

Abstract
Cinnamic acid and its analogues (pyragrel and ozagrel) undergo chain-shortened (β-oxidative) and reductive metabolism on acyl side chain. In this study, we characterized the β-oxidative and reductive metabolism on acyl side chain of cinnamic acid and its analogues using primary rat hepatocytes, hepatic mitochondrial, and microsomal systems. A compartmental model including parent compounds and metabolites was developed to characterize in vivo β-oxidative and reductive metabolism following an intravenous dose of parent compounds to rats. The fitted total in vivo clearance values were further compared with the in vitro values predicted by the well-stirred model. We showed that hepatic microsomal CYP450s did not catalyze β-oxidative or reductive metabolism of the three compounds. Similar to β-oxidation of fatty acids, β-oxidative metabolism on their acyl side chain occurred mainly in mitochondria, which was highly dependent on ATP, CoA and NAD+. Fatty acids and NADH inhibited the β-oxidative metabolism. Reductive metabolism occurred in both mitochondria and microsomes. Reduction in mitochondria was ATP-, CoA-, and NAD(P)H-dependent and reversible, which was suppressed by enoyl reductase inhibitor triclosan. Reduction in microsomes was ATP-, CoA-, and NADPH-dependent but little affected by triclosan. Both plasma concentrations of β-oxidative metabolites and reductive metabolites were successfully fitted using the compartmental model. The estimated total in vivo clearance values were consistent with those predicted from hepatocytes and organelles, implicating significance of in vitro kinetics. These findings demonstrate the roles of hepatic mitochondria and microsomes in β-oxidative and reductive metabolism on acyl side chain of cinnamic acid and its analogues along with their metabolic characteristics.
Keywords: antiplatelet drug, cinnamic acid, pyragrel, ozagrel, drug metabolism, hepatocytes, mitochondria, microsomes, pharmacokinetics, in vitro-in vivo correlation
Introduction
Pyragrel sodium, a cinnamic acid analogue, is in development in phase I clinical trials as a potential anticoagulant [1]. Pharmacological studies have shown that pyragrel possesses antiplatelet aggregative activity by inhibiting thromboxane A2 formation and it alleviates inflammatory injury to endothelial cells via the NF-κB signal pathway [1]. Chain-shortening (pyragrel-M1) and reduction (pyragrel-M2) of the double bond in its acyl side chain followed by glucuronidation is the main pathway of pyragrel metabolism in humans [2, 3] (Fig. 1). Ozagrel, another cinnamic acid analogue, is commonly used in Japan for acute ischemic stroke as an inhibitor of thromboxane A2 synthase [4, 5]. In rats, ozagrel also undergoes chain-shortening (ozagrel-M1) and reductive (ozagrel-M2) metabolism on its acyl side chain [6] (Fig. 1a). Additionally, ozagrel-M2 is converted to ozagrel-M1 in rats [6]. Pyragrel and ozagrel share the similar structure of cinnamic acid, and both contain an acrylic acid in their side chain. Animal experiments have shown that the main metabolite in the urine of rats and mice is hippuric acid (Fig. 1b) following the oral administration of cinnamic acid [4, 6]. The double bond of cinnamic acid can also be reduced to form phenylpropionic acid in rats [7] (Fig. 1a). These results indicate that pyragrel, ozagrel and cinnamic acid have similar metabolic routes in their acyl side chain. However, the organelles where these reactions occur have not been fully identified.
Fig. 1.

a Structures of pyragrel, ozagrel, cinnamic acid, their β-oxidative metabolites M1 and reductive metabolites M2. b Structure of hippuric acid. c Metabolic stability of pyragrel, ozagrel, and cinnamic acid for the 1-h incubation in microsomes (1 mg protein/mL) with an NADPH-regenerating system
Generally, the metabolism of most compounds that occurs in hepatic microsomes is mediated by cytochrome P450 enzymes (CYP450s). However, our pre-experiments showed that following a 1-h incubation with rat hepatic microsomes (1 mg/mL) in the presence of an NADPH-regenerating system, pyragrel, ozagrel, and cinnamic acid demonstrated metabolic stability (Fig. 1c). Furthermore, neither β-oxidative nor reductive metabolism occurred in the incubation system. These results indicated that hepatic CYP450s did not catalyze metabolism on the acyl side chain of cinnamic acid or its analogues. In the β-oxidative metabolism of their acyl side chains, two carbon atoms were liberated from the parent compounds, and this resembled the β-oxidative metabolism of fatty acids that occurs in the mitochondria, indicating that these compounds were substrates of the mitochondrial β-oxidation enzyme system in the rat liver. The reductive metabolism, which is the hydrogenation of the double bond in the acyl side chain, was similar to the step in fatty acid chain elongation, which occurs in both microsomes and mitochondria [8]. β-Oxidative and reductive metabolism of p-hydroxycinnamic acid, another cinnamic acid analogue, was also reported to occur in isolated rat liver mitochondria [9].
The present studies were undertaken to characterize β-oxidative and reductive metabolism on the acyl side chain of cinnamic acid and its analogues (pyragrel and ozagrel) using primary rat hepatocytes, rat hepatic mitochondria, and microsomes. A compartmental model involving the parent compounds and their two metabolites was developed to characterize the pharmacokinetics of cinnamic acid and its analogues in rats. The estimated in vivo clearance values were further compared with the in vitro data that was predicted by the well-stirred model.
Materials and methods
Chemicals and materials
Pyragrel sodium (purity 99.79%), its β-oxidative metabolite (pyragrel-M1, purity 99.66%) and its reductive metabolite (pyragrel-M2, purity 99.12%) were from Hefei Industrial Pharmaceutical Institute Co., Ltd. (Hefei, China). Ozagrel sodium (purity > 99%) and phenacetin (internal standard, purity > 99%) were from National Institutes for Food and Drug Control (Beijing, China). The β-oxidative metabolite of ozagrel (ozagrel-M1, purity 95%) was from Enamine Ltd. (Kiev, Ukraine), and the reductive metabolite (ozagrel-M2, purity 95.5%) was from the Department of Medicinal Chemistry of China Pharmaceutical University (Nanjing, China). Trans-cinnamic acid (purity 99%), 3-phenylpropionic acid (purity 99%), sodium benzoate (purity 99%) and theophylline (internal standard, purity 99%) were from Aladdin Co., Ltd. (Shanghai, China). Adenosine 5’-triphosphatedisodium salt hydrate (ATP) was from J&K Scientific Ltd. (Beijing, China). β-Nicotinamide adenine dinucleotide hydrate (NAD+), flavin adenine dinucleotide disodium salt hydrate (FAD+), β-nicotinamide adenine dinucleotide, reduced disodium salt hydrate (NADH), β-nicotinamide adenine dinucleotide 2’-phosphate reduced tetra-sodium salt (NADPH), β-nicotinamide adenine dinucleotide phosphate disodium salt (NADP+), coenzyme A sodium salt hydrate (CoA), L-carnitine, D-glucose-6-phosphate (G-6-P), D-glucose-6-phosphate dehydrogenase (G-6PDH), palmitic acid, stearic acid, oleic acid and linoleic acid were from Sigma-Aldrich (St Louis, MO, USA). Triclosan was from Macklin Biochemical Co., Ltd. (Shanghai, China). All other reagents were of analytical grade and commercially available.
Animals
Male Sprague-Dawley rats, weighing 180–220 g (6-week-old), were from SIPPR/BK Experimental Animal Co., Ltd. (Shanghai, China). They were kept in an environmentally controlled room with ambient temperature (25 ± 2 °C), humidity (50 ± 5%) and a 12-h light/dark cycle. The animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals. The animal project was approved by Animal Ethics Committee of China Pharmaceutical University (No. CPU-PCPK-1631010071).
Cultures of primary rat hepatocytes and drug metabolism studies
Rat hepatocytes were isolated with a two-step perfusion method that has been described previously [10, 11]. A 250-μL aliquot of hepatocytes suspension was seeded in each well of collagen-coated 48-well plates (105 cells/well). Following a 4-h incubation, the medium was removed and then hepatocytes were exposed to pyragrel (7.81–500 μM), ozagrel (31.25–1000 μM) or cinnamic acid (0.49–62.5 μM). The reactions were terminated using a 25-μL aliquot of NaOH solution (1 M) following the 1-h incubation for cinnamic acid or 2-h incubation for pyragrel and ozagrel. The cultured hepatocytes were also incubated with 100 μM pyragrel-M2, ozagrel-M2 or phenylpropionic acid to investigate whether the reverse-conversion from the reductive metabolites to parent compounds occurred. Enhanced cell counting kit-8 (CCK-8) assays showed that the compounds had no influence on cell viability at the concentration that were used.
Preparation of hepatic mitochondria and microsomes
Rat hepatic mitochondria were prepared by employing differential centrifugation in 250 mM sucrose according to a method that has previously been described [12], and the samples were re-suspended in 30 mM phosphate buffer solution (PBS, pH 7.4). Rat hepatic microsomes were also prepared by differential ultracentrifugation as described previously and re-suspended in 100 mM PBS containing 20% glycerol [13]. Mitochondrial β-oxidation activity was assessed by the rate of consumption of octanoic acid (a substrate for β-oxidation, 1.0 mM as described) [12]. Both the mitochondrial and microsomal protein levels were measured using a BCA protein assay kit. All of the procedures were performed at 4 °C before their incubation. All of the mitochondrial metabolic experiments were performed immediately after the mitochondrial preparation.
β-Oxidative metabolism on the acyl side chain of pyragrel, ozagrel and cinnamic acid in the mitochondrial β-oxidation incubation system
The mitochondrial β-oxidation incubation system consisted of mitochondrial protein (approximately 1 mg/mL for pyragrel and ozagrel and 0.16 mg/mL for cinnamic acid), CoA (1 mM), ATP (5 mM), l-carnitine (2 mM), MgCl2 (5 mM), NAD+ or NADP+ (2 mM) and different levels of substrates (pyragrel, 3.91–500 μM; ozagrel, 15.63–2000 μM; cinnamic acid, 0.98–31.25 μM) (final volume: 0.2 mL). Following a 20-min pre-incubation at 37 °C, the reactions were started with the addition NAD+, continued at 37 °C for 10 min and quenched with 10 μL of NaOH solution (1 M). The CoA or l-carnitine ester intermediates of the analytes in the samples were then hydrolyzed under NaOH conditions [12] in a water bath at 70 °C for 0.5 h. Mitochondrial protein levels and incubation time were optimized beforehand to ensure the linear formation of metabolites in all incubation systems.
The effects of CoA, ATP, L-carnitine and NAD+ on the β-oxidative metabolism of pyragrel, ozagrel and cinnamic acid in the mitochondrial incubation system were determined in the absence of each of the above cofactors. The concentrations of pyragrel, ozagrel, and cinnamic acid were set at 25 μM, 100 μM, and 3.9 μM, respectively, which were all lower than or close to their Km values.
Effects of fatty acids and the cofactor NADH on the β-oxidative metabolism of the acyl side chain of pyragrel, ozagrel, and cinnamic acid
The fatty acid inhibition study was performed by adding fatty acids into the mitochondrial β-oxidation incubation system. The fatty acids that were tested include butyric acid, decanoic acid, palmitic acid, stearic acid, oleic acid, linoleic acid, and two irreversible inhibitors of fatty acid β-oxidation, 4-pentenoic acid and 2-bromooctanoic acid [12]. The final concentrations of the tested fatty acids were 100 μM, and the concentrations of pyragrel, ozagrel and cinnamic acid were 25 μM, 100 μM, and 3.9 μM, respectively.
NADH was reported to inhibit NAD+-dependent dehydrogenase activity [14]. The effects of NADH on the β-oxidative metabolism of cinnamic acid and its analogues were evaluated by adding different levels of NADH into the hepatic mitochondrial β-oxidation incubation system (2 mM NAD+). The concentrations of the substrates pyragrel, ozagrel and cinnamic acid were 125 μM, 1000 μM, and 3.9 μM, respectively, which were close to their Km values. The concentrations for the 50% of inhibition effect (IC50) of NADH were calculated. The inhibition assays were performed by using the same hepatic mitochondrial incubation system and experimental procedures as described above.
Reductive metabolism of the acyl side chain of pyragrel, ozagrel, and cinnamic acid in both mitochondria and microsomes
In mitochondria, NADH (2 mM) or an NADPH-regenerating system (10 mM G-6-P, 0.5 mM NADP+, 5 mM MgCl2 and 1 U/mL G-6-PDH) were used as hydrogen donors for reductive metabolism. Based on the results of the pre-experiment, the substrate concentrations were set at 3.91–500 μM for pyragrel and 0.98–31.25 μM for cinnamic acid. The ozagrel levels were set at 7.81–1000 μM and 125–16000 μM when NADPH and NADH were used as cofactors, respectively. Mitochondrial protein concentrations were set at 0.4 mg/mL for pyragrel, 1 mg/mL for ozagrel and 0.16 mg/mL for cinnamic acid when NADPH served as a cofactor; when NADH served as the cofactor, the concentrations were 1 mg/mL for pyragrel, 1 mg/mL for ozagrel and 0.32 mg/mL for cinnamic acid. The reaction mixtures, incubation procedures and sample preparation procedures were the same as those of the mitochondrial β-oxidation incubation system, except that NAD+ was replaced by NADH or an NADPH-regenerating system.
The pre-experiments showed that reductive metabolism on the acyl side chain of cinnamic acid and its analogues did not occur without CoA and ATP in rat hepatic microsomes in the presence of an NADPH-regenerating system. Here, the microsomal reductive system consisted of microsomal protein (1 mg/mL for pyragrel and ozagrel and 0.4 mg/mL for cinnamic acid), CoA (1 mM), ATP (5 mM), an NADPH-regenerating system and substrates (pyragrel, 3.91–500 μM; ozagrel, 7.81–1000 μM; or cinnamic acid, 0.98–31.25 μM) (final volume: 0.2 mL). The NADPH-regenerating system was also replaced by NADH (2 mM) to investigate the roles of these cofactors in reductive metabolism.
The effects of the enoyl reductase inhibitor triclosan on the reductive metabolism on the acyl side chain of pyragrel (25 μM), ozagrel (100 μM) and cinnamic acid (3.9 μM) were determined in both hepatic mitochondria and microsomes. Triclosan blocks lipid synthesis by specifically inhibiting the enoyl-acyl carrier protein reductase of microbes [15] and humans in vitro [16, 17]. Here, it was shown to be involved in the in vitro incubation systems. The concentrations of triclosan in mitochondria and microsomes were 12.5–100 μM and 100–200 μM, respectively.
Conversion of reductive metabolites to parent compounds in mitochondria
The mitochondrial incubation system consisted of mitochondrial protein (approximately 1 mg/mL for pyragrel-M2 and ozagrel-M2 and 0.05 mg/mL for phenylpropionic acid), CoA (1 mM), ATP (5 mM), l-carnitine (2 mM), MgCl2 (5 mM), FAD+ (2 mM) and different levels of substrates (pyragrel-M2, 7.81–500 μM; ozagrel-M2, 15.63–1000 μM; phenylpropionic acid, 3.91–125 μM) (final volume: 0.2 mL).
Effects of the NAD+/NADH or NAD+/NADPH ratios on β-oxidative and reductive metabolism
As described above, β-oxidative and reductive metabolism might simultaneously occur in rat liver mitochondria if both NAD+ and NAD(P)H are present. Some diseases alter NAD+/NADH and NADP+/NADPH ratios [18, 19]. Therefore, the effects of the NAD+/NADH and NAD+/NADPH ratios on the β-oxidative and reductive metabolism of pyragrel (25 μM) in rat liver mitochondria were documented. The total concentration of cofactors (NAD+ + NADH or NAD+ + NADPH) was 2 mM, while the NAD+/NADH and NAD+/NADPH ratios were 1:4, 1:2, 1:1, 2:1, and 4:1, respectively. Other incubation conditions and experimental procedures have been described above.
Pharmacokinetics of pyragrel, ozagrel and cinnamic acid in rats
Eighteen male Sprague-Dawley rats were randomly divided into three groups with 6 rats in each group. The rats were fasted overnight and received a single intravenous dose of pyragrel sodium (15 mg/kg, dissolved in normal saline), ozagrel sodium (6 mg/kg, dissolved in normal saline) or cinnamic acid (2 mg/kg, dissolved in normal saline containing 30% PEG400). The doses of pyragrel sodium, ozagrel sodium and cinnamic acid that were employed in our pharmacokinetic experiments were similar to those used in previous reports [6, 20, 21]. Blood samples (approximately 200 μL for pyragrel and cinnamic acid or 50 μL for ozagrel) were collected from the oculi chorioideae vein under light ether anesthesia into heparinized tubes prior to dosing and at the designated postdose time. The designated time points were as follows: 5, 10, 20, 40, 60, 90, 120, 180, 240, 360, 480, and 720 min for pyragrel; 2, 5, 10, 20, 30, 45, 60, 75, and 90 min for ozagrel; and 2, 5, 10, 20, 30, and 45 min for cinnamic acid. After every 3-4 samplings, the appropriate amount of 0.9% saline was given to the rats via tail vein to compensate for blood loss. Plasma samples were immediately separated by centrifugation and stored at −70 °C until analysis.
In vitro-in vivo correlations
In vitro kinetic parameters were estimated by the Michaelis–Menten (M-M) equation v = Vmax × S/(Km + S) or sigmoidal equation v = Vmax × SN/(KmN + SN) based on the Eadie-Hofstee plots, where v, S, KmVmax and N represented the velocity of metabolic reaction, the drug concentration, Michaelis constant, the maximum reaction velocity and the Hill coefficient, respectively. For the M-M kinetics, the in vitro intrinsic clearance (CLint,invitro) was defined as CLint,invitro = Vmax/Km. For the sigmoidal kinetics, the maximum clearance (CLmax,invitro) was calculated as CLmax,invitro = Vmax × (N−1)/(Km × N × (N−1)1/N). If the Hill coefficient N was greater than 1.2, a sigmoidal equation was fitted to the kinetic data [22]. The CLint,invitro (or CLmax,invitro) was further scaled to the in vivo intrinsic clearance (CLint,invivo) using CLint,invitro (or CLmax,invitro) × scaling factor (SF). SF was the total hepatic mitochondrial (or microsomal) protein content or hepatocyte number per kilogram of body weight.
The well-stirred model was used to predict the total hepatic clearance (CLH) of the M1 and M2 formations as below,
where QH, fu and Rbp are the liver blood flow rate, the unbound fractions in plasma and blood-to-plasma concentration ratio, respectively. The fu and Rbp values were calculated as 17.91% and 0.54 for pyragrel, 14.22% and 0.71 for ozagrel and 17.22% and 0.61 for cinnamic acid, respectively, using the method described in “Supplementary information”. The QH value was set at 55.2 mL min−1 kg−1 body weight [23]. The CLint,invivo value is the total CLint,invivo of the β-oxidation and reduction processes in the organelles or hepatocytes.
A compartmental model involving both the parent compound and the metabolites was constructed to fit the individual pharmacokinetic data to estimate the in vivo clearance rate of M1 and M2 formation (Fig. 2). The essential structure of the model consisted of the central compartment of the parent and its two metabolites as well as the peripheral compartments based on their pharmacokinetic characteristics, if necessary.
Fig. 2.
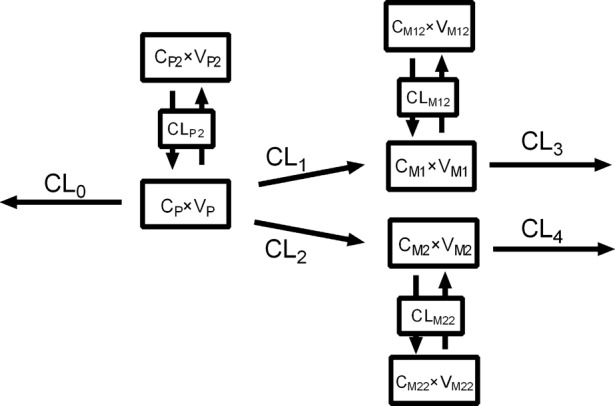
Schematic diagram of the compartmental model involving the parent compound and the β-oxidative and reductive metabolites
The differential equations for all the compounds with two compartments are described as follows:
for the parent compound,
for metabolite M1,
for metabolite M2,
where the subscripts P, M1 and M2 refer to the parent compound, M1 and M2, respectively, in central compartment. The subscripts P2, M12 and M22 refer to the parent compound, M1 and M2, respectively, in the peripheral compartment. The symbols C and V are the concentration and distribution volume of these compounds in their corresponding compartments. CLP and CLP2 are defined as the total clearance of the parent compound in the central and peripheral compartments, respectively. CLM1 and CLM2 represent the clearance of M1 and M2, respectively, in the peripheral compartment. CL1 and CL2 represent the clearance of M1 and M2 formation, respectively. CL3 and CL4 are defined as the clearance of M1 and M2 elimination, respectively. The value CL0 in Fig. 2 represents the clearance of the parent compound via other ways, where CL0 = CLP−CL1−CL2.
The distribution volumes of the metabolites in the central compartment were assumed to be the same as that of their parent compounds. The pharmacokinetic profiles were fitted, and their corresponding parameters were estimated. The fitted total blood clearance of β-oxidative and reductive metabolism was defined as (CL1 + CL2)/Rbp, and it was compared with the CLH that was predicted from the in vitro data. The accuracy of the predictions was assessed by fold-error, and the predicted values that were within 2-fold error were considered acceptable [24, 25].
Drug analysis
The concentrations of the parent compounds and their metabolites in the biological matrix were measured according to the methods in “Supplementary information”. The linear ranges of the analytes in the incubation system were as follows: 12.21–6250 ng/mL for pyragrel, 3.91–2000 ng/mL for pyragrel-M1 and pyragrel-M2; 3.91–2000 ng/mL for ozagrel, ozagrel-M1 and ozagrel-M2; 19.53–625 ng/mL for cinnamic acid, benzoic acid and phenylpropionic acid; and 39.06–10000 ng/mL for hippuric acid. The linear ranges of the analytes in rat plasma were as follows: 6.10–781.25 ng/mL for pyragrel, 1.95–250 ng/mL for pyragrel-M1 and 3.91–500 ng/mL for pyragrel-M2; 39.06–10,000 ng/mL for ozagrel, ozagrel-M1 and ozagrel-M2; 78.13–5000 ng/mL for hippuric acid and 9.77–1250 ng/mL for cinnamic acid. Based on the pre-experimental results, if the analyte concentrations exceeded the upper limit of quantification, the analysis would be performed following dilution with the blank matrix.
Data analysis
The kinetic parameters and mathematical models were coded and solved by Phoenix WinNonlin software (version 7.0, Certara, Co., Princeton, NJ, USA). The pharmacokinetic parameters and IC50 values of NADH were also estimated with this software. The relationship between two variables was analyzed by linear regression analyses.
Results were presented as the means ± standard deviation (SD). The significant difference was determined by a one-way analysis of variance (ANOVA) followed by a post hoc Tukey’s test. A P-value less than 0.05 was considered statistically significant.
Results
Metabolism of pyragrel, ozagrel and cinnamic acid in primary rat hepatocytes
The results demonstrated that both β-oxidative and reductive metabolism on the acyl side chain of pyragrel, ozagrel and cinnamic acid occurred in primary rat hepatocytes (Fig. 3a–c). It is worth mentioning that in hepatocytes the final β-oxidative metabolite of cinnamic acid was hippuric acid instead of benzoic acid. It was also found that following a 2-h (for pyragrel and ozagrel) or 1-h incubation (for cinnamic acid) with pyragrel (31.25 μM), ozagrel (62.50 μM) or cinnamic acid (3.91 μM) in primary rat hepatocytes, approximately 2.52%, 6.53%, and 44.17% of pyragrel, ozagrel, and cinnamic acid, respectively, were metabolized to their corresponding β-oxidative metabolites. Approximately 2.81%, 6.81%, and 5.28% of pyragrel, ozagrel, and cinnamic acid, respectively, were metabolized to corresponding reductive metabolites. Formation of all of these metabolites except for ozagrel-M2 was better fitted by the M-M equation, and all of the kinetic parameters were estimated (Table 1) [23, 26, 27]. Despite the lower Vmax values for β-oxidative metabolism, the CLint,invitro values for β-oxidative and reductive metabolism were similar for pyragrel and ozagrel due to the higher affinity of the metabolism enzymes for these compounds. However, the CLint,invitro of hippuric acid formation was much higher than that of phenylpropionic acid formation, which was consistent with the in vivo data showing that hippuric acid was the main metabolite of cinnamic acid [4, 28]. These findings indicated that the rat liver could be used to further probe the organelles that are involved in metabolism of the acyl side chains of cinnamic acid and its analogues.
Fig. 3.

Kinetic profiles of the formation of β-oxidative and reductive metabolites in primary rat hepatocytes (a–c), the formation of β-oxidative metabolites in rat hepatic mitochondria (d–f) and reductive metabolites in rat hepatic mitochondria (NADH-dependent and NADPH-dependent) and microsomes (mic, NADPH-dependent) (g–i) from pyragrel, ozagrel and cinnamic acid. The kinetic profiles of the formation of pyragrel, ozagrel and cinnamic acid from pyragrel-M2, ozagrel-M2, and phenylpropionic acid in mitochondria (FAD+-dependent) (j–l). Data are expressed as the means ± SD of 3 rats
Table 1.
Kinetic parameters (mean ± SD of 3 rats) for formations of β-oxidative and reductive metabolites on acyl side chain of pyragrel (Pyr), ozagrel (Oza) and cinnamic acid (Cin) in primary hepatocytes (Hep), isolated hepatic mitochondria (Mit) and microsomes (Mic) of rats
| Compounds | System | Reaction | Cofactor | Km (μM) | Vmax (pmol min−1 mg−1 or pmol min−1 10−5 cells) | N | aCLint, in vitro (μL min−1 mg−1 or μL min−1 10−5 cells) | bCLint, in vivo (mL min−1 kg−1) | cCLH (mL min−1 kg−1) |
|---|---|---|---|---|---|---|---|---|---|
| Pyr | Hep | Oxidation | - | 37.65 ± 6.96 | 12.38 ± 1.07 | - | 0.33 ± 0.04 | 6.70 ± 0.83 | 3.95 |
| Hep | Reduction | - | 139.29 ± 11.93 | 43.08 ± 1.06 | - | 0.31 ± 0.02 | 6.23 ± 0.36 | ||
| Mit | Oxidation | NAD+ | 145.55 ± 10.42 | 275.15 ± 40.23 | - | 1.90 ± 0.35 | 3.04 ± 0.56 | 3.15 | |
| Mit | Reduction | NADH | 100.29 ± 9.15 | 81.98 ± 6.29 | - | 0.82 ± 0.02 | 1.31 ± 0.02 | ||
| Mit | Reduction | NADPH | 152.85 ± 44.68 | 321.73 ± 27.73 | - | 2.25 ± 0.80 | 3.06 ± 1.28 | ||
| Mic | Reduction | NADPH | 534.81 ± 140.17 | 805.07 ± 150.23 | - | 1.53 ± 0.14 | 2.75 ± 0.26 | ||
| Oza | Hep | Oxidation | - | 78.21 ± 3.09 | 64.71 ± 1.69 | - | 0.83 ± 0.01 | 16.62 ± 0.23 | 5.03 |
| Hep | Reduction | - | 188.10 ± 43.94 | 186.98 ± 28.13 | 1.51 ± 0.33 | 0.55 ± 0.02 | 11.07 ± 0.57 | ||
| Mit | Oxidation | NAD+ | 1119.61 ± 281.72 | 647.04 ± 25.68 | - | 0.61 ± 0.18 | 0.97 ± 0.28 | 1.32 | |
| Mit | Reduction | NADH | 2386.90 ± 838.21 | 674.45 ± 139.69 | 1.31 ± 0.20 | 0.18 ± 0.01 | 0.28 ± 0.02 | ||
| Mit | Reduction | NADPH | 203.57 ± 53.07 | 731.56 ± 29.51 | 1.31 ± 0.09 | 2.19 ± 0.58 | 3.51 ± 0.93 | ||
| Mic | Reduction | NADPH | 1889.86 ± 297.76 | 2078.98 ± 222.47 | - | 1.11 ± 0.06 | 1.99 ± 0.12 | ||
| Cin | Hep | Oxidation | - | 0.77 ± 0.12 | 18.02 ± 1.19 | - | 23.56 ± 1.91 | 473.13 ± 959.40 | 39.71 |
| Hep | Reduction | - | 28.21 ± 7.24 | 39.68 ± 4.34 | - | 1.44 ± 0.22 | 28.98 ± 4.46 | ||
| Mit | Oxidation | NAD+ | 2.98 ± 0.15 | 577.87 ± 20.72 | - | 194.57 ± 16.33 | 311.31 ± 26.12 | 42.14 | |
| Mit | Reduction | NADH | 6.79 ± 0.98 | 144.03 ± 7.97 | - | 21.48 ± 2.89 | 34.36 ± 4.62 | ||
| Mit | Reduction | NADPH | 5.89 ± 0.59 | 353.78 ± 57.78 | - | 60.26 ± 10.47 | 96.42 ± 16.75 | ||
| Mic | Reduction | NADPH | 4.41 ± 0.09 | 464.45 ± 7.59 | - | 105.39 ± 2.41 | 189.71 ± 4.35 |
Km, Michaelis constant, Vmax the maximum reaction velocity, N the Hill coefficient
aCLint, in vitro (μL min−1 mg−1 or μL min−1 10−5 cells) was defined as Vmax/Km or Vmax × (N−1)/(Km × N × (N−1)1/N).
bCLint, in vivo (mL min−1 kg−1) was estimated by CLint, in vitro × SF, where SF was the product of protein yield (40 mg mitochondrial protein/g liver [26] or 45 mg microsomal protein/g liver [27]) or hepatocyte yield (5.02 × 107 cells/g liver) [23] and the liver weight per kilogram of body weight (40 g/kg) [23]
cCLH (mL min−1kg−1) was predicted from the well-stirred model
β-Oxidative metabolism of the acyl side chain of pyragrel, ozagrel, and cinnamic acid in mitochondrial β-oxidation incubation system
The consumption of octanoic acid in our prepared mitochondrial β-oxidation incubation system (6.73 ± 2.02 nmol min−1·mg−1 protein in 3 rats) was comparable to the data (approximately 5.5 nmol min−1 mg−1 protein) that has previously been reported [12] that demonstrated the existence of mitochondrial β-oxidation activity. Thus, the mitochondrial β-oxidation incubation system was used to characterize β-oxidative metabolism on the acyl side chains of pyragrel, ozagrel, and cinnamic acid. Consistent with our expectations, β-oxidative metabolism of the three compounds occurred in the mitochondria. These findings were different from those observed in primary rat hepatocytes, which showed that the primary β-oxidative metabolite of cinnamic acid was benzoic acid in the mitochondria. The kinetics of β-oxidative metabolism for the three compounds were characterized (Fig. 3d–f) by the M-M equation (Table 1). The results showed that cinnamic acid possessed the lowest Km value for β-oxidative metabolism, followed by those of pyragrel and ozagrel. The orders of the estimated CLint,invitro values were cinnamic acid (194.57 ± 16.33 μL min−1 mg−1) > pyragrel (1.90 ± 0.35 μL min−1 mg−1) > ozagrel (0.61 ± 0.18 μL min−1 mg−1), indicating that the β-oxidative metabolism of cinnamic acid was the process that occurred the easiest.
The effects of several cofactors including CoA, ATP, NAD+, and l-carnitine, on the β-oxidative metabolism of the three compounds in mitochondria were investigated (Fig. 4a). The results demonstrated that the lack of ATP or NAD+ completely abolished the β-oxidative metabolism of pyragrel and ozagrel. The formation of benzoic acid from cinnamic acid was also remarkably suppressed due to a lack of ATP or NAD+. Notably, the substitution of NADP+ for NAD+, abolished the β-oxidative metabolism of both pyragrel and ozagrel. The β-oxidative metabolism of cinnamic acid was also decreased significantly. A deficiency in CoA also remarkably inhibited the β-oxidative metabolism of these three compounds. These results indicated that similar to the β-oxidative metabolism of fatty acids, the cofactors CoA, ATP, and NAD+ were also essential for the β-oxidative metabolism of cinnamic acid and its analogues. Interestingly, the absence of L-carnitine greatly decreased the β-oxidative metabolism of pyragrel and ozagrel, but it notably increased the β-oxidative metabolism of cinnamic acid by approximately 60%.
Fig. 4.

Effects of cofactors (a), fatty acids (b–d) and NADH (e) on the β-oxidation of three cinnamic acid analogues in rat hepatic mitochondria. “−” means the lack of corresponding components and “ + NADP” means that 2 mM NADP+ were added to the reaction mixture instead of 2 mM NAD+. Abbreviations: But, butyric acid; DA, decanoic acid; Pal, palmitic acid; SA, stearic acid; OA, oleic acid; LOA, linoleic acid; 4-PA, 4-pentenoic acid and 2Br-Oct, 2-bromooctanoic acid. The absolute data for pyragrel-M1, ozagrel-M1 and benzoic acid formation in the control group of (a) are 44.62 ± 2.11, 34.58 ± 1.44 and 337.34 ± 26.15 pmol min−1 mg−1, respectively. The absolute data for the formation of the β-oxidative metabolites in the control groups of (b)–(d) are 40.72 ± 2.87, 30.86 ± 2.92 and 312.57 ± 9.87 pmol min−1 mg−1, respectively. The absolute data for pyragrel-M1, ozagrel-M1 and benzoic acid formation in control group of (e) are 138.56 ± 3.95, 280.11 ± 1.44 and 322.91 ± 55.02 pmol min−1 mg−1, respectively. Data are expressed as the means ± SD of 3 rats, *P < 0.05, **P < 0.01 vs. control
Effects of fatty acids and the cofactor NADH on β-oxidative metabolism of the acyl side chains of cinnamic acid and its analogues
The effects of two irreversible fatty acid β-oxidative inhibitors (4-pentenoic acid and 2-bromooctanoic acid) and six fatty acids (butyric acid, decanoic acid, palmitic acid, stearic acid, oleic acid, and linoleic acid) on the β-oxidative metabolism of the acyl side chains of cinnamic acid and its analogues were examined in the mitochondrial incubation system (Fig. 4b–d). It was found that 2-bromooctanoic acid was a very effective inhibitor of the β-oxidation of the three compounds. 4-Pentenoic acid also remarkably decreased the β-oxidation of pyragrel and cinnamic acid but not ozagrel. The six fatty acids that were tested considerably inhibited the β-oxidative metabolism of cinnamic acid; however, the four long-chain fatty acids (palmitic acid, stearic acid, oleic acid, and linoleic acid) had stronger inhibitory effects on pyragrel and ozagrel than the short- and medium-chain fatty acids.
The formation of the β-oxidative metabolites was also found to decrease as the NADH concentration increased (Fig. 4e), demonstrating that NADH possessed strong inhibitory effects on the β-oxidative metabolism of cinnamic acid and its analogues. The order of the inhibitory effect of NADH was cinnamic acid > pyragrel > ozagrel, and the IC50 values in 3 rats were estimated to be 27.03 ± 13.07, 232.67 ± 9.38 and 367.96 ± 25.24 μM, respectively.
Reductive metabolism of the acyl side chain of cinnamic acid and its analogues in both mitochondria and microsomes
We tried to identify the reductive metabolism of pyragrel, ozagrel, and cinnamic acid in both mitochondria and microsomes. Reductive metabolism barely occurred in the mitochondrial β-oxidation incubation system. However, when NAD+ was replaced by an NADPH-regenerating system or NADH in the mitochondrial incubation system, reductive metabolism occurred, indicating that reductase used NADH or NADPH as a cofactor in the mitochondria to catalyze the reductive metabolism of the acyl side chains of cinnamic acid and its analogues. The kinetics of reductive metabolism for pyragrel and cinnamic acid in mitochondria were characterized by the M-M equation, while the kinetics of reductive metabolism of ozagrel was characterized by the sigmoidal model (Fig. 3g–i) (Table 1). The results uncovered that the enzymes that perform the reductive metabolism of the acyl side chains of cinnamic acid and its analogues preferred NADPH in the mitochondria, as evidenced by the higher CLint,invitro values.
The reductive metabolism of cinnamic acid and its analogues also occurred in the hepatic microsomal systems that contained an NADPH-regenerating system with both CoA and ATP. All of the kinetics were characterized by the M-M equation. If the NADPH-regenerating system was replaced with 2 mM NADH or the incubation system did not contain CoA and ATP, reductive metabolism did not occur, indicating that the reductive metabolism in hepatic microsomes was highly dependent on CoA, ATP, and NADPH.
Effects of triclosan on reductive metabolism
The effects of the enoyl reductase inhibitor triclosan on the acyl side chain reduction of pyragrel, ozagrel and cinnamic acid were assessed in both hepatic mitochondrial and microsomal incubation systems (Fig. 5a–i). The results showed that triclosan substantially suppressed the reductive metabolism of cinnamic acid and its analogues in the mitochondrial incubation system using NADPH or NADH as a cofactor in a concentration-dependent manner, and a stronger inhibitory effect was found when NADH served as a cofactor. For example, 50 μM triclosan almost completely abolished the reductive metabolism of cinnamic acid when NADH was used as a cofactor. These results indicated that enoyl reductase catalyzed the reductive metabolism of cinnamic acid and its analogues in hepatic mitochondria. However, up to 200 μM triclosan did not inhibit the reductive metabolism of pyragrel or ozagrel in the hepatic microsomes. Although 100 and 200 μM triclosan notably inhibited the formation of phenylpropionic acid in hepatic microsomes, the extent of this inhibition was far less than that in the hepatic mitochondrial reaction.
Fig. 5.

Effects of triclosan on the reduction of pyragrel (a, d and g), ozagrel (b, e and h) and cinnamic acid (c, f and i) in rat hepatic mitochondria with NADPH (a–c) or NADH (d–f), and in rat hepatic microsomes with NADPH as a cofactor (g–i). The effects of different NAD+/NADPH (j) and NAD+/NADH ratios (k) on the formation of pyragrel-M1/2 in rat hepatic mitochondria. Data are expressed as the means ± SD of 3 rats, *P < 0.05, **P < 0.01 vs control (a–i); $P < 0.05, $$P < 0.01 vs 1:4 group for pyragrel-M1, ##P < 0.01 vs 1:4 group for pyragrel-M2 (j and k)
Conversion of reductive metabolites to parent compounds
The results showed that the reductive metabolites of cinnamic acid and its analogues were metabolized to the parent compounds and their β-oxidative metabolites in primary rat hepatocytes. In primary rat hepatocytes, following a 2-h incubation with pyragrel-M2/ozagrel-M2 or a 1-h incubation with phenylpropionic acid, approximately 1.09% of pyragrel-M2, 0.25% of ozagrel-M2 and 4.80% of phenylpropionic acid were converted to their corresponding parent compounds. Approximately 0.21% of pyragrel-M2, 0.34% of ozagrel-M2 and 1.06% of phenylpropionic acid were converted to pyragrel-M1, ozagrel-M1 and hippuric acid, respectively. Dehydrogenation occurred in the hepatic mitochondria when M2 was the substrate (Fig. 3j–l). Due to solubility limitations, the clearances of pyragrel and ozagrel formation were calculated as the slopes of their reaction velocity-concentration curves. Compared with the formation of the reductive metabolites, the effects of the reverse reaction on the formation of pyragrel-M2 (0.16 ± 0.02 μL min−1 mg−1 protein from 3 rats) and ozagrel-M2 (0.11 ± 0.01 μL min−1 mg−1 protein from 3 rats) were slight, while phenylpropionic acid was efficiently converted to cinnamic acid, with a clearance of 93.32 ± 19.93 μL min−1 mg−1 protein from 3 rats, which were approximately half of the clearance of phenylpropionic acid formation from cinnamic acid.
Effects of the NAD+/NAD(P)H ratios on formations of pyragrel-M1 and pyragrel-M2
The effect of the NAD+/NAD(P)H ratio on the formations of pyragrel-M1 and pyragrel-M2 was determined (Fig. 5j, k). The results showed that pyragrel-M1 formation gradually increased along with an NAD+/NAD(P)H ratio that increased from 1:4 to 2:1, whereas a remarkable increase occurred at the ratio of 4:1. In contrast, pyragrel-M2 formation was gradually diminished with the increase in the NAD+/NAD(P)H ratio and a remarkable decrease was found at a NAD+/NAD(P)H ratio of 4:1. These results suggested that the formation of pyragrel-M1 and pyragrel-M2 was highly dependent on the NAD+/ NAD(P)H ratio.
In vitro-in vivo correlations
The estimated CLint,invitro for pyragrel, ozagrel, and cinnamic acid metabolism in the organelles or primary rat hepatocytes were scaled up to their respective CLint,invivo, which were further scaled up to the total hepatic clearance CLH (Table 1). The predicted CLH of pyragrel and cinnamic acid from the organelles was in line with data from primary rat hepatocytes (3.15 mL min−1 kg−1 from organelles vs 3.95 mL min−1 kg−1 from hepatocytes for pyragrel; 42.14 mL min−1 kg−1 from organelles vs 39.71 mL min−1 kg−1 from hepatocytes for cinnamic acid), and the fold-errors were less than 2. However, the predicted CLH of ozagrel from the organelles (1.32 mL min−1 kg−1) was far lower than that from primary rat hepatocytes (5.03 mL min−1 kg−1).
A compartmental model involving the parent compounds and their metabolites was fitted to the plasma concentration-time profiles of cinnamic acid and its analogues and their metabolites following an intravenous administration to rats (Fig. 2 and Fig. 6a–c). The corresponding pharmacokinetic parameters were estimated and are listed in Table 2. The model that was developed well-characterized the pharmacokinetics of these tested compounds, and all of the parameters were in good agreement and all the coefficients of variation were less than 25%. Notably, benzoic acid and phenylpropionic acid could only be detected at one or two time points, so only the plasma concentrations of hippuric acid and cinnamic acid were used. The two-compartmental model was fitted to the pharmacokinetics of pyragrel, pyragrel-M1, pyragrel-M2, ozagrel and cinnamic acid, while a one-compartmental model displayed a better fitness for ozagrel-M1, ozagrel-M2 and hippuric acid. The estimated total plasma clearance of β-oxidative and reductive metabolism (CL1 + CL2) for cinnamic acid (29.53 ± 6.27 mL min−1 kg−1) was close to the total plasma clearance (CLP: 32.96 ± 4.80 mL min−1 kg−1), while the sum of estimated clearance (CL1 and CL2) of β-oxidative and reductive metabolism for pyragrel and ozagrel was less than the respective total clearance CLp.
Fig. 6.

The observed (dots and error bars, means ± SD of 6 rats) and fitted (line) plasma concentrations of pyragrel (a), ozagrel (b) and cinnamic acid (c) and their corresponding metabolites following an intravenous administration of pyragrel (15 mg/kg), ozagrel (6 mg/kg) or cinnamic acid (2 mg/kg) to rats. d The relationship of the estimated total clearance of M1 and M2 in vivo and the predicted clearance of M1 and M2 in vitro from the organelles (open square) or hepatocytes (solid square), in which the solid and dashed lines indicate unity and two-fold errors between the fitted and observed data, respectively
Table 2.
The comparison of observed and fitted plasma pharmacokinetic parameters in rats
| aCL1 (mL min−1 kg−1) | aCL2 (mL min−1 kg−1) | a,bCL0 (mL min−1 kg−1) | aCLP (mL min−1 kg−1) | ct1/2 (min) | cAUC0-tn (μg min/mL) | cAUC0-∞ (μg min/mL) | cCL (mL min−1 kg−1) | ||
|---|---|---|---|---|---|---|---|---|---|
| Pyragrel | Observed | - | - | - | - | 68.23 ± 16.9 | 1697.89 ± 155.21 | 1699.12 ± 155.41 | 8.89 ± 0.85 |
| Fitted | 1.18 ± 0.30 | 1.31 ± 0.42 | 6.74 ± 0.98 | 9.23 ± 0.87 | 81.08 ± 26.87 | 1636.52 ± 671.62 | 1638.05 ± 671.98 | 9.22 ± 0.87 | |
| Pyragrel-M1 | Observed | - | - | - | - | 199.11 ± 69.91 | 127.79 ± 23.20 | 132.21 ± 25.92 | - |
| Fitted | - | - | - | - | 215.62 ± 92.71 | 126.97 ± 57.39 | 131.74 ± 60.52 | - | |
| Pyragrel-M2 | Observed | - | - | - | - | 164.18 ± 55.49 | 252.32 ± 74.10 | 257.92 ± 77.40 | - |
| Fitted | - | - | - | - | 185.72 ± 78.66 | 248.57 ± 125.88 | 255.46 ± 130.31 | - | |
| Ozagrel | Observed | - | - | - | - | 16.54 ± 3.05 | 358.72 ± 46.74 | 365.29 ± 50.64 | 16.75 ± 2.37 |
| Fitted | 1.96 ± 0.45 | 6.13 ± 1.67 | 9.66 ± 2.59 | 17.76 ± 3.12 | 17.03 ± 3.33 | 339.74 ± 150.75 | 346.72 ± 155.00 | 17.76 ± 3.11 | |
| Ozagrel-M1 | Observed | - | - | - | - | 23.45 ± 4.40 | 87.86 ± 40.49 | 99.65 ± 48.41 | - |
| Fitted | - | - | - | - | 27.08 ± 10.57 | 83.00 ± 49.16 | 102.96 ± 75.66 | - | |
| Ozagrel-M2 | Observed | - | - | - | - | 29.90 ± 9.06 | 268.60 ± 61.68 | 325.27 ± 78.66 | - |
| Fitted | - | - | - | - | 28.31 ± 5.3 | 271.23 ± 134.64 | 325.19 ± 169.2 | - | |
| - | - | ||||||||
| Cinnamic acid | Observed | - | - | - | - | 6.63 ± 0.99 | 65.20 ± 9.06 | 65.58 ± 9.06 | 30.97 ± 4.17 |
| Fitted | 29.53 ± 6.27 | - | 3.42 ± 2.70 | 32.96 ± 4.80 | 7.29 ± 1.45 | 61.68 ± 24.41 | 62.09 ± 24.56 | 32.83 ± 4.79 | |
| Hippuric acid | Observed | - | - | - | - | 6.38 ± 1.33 | 80.98 ± 10.59 | 82.08 ± 10.46 | - |
| Fitted | - | - | - | - | 7.24 ± 0.87 | 80.32 ± 36.63 | 81.90 ± 37.12 | - |
CL1 clearance of M1 formation in vivo, CL2 clearance of M2 formation in vivo, CLP clearance of parent compound in central compartment, t1/2 terminal half-life, AUC0-tn area under the concentration-time curve from time zero to last time, AUC0-∞ area under the concentration-time curve from time zero to infinite, CL clearance of parent compound
aData were obtained from the compartmental model
bCL0 represents the clearance of the parent compound via other ways and was calculated by CLP–CL1–CL2
cData were estimated by noncompartmental analysis (NCA) methods
The fitted in vivo data (CL1 + CL2)/Rbp were also compared with the predicted data from the organelles or primary rat hepatocytes (Fig. 6d). The results showed that the fitted clearance values for β-oxidative and reductive metabolism were consistent with the data from the organelles (except ozagrel) and primary hepatocytes, and the fold-errors were less than 2, indicating successful predictions.
Discussion
Hepatic CYP450s are considered to be the major enzymes involved in drug metabolism, contributing to approximately 75% of the metabolism of marketed drugs [29]. The present study showed that hepatic CYP450s did not catalyze the β-oxidative or reductive metabolism of the acyl side chains of cinnamic acid and its analogues (ozagrel and pyragrel). The primary goal of this study was to characterize the metabolism of the three compounds by rats, probe the factors that modulate metabolism, and evaluate the relevance of extrapolating the in vitro kinetics to in vivo pharmacokinetics.
The data from the primary rat hepatocytes proved the role of the rat liver in β-oxidative and reductive metabolism of pyragrel, ozagrel and cinnamic acid. 4-Pentenoic acid was considered to inhibit fatty acid β-oxidation by directly competing with fatty acids for the acyl-CoA synthetase and cofactors (such as CoA) in hepatic mitochondria [30]. These results indicated that cinnamic acid and its analogues first formed the substrate-CoA complex; similarly to fatty acids, this process was mediated by acyl-CoA synthetase. Acyl-CoA synthetase was not added in the mitochondrial/microsomal incubation systems, but β-oxidative and reductive metabolism still occurred. Several studies also demonstrated that the activity of acyl-CoA synthetase could be detected in isolated rat liver mitochondria and microsomes [31–34]. Moreover, the addition of acyl-CoA synthetase did not increase the reaction velocity in our study (data not shown). The pre-experimental data also showed that the β-oxidative metabolites were only generated following hydrolysis, indicating that these metabolites were in the form of their CoA or l-carnitine esters, which further demonstrated the existence of acyl-CoA synthetase activity in the isolated rat liver mitochondria and microsomes.
The fatty acids that were tested inhibited the β-oxidative metabolism of the acyl side chains of cinnamic acid and its analogues, indicating that these compounds might share the same metabolic route and mechanism. Generally, fatty acid β-oxidation occurs in both mitochondria and peroxisomes. We found that L-carnitine stimulated the β-oxidative metabolism of ozagrel and pyragrel (Fig. 4a). An l-carnitine-dependent transport system was determined to exist in mitochondria rather than peroxisomes [35], inferring that the β-oxidation of ozagrel and pyragrel occurred primarily in the mitochondria, although a report demonstrated the involvement of peroxisomes in the β-oxidation of ozagrel [36]. The β-oxidation of cinnamic acid was also found to occur mainly in the mitochondria [7], and this finding was consistent with those of the present study. Unlike ozagrel and pyragrel, the removal of l-carnitine from the reaction system increased rather than decreased the β-oxidation of cinnamic acid, implying that the β-oxidation of cinnamic acid was independent of l-carnitine, similarly to the short- and medium-chain fatty acids [37, 38]. The fact that both butyric acid and decanoic acid tremendously inhibited the β-oxidative metabolism of cinnamic acid further supported this hypothesis. In addition, molecular weight seemed to be negatively correlated to β-oxidative activity in the l-carnitine deficient incubation system (Fig. 7). The β-oxidation of the three compounds was concentration-dependently suppressed by NADH, thus exhibiting the characteristics of NAD+-dependent dehydrogenase activity [14]. The results indicated that NAD+-dependent dehydrogenase might be involved in the β-oxidation of these compounds.
Fig. 7.
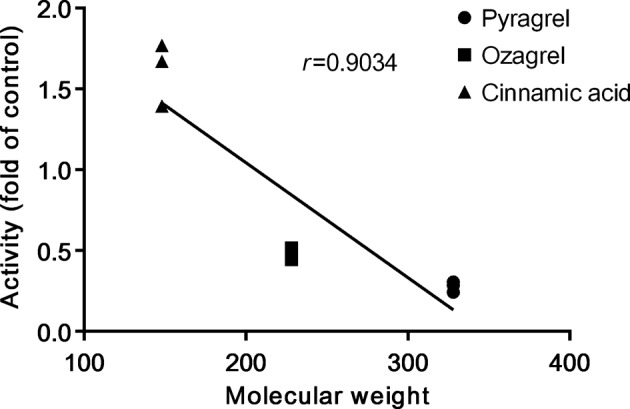
Correlation between β-oxidative activity and molecular weights of cinnamic acid, pyragrel and ozagrel
Generally, double bonds of 2-trans-enoyl-CoAs are reduced to form saturated acyl-CoAs by 2-trans-enoyl-CoA reductase [39] rather than CYP450s [40]. NADPH-dependent or NADH-dependent 2-trans-enoyl-CoA reductases were reported to exist in mitochondria [41–44], and these enzymes were reported to prefer NADPH [40]. We consistently reported that the reduction of cinnamic acid and its analogues in mitochondria was dependent on NADH, and especially NADPH, as evidenced by the higher CLint,invitro values. Triclosan, an inhibitor of enoyl-CoA reductase, inhibited both the NADPH- and NADH-dependent reduction process and especially NADH-dependent reduction in the rat hepatic mitochondria. These findings showed that 2-trans-enoyl-CoA reductase also catalyzed the reduction of the acyl side chains of cinnamic acid and its analogues, which shared the same process of fatty acid elongation [42].
Since NADPH-specific enoyl-CoA reductase was reported to exist in microsomes [43, 44], the reduction of cinnamic acid analogues might be mediated by this enzyme independently of CYP450s. Unlike in the mitochondria, reduction of pyragrel and ozagrel in the microsomes was hardly suppressed by triclosan, indicating that the mechanisms of reduction of cinnamic acid and its analogues were mediated by enoyl reductase in microsomes and were different from those processes in the mitochondria.
The in vitro kinetic study indicated that phenylpropionic acid formation in both mitochondria and microsomes was much greater than benzoic acid formation, while the opposite was true in the hepatocytes and in vivo; this difference may be due to the fast conversion of phenylpropionic acid to cinnamic acid and then to hippuric acid. In contrast to cinnamic acid, the conversion of pyragrel-M2 and ozagrel-M2 to their parent compounds was slight.
A compartmental model involving the parent compounds and their metabolites was successfully developed to characterize the pharmacokinetics of cinnamic acid and its analogues as well as their metabolites in rats. Ozagrel, ozagrel-M1 and ozagrel-M2 were administered (i.v.) to rats according to a previous report [6]. The conversion rates of ozagrel to ozagrel-M1 and ozagrel-M2 could thus be calculated (13.73% to M1 and 33.20% to M2), which were in concordance with the data from the compartmental model (11.04% to M1 and 34.52% to M2), confirming the high accuracy of our model. The in vivo data that was calculated from the model demonstrated that both β-oxidative and reductive metabolism of the acyl side chains of pyragrel and ozagrel only accounted for approximately 26.98% and 45.55% of total metabolism, respectively, indicating the existence of other elimination pathways. Five metabolites of pyragrel were detected in human urine, in which the β-oxidative and reductive metabolites were the major metabolites [2], suggesting the existence of species differences between humans and rats. However, the contribution of β-oxidative metabolism of the acyl side chain of cinnamic acid to the total clearance of cinnamic acid was high (up to 89.59%), indicating that cinnamic acid was mainly eliminated via β-oxidation, which was consistent with the reported in vivo studies of the metabolic fate of cinnamic acid [7].
The novelty of our compartmental model was that the clearance of each part could be calculated from a pharmacokinetic experiment. The results were consistent with our expectation that the predicted in vivo clearances via both β-oxidation and reduction from the cellular organelles (except for ozagrel) or hepatocytes were comparable to the in vivo observations. Peroxisomes were reported to be responsible for the β-oxidation of ozagrel [36], but our preliminary experiment showed that the contribution of peroxisomes to β-oxidative metabolism was approximately 12.86% of mitochondrial contribution. For pyragrel and cinnamic acid, peroxisomes contributed approximately 4.97% and 0.14% of the mitochondrial contribution, respectively, which was much less than the mitochondrial contribution and could be neglected. Other tissues containing mitochondria might also be involved in the β-oxidative metabolism of ozagrel. All of these factors might contribute to the underestimation of the hepatic clearance of ozagrel from hepatic cellular organelles, which needs further investigation.
Carboxylic acid drugs such as valproic acid and tianeptine have been reported to cause hepatotoxicity due to their inhibition of mitochondrial fatty acid β-oxidation [45, 46]. Some fragrances contain cinnamyl phenylpropyl materials, which participate in the same β-oxidative pathways as cinnamic acid analogues [47]. Since these compounds and fatty acids share the same metabolic pathways, the administration of these compounds might affect fatty acids β-oxidation or interfere with endogenous lipid metabolism, leading to hepatotoxicity, and this process thus deserves more attention.
Moreover, some potent G protein-coupled receptor 40 (GPR40)/free fatty acid receptor 1 (FFA1) agonists are phenylpropanoid acid derivatives that can be metabolized to their cinnamic acid and benzoic acid derivatives [48]. To design novel GPR40/FFA1 agonists with low clearance and high plasma exposure, small substituted groups such as a fused-ring structures were introduced to the α- or β-position of the propanoic acid moiety to block the β-oxidation of the phenylalkanoic acid moiety [49]. Our study provided the detailed metabolic information of compounds containing acrylic acid or propanoic acid moiety, and these findings will be useful for the structural optimization of new compounds and related drug-drug interaction studies.
In summary, cinnamic acid and its analogues underwent β-oxidative metabolism and reductive metabolism on their acyl side chain (Fig. 8). Similarly to fatty acids, the β-oxidation of cinnamic acid and its analogues mainly occurred in the mitochondria and was inhibited by fatty acids and NADH. The reduction of cinnamic acid and its analogues, independent of CYP450s, occurred both in mitochondria and microsomes. These results provided detailed metabolic information and highlighted the important roles of the mitochondria and microsomes in the β-oxidative and reductive metabolism of cinnamic acid analogues.
Fig. 8.

Proposed schemes for the β-oxidative and reductive metabolism of cinnamic acid and its analogues in rat hepatocytes
Supplementary Information
Acknowledgements
The project was supported by the National Natural Science Foundation of China (No. 81573490, 81872930, 81473273 and 81673505); Natural Science Foundation of Jiangsu Province of China (BK20161457); and the “333”, “Six Talent Peaks” and “Cyan Blue” Projects of Jiangsu Province. We thank Hefei Industrial Pharmaceutical Institute Co., Ltd. (Hefei, China) for kindly providing the pyragrel and its two metabolites and the Department of Medicinal Chemistry of China Pharmaceutical University (Nanjing, China) kindly providing the ozagrel-M2.
Author contributions
K-jZ and X-dL designed the experiments, analyzed the data, and wrote the manuscript; K-jZ, YC, S-jH, Y-tY, JX, H-yY, LZ, X-gT, Q-sX, T-tY and Y-qZ conducted the experiments; K-jZ and YC performed the data analysis and ML and LL reviewed the manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Li Liu, Phone: +86 25 8327 1006, Email: liulee@cpu.edu.cn.
Xiao-dong Liu, Phone: +86 25 8327 1006, Email: xdliu@cpu.edu.cn.
Electronic supplementary material
The online version of this article (10.1038/s41401-019-0218-8) contains supplementary material, which is available to authorized users.
References
- 1.Fang W, Wei J, Han D, Chen X, He G, Wu Q, et al. MC-002 exhibits positive effects against platelets aggregation and endothelial dysfunction through thromboxane A2 inhibition. Thromb Res. 2014;133:610–5. doi: 10.1016/j.thromres.2014.01.029. [DOI] [PubMed] [Google Scholar]
- 2.Zhao X, Jiang J, Yang G, Huang J, Yang G, He G, et al. Profiling and preparation of metabolites from pyragrel in human urine by online solid-phase extraction coupled with high performance liquid chromatography tandem mass spectrometry followed by a macroporous resin-based purification approach. Molecules. 2017;22:E494. doi: 10.3390/molecules22030494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang G, Pei Q, Fu C, Huang J, Chen J, Yang S, et al. Simultaneous determination of Pyragrel, a novel anti-thrombotic agent, and its two primary metabolites in plasma by HPLC-MS/MS. J Pharm Biomed Anal. 2018;156:199–205. doi: 10.1016/j.jpba.2018.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Nutley BP, Farmer P, Caldwell J. Metabolism of trans-cinnamic acid in the rat and the mouse and its variation with dose. Food Chem Toxicol. 1994;32:877–86. doi: 10.1016/0278-6915(94)90085-X. [DOI] [PubMed] [Google Scholar]
- 5.Oguro H, Mitaki S, Takayoshi H, Abe S, Onoda K, Yamaguchi S. Retrospective analysis of argatroban in 353 patients with acute noncardioembolic stroke. J Stroke Cerebrovasc Dis. 2018;27:2175–81. doi: 10.1016/j.jstrokecerebrovasdis.2018.03.016. [DOI] [PubMed] [Google Scholar]
- 6.Ogiso T, Iwaki M, Hara Y, Tanino T. Pharmacokinetics of ozagrel and its metabolites after intravenous and oral administrations. J Pharm Sci. 1997;86:1111–4. doi: 10.1021/js970013j. [DOI] [PubMed] [Google Scholar]
- 7.Hoskins JA. The occurrence, metabolism and toxicity of cinnamic acid and related compounds. J Appl Toxicol. 1984;4:283–92. doi: 10.1002/jat.2550040602. [DOI] [PubMed] [Google Scholar]
- 8.Seubert W, Podack ER. Mechanisms and physiological roles of fatty acid chain elongation in microsomes and mitochondria. Mol Cell Biochem. 1973;1:29–40. doi: 10.1007/BF01659936. [DOI] [PubMed] [Google Scholar]
- 9.Ranganathan S, Ramasarma T. The metabolism of phenolic acids in the rat. Biochem J. 1974;140:517–22. doi: 10.1042/bj1400517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li F, Zhang M, Xu D, Liu C, Zhong ZY, Jia LL, et al. Co-administration of paroxetine and pravastatin causes deregulation of glucose homeostasis in diabetic rats via enhanced paroxetine exposure. Acta Pharmacol Sin. 2014;35:792–805. doi: 10.1038/aps.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kemp DC, Brouwer KL. Viability assessment in sandwich-cultured rat hepatocytes after xenobiotic exposure. Toxicol Vitr. 2004;18:869–77. doi: 10.1016/j.tiv.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 12.Bjorge SM, Baillie TA. Studies on the beta-oxidation of valproic acid in rat liver mitochondrial preparations. Drug Metab Dispos. 1991;19:823–9. [PubMed] [Google Scholar]
- 13.Hu N, Xie S, Liu L, Wang X, Pan X, Chen G, et al. Opposite effect of diabetes mellitus induced by streptozotocin on oral and intravenous pharmacokinetics of verapamil in rats. Drug Metab Dispos. 2011;39:419–25. doi: 10.1124/dmd.110.035642. [DOI] [PubMed] [Google Scholar]
- 14.Lenartowicz E, Winter C, Kunz W, Wojtczak AB. The inhibition of isocitrate oxidation by palmitoyl-l-carnitine and palmitoyl-CoA in rat liver mitochondria. Eur J Biochem. 1976;67:137–44. doi: 10.1111/j.1432-1033.1976.tb10642.x. [DOI] [PubMed] [Google Scholar]
- 15.Yang X, Lu J, Ying M, Mu J, Li P, Liu Y. Docking and molecular dynamics studies on triclosan derivatives binding to FabI. J Mol Model. 2017;23:25. doi: 10.1007/s00894-016-3192-9. [DOI] [PubMed] [Google Scholar]
- 16.Sippel KH, Vyas NK, Zhang W, Sankaran B, Quiocho FA. Crystal structure of the human fatty acid synthase enoyl-acyl carrier protein-reductase domain complexed with triclosan reveals allosteric protein-protein interface inhibition. J Biol Chem. 2014;289:33287–95. doi: 10.1074/jbc.M114.608547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu B, Wang Y, Fillgrove KL, Anderson VE. Triclosan inhibits enoyl-reductase of type I fatty acid synthase in vitro and is cytotoxic to MCF-7 and SKBr-3 breast cancer cells. Cancer Chemother Pharmacol. 2002;49:187–93. doi: 10.1007/s00280-001-0399-x. [DOI] [PubMed] [Google Scholar]
- 18.Wu J, Jin Z, Zheng H, Yan LJ. Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. Diabetes Metab Syndr Obes. 2016;9:145–53. doi: 10.2147/DMSO.S106087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diaz-Flores M, Ibanez-Hernandez MA, Galvan RE, Gutierrez M, Duran-Reyes G, Medina-Navarro R, et al. Glucose-6-phosphate dehydrogenase activity and NADPH/NADP+ratio in liver and pancreas are dependent on the severity of hyperglycemia in rat. Life Sci. 2006;78:2601–7. doi: 10.1016/j.lfs.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 20.Dou YH, Wang L, He GW, Wu Q, Fan GR, Hang TJ. Determination of (E)-3-(3-methoxy-4-((3,5,6-trimethylpyrazin-2-yl)methoxy)phenyl)acrylate in rat plasma by RP-HPLC. J Pharm Pract. 2012;30:42–4, 48. [Google Scholar]
- 21.Yang BK, Wang SJ, Mo LL, Zeng J, Zhong YM, Zang LQ. Studies on the absolute bioavailability of cinnamic acid in rats and its absorption properties. PharmJChinPLA. 2013;29:13–6. [Google Scholar]
- 22.Xu D, Li F, Zhang M, Zhang J, Liu C, Hu MY, et al. Decreased exposure of simvastatin and simvastatin acid in a rat model of type 2 diabetes. Acta Pharmacol Sin. 2014;35:1215–25. doi: 10.1038/aps.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naritomi Y, Terashita S, Kimura S, Suzuki A, Kagayama A, Sugiyama Y. Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metab Dispos. 2001;29:1316–24. [PubMed] [Google Scholar]
- 24.Guest EJ, Aarons L, Houston JB, Rostami-Hodjegan A, Galetin A. Critique of the two-fold measure of prediction success for ratios: application for the assessment of drug-drug interactions. Drug Metab Dispos. 2011;39:170–3. doi: 10.1124/dmd.110.036103. [DOI] [PubMed] [Google Scholar]
- 25.Parrott N, Paquereau N, Coassolo P, Lave T. An evaluation of the utility of physiologically based models of pharmacokinetics in early drug discovery. J Pharm Sci. 2005;94:2327–43. doi: 10.1002/jps.20419. [DOI] [PubMed] [Google Scholar]
- 26.Bustamante E, Soper JW, Pedersen PL. A high-yield preparative method for isolation of rat liver mitochondria. Anal Biochem. 1977;80:401–8. doi: 10.1016/0003-2697(77)90661-3. [DOI] [PubMed] [Google Scholar]
- 27.Poet TS, Wu H, English JC, Corley RA. Metabolic rate constants for hydroquinone in F344 rat and human liver isolated hepatocytes: application to a PBPK model. Toxicol Sci. 2004;82:9–25. doi: 10.1093/toxsci/kfh229. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Ma Y, Ma W. Pharmacokinetics and bioavailability of cinnamic acid after oral administration of Ramulus Cinnamomi in rats. Eur J Drug Metab Pharmacokinet. 2009;34:51–6. doi: 10.1007/BF03191384. [DOI] [PubMed] [Google Scholar]
- 29.Guengerich FP. Cytochrome p450 and chemical toxicology. Chem Res Toxicol. 2008;21:70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- 30.Fukami MH, Williamson JR. On the mechanism of inhibition of fatty acid oxidation by 4-pentenoic acid in rat liver mitochondria. J Biol Chem. 1971;246:1206–12. [PubMed] [Google Scholar]
- 31.Suzue G, Marcel YL. Kinetic studies on the chain length specificity of long chain acyl coenzyme A synthetase from rat liver microsomes. J Biol Chem. 1972;247:6781–3. [PubMed] [Google Scholar]
- 32.Krisans SK, Mortensen RM, Lazarow PB. Acyl-CoA synthetase in rat liver peroxisomes. Computer-assisted analysis of cell fractionation experiments. J Biol Chem. 1980;255:9599–607. [PubMed] [Google Scholar]
- 33.Lageweg W, Wanders RJ, Tager JM. Long-chain-acyl-CoA synthetase and very-long-chain-acyl-CoA synthetase activities in peroxisomes and microsomes from rat liver. An enzymological study. Eur J Biochem. 1991;196:519–23. doi: 10.1111/j.1432-1033.1991.tb15844.x. [DOI] [PubMed] [Google Scholar]
- 34.Lewin TM, Van Horn CG, Krisans SK, Coleman RA. Rat liver acyl-CoA synthetase 4 is a peripheral-membrane protein located in two distinct subcellular organelles, peroxisomes, and mitochondrial-associated membrane. Arch Biochem Biophys. 2002;404:263–70. doi: 10.1016/S0003-9861(02)00247-3. [DOI] [PubMed] [Google Scholar]
- 35.Mannaerts GP, Debeer LJ. Mitochondrial and peroxisomal beta-oxidation of fatty acids in rat liver. Ann N Y Acad Sci. 1982;386:30–9. doi: 10.1111/j.1749-6632.1982.tb21405.x. [DOI] [PubMed] [Google Scholar]
- 36.Yamada J, Horie S, Watanabe T, Suga T. Participation of peroxisomal beta-oxidation system in the chain-shortening of a xenobiotic acyl compound. Biochem Biophys Res Commun. 1984;125:123–8. doi: 10.1016/S0006-291X(84)80343-5. [DOI] [PubMed] [Google Scholar]
- 37.Schonfeld P, Wojtczak L. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res. 2016;57:943–54. doi: 10.1194/jlr.R067629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bremer J. Carnitine--metabolism and functions. Physiol Rev. 1983;63:1420–80. doi: 10.1152/physrev.1983.63.4.1420. [DOI] [PubMed] [Google Scholar]
- 39.Smeland TE, Nada M, Cuebas D, Schulz H. NADPH-dependent beta-oxidation of unsaturated fatty acids with double bonds extending from odd-numbered carbon atoms. Proc Natl Acad Sci USA. 1992;89:6673–7. doi: 10.1073/pnas.89.15.6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagi MN, Prasad MR, Cook L, Cinti DL. Biochemical properties of short- and long-chain rat liver microsomal trans-2-enoyl coenzyme A reductase. Arch Biochem Biophys. 1983;226:50–64. doi: 10.1016/0003-9861(83)90270-9. [DOI] [PubMed] [Google Scholar]
- 41.Prasad MR, Chiang CF, Cook L, Cinti DL. Solubilization and purification of hepatic microsomal trans-2-enoyl-CoA reductase: evidence for the existence of a second long-chain enoyl-CoA reductase. Arch Biochem Biophys. 1985;237:535–44. doi: 10.1016/0003-9861(85)90306-6. [DOI] [PubMed] [Google Scholar]
- 42.Hinsch W, Klages C, Seubert W. On the mechanism of malonyl-CoA-independent fatty-acid synthesis. Different properties of the mitochondrial chain elongation and enoylCoA reductase in various tissues. Eur J Biochem. 1976;64:45–55. doi: 10.1111/j.1432-1033.1976.tb10273.x. [DOI] [PubMed] [Google Scholar]
- 43.Prasad MR, Nagi MN, Cook L, Cinti DL. Kinetic evidence for two separate trans-2-enoyl CoA reductases in rat hepatic microsomes: NADPH-specific short chain- and NAD(P)H-dependent long chain-reductase. Biochem Biophys Res Commun. 1983;113:659–65. doi: 10.1016/0006-291X(83)91777-1. [DOI] [PubMed] [Google Scholar]
- 44.Cinti DL, Nagi MN, Cook L, White RE. Evidence for a second microsomal trans-2-enoyl coenzyme A reductase in rat liver. NADPH-Specif Short chain reductase J Biol Chem. 1982;257:14333–40. [PubMed] [Google Scholar]
- 45.Farinelli E, Giampaoli D, Cenciarini A, Cercado E, Verrotti A. Valproic acid and nonalcoholic fatty liver disease: A possible association? World J Hepatol. 2015;7:1251–7. doi: 10.4254/wjh.v7.i9.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fromenty B, Freneaux E, Labbe G, Deschamps D, Larrey D, Letteron P, et al. Tianeptine, a new tricyclic antidepressant metabolized by beta-oxidation of its heptanoic side chain, inhibits the mitochondrial oxidation of medium and short chain fatty acids in mice. Biochem Pharmacol. 1989;38:3743–51. doi: 10.1016/0006-2952(89)90580-7. [DOI] [PubMed] [Google Scholar]
- 47.Panel RE, Belsito D, Bickers D, Bruze M, Calow P, Dagli M, et al. A toxicologic and dermatologic assessment of cinnamyl phenylpropyl materials when used as fragrance ingredients. Food Chem Toxicol. 2011;49(Suppl 2):S256–67. doi: 10.1016/j.fct.2011.07.053. [DOI] [PubMed] [Google Scholar]
- 48.Sasaki S, Kitamura S, Negoro N, Suzuki M, Tsujihata Y, Suzuki N, et al. Design, synthesis, and biological activity of potent and orally available G protein-coupled receptor 40 agonists. J Med Chem. 2011;54:1365–78. doi: 10.1021/jm101405t. [DOI] [PubMed] [Google Scholar]
- 49.Negoro N, Sasaki S, Ito M, Kitamura S, Tsujihata Y, Ito R, et al. Identification of fused-ring alkanoic acids with improved pharmacokinetic profiles that act as G protein-coupled receptor 40/free fatty acid receptor 1 agonists. J Med Chem. 2012;55:1538–52. doi: 10.1021/jm2012968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.