AgUricase tetramer. The C-terminal truncation destroyed the C-loop interaction between the subunits, thus decreasing the stability of the intact AgUricase. These results suggest that the C-terminal residues 298–302 are required for the formation of a stable AgUricase tetramer and confirm that the K12C–E286C mutations increase the stability of the enzyme.
Fig. 6.

A C-terminal flexible loop is required for the stability of AgUricase protein. a The 2│Fo│−│Fc│density map of residues 298–302 in AgUricase. b Structural comparison of the C-terminal residues 286–302 in wild-type and K12C–E286 mutant AgUricase. Wild-type AgUricase is gray, and the K12C–E286C mutant AgUricase is light blue, and its C-terminal residues (286–302) are green and the disulfide bonds are yellow. c Mean B-factors of wild-type and K12C–E286C AgUricase. The mean B-factors of wild-type and K12C–E286C AgUricase were calculated according to the crystal structures of wild-type AgUricase (PDB ID 2YZB) and K12C–E286C AgUricase (PDB ID 6OE8). d Results of the thermal shift assay (TSA) of the △C5 deletion mutant of the K12C/E286C AgUricase compared with those of the K12C/E286C AgUricase
K244C forms disulfides with C302 and improves the thermostability of AgUricase
According to the solved K12C–E286C mutant structure, the Cα–Cα distances between K244 and S246 in one subunit and C302 in the other subunit in a dimer were 6.7 Å and 6.6 Å, respectively, which indicates that K244 and S246 have the potential to form intersubunit disulfide bonds with C302 if they are substituted with cysteine (Fig. 7a). SDS–PAGE analysis showed that the K244C mutation successfully introduced disulfide bonds into both wild-type AgUricase and the K12C–E286C mutant, whereas S246C did not (Fig. 7b). The TSA results showed that K244C increased the Tm1 value by 5.9 °C (50.6–56.5 °C) and the Tm2 value by 6.6 °C (72–78.6 °C) compared with WT AgUricase and increased the Tm1 value by 5.3 °C (52.3–57.6 °C) and the Tm2 value by 3.6 °C (83.3–86.9 °C) compared with the K12C–E286C AgUricase mutant (Fig. 7c). A heat resistance assay showed that the K244C mutation increased the Tm value of WT AgUricase by 4.6 °C (61–65.6 °C) and that of the K12C–E286C mutant by 3.5 °C (75–78.5 °C) (Fig. 7d). Our structure indicated that K244C–C302 cross-linking would fix the flexible C-loop in the AgUricase and hinder the disassociation of the dimer, thereby improving the stability of the enzyme. Our TSA and heat resistance data confirmed these hypotheses.
Fig. 7.

K244C forms disulfide bonds with C302 and improves the thermostability of AgUricase. a Cα–Cα distance between K244 and S246 in subunit A and C302 in subunit D. b SDS–PAGE results showing that mutation K244C strengthened dimer formation between the subunits in both the WT and K12C/E286C mutant AgUricases, whereas S246C did not. c TSA results showing that the K244C mutation shifted the Tm peaks of both the WT and K12C–E286C mutant AgUricases to higher temperatures. d The K244C mutation increased the thermostability of the WT and K12C–E286C mutant AgUricase
Disulfide cross-linking protects AgUricase from protease digestion
To test whether disulfide cross-linking protected AgUricase from protease digestion, we tested the activity of AgUricases treated with Proteinase K at various times. Wild-type AgUricase retained 59.4% activity after 5 min of Proteinase K treatment, 30.1% activity after 10 min and 0% activity after 15 min. The K12C–E286C mutant retained 78.3% activity after 5 min of Proteinase K treatment, 52.6% activity after 10 min and 6.6% activity after 15 min, whereas the K12C–E286C-K244C mutant retained 82.2% activity after 5 min of Proteinase K treatment, 54.7% activity after 10 min and 8.1% activity after 15 min (Fig. 8). These results suggested that disulfide cross-linking could protect AgUricase from protease digestion. Our structure analysis showed that K12C–E286C disulfide cross-linking fixed the flexible C-terminal loop and decreased the accessible surface area of intact AgUricase, which reduced the surface area exposed to Proteinase K, thus protecting AgUricase from protease digestion.
Fig. 8.
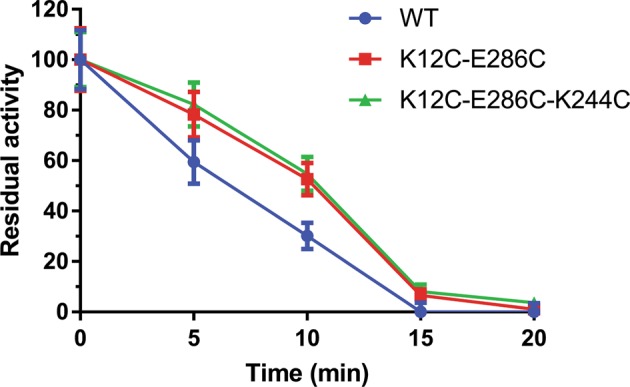
Disulfide cross-linking protects AgUricase from protease digestion. Wild-type (circle), K12C–E286C (square), and K12C–E286C-K244C (triangle) mutant AgUricase (80 μg) were treated with 20 μg Proteinase K at room temperature, and the residual activity of AgUricase was measured at various times. The data are presented as the mean ± standard error of the mean (SEM) of at least three independent experiments
Discussion
Thermostability is a key issue for protein drugs [5]. The disassociation of multimeric proteins into subunits is a key factor contributing to the thermostability of those proteins [28]. AgUricase is a tetramer composed of four identical subunits, and thermal unfolding of AgUricase involves multiple dissociation steps: the tetramer dissociates into dimers, the dimers dissociate into monomers, and finally the unfolding of the monomers occurs [6]. Introducing disulfide cross-linking has been proven to be a useful method to improve the thermostability and heat resistance of proteins [6, 25, 26, 29]. In this study, we introduced intersubunit cysteine pair mutations into AgUricase based on its WT crystal structure (Fig. 1). The formation of the K12C–E286C and S296C–S296C monomer–monomer disulfide bonds (Fig. 2a) prevented the dimer from dissociating, resulting in an increased unfolding temperature in the second step of dissociation (Fig. 2b). The formation of these intersubunit disulfide bonds significantly enhanced the thermostability and heat resistance of this multimeric enzyme (Figs. 2 and 3), whereas avoiding any observable reduction of the enzymatic activity (Fig. 4). These observations were further confirmed by the structure of the K12C–E286C mutant. Disulfide bonds covalently cross-linked subunits A and D and B and C (Fig. 5), which increased the ΔGdiss values and the buried surface areas of the dimers (A–D and B–C) (Table 1) and resulted in strong dimer interfaces that required more energy to dissociate the interactions between the subunits. Disulfide cross-linking also protected AgUricase from protease digestion through fixing the flexible loops and decreasing the surface area exposed to protease (Fig. 8).
Many proteins have variable loop regions, the flexibility of which may lead to low protein stability [30]. Our structure analysis showed that the C-terminal residues 298–302 in AgUricase form a loop region. This loop region is flexible and therefore disordered in the crystal structure of wild-type AgUricase, which has no intersubunit disulfide bonds (Fig. 6a, b). The crystal structure of the K12C–E286C AgUricase mutant shows that this loop region is involved in the intersubunit interface and is thus required for the stability of the enzyme (Fig. 6c, d). The introduction of a K244C–C302 disulfide bond, which was designed based on the crystal structure, improved the thermostability of the enzyme. The SDS–PAGE assay (Fig. 7b), TSA (Fig. 7c) and the heat resistance assay (Fig. 7d) confirmed the formation of the disulfide bond and the improvement of thermostability.
Our studies demonstrated that introducing disulfide bonds that cross-linked subunits stabilized the flexible loops and prevented the disassociation of the multisubunit protein, resulting in a mutant enzyme with significantly enhanced thermostability and heat resistance.
Over the years, a number of uricase preparations from a variety of sources have been developed for therapeutic use in the treatment of gout and hyperuricemia associated with tumor lysis in patients with malignant disease [1, 31]. To date, two recombinant uricase preparations, rasburicase [32, 33], and pegloticase [34–37], have been approved for the management of uric acid levels in plasma. Pegloticase, a PEGylated chimeric porcine–baboon uricase, has increased thermostability, an extended serum half-life and reduced immunogenicity [38], but it still induces an immune response, which results in a loss of activity and an increased risk of infusion reactions [39]. Many of the adverse side effects of protein drugs are associated with protein stability and immunogenicity, which are considered to be the major challenges for the development of therapeutic proteins [39, 40]. Immunogenicity is caused by various factors, including intrinsic causes, such as specific amino-acid sequences and posttranslational modifications, and extrinsic factors, including the route of administration, the dosage and, in particular, the patients’ genetic profile [39–43]. The instability of proteins is also regarded as one of the highest risk factors for the immunogenicity of therapeutic proteins [40]. Improving stability can reduce the immunogenicity of therapeutic proteins. The solvent-exposed flexible loops of AgUricase are sensitive to proteases such as Proteinase K under in vivo conditions, resulting in the loss of enzyme activity and immune reactions. In this study, we fixed the C-terminal flexible loop of AgUricase by disulfide cross-linking to generate a mutant enzyme with increased thermal thermostability and enhanced resistance to protease digestion (Fig. 8), which may reduce the in vivo immunogenicity of the enzyme. This engineered enzyme provides a potential therapeutic uricase, but further animal model studies and clinical trials will be needed before it is used as a pharmaceutical agent. The method reported in this paper provides a common strategy that could be used for the development of protein drugs with high stability and low immunogenicity.
In summary, we have obtained two highly stable AgUricase mutants with activity equivalent to that of the full-length WT enzyme by introducing intersubunit disulfide bonds. The disulfide cross-linking significantly increased the thermostability and heat resistance of AgUricase by preventing the disassociation of the multimeric enzyme assembly and stabilizing the flexible C-terminal loops in each subunit of the enzyme. These heat-resistant AgUricase mutants provide promising therapeutic options for the treatment of hyperuricemia-related diseases. The introduction of intersubunit disulfide bonds in multimeric protein enzymes has been shown to be a promising protein engineering strategy to increase protein stability for therapeutic uses.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31770796 to YJ); the Strategic Priority Research Program of CAS (XDB08020303 to HEX); the National Science and Technology Major Project (2018ZX09711002–002–002 to YJ); the K.C. Wong Education Foundation (to YJ).
Author contributions
HEX and YS designed the study. YS, TW, and QFL performed the experiments and analyzed the data. YS and XEZ solved the structure. YS and TW wrote the paper. HEX, XEZ, and YJ revised the paper. All authors reviewed the results and approved the final version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yi Shi, Ting Wang
Contributor Information
Yi Shi, Phone: +(86)21-20231000-5523, Email: shiyi@simm.ac.cn.
H. Eric Xu, Phone: +(86)21-20231000-5523, Email: eric.xu@simm.ac.cn.
Supplementary information
The online version of this article (10.1038/s41401-019-0269-x) contains supplementary material, which is available to authorized users.
References
- 1.Nuki G. Uricase therapy of gout. In Gout and Other Crystal Arthropathies. Elsevier Inc.; 2012. 174–86.
- 2.Kratzer JT, Lanaspa MA, Murphy MN, Cicerchi C, Graves CL, Tipton PA, et al. Evolutionary history and metabolic insights of ancient mammalian uricases. Proc Natl Acad Sci USA. 2014;111:3763–8. doi: 10.1073/pnas.1320393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abeles AM. Hyperuricemia, gout, and cardiovascular disease: an update. Curr Rheumatol Rep. 2015;17:13. doi: 10.1007/s11926-015-0495-2. [DOI] [PubMed] [Google Scholar]
- 4.Nyborg AC, Ward C, Zacco A, Chacko B, Grinberg L, Geoghegan JC, et al. A therapeutic uricase with reduced immunogenicity risk and improved development properties. PLoS ONE. 2016;11:e0167935. doi: 10.1371/journal.pone.0167935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiskoot W, Randolph TW, Volkin DB, Middaugh CR, Schoneich C, Winter G, et al. Protein instability and immunogenicity: roadblocks to clinical application of injectable protein delivery systems for sustained release. J Pharm Sci. 2012;101:946–54. doi: 10.1002/jps.23018. [DOI] [PubMed] [Google Scholar]
- 6.Hibi T, Kume A, Kawamura A, Itoh T, Fukada H, Nishiya Y. Hyperstabilization of tetrameric Bacillus sp. TB-90 urate oxidase by introducing disulfide bonds through structural plasticity. Biochemistry. 2016;55:724–32. doi: 10.1021/acs.biochem.5b01119. [DOI] [PubMed] [Google Scholar]
- 7.Eijsink VG, Bjork A, Gaseidnes S, Sirevag R, Synstad B, van den Burg B, et al. Rational engineering of enzyme stability. J Biotechnol. 2004;113:105–20. doi: 10.1016/j.jbiotec.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 8.Marshall SA, Lazar GA, Chirino AJ, Desjarlais JR. Rational design and engineering of therapeutic proteins. Drug Discov Today. 2003;8:212–21. doi: 10.1016/S1359-6446(03)02610-2. [DOI] [PubMed] [Google Scholar]
- 9.Yainoy S, Phuadraksa T, Wichit S, Sompoppokakul M, Songtawee N, Prachayasittikul V, et al. Production and characterization of recombinant wild type uricase from indonesian coelacanth (L. menadoensis) and improvement of its thermostability by in silico rational design and disulphide bridges engineering. Int J Mol Sci. 2019;20:E1269. doi: 10.3390/ijms20061269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.HHD HoSN, Horton RM, et al. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–9. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 11.Liu P, Jia MZ, Zhou XE, De Waal PW, Dickson BM, Liu B, et al. The structural basis of the dominant negative phenotype of the Galphai1beta1gamma2 G203A/A326S heterotrimer. Acta Pharm Sin. 2016;37:1259–72. doi: 10.1038/aps.2016.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi Y, Zheng D, Xie J, Zhang Q, Zhang H. Thermal stability of thermoanaerobacter tengcongensis ribosome recycling factor. Protein Pept Lett. 2014;21:285–91. doi: 10.2174/09298665113206660118. [DOI] [PubMed] [Google Scholar]
- 13.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 14.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 16.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–42. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pikal-Cleland KA, Rodríguez-Hornedo N, Amidon GL, Carpenter JF. Protein denaturation during freezing and thawing in phosphate buffer systems: monomeric and tetrameric β-galactosidase. Arch Biochem Biophys. 2000;384:398–406. doi: 10.1006/abbi.2000.2088. [DOI] [PubMed] [Google Scholar]
- 19.Mn González, Argaraña CE, Fidelio GD. Extremely high thermal stability of streptavidin and avidin upon biotin binding. Biomol Eng. 1999;16:67–72. doi: 10.1016/S1050-3862(99)00041-8. [DOI] [PubMed] [Google Scholar]
- 20.Kurganov BI, Kornilaev BA, Chebotareva NA, Malikov VP, Orlov VN, Lyubarev AE, et al. Dissociative mechanism of thermal denaturation of rabbit skeletal muscle glycogen phosphorylase b. Biochemistry. 2000;39:13144–52. doi: 10.1021/bi000975w. [DOI] [PubMed] [Google Scholar]
- 21.Reddy GB, Bharadwaj S, Surolia A. Thermal stability and mode of oligomerization of the tetrameric peanut agglutinin: a differential scanning calorimetry study. Biochemistry. 1999;38:4464–70. doi: 10.1021/bi982828s. [DOI] [PubMed] [Google Scholar]
- 22.Bayol A, Dupin P, Boe JF, Claudy P, Létoffé JM. Study of pH and temperature-induced transitions in urate oxidase (Uox-EC1.7.3.3) by microcalorimetry (DSC), size exclusion chromatography (SEC) and enzymatic activity experiments. Biophys Chem. 1995;54:229–35. doi: 10.1016/0301-4622(94)00150-I. [DOI] [PubMed] [Google Scholar]
- 23.Rastegari AA, Buzari B, Bordbar AK. Thermal stability of pepsin: a predictive thermodynamic model of a multi-domain protein. Biochem Biophys Rep. 2017;9:295–301. doi: 10.1016/j.bbrep.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muñoz V, Cronet P, López-Hernéndez E, Serrano L. Analysis of the effect of local interactions on protein stability. Fold Des. 1996;1:167–78. doi: 10.1016/S1359-0278(96)00029-6. [DOI] [PubMed] [Google Scholar]
- 25.Jeong M-Y, Kim S, Yun C-W, Choi Y-J, Cho S-G. Engineering a de novo internal disulfide bridge to improve the thermal stability of xylanase from Bacillus stearothermophilus No. 236. J Biotechnol. 2007;127:300–9. doi: 10.1016/j.jbiotec.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Nordlund HR, Laitinen OH, Uotila ST, Nyholm T, Hytonen VP, Slotte JP, et al. Enhancing the thermal stability of avidin. Introduction of disulfide bridges between subunit interfaces. J Biol Chem. 2003;278:2479–83. doi: 10.1074/jbc.M210721200. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Lafuente R. Stabilization of multimeric enzymes: strategies to prevent subunit dissociation. Enzyme Microbiol Technol. 2009;45:405–18. doi: 10.1016/j.enzmictec.2009.08.009. [DOI] [Google Scholar]
- 28.Donovan JW, Beardslee RA. Heat stabilization produced by protein-protein association. A differential scanning calorimetric study of the heat denaturation of the trypsin-soybean trypsin inhibitor and trypsin-ovomucoid complexes. J Biol Chem. 1975;250:1966–71. [PubMed]
- 29.Pecher P, Arnold U. The effect of additional disulfide bonds on the stability and folding of ribonuclease A. Biophys Chem. 2009;141:21–8. doi: 10.1016/j.bpc.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Nagi AD, Regan L. An inverse correlation between loop length and stability in a four-helix-bundle protein. Fold Des. 1997;2:67–75. doi: 10.1016/S1359-0278(97)00007-2. [DOI] [PubMed] [Google Scholar]
- 31.Burns CM, Wortmann RL. Gout therapeutics: new drugs for an old disease. Lancet. 2011;377:165–77. doi: 10.1016/S0140-6736(10)60665-4. [DOI] [PubMed] [Google Scholar]
- 32.Vogt B. Urate oxidase (rasburicase) for treatment of severe tophaceous gout. Nephrol Dial Transpl. 2005;20:431–3. doi: 10.1093/ndt/gfh629. [DOI] [PubMed] [Google Scholar]
- 33.Coiffier B, Mounier N, Bologna S, Ferme C, Tilly H, Sonet A, et al. Efficacy and safety of rasburicase (recombinant urate oxidase) for the prevention and treatment of hyperuricemia during induction chemotherapy of aggressive non-Hodgkin’s lymphoma: results of the GRAAL1 (Groupe d’Etude des Lymphomes de l’Adulte Trial on Rasburicase Activity in Adult Lymphoma) study. J Clin Oncol. 2003;21:4402–6. doi: 10.1200/JCO.2003.04.115. [DOI] [PubMed] [Google Scholar]
- 34.Schlesinger N, Yasothan U, Kirkpatrick P. Pegloticase. Nat Rev Drug Discov. 2010;10:17. doi: 10.1038/nrd3349. [DOI] [PubMed] [Google Scholar]
- 35.Sundy JS, Baraf HB, Yood RA, et al. Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: Two randomized controlled trials. JAMA. 2011;306:711–20. doi: 10.1001/jama.2011.1617. [DOI] [PubMed] [Google Scholar]
- 36.Sundy JS, Becker MA, Baraf HS, Barkhuizen A, Moreland LW, Huang W, et al. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol-conjugated uricase) in patients with treatment-failure gout: results of a phase II randomized study. Arthritis Rheum. 2008;58:2882–91. doi: 10.1002/art.23810. [DOI] [PubMed] [Google Scholar]
- 37.Becker MA, Baraf HS, Yood RA, Dillon A, Vazquez-Mellado J, Ottery FD, et al. Long-term safety of pegloticase in chronic gout refractory to conventional treatment. Ann Rheum Dis. 2013;72:1469–74. doi: 10.1136/annrheumdis-2012-201795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mandell BF. Clinical manifestations of hyperuricemia and gout. Cleve Clin J Med. 2008;75:S5–8. [DOI] [PubMed]
- 39.Tao L, Li D, Li Y, Shi X, Wang J, Rao C, et al. Designing a mutant Candida uricase with improved polymerization state and enzymatic activity. Protein Eng Des Sel. 2017;30:753–9. doi: 10.1093/protein/gzx056. [DOI] [PubMed] [Google Scholar]
- 40.Deehan M, Garces S, Kramer D, Baker MP, Rat D, Roettger Y, et al. Managing unwanted immunogenicity of biologicals. Autoimmun Rev. 2015;14:569–74. doi: 10.1016/j.autrev.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 41.Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J. 2012;14:296–302. doi: 10.1208/s12248-012-9340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wadhwa M, Knezevic I, Kang HN, Thorpe R. Immunogenicity assessment of biotherapeutic products: an overview of assays and their utility. Biologicals. 2015;43:298–306. doi: 10.1016/j.biologicals.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 43.Kuriakose A, Chirmule N, Nair P. Immunogenicity of biotherapeutics: causes and association with posttranslational modifications. J Immunol Res. 2016;2016:1298473. doi: 10.1155/2016/1298473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.