

Abstract
The manipulation of bile acid (BA) homeostasis by blocking the ileal apical Na+-dependent bile salt transporter (ASBT/SLC10A2) may have therapeutic effects in nonalcoholic fatty liver disease. We developed a novel ASBT inhibitor, an N-(3,4-o-dichlorophenyl)−2-(3-trifluoromethoxy) benzamide derivative referred to as IMB17–15, and investigated its therapeutic effects and the molecular mechanisms underlying the effects. Syrian golden hamsters were challenged with high-fat diet (HFD) to induce NAFLD and were subsequently administered 400 mg/kg IMB17–15 by gavage daily for 21 days. Serum, liver, and fecal samples were collected for further analysis. Plasma concentration-time profiles of IMB17–15 were also constructed. The human hepatocyte cell line HL-7702 was treated with Oleic acid (OA) with or without IMB17–15. Western blotting and real-time PCR were used to study the molecular mechanisms of IMB17–15. We found that IMB17–15 inhibited ASBT and subsequently suppressed ileal farnesoid X receptor (FXR) and FXR-activated fibroblast growth factor15/19 (FGF15/19) expression, which reduced the hepatic phosphorylated extracellular regulated protein kinase (ERK) and c-Jun N-terminal kinase (JNK) levels and upregulated the cholesterol 7α-hydroxylase (CYP7A1) activity. Additionally, IMB17–15 stimulated adenosine monophosphate (AMP)-activated protein kinase (AMPKα) phosphorylation and enhanced peroxisome proliferator activated receptor α (PPARα) expression and thus promoted triglyceride (TG) oxidation and high-density lipoprotein cholesterol (HDL-c) metabolism through an ASBT-independent mechanism. In conclusion, a novel ASBT inhibitor known as IMB17–15 protected hamsters against HFD-induced NFALD by manipulating BA and lipid homeostasis. IMB17–15 also reduced lipid deposition in human hepatic cell lines, indicating that it may be useful as a therapy for NAFLD patients.
Keywords: ASBT inhibitor, NAFLD, CYP7A1, AMPKα, PPARα
Introduction
Nonalcoholic fatty liver disease (NAFLD) comprises a cluster of clinicopathological syndromes ranging from simple lipid accumulation (steatosis) [nonalcoholic fatty liver (NAFL)] to intralobular inflammation and hepatocyte ballooning, as well as hepatic steatosis [non-alcoholic steatohepatitis (NASH)] [1]. NAFL is traditionally regarded as a benign disease because of the reversibility of the steatosis, whereas NASH is generally considered a progressive disease that sets the stage for further liver damage and additional diseases, including fibrosis, cirrhosis and hepatocellular carcinoma [2]. Sustained hepatic inflammation marks the transition toward NASH. However, the actual risk factors that initiate the inflammatory response during the progression to NASH remain largely unknown. Although lifestyle changes and/or treatment with lipid-regulating agents have improved NAFLD conditions in some patients, these strategies tend to fail more often than they succeed [3]. A recent study indicated that the cholesterol content in hepatocytes plays a critical role in NAFLD pathology because cholesterol-loaded hepatocytes are the major source of inflammatory cytokines and induce inflammation [4]. These findings suggest that pharmacologically lowering hepatic cholesterol may be a new option for treating NAFLD.
The hepatic level of cholesterol can be modulated by manipulating the BA pool size because cholesterol is used for BA synthesis. Two major components maintain the BA pool size, i.e., de novo BA synthesis in the liver and reclaimed BAs from the ileum. In the liver, CYP7A1 is the rate-limiting enzyme in converting cholesterol to BA. Dysfunction of CYP7A1 causes hypercholesterolemia [5]. In contrast, stimulation of CYP7A1 expression lowers the level of cholesterol [6]. CYP7A1 expression is negatively regulated by BA via the FXR. This FXR-dependent regulation is mediated by two separate pathways. In the liver, BA activates FXR and stimulates the expression of the short heterodimer partner (SHP), which, in turn, inhibits the transactivation of liver receptor homolog 1 and hepatocyte nuclear factor 4α (HNF4α), two transactivators of CYP7A1 [7]. In the ileum and possibly the liver, BA induces the expression of FGF19 in humans or FGF15 in rodents through activation of FXR. FGF19/15 in turn activates its receptor FGFR4 and represses hepatic CYP7A1 expression through signal transduction [6].
In the ileum, BAs are reclaimed through the ASBT and the organic solute transporters OSTα and β (SLC51A/B), a pair of membrane transporters responsible for BA uptake and efflux from enterocytes and return to the liver through the portal veins [8]. Loss of ASBT or OSTα/β results in a reduced BA pool size and triggers de novo BA synthesis. Therefore, inhibition of ASBT conceivably reduces the BA pool size and thus lowers hepatic cholesterol. Moreover, recent studies that used non-absorbable ASBT inhibitors demonstrated improved liver conditions in rodent models of NAFLD [9, 10]. However, these drug candidates are in the early stage of their clinical trials. Thus, novel drugs are needed for NAFLD.
Recently, we synthesized a series of novel potent ASBT inhibitors using cell-based in vitro assays [11], including N-(3,4-dichlorophenyl)−2-(3-trifluoromethoxy) benzamide, which is also referred to as IMB17–15 (Fig. 1a). In this report, we assess the hepatoprotective effects of IMB17–15 in a high-fat diet-fed hamster NAFLD model because its lipid profile is more similar to that in humans than that in mice and rats. We found that this absorbable ASBT inhibitor inhibited bile acid absorption in the ileum, stimulated hepatic CYP7A1 expression and prevented liver injury in the high-fat diet-fed hamster model of NAFLD. Our data suggest that IMB17–15 may warrant a clinical trial for the treatment of patients with NAFLD.
Fig. 1.

IMB17–15 showed no significant toxicity in KM mice. a The structure of IMB17–15. b KM mice were treated with different concentrations of IMB17–15 once by gavage and were continuously observed for 14 days. The relative body weight was calculated by the ratio of the final weight and the initial weight. This figure shows that IMB17–15 was not toxic in KM mice. c Histopathological examination of various organs (H&E staining, ×40) from KM mice with or without IMB17–15 treatment at a dose of 2000 mg/kg. No toxicopathological changes were found in the heart, liver, spleen, lung, and kidney. d Plasma concentration-time profiles of IMB17–15 after gavage administration at a single dose of 400 mg/kg in hamsters. KM mice Kunming mice. The values are expressed as the mean ± SD (n = 4 in each group for the acute oral toxicity study, n = 6 in each group for the pharmacokinetics in vivo study)
Material and methods
Animal experiments
All animal experiments were approved by the local IACUC (Institutional Animal Care and Use Committee, Beijing, China) and were conducted according to the protocols described by the guidelines for drug testing (China Food and Drug Administration, 2014a). Adult Kunming (KM) mice (weighing 18–22 g) of both genders and male Syrian golden hamsters (weighing 80–100 g) were purchased from the Institute of Laboratory Animal Science (Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China). The animals were maintained under pathogen-free conditions and were housed at a temperature of 20–24 °C and a humidity of 40–60% under a 12 h/12 h alternating light/dark cycle.
Acute oral toxicity study
A total of 20 KM mice of both genders were randomly divided into 5 groups with 4 animals (two females and two males) in each group. After the mice were fasted for 12 h, increasing concentrations of IMB17–15 (50, 500, 1000 and 2000 mg/kg) were orally administered once to the mice. The control group received an equal volume of water. The animals were observed for general behavioral changes and signs of toxicity during the entire experiment for 14 days. The mice were weighed on the first and last days. At the end of the experiment, the major organs, including the heart, lung, liver and kidney, were collected for histological analysis.
Hamster NAFLD model establishment
After three days of adaption to their environment, the hamsters were weighed and randomly divided into the following three groups: (1) the normal chow diet group (control group); (2) the high-fat diet (HFD) group treated with normal saline (NS) solution daily for 21 days (HFD+NS group) and (3) the HFD group treated with 400 mg/kg IMB17–15 by gavage daily for 21 days (HFD+IMB17–15 group). The normal chow diet (NCD) group was fed standard laboratory chow. As described previously, compared with HFD treatment alone, cholic acid supplement enhances the severity of NAFLD [12]. Thus, in this study, the HFD consisted of 15% lard, 10% saccharose, 1% cholesterol and 0.1% cholic acid. At the end of the study, the animals were anaesthetized with isoflurane after an overnight fast. Blood, liver and ileum samples were collected for analysis.
In vivo study of the pharmacokinetics (PKs) of IMB17–15
After environmental adaption and a 12 h fast, six male hamsters were administered 400 mg/kg IMB17–15 by gavage once. Blood samples (200 μL) were collected from the retro-orbital venous plexus using vacuum tubes containing heparin sodium at 0, 0.25, 0.5, 1, 2, 4, 8, 12, 24 and 36 h after intragastric administration of IMB17–15. The samples were then subjected to centrifugation at 1200 × g for 10 min to isolate the plasma. One-hundred-microliter plasma samples were mixed with 900 μL ethyl acetate. After vortexing for 1 min and centrifuging at 13,400 × g for 10 min, the supernatants were concentrated using a vacuum concentrating apparatus (Concentrator 5301 Eppendorf, Hamburg, Germany). Two hundred microliters of methyl alcohol were added to the residue and filtered through a 0.22 μm membrane. A linear range of 100, 50, 25, 12.5, 6.25, 3.125 and 1.5625 μg/mL IMB17–15 standards were dissolved in blank plasma samples before being subjected to the above-described procedures to establish a standard curve, which was y = 13.928x−57.404 (R2 = 0.9993) (y indicates the plasma IMB17–15 concentration, and x indicates the peak area in the HPLC chromatogram).
Serum biochemistry parameter and BA detection
The plasma alanine aminotransferase (ALT), aspartate aminotransferase (AST), total cholesterol (TC), triglyceride (TG), low-density lipoprotein (LDL-C), high-density lipoprotein (HDL-C) and total BA (TBA) concentrations were analyzed with a Hitachi 7100 Analyzer using kits obtained from Zhongsheng Beikong Biotechnology (Beijing, China).
Measurement of hepatic TBA, TC and TG levels and fecal BA content
For the measurement of the BA levels, liver tissues were homogenized in 9-fold diluted normal saline and centrifuged at 13,400 × g for 10 min. The fecal samples were oven-dried and suspended in tertiary butanol/water (1:1, v/v) [13, 14]. After vortexing and sonication for 10 min, the resulting suspensions were centrifuged twice at 18,000 × g for 10 min. Both supernatants were collected for analysis. To measure the TC and TG levels, the liver tissue samples were homogenized in 9-fold diluted absolute ethyl alcohol, and the homogenates were centrifuged at 600 × g for 10 min to collect the supernatants for analysis. The TBA, TC and TG levels were determined using commercially available kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), according to the manufacturer’s instructions.
Histological analysis of tissue samples
The tissue samples were fixed in 10% formaldehyde, embedded in paraffin and subsequently stained with hematoxylin and eosin (H&E). Liver inflammation was quantified on a scale of 1 to 5 in a blinded manner with a Leica DM1000 microscope (×10 objective lens).
PKs analysis
The plasma IMB17–15 concentrations were determined using an HPLC-MS/MS system (Agilent, Palo Alto, CA, USA). The injection volume was 20 μL, and the column temperature was 25 °C. Methanol was used as the mobile phase. The run time was 10 min, and the gradient increased from 50% organic to 100% organic over 5 min at a flow rate of 1 mL/min. Ionization was performed using an electrospray ionization source with a gas flow rate of 12 L/min. The nebulizer pressure was set to 35 psi, the source temperature was set to 300 °C, and the capillary voltage was set to 4000 V. Data acquisition was performed using Mass Hunter Data Acquisition B.04.01 Software (Agilent) under positive polarity.
The two-compartment method was applied for the estimation of the PK parameters for the extravenous administration of IMB17–15 using PKSolver 2.0 [15]. The time-concentration curve of the intragastric route was best-matched in the PKSolver program as follows: Concentration (T)=Ae−α∙T+Be−β∙T. The PK parameters were calculated via PKSolver.
BA assessment
Liver sample preparation: Each 10 mg liver tissue sample was homogenized in acetonitrile/methanol (8:2, containing the ten internal standards DCA-d4, GCA-d4, TCA-d4, TCDCA-d9, CA-d4, LCA-d4, UDCA-d4, GDCA-d4, GCDCA-d4 and β-CA-d5, 150 nM each). The samples were centrifuged (17500 × g, 4 °C for 20 min), and the supernatant was lyophilized. The sample residue was further extracted using a 100 μL aliquot of methanol/acetonitrile (2:8, containing the same ten internal standards as the first extraction solvent) and centrifuged (17500 × g, 4 °C for 20 min). The supernatant was then transferred to a sampling vial for analysis, and the injection volume was 5 μL.
Serum sample preparation: A 180 μL aliquot of acetonitrile/methanol (8:2) containing the same ten internal standards was added to 20 μL of serum. After centrifuging at 200 × g and 10 °C for 20 min, the supernatant was lyophilized. The dried samples were reconstituted with 1:1 (v/v) of methanol/acetonitrile (2:8) and centrifuged at 17,500 × g and 4 °C for 20 min. The supernatant was used for LC-MS analysis, and the injection volume was 5 μL.
BA analysis was performed on the UPLC-MS/MS system (ACQUITY UPLC-Xevo TQ-S, Waters Corp., Milford, MA, USA). The column temperature was 30 °C. The elution solvents were water+formic acid (pH = 3.25) (A) and acetonitrile/methanol (80:20) (B). The gradient conditions at a flow rate of 400 μL/min were as follows: 0–1 min (5% B), 1–3 min (5–30% B), 3–15 min (30–100% B), and 16–17 min (5% B). MS was conducted in the negative electrospray ionization mode. The raw data files generated by UPLC-MS/MS were processed using QuanMET software (v1.0, Metabo-Profile, Shanghai, China) to perform calibration and quantitation for each metabolite.
Hepatocyte culture, steatosis induction and treatment
The human hepatocyte cell line HL-7702 was obtained from the Institute of Biochemistry and Cell Biology (SIBS, CAS, Shanghai, China). The cells were maintained in RPMI-1640 medium with 20% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S, Invitrogen, Carlsbad, CA, USA) and were grown at 37 °C in a 5% CO2 humidified atmosphere [16].
As previously described [17], 600 μM oleic acid (OA) was used to establish the in vitro hepatic steatosis model. HL-7702 cells were seeded in 6-well plates and incubated with OA for 24 h. The cells underwent different treatments for an additional 24 h as shown in the results section. In the blocking experiments, serum-starved HL-7702 cells were pretreated with 10 μM compound C (CC, Selleck, S7840) for 1 h; IMB17–15 or OA were subsequently added for 24 h.
Oil Red O staining and intracellular TG assay
At the end of the treatment, HL-7702 cells were fixed with 4% paraformaldehyde for 15 min at room temperature and then washed thrice with PBS. ORO working solution (0.5% ORO saturated solution: distilled water = 3:2) was added and incubated for 15 min at room temperature. After 60% isopropanol rinsing and PBS washing, the intracellular lipid droplets were stained and subsequently observed with a light microscope (Leica DM1000). In parallel experiments, cells were lysed, and the intracellular TG contents were evaluated by an Intracellular TG Assay Kit and normalized to the protein concentrations.
Analysis of gene expression by real-time quantitative PCR
Total RNA from cultured cells and tissue samples was extracted and reverse-transcribed into cDNAs according to the supplier’s protocols. Real-time quantitative PCR was performed in an ABI 7500 Fast Real-Time PCR System. The ACTB gene was employed as an internal reference. The gene-specific primers used for these experiments are listed in Tables 1 and 2.
Table 1.
Primer sets for real-time PCR analysis of hamster gene expression
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| Fxr | TCAGTTGCTGGGAATAGACTAAGG | GCTAACCTGAACCGGATTTTTCT |
| Fgf15 | ACCCGCCTGCAGTACCTGT | GCCGAGTAATGAATCAGCC |
| Cyp7a1 | ACTGCTAAGGAGGATTTCACTCT | CTCATCCAGGTATCGATCATATT |
| Ldlr | TGAGGAACATCAACAGCATAAAC | ATCCTCCAGGCTGACCATCTGT |
| Pparα | CTCCACCTGCAGAGCAACCA | CGTCAGACTCGGTCTTCTTGAT |
| Fas | AGCCCCTCAAGTGCACAGTG | TGCCAATGTGTTTTCCCTGA |
| Acc | CTGCTGGAGACCGAAAGCTT | CAACATGGTGTCAGGACGTTCT |
| Srebp1c | GCTGTTGGCATCCTGCTATC | TAGCTGGAAGTGACGGTGGT |
| Scd1 | TGGGTTGGCTGCTTGTG | GCGTGGGCAGGATGAAG |
| Asbt | GTAGATGGCGACATGGACCT | GAGAAGTGCCAATGCTGTCA |
| Il-6 | GGACAATGACTATGTGTTGTTAGAA | AGGCAAATTTCCCAATTGTATCCAG |
| Il-1β | GCAGGTGGTGTCAGTCATTG | AGACAGCACGAGGCATTTCT |
| Tnf-α | TGAGCCATCGTGCCAATG | AGCCCGTCTGCTGGTATCAC |
| Actb | CAGCCTTCCTTCCTGGGTATGG | CAGGAGGAGCAATGATCTTGAT |
ACC acetyl-CoA carboxylase, ACTB Beta-actin, ASBT/SLC10A2 apical Na+-dependent bile salt transporter, CYP7A1 cholesterol 7α-hydroxylase, FAS fatty acid synthase, FGF fibroblast growth factor, FXR farnesoid X receptor, IL-1β interleukin 1β, IL-6 interleukin 6, LDLR low-density lipoprotein receptor, PPARα peroxisome proliferator activated receptor α, SCD1 stearoyl-CoA desaturase, SREPB1C sterol regulatory element-binding protein 1c, TNFα tumor necrosis factor α
Table 2.
Primer sets for real-time PCR analysis of human gene expression
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| PPARα | CGCAATCCATCGGCGAGGAT | ACCACAGGATAAGTCACCGAG |
| CPT1A | CAAAACCATGTTGTACAGCTTCCA | GCTTCCTTCATCAGAGGCTTCA |
| SCP2 | TGGCCTGACACCAAATGATA | CCTAGTGGGTGTCCCTTTGA |
| ApoA-I | AGCTTGCTGAAGGTGGAGGT | ATCGAGTGAAGGACCTGGC |
| ApoA-V | CCATCGTGTAGGGCTTCAGT | CCTGAAAGACAGCCTTGAGC |
| ACTB | TGGCACCCAGCACAATGAA | CTAAGTCATAGTCCGCCTAGAAGCA |
ACTB Beta-actin, ApoA-I apolipoprotein A-I, ApoA-V Apolipoprotein A-V, CPT1A carnitine palmitoyltransferase I, PPARα peroxisome proliferator activated receptor α, SCP-2 sterol carrier protein 2
Analysis of protein expression by Western blotting
The cells and homogenized tissues were lysed in RIPA buffer. The protein concentrations were determined by an enhanced BCA Protein Assay Kit. Equal amounts of lysate were mixed with 2× SDS loading buffer. The samples were separated by SDS-PAGE and transferred to PVDF membranes (Millipore, Bedford, MA, USA), which were blocked with 5% skim milk in PBS-T buffer for 1 h at room temperature. Protein detection was performed using polyclonal rabbit antibodies to total p44/42 MAPK (Erk1/2) (Rabbit mAb #4695), phosphorylated p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (Rabbit mAb #4370), total SAPK/JNK (#9252), phosphorylated SAPK/JNK (Thr183/Tyr185) (Rabbit mAb #4668), total AMPKα (Rabbit mAb #5831), phosphorylated AMPKα (Thr172) (Rabbit mAb #2535), IL-6 (Rabbit mAb #12153), IL-1β (Rabbit mAb #12703), TNF-α (Rabbit mAb #6945) (Cell Signaling Technology, Danvers, MA, USA), LDLR (ab30532), PPARα (ab24509) (Abcam, Cambridge, MA, USA), CYP7A1 (MABD42, Millipore, Bedford, MA, USA) and β-actin (sc-58673, Santa Cruz Biotechnology). Bound antibodies were detected using the appropriate horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology, Danvers, MA, USA) and were developed using a ProteinSimple FluorChem HD2 imaging system. Semiquantitation of the changes in the protein expression was performed by grayscale imaging using ImageJ (https://imagej.nih.gov).
Statistical analysis
All data are expressed as the mean ± SD. The data were analyzed by one-way ANOVA, followed by Duncan’s multiple range tests (SPSS, version 23.0, SPSS Inc., Chicago, IL, USA). The results were considered statistically significant at P < 0.05 and extremely significant at P < 0.01.
Results
Acute oral toxicity
As shown in Fig. 1b, even at a dose of 2000 mg/kg, IMB17–15 did not change the body weights of the mice. No behavioral changes were observed, including the respiratory rate, convulsions, analgesia, uroclepsia, hygrostomia and erythema and local skin edema, in the treated groups. Examination of the tissue histology did not show abnormalities in the heart, liver, lung and kidney (Fig. 1c). These observations indicate that IMB17–15 was well tolerated in mice, even at a dose of 2000 mg/kg and did not cause liver or other organ injury.
Evaluation of IMB17–15 PKs in hamsters
As it has been reported that classical ASBT inhibitors can hardly be detected in the plasma, pharmacokinetic analysis was performed to evaluate the in vivo process of the novel ASBT inhibitor IMB17–15. No side effects or signs of intolerance were observed during the experiment with 400 mg/kg IMB17–15. Interestingly, IMB17–15 was detected in the plasma of these animals at a maximum concentration of 37.57 ± 3.39 μg/mL 0.87 h after oral administration. The plasma concentration-time profile of IMB17–15 is shown in Fig. 1d. The drug remained in the circulation for more than 24 h in these animals. The corresponding PK parameters are summarized in Table 3. These results indicate that IMB17–15 was an absorbable ASBT inhibitor in contrast to the previously reported ASBT inhibitors.
Table 3.
PK parameters after treatment with 400 mg/kg IMB17–15 by gavage in hamsters (n = 6)
| Parameter | Value |
|---|---|
| Ka (1/h) | 2.19 ± 0.57 |
| K10 (1/h) | 0.12 ± 0.04 |
| K12 (1/h) | 0.86 ± 0.57 |
| K21 (1/h) | 0.50 ± 0.33 |
| t1/2(α) (h) | 0.78 ± 0.65 |
| t1/2(β) (h) | 17.97 ± 5.11 |
| t1/2(Ka) (h) | 0.35 ± 0.13 |
| Cmax (μg/mL) | 37.57 ± 3.39 |
| Tmax (h) | 0.87 ± 0.32 |
| AUC0-t (μg/mL*h) | 455.85 ± 82.76 |
| MRT (h) | 24.44 ± 6.07 |
PK parameters were generated using two-compartment model analysis, Ka absorption rate constant, K10 elimination rate constant, K12 and K21 transfer rate constant, t1/2(α) distribution half-life, t1/2(β) elimination half-life, t1/2(Ka) absorption half-life, Cmax peak plasma concentration, Tmax time to Cmax, AUC0-t area under the curve until the last time point, MRT mean residence time
The values are expressed as the mean ± SD (n = 6 in each group)
IMB17–15 ameliorated serum biochemistry parameter abnormalities and liver steatosis in hamsters
As shown in Fig. 2a, b, 21 days of HFD feeding markedly increased the body weight and the liver weight to body weight ratio when the HFD-NS group was compared with the control diet group. However, IMB17–15 treatment significantly prevented this weight gain and the increase in the liver weight to body weight ratio due to HFD feeding. The serum biochemistry measurements also revealed that HFD feeding markedly increased the serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and lipid parameters in these hamsters. Again, IMB17–15 treatment significantly decreased the serum levels of ALT, AST, total cholesterol (TC), triglyceride (TG) and low-density lipoprotein cholesterol (LDL-c) and increased the serum high-density lipoprotein cholesterol (HDL-c) compared to those in the HFD-NS group (Table 3). Liver histology revealed that the livers from the NCD group had a normal histological structure. However, the livers from the HFD-NS group displayed inflammation, severe steatosis, and diffuse and extensive lipid-enriched vacuole formation. The administration of IMB17–15 in HFD-fed animals resulted in marked reductions in inflammation and fatty accumulation and the reversal of hepatic steatosis (Fig. 2d). A blinded assessment of the liver histology further confirmed that IMB17–15 supplementation significantly alleviated inflammation compared with the HFD-fed hamsters (Fig. 2c). Together, these findings indicate that IMB17–15 prevented liver injury in the HFD-induced NAFLD model.
Fig. 2.

IMB17–15 markedly prevented weight gain due to HFD feeding and improved liver histology in HFD-fed hamsters. a Relative body weight gain in the control, HFD + NS and HFD + IMB17–15 groups. b The liver weight to body weight ratio after 21 days. Double-blinded quantitative assessment of (c) Hepatic inflammation showed that IMB17–15 ameliorated HFD-induced histopathological changes. d Liver pathological changes were detected by H&E staining (magnification × 400). H&E hematoxylin and eosin, HFD high fat diet. The values are expressed as the mean ± SD (n = 6 in each group), ##P < 0.01 versus the control group, *P < 0.05, **P < 0.01 versus the HFD + NS group; ANOVA followed by Duncan's multiple range tests
IMB17–15 treatment altered BA composition and increased fecal BA excretion in hamsters
As demonstrated in previous studies [11], IMB17–15 is a novel and potent ASBT inhibitor. Consistent with the results of other ASBT inhibitor-related studies [9], our results showed that IMB17–15 increased the blood and hepatic BA levels and impaired the ileal BA absorption. Specifically, IMB17–15 elicited a statistically significant increase in BA excretion in the feces (Table 4). No discernible impact on the hepatic BA composition was found among the three groups. HFD feeding increased most kinds of BA in the serum, particularly the FXR agonistic BA species (non–6-hydroxylated BAs), such as TCA, TCDCA, TDCA, CDCA and TLCA, compared to the standard chow. IMB17–15 administration reduced the levels of CDCA, the most potent FXR agonist, and other FXR endogenous agonists (TCDCA, TDCA and TLCA) in the serum (Fig. 3a, b). Furthermore, IMB17–15 treatment also suppressed Fxr and Fxr-activated Fgf15 expression; however, it did not alter the ASBT expression in the ileum compared to the untreated HFD-fed group (Fig. 4a, b). These results indicate that IMB17–15 treatment resulted in antagonism of the FXR in the ileum.
Table 4.
Hamster serum and liver biochemical parameters
| Control | HFD+NS | HFD+IMB17–15 | |
|---|---|---|---|
| Serum ALT (U/L) | 89.00 ± 20.55 | 169.21 ± 36.51## | 121.03 ± 13.67* |
| Serum AST (U/L) | 91.88 ± 20.03 | 154.63 ± 40.12# | 92.61 ± 16.07* |
| Serum TC (mmol/L) | 4.16 ± 0.52 | 27.41 ± 7.74## | 17.24 ± 3.65* |
| Serum TG (mmol/L) | 1.77 ± 0.52 | 16.75 ± 9.02## | 3.94 ± 1.95* |
| Serum LDL-C (mmol/L) | 0.94 ± 0.19 | 11.37 ± 3.56## | 5.69 ± 1.60* |
| Serum HDL-C (mmol/L) | 2.66 ± 0.37 | 3.95 ± 1.24## | 6.99 ± 0.65** |
| Serum TBA (μmol/L) | 1.50 ± 0.55 | 4.67 ± 1.37## | 9.17 ± 3.37* |
| Liver TC (mmol/mg liver tissue) | 5.15 ± 0.45 | 12.93 ± 0.88## | 10.84 ± 1.56* |
| Liver TG (mmol/mg liver tissue) | 4.38 ± 0.91 | 8.37 ± 2.31## | 5.03 ± 0.99** |
| Liver TBA (nmol/mg liver tissue) | 141.33 ± 9.60 | 145.67 ± 7.35## | 160.33 ± 12.21** |
| Fecal Bile Acid (nmol/mg feces) | 46.45 ± 10.70 | 109.29 ± 16.09## | 134.56 ± 11.24** |
ALT alanine aminotransferase, AST aspartate aminotransferase, HDL-c high density lipoprotein cholesterol, LDL-c low density lipoprotein cholesterol, TBA total bile acid, TC total cholesterol, TG triglyceride
The values are expressed as the mean ± SD (n = 6 in each group), #P < 0.05, significantly different from the control group; ##P < 0.01, significantly different from the control group; *P < 0.05, significantly different from the HFD group; **P < 0.01, significantly different from the HFD group; ANOVA followed by Duncan's multiple range tests
Fig. 3.

IMB17–15 administration shifted the hepatic and serum BA compositions in hamsters. Hepatic BA composition (a) and serum BA composition (b) (ACA Allocholic acid, CA cholic acid, CDCA chenodeoxycholic acid, DCA deoxycholic acid, GCA glycocholic acid, HDCA α-hyodeoxycholic acid, LCA lithocholic acid, TCA taurocholic acid, TCDCA taurocheno-deoxycholic acid, TDCA taurodeoxycholic acid, THDCA taurohyodeoxycholic acid, TLCA taurolithocholic acid, TUDCA tauroursodeoxycholic acid, TαMCA tauro α-muricholate, TβMCA tauro β-muricholate, UDCA ursodeoxycholic acid, αMCA α-muricholic acid, βMCA β-muricholic acid). The labeling scheme for each group is indicated in the embedded legend. The means ± SD are shown. #P < 0.05 versus the control group, *P < 0.05 versus the HFD+NS group; ANOVA followed by Tukey’s test
Fig. 4.

IMB17–15 upregulated Cyp7a1 expression through FGF15/19-mediated ERK and JNK signaling pathway inhibition. a mRNA expression levels of hepatic Fxr, Shp and Cyp7a1 and ileal Asbt, Fxr and Fgf15 in the control, HFD + NS and HFD + IMB17–15 samples were normalized against Actb. Western blot analysis of ileal FXR and SHP (b) and hepatic total and phosphorylated ERK and JNK and CYP7A1 (c) expression in hamster liver and ileum tissue samples. β-actin served as the loading control. Western blot results of each animal are quantified. ASBT/SLC10A2 apical Na+-dependent bile salt transporter, CYP7A1 cholesterol 7α-hydroxylase, ERK extracellular regulated protein kinases, FGF fibroblast growth factor, FXR farnesoid X receptor, JNK c-Jun N-terminal kinase, SHP short heterodimer partner. The values are expressed as the mean ± SD (n = 6 in each group), ##P < 0.01 versus control group, *P < 0.05 versus HFD + NS group, **P < 0.01 versus HFD + NS group; ANOVA followed by Duncan's multiple range tests
IMB17–15 treatment altered the hepatic expression of genes involved in BA and lipid metabolism
As shown in Fig. 4a–c, Cyp7a1 mRNA expression was significantly suppressed after HFD feeding for 21 days, which was reversed by IMB17–15 treatment. In contrast, hepatic mRNA and protein expression levels of FXR and SHP were unchanged in these animals (Fig. 4a and Supplemental Fig. 1). Extracellular regulated protein kinases (ERK) and c-Jun N-terminal kinase (JNK) signaling are reportedly involved in FGF15/19-induced repression of hepatic CYP7A1 expression [18, 19]. We determined the hepatic expression levels of these proteins and found that HFD feeding upregulated ERK and JNK phosphorylation. Interestingly, IMB17–15 treatment abrogated ERK and JNK phosphorylation (Fig. 4c). These findings indicate that IMB17–15-induced promotion of CYP7A1 expression was mediated through the FGF15/19 and MAPK signaling pathways rather than the classical hepatic FXR-SHP pathway.
Analysis of the genes involved in fatty acid and TG metabolism revealed that the hepatic mRNA expression levels of sterol regulatory element-binding protein 1c (Srebp1c), stearoyl-CoA desaturase (Scd1) and fatty acid synthase (Fas) were increased by HFD feeding. IMB17–15 treatment reduced Fas expression, whereas the Srebp1c and Scd1 expression levels were not statistically changed. In addition, the expression levels of fatty acid oxidation and transportation-related genes, such as acetyl-CoA carboxylase (Acc), peroxisome proliferator-activated receptor α (Ppara) and low-density lipoprotein receptor (Ldlr), were substantially upregulated by IMB17–15 treatment (Fig. 5a). Based on these results, IMB17–15 decreased the serum and hepatic lipid contents and blocked NAFLD progression by accelerating ileal BA excretion and repressing hepatic BA biosynthesis and lipid metabolism.
Fig. 5.

IMB17–15 significantly activated AMPK pathway and regulated various genes involved in BA and lipid metabolism in hamster liver tissues. a mRNA expressions of Fas, Acc, Ldlr, Srebp1c, Scd1 and Pparα in the control, HFD + NS and HFD + IMB17–15 livers were normalized against Actb. b The protein expression levels of total and phosphorylated P-AMPKα and PPARα in hamster liver tissues, β-actin served as the loading control. ACC acetyl-CoA carboxylase, AMPKα adenosine monophosphate (AMP)-activated protein kinase, FAS fatty acid synthase, LDLR low-density lipoprotein receptor, PPARα peroxisome proliferator activated receptor α, SCD1 stearoyl-CoA desaturase, SREPB1C sterol regulatory element-binding protein 1c. The values are expressed as the mean ± SD (n = 6 in each group), #P < 0.05 versus the control group, *P < 0.05 versus the HFD + NS group, **P < 0.01 versus the HFD + NS group; ANOVA followed by Duncan's multiple range tests
IMB17–15 supplementation inhibited the hepatic inflammatory response in hamsters
Repression of CYP7A1 expression is a feature of the hepatic inflammatory response induced by HFDs. Activation of inflammatory cytokines, such as interleukin 6 (IL-6), interleukin 1β (IL-1β) and tumor necrosis factor α (TNFα), contributed to the induction of liver injury and had an inhibitory effect on the hepatic CYP7A1 expression. HFD-induced hepatic steatosis promotes the release of TNFα and inhibits adiponectin signaling, which leads to increased inflammation in liver tissue. Hamsters fed an HFD for 21 days experienced a more potent inflammatory response than normal chow diet-fed hamsters, a change signified by the significant upregulation of the hepatic Il-6, Il-1β and TNFα mRNA and protein levels in the HFD-fed hamsters compared with the normal chow-fed hamsters. Significant reductions in the expression levels of these inflammation-related cytokines were observed when the HFD-fed hamsters were treated with IMB17–15 (Fig. 6a, b), which is consistent with the inflammatory repression observed with H&E staining (Fig. 2c). The anti-inflammatory effect of IMB17–15 is attributable to improvements in hepatic lipid deposition and the induction of CYP7A1 expression.
Fig. 6.

IMB17–15 significantly inhibited the hepatic inflammation response in hamsters fed with the HFD. a Hepatic mRNA expression levels of IL-6, IL-1β and TNFα in the control, HFD + NS and HFD + IMB17–15 groups. ACTB served as the loading control. b The protein expression levels of IL-6, IL-1β and TNFα in each group of hamsters. β-actin served as the loading control. Western blot results for each animal are quantified in the three groups. IL-1β interleukin 1β, IL-6 interleukin 6, TNFα tumor necrosis factor α. The values are expressed as the mean ± SD (n = 6 in each group), #P < 0.05 versus the control group, ##P < 0.01 versus the control group, *P < 0.05 versus the HFD + NS group, **P < 0.01 versus the HFD + NS group; ANOVA followed by Duncan's multiple range tests
IMB17–15 also repressed NAFLD development via ASBT-independent mechanisms
We detected IMB17–15 in the circulation, which indicated that ASBT-independent mechanisms may exist in suppressing NAFLD. As shown in Fig. 5b, IMB17–15 treatment significantly enhanced hepatic adenosine monophosphate AMPK phosphorylation in hamster liver tissues. The mRNA and protein expression levels of PPARα, which is involved in fatty acid β-oxidation and HDL metabolism, were also simultaneously increased in the IMB17–15-treated HFD-fed hamsters compared with those in the untreated HFD-fed hamsters (Fig. 5a, b).
To verify whether IMB17–15 can directly exert anti-NAFLD effects in hepatocytes, we used OA treatment to induce steatosis in HL-7702 cells. As shown in Fig. 7a, 0.6 mM OA induced steatosis in HL-7702 cells. Western blot analysis demonstrated that IMB17–15 treatment increased the ratios of p-AMPKα/AMPKα and p-ACC/ACC in a dose-dependent manner in OA-treated HL-7702 cells. Treatment with 10 μM IMB17–15 significantly stimulated the AMPK signaling pathway. IMB17–15 treatment also upregulated the PPARα mRNA and protein expression, which resulted in the stimulation of downstream lipid oxidation- and lipoprotein metabolism-related genes, including carnitine palmitoyltransferase I (CPT1A), sterol carrier protein 2 (SCP-2), apolipoprotein A-I (ApoA-I) and Apolipoprotein A-V (ApoA-V) (Fig. 7a, b). We also found that the intracellular TG levels and oil red O-positive lipid droplet formation in HL-7702 cells were substantially decreased in the IMB17–15-treated group compared with those in the HFD group following IMB17–15 treatment (Fig. 7c, d). Furthermore, the stimulating activities of IMB17–15 on the phosphorylation of AMPK and ACC can be completely blocked by compound C, a specific inhibitor of AMPK (Fig. 8a). Compound C pretreatment also interdicted the ability of IMB17–15 to reduce hepatocyte TG accumulation and alleviate hepatic steatosis (Fig. 8b, c). Taken together, these results demonstrate that the anti-NAFLD effects of IMB17–15 are also dependent on AMPK signaling pathway activation and PPARα upregulation, indicating that IMB17–15 exerts ASBT-independent effects to ameliorate NAFLD.
Fig. 7.

IMB17–15 alleviated OA-induced steatosis and TG accumulation by stimulating the AMPK pathway and upregulating PPARα expression in HL-7702 cells. Cells were left untreated or treated with OA and different concentrations of IMB17–15, as indicated, for 24 h. a Cell total proteins were extracted. P-AMPKα, AMPKα, p-ACC and ACC protein expression levels were determined by western blotting and were normalized against β-actin protein expression levels. b IMB17–15 enhanced the mRNA expression of PPARα and its downstream lipid oxidation- and lipoprotein metabolism-related genes, includingPPARA, CPT1A, SCP-2, ApoA-I and ApoA-V, in a dose-dependent manner. ACTB served as the loading control. After treatment, the cells were also subjected to ORO staining (c) and harvested for intracellular TG assay (d) to investigate the influence of IMB17–15 on lipid metabolism (magnification ×400). ACC acetyl-CoA carboxylase, AMPKα adenosine monophosphate (AMP)-activated protein kinase, ApoA-I apolipoprotein A-I, ApoA-V Apolipoprotein A-V, CPT1A carnitine palmitoyltransferase I, OA oleic acid, PPARα peroxisome proliferator activated receptor α, SCP-2 sterol carrier protein 2. The values are expressed as the mean ± SD of five independent assays, #P < 0.05 versus the control group, ##P < 0.01 versus the control group, **P < 0.01 versus the OA-induced group, ΔΔP < 0.01; indicates that the control group was significantly different from the OA-induced group; ANOVA followed by Duncan's multiple range tests
Fig. 8.

AMPK inhibition can block the effects of IMB17–15 in alleviating OA-induced HL-7702 cell steatosis and TG accumulation. HL-7702 cells were pretreated with compound C for 1 h and then with OA and different concentrations of IMB17–15 for 24 h. a The protein expression levels of total and phosphorylated AMPK and total and phosphorylated ACC were analyzed by western blot, and β-actin served as the loading control. The cells were used for intracellular TG detection (b) or oil red O staining (magnification ×400) (c). ACC acetyl-CoA carboxylase, AMPKα adenosine monophosphate (AMP)-activated protein kinase, OA oleic acid, PPARα peroxisome proliferator activated receptor α. The values are expressed as the mean ± SD, ##P < 0.01; significantly different from the control group, **P < 0.01; significantly different from the OA-treated group; ΔΔP < 0.01; significantly different from the OA and CC-treated group; ANOVA followed by Duncan's multiple range tests
Discussion
Lowering the hepatic lipid content has been a therapeutic strategy for treating NFALD and NASH. In this report, we assessed the therapeutic effects of a novel ASBT inhibitor, IMB17–15, in a hamster model of NAFLD. We demonstrated that IMB17–15: (1) was well tolerated without tissue injury at a high dose in rodents (Fig. 1); (2) markedly prevented the weight gain induced by HFD feeding (Fig. 2); (3) substantially improved the liver histology as indicated by reduced liver steatosis and inflammation (Fig. 2 and Fig. 6); and (4) significantly improved the serum and liver biochemistry, e.g., the levels of liver enzymes, TC and TGs (Table 3). Further mechanistic studies revealed that IMB17–15 not only blocked ASBT activity in the ileum, which lowered cholesterol by stimulating bile acid synthesis in the liver (Fig. 4), but also activated AMPK and PPAR in the liver to accelerate fatty acid metabolism (Fig. 5 and Fig. 7). Therefore, we propose that the therapeutic beneficial effects of IMB17–15 in the NAFLD model are mediated through both ASBT-dependent and -independent mechanisms (Fig. 9).
Fig. 9.
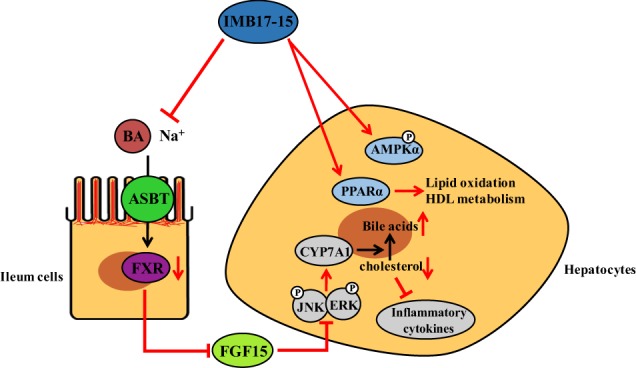
IMB17–15 has two pharmacological effects on NAFLD. First, IMB17–15 inhibits ASBT and subsequently suppresses ileal FXR and FXR-activated FGF15/19 expression, changes that reduce ERK and JNK phosphorylation in the liver and upregulate CYP7A1 activity. CYP7A1 is the key enzyme in BA biosynthesis, and its stimulation accelerates the transformation of cholesterol into BAs and leads to the inhibition of the inflammatory response. Second, IMB17–15 stimulates the phosphorylation of AMPKα and enhances the expression of PPARα and thus promotes TG oxidation and HDL metabolism through an ASBT-independent mechanism. The potent anti-NAFLD activity of IMB17–15 is attributable to these two synergistic effects. AMPKα adenosine monophosphate (AMP)-activated protein kinase, ASBT/SLC10A2 apical Na+-dependent bile salt transporter, BA bile acid, CYP7A1 cholesterol 7α-hydroxylase, ERK extracellular regulated protein kinases, FGF fibroblast growth factor, FXR farnesoid X receptor, HDL-c high density lipoprotein cholesterol, JNK c-Jun N-terminal kinase, NAFLD Nonalcoholic fatty liver disease, PPARα peroxisome proliferator activated receptor α, TG triglyceride
Previous studies have shown that treatment with the ASBT inhibitors SC-435 [20], 264W94 [21] and S-8921 [22] increased the hepatic CYP7A1 expression in guinea pigs and rats. Consistent with these observations, IMB17–15 treatment in hamsters significantly stimulated liver CYP7A1 expression and resulted in increased BA contents in both the feces and liver, confirming that the drug blocked intestinal BA reabsorption and enhanced hepatic BA synthesis. However, in contrast to the previously reported ASBT inhibitors that are presumed to be unabsorbable, IMB17–15 is partially absorbable. Because of this unique property, IMB17–15 was also able to activate AMPK and PPARα in hepatocytes, while SC-435 has been shown to only minimally affect the expression of PPARα [23]. AMPK, a pivotal regulator of energy homeostasis, harmonizes metabolic pathways and maintains the balance between the nutrient supply and energy demand [24]. PPARα plays crucial roles in decreasing serum lipid levels and inducing enzymes involved in lipid β-oxidation and TG catabolism [25]. Since these two genes play very important roles in maintaining hepatic lipid homeostasis and the development of NAFLD, stimulation of their activation would reduce lipid accumulation, which may explain why IMB17–15 substantially decreased the serum and liver TG contents and improved the liver conditions in the HFD-fed hamsters. However, how this drug stimulated AMPK and PPARα phosphorylation remains to be determined, which will be addressed in a future study.
Epidemiological and experimental studies have documented that secondary bile acids (such as LCA and DCA) play a significant role in the development of colorectal cancer by affecting the ERK, AKT and EGFR pathways [26, 27]. Ursodeoxycholic acid intervention to reduce the proportion of fecal DCA can inhibit colon cancer formation in both rodents and inflammatory bowel disease patients [28–30]. One of the greatest concerns in ASBT inhibitor development is that ASBT inhibition causes increasing colonic epithelial exposure to secondary BAs and stimulates colonic carcinogenesis, although epidemiological data are lacking [31]. IMB17–15, a novel ASBT inhibitor, increased the fecal BA excretion compared to that observed in the untreated HFD-fed group, and hepatic and serum BA composition analyses revealed a nonsignificant elevation in DCA after IMB17–15 treatment. Moreover, ASBT-deficient mice have been reported to exhibit a greater than 10-fold increase in fecal bile acids and a 59% increase in colon tumor number and size compared to wild-type mice, revealing that increased fecal bile acids stimulate tumor promotion rather than initiation [32]. IMB17–15-treated hamsters only showed a 25% higher fecal bile acids than HFD-fed hamsters. Abnormal activation of FGF19 facilitated colon cancer formation and other malignancies [33]. However, in our study, IMB17–15 administration did not upregulate FGF19 mRNA expression compared with the HFD group. Based on these results, IMB17–15 showed low probability in causing colon neoplasia.
Recent evidence has revealed the pathogenic nexus between gut microbiota dysbiosis and NAFLD development [34]. Compared to healthy subjects, higher abundance levels of Streptococcaceae (Gram-positive) and Enterobacteriaceae (Gram-negative) were detected in NAFLD patients who consumed a Western diet, which both contributed to persistent inflammation [34, 35]. With an increased percentage of Enterobacteriaceae after CA administration in rats, the serum concentration of adiponectin, a protein secreted from white adipose tissue that participates in the suppression of inflammation, was significantly decreased [36]. Furthermore, it has been demonstrated that HFD-fed mice exhibited a substantial upregulation in Gram-positive gut microbiota in their feces and increased hepatic translocation of the Gram-positive bacteria cell wall component, lipoteichoic acid (LTA). LTA can bind to the innate immunity receptor Toll-like receptor 2, induce the expression of IL1β and IL6 and subsequently aggravate hepatic inflammation [37]. As IMB17–15 led to decreased IL1β and IL6 expression and lower CA levels in the liver, we speculated that its anti-inflammatory effects were relevant to the shift of the gut microbiome composition toward fewer Gram-positive species. However, the specific mechanism requires further exploration.
In summary, IMB17–15 is a well-tolerated novel drug that elicited significant improvements in HFD-fed NAFLD hamster models by both inhibiting the ileal bile acid transporter ASBT and stimulating the activities of hepatic AMPK and PPARα. Because hamsters have a very similar lipid profile to that of humans, we propose that a clinical trial for IMB17–15 treatment in patients with NAFLD is warranted.
Supplementary information
Acknowledgements
This study was supported by the National Nature Scientific Foundation of China (81673497 and 81473098), the CAMS Initiative for Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-1–001), and the Beijing Nature Scientific Foundation (7162130). We thank the Liver Meeting 2017 held by the American Association for the Study of Liver Diseases for receiving and publishing this document as a meeting abstract.
Author contributions
MG, HH, RS and SC designed the research project; MG, WN, JR, NZ and YZ performed the experiment; DY and HL analyzed the data; JW, YW and RS contributed the reagents and materials; and MG, HH and SC wrote the manuscript.
Competing interests
The authors report no competing interests.
Contributor Information
Ju-xian Wang, Phone: +86-10-63131053, Email: imbjxwang@163.com.
Hong-wei He, Phone: +86-10-83166673, Email: hehwei@imb.pumc.edu.cn.
Electronic supplementary material
The online version of this article (10.1038/s41401-018-0195-3) contains supplementary material, which is available to authorized users.
References
- 1.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145–71. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- 2.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–46. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 3.Alli V, Rogers AM. Gastric bypass and influence on improvement of NAFLD. Curr Gastroenterol Rep. 2017;19:25. doi: 10.1007/s11894-017-0567-8. [DOI] [PubMed] [Google Scholar]
- 4.Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lutjohann D, et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008;48:474–86. doi: 10.1002/hep.22363. [DOI] [PubMed] [Google Scholar]
- 5.Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110:109–17. doi: 10.1172/JCI0215387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding L, Yang L, Wang Z, Huang W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm Sin B. 2015;5:135–44. doi: 10.1016/j.apsb.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim SG, Kim BK, Kim K, Fang S. Bile acid nuclear receptor farnesoid X receptor: therapeutic target for nonalcoholic fatty liver disease. Endocrinol Metab. 2016;31:500–4. doi: 10.3803/EnM.2016.31.4.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doring B, Lutteke T, Geyer J, Petzinger E. The SLC10 carrier family: transport functions and molecular structure. Curr Top Membr. 2012;70:105–68. doi: 10.1016/B978-0-12-394316-3.00004-1. [DOI] [PubMed] [Google Scholar]
- 9.Rao A, Kosters A, Mells JE, Zhang W, Setchell KD, Amanso AM, et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci Transl Med. 2016;8:357ra122. doi: 10.1126/scitranslmed.aaf4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Yao X, Young A, McNulty J, Anderson D, Liu Y, et al. Inhibition of apical sodium-dependent bile acid transporter as a novel treatment for diabetes. Am J Physiol Endocrinol Metab. 2012;302:E68–76. doi: 10.1152/ajpendo.00323.2011. [DOI] [PubMed] [Google Scholar]
- 11.Liu HT, He HW, Bai XG, Wang JX, Xu CL, Cai SY, et al. Arylsulfonylamino-benzanilides as inhibitors of the apical sodium-dependent bile salt transporter (SLC10A2) Molecules. 2013;18:6883–97. doi: 10.3390/molecules18066883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamada S, Kawaguchi H, Yamada T, Guo X, Matsuo K, Hamada T, et al. Cholic acid enhances visceral adiposity, atherosclerosis and nonalcoholic fatty liver disease in microminipigs. J Atheroscler Thromb. 2017;24:1150–66. doi: 10.5551/jat.39909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Deng Y, Wang X, Xu J, Li Z. Conformational and bioactivity analysis of insulin: freeze-drying TBA/water co-solvent system in the presence of surfactant and sugar. Int J Pharm. 2009;371:71–81. doi: 10.1016/j.ijpharm.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 14.Benson GM, Haskins NJ, Eckers C, Moore PJ, Reid DG, Mitchell RC, et al. Polydeoxycholate in human and hamster feces: a major product of cholate metabolism. J Lipid Res. 1993;34:2121–34. [PubMed] [Google Scholar]
- 15.Zhang Y, Huo M, Zhou J, Xie S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Prog Biomed. 2010;99:306–14. doi: 10.1016/j.cmpb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 16.Shan YQ, Ren G, Wang YX, Pang J, Zhao ZY, Yao J, et al. Berberine analogue IMB-Y53 improves glucose-lowering efficacy by averting cellular efflux especially P-glycoprotein efflux. Metabolism. 2013;62:446–56. doi: 10.1016/j.metabol.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 17.Jian T, Wu Y, Ding X, Lv H, Ma L, Zuo Y, et al. A novel sesquiterpene glycoside from Loquat leaf alleviates oleic acid-induced steatosis and oxidative stress in HepG2 cells. Biomed Pharmacother. 2017;97:1125–30. doi: 10.1016/j.biopha.2017.11.043. [DOI] [PubMed] [Google Scholar]
- 18.Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. 2012;56:1034–43. doi: 10.1002/hep.25740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56:1085–99. doi: 10.1194/jlr.R054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.West KL, McGrane M, Odom D, Keller B, Fernandez ML. SC-435, an ileal apical sodium-codependent bile acid transporter inhibitor alters mRNA levels and enzyme activities of selected genes involved in hepatic cholesterol and lipoprotein metabolism in guinea pigs. J Nutr Biochem. 2005;16:722–8. doi: 10.1016/j.jnutbio.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Root C, Smith CD, Sundseth SS, Pink HM, Wilson JG, Lewis MC. Ileal bile acid transporter inhibition, CYP7A1 induction, and antilipemic action of 264W94. J Lipid Res. 2002;43:1320–−30. [PubMed] [Google Scholar]
- 22.Sakamoto S, Kusuhara H, Horie K, Takahashi K, Baba T, Ishizaki J, et al. Identification of the transporters involved in the hepatobiliary transport and intestinal efflux of methyl 1-(3,4-dimethoxyphenyl)-3-(3-ethylvaleryl)-4-hydroxy-6,7,8-trimethoxy-2-naphthoate (S-8921) glucuronide, a pharmacologically active metabolite of S-8921. Drug Metab Dispos. 2008;36:1553–61. doi: 10.1124/dmd.108.020511. [DOI] [PubMed] [Google Scholar]
- 23.Chen X, Gong Q, Wang CY, Zhang K, Ji X, Chen YX, et al. High-fat diet induces distinct metabolic response in interleukin-6 and tumor necrosis factor-alpha knockout mice. J Interferon Cytokine Res. 2016;36:580–8. doi: 10.1089/jir.2016.0022. [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Yang G, Kim Y, Kim J, Ha J. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med. 2016;48:e224. doi: 10.1038/emm.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest. 2006;116:571–80. doi: 10.1172/JCI27989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farhana L, Nangia-Makker P, Arbit E, Shango K, Sarkar S, Mahmud H, et al. Bile acid: a potential inducer of colon cancer stem cells. Stem Cell Res Ther. 2016;7:181. doi: 10.1186/s13287-016-0439-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dent P, Fang Y, Gupta S, Studer E, Mitchell C, Spiegel S, et al. Conjugated bile acids promote ERK1/2 and AKT activation via a pertussis toxin-sensitive mechanism in murine and human hepatocytes. Hepatology. 2005;42:1291–9. doi: 10.1002/hep.20942. [DOI] [PubMed] [Google Scholar]
- 28.Pardi DS, Loftus EV, Jr, Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889–93. doi: 10.1053/gast.2003.50156. [DOI] [PubMed] [Google Scholar]
- 29.Kohno H, Suzuki R, Yasui Y, Miyamoto S, Wakabayashi K, Tanaka T. Ursodeoxycholic acid versus sulfasalazine in colitis-related colon carcinogenesis in mice. Clin Cancer Res. 2007;13:2519–25. doi: 10.1158/1078-0432.CCR-06-2727. [DOI] [PubMed] [Google Scholar]
- 30.Saeki T, Yui S, Hirai T, Fujii T, Okada S, Kanamoto R. Ursodeoxycholic acid protects colon cancer HCT116 cells from deoxycholic acid-induced apoptosis by inhibiting apoptosome formation. Nutr Cancer. 2012;64:617–26. doi: 10.1080/01635581.2012.669876. [DOI] [PubMed] [Google Scholar]
- 31.Zheng X, Ekins S, Raufman JP, Polli JE. Computational models for drug inhibition of the human apical sodium-dependent bile acid transporter. Mol Pharm. 2009;6:1591–603. doi: 10.1021/mp900163d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raufman JP, Dawson PA, Rao A, Drachenberg CB, Heath J, Shang AC, et al. Slc10a2-null mice uncover colon cancer-promoting actions of endogenous fecal bile acids. Carcinogenesis. 2015;36:1193–200. doi: 10.1093/carcin/bgv107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pai R, Dunlap D, Qing J, Mohtashemi I, Hotzel K, French DM. Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating beta-catenin signaling. Cancer Res. 2008;68:5086–95. doi: 10.1158/0008-5472.CAN-07-2325. [DOI] [PubMed] [Google Scholar]
- 34.Bibbo S, Ianiro G, Dore MP, Simonelli C, Newton EE, Cammarota G. Gut microbiota as a driver of inflammation in nonalcoholic fatty liver disease. Mediat Inflamm. 2018;2018:9321643. doi: 10.1155/2018/9321643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiu Chien-Chao, Ching Yung-Hao, Li Yen-Peng, Liu Ju-Yun, Huang Yen-Te, Huang Yi-Wen, Yang Sien-Sing, Huang Wen-Ching, Chuang Hsiao-Li. Nonalcoholic Fatty Liver Disease Is Exacerbated in High-Fat Diet-Fed Gnotobiotic Mice by Colonization with the Gut Microbiota from Patients with Nonalcoholic Steatohepatitis. Nutrients. 2017;9(11):1220. doi: 10.3390/nu9111220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Islam KB, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141:1773–81. doi: 10.1053/j.gastro.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 37.Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y, et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov. 2017;7:522–38. doi: 10.1158/2159-8290.CD-16-0932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.