

Abstract
Pyrotinib is a novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor that is used to treat HER2-positive breast cancer. In this study we investigated the metabolism and disposition of pyrotinib in six healthy Chinese men after a single oral dose of 402 mg of [14C]pyrotinib. At 240 h postdose, the mean cumulative excretion of the dose radioactivity was 92.6%, including 1.7% in urine and 90.9% in feces. In feces, oxidative metabolites were detected as major drug-related materials and the primary metabolic pathways were O-depicoline (M1), oxidation of pyrrolidine (M5), and oxidation of pyridine (M6-1, M6-2, M6-3, and M6-4). In plasma, the major circulating entities identified were pyrotinib, SHR150980 (M1), SHR151468 (M2), and SHR151136 (M5), accounting for 10.9%, 1.9%, 1.0%, and 3.0%, respectively, of the total plasma radioactivity based on the AUC0–∞ ratios. Approximately 58.3% of the total plasma radioactivity AUC0–∞ was attributed to covalently bound materials. After incubation of human plasma with [14C]pyrotinib at 37 °C for 2, 5, 8, and 24 h, the recovery of radioactivity by extraction was 97.4%, 91.8%, 69.6%, and 46.7%, respectively, revealing covalent binding occurred independently of enzymes. A group of pyrotinib adducts, including pyrotinib-lysine and pyrotinib adducts of the peptides Gly-Lys, Lys-Ala, Gly-Lys-Ala, and Lys-Ala-Ser, was identified after HCl hydrolysis of the incubated plasma. Therefore, the amino acid residue Lys190 of human serum albumin was proposed to covalently bind to pyrotinib via Michael addition. Finally, the covalently bound pyrotinib could dissociate from the human plasma protein and be metabolized by oxidation and excreted via feces.
Keywords: pyrotinib, EGFR/HER2 dual tyrosine kinase inhibitor, breast cancer, drug metabolism, drug disposition, human plasma, covalent binding
Introduction
Pyrotinib (SHR1258) is a novel irreversible epidermal growth factor receptor/human epidermal growth factor receptor 2 (EGFR/HER2) dual tyrosine kinase inhibitor. Recently, pyrotinib was developed for the treatment of HER2-positive breast cancer in China and the United States [1]. In the first-in-patient study, the most common adverse effects were grade I/II gastrointestinal symptoms, such as diarrhea. The incidence and severity of diarrhea were dose-dependent and clinically manageable up to 400 mg. The majority of incidences were transient and occurred in the first cycle. In Chinese patients with HER2-positive metastatic breast cancer, the best objective response rates were 55.6 and 87.5%, and the median progression-free survival rates were 31.9 and 59.7 weeks, respectively, for the 320-mg and 400-mg dose cohorts [2]. Data from subsequent clinical investigations demonstrated that 400 mg pyrotinib once daily in combination with capecitabine was generally safe and well tolerated with an encouraging antitumor response when administered to HER2-positive breast cancer patients [1]. Considering its significant clinical value, pyrotinib received a priority review and was recently approved by the China Food and Drug Administration.
The plasma pharmacokinetics of pyrotinib were characterized in patients during a phase I clinical study [2]. After the first dose of pyrotinib at 400 mg, the absorption of pyrotinib was relatively slow with a median tmax of 4.00 h. The multiple-dose plasma concentration reached a steady state on day 8. The exposure on study day 28 was 1.22-fold increased compared with that of the single-dose exposure. These results suggest that pyrotinib does not significantly accumulate after repeated daily administration. Our previous study demonstrated that pyrotinib was extensively metabolized in humans, and the principle metabolites were SHR150980 (O-depicolyl pyrotinib, M1), SHR151468 (O-depicolyl and pyrrolidine lactam pyrotinib, M2), and SHR151136 (pyrrolidine lactam pyrotinib, M5) (Fig. 1). Phenotyping experiments demonstrated that CYP3A4 is the most active enzyme responsible for the biotransformation of pyrotinib [3]. As an irreversible inhibitor, pyrotinib has an α,β-unsaturated amide moiety that acts as a pharmacological warhead and has the potential to covalently bind to protein via Michael addition [4–9]. This feature could make the disposition of pyrotinib more complicated. The rational development of pyrotinib relies on deeply understanding the dispositional characteristics of pyrotinib in humans. Therefore, the objective of the present study was to investigate the pharmacokinetics, metabolism, and excretion of pyrotinib in humans after the administration of a single oral dose of [14C]pyrotinib with a focus on the mechanism of the covalent binding of pyrotinib with human plasma protein.
Fig. 1.

Principal metabolic pathways of pyrotinib in humans reported in a previous study [3]
Materials and methods
Study participants
Healthy male Chinese volunteers aged 25–38 years old with a body mass index between 21 and 26 kg/m2 and a body weight of 55–76 kg were enrolled in the study. The use of prescribed or non-prescribed concomitant medications was not permitted 14 d before and during the study. In addition, the use of medications with CYP450 enzyme-inducing properties was not permitted 30 d before the administration of pyrotinib [10]. Volunteers were also excluded if they had any intake of any product containing grapefruit, irregular bowel movements or abnormal values for clinical laboratory tests (hematology, serum chemistry, urinalysis, and stool analysis) or twelve-lead electrocardiography. The Independent Ethics Committee approved the protocol, and the study was conducted in accordance with the ethical principles that originate in the Declaration of Helsinki and are consistent with good clinical practices and applicable regulatory requirements. All patients provided written informed consent before enrollment.
Study design
This study was an open-label, single-center, single-dose study conducted at the First Affiliated Hospital of Soochow University (Suzhou, China). Plasma, urine, and feces were collected for 10 d after dosing, and the volunteers were discharged from the clinic when the following criteria were satisfied: the accumulated excreted radioactivity was greater than 80% of the administered radioactivity; the excreted radioactivity over 2 consecutive d was less than 1% of the administered radioactivity; and the measured radioactivity in plasma collected over 2 continuous d was less than two times that of the background. After the study, volunteers with clinically significant abnormal values were retained in the clinical facility or followed until normal levels were obtained.
Study medication
An oral suspension of [14C]pyrotinib was prepared by the dilution of high specific activity [14C]pyrotinib (WuXi AppTec, Shanghai, China) with unlabeled pyrotinib and milled in a 0.5% carboxymethylcellulose sodium solution. The 14C label was located on the nitrile group.
On day 1, following an overnight fast, each of the six volunteers received a single oral dose of 402 mg of the pyrotinib suspension containing 5.55 × 106 Bq of [14C]pyrotinib. The vial used for drug administration was rinsed 3× with plain noncarbonated water, and the volunteers consumed the rinsing fluids along with additional plain noncarbonated water (200 mL). The volunteers were prohibited from drinking water for 1 h and fasted for 4 h.
Pharmacokinetic evaluation
Blood samples (~4 mL) were collected predose and at 1, 2, 3, 4, 5, 6, 8, 12, 24, 48, 72, 96, 120, 144, 168, 192, 216, and 240 h postdose into tubes containing K2EDTA anticoagulant and centrifuged to determine the total radioactivity and the plasma pyrotinib, SHR150980, SHR151468, and SHR151136 concentrations (Jiangsu Hengrui Medicine Co., Ltd., Lianyungang, China). Extra blood samples (~10 mL) were collected at 2, 5, 8, 12, 24, and 48 h postdose for metabolite identification. Plasma pellets generated during the preparation of plasma (for metabolite identification) were used to further investigate the possible covalent binding of radioactive compounds to proteins. Plasma pellets were combusted using a Harvey OX-501 Biological Oxidizer (R. J. Harvey Instrument Corp., Tappan, New York, USA), and the total radioactivity was determined using a Packard Tri-Carb 3100TR liquid scintillation counter (PerkinElmer, Waltham, MA, USA). Urine and feces were collected predose and at 0–4, 4–8, 8–24, 24–48, 48–72, 72–96, 96–120, 120–144, 144–168, 168–192, 192–216, and 216–240 h postdose. All urine and feces samples were used for total radioactivity analysis. However, urine (0–4, 4–8, and 8–24 h postdose) and feces (with radioactivity greater than 1% of the administered radioactivity) samples were selected and pooled for metabolic profiling. All samples collected were stored at −15 °C or less until analysis.
Bioanalytical procedures
Plasma samples were analyzed to determine the concentrations of pyrotinib, SHR150980, SHR151468, and SHR151136 using a validated liquid chromatography tandem mass spectrometry (LC-MS/MS) method. Total radioactivity was measured in plasma, urine, and feces using a Packard Tri-Carb 3100 TR liquid scintillation counter. Fecal residues were combusted using a Harvey OX-501 Biological Oxidizer prior to liquid scintillation counting. Metabolite profiling was performed using an 1100 HPLC system (Agilent Technologies, Inc., Santa Clara, CA, USA) coupled with an off-line TopCount NXT microplate scintillation and luminescence counter (PerkinElmer, Waltham, MA, USA). Separation was conducted using a Venusil C18 column (250 mm × 4.6 mm I.D., 5 μm; Agela Corporation, Tianjin, China). A mobile phase gradient of 5 mmol/L ammonium acetate (containing 0.5% formic acid, A) and acetonitrile (B) was programmed linearly as follows: initiated with 20% B, changed to 35% B over 20 min, changed to 50% B over 30 min, changed to 95% B over 5 min, changed to 100% B over 10 min and held at 100% B for 5 min. Then, the system was allowed to equilibrate for 15 min before the next injection. The flow rate of the mobile phase was 0.6 mL/min. HPLC fractions collected in a LumaPlate 96-well plate were dried and counted using the TopCount NXT counter. Data were applied to construct radiochromatograms using the ARC data system (AIM Research Company, Hockessin, DE, USA). Metabolite identification was conducted on a Triple TOF 5600+ MS system (AB SCIEX, Concord, Ontario, Canada) with a DuoSpray ion source in positive ion mode. Mass calibration was performed after every injection with a set of standards. Data were acquired using Analyst® TF 1.6 software (AB SCIEX) and processed using MetabolitePilotTM 1.5 software (AB SCIEX).
Sample preparation for metabolic profiling
Feces
A 5-g portion of the pooled fecal homogenates was extracted using a three-step extraction procedure with 15 mL of acetonitrile/methanol (50/50) as the extraction solvent, and the supernatant was reduced to dryness under a stream of nitrogen at 40 °C. Dried fecal extracts were reconstituted in acetonitrile/water (50/50, 1000 µL). An 100-µL aliquot was injected into the HPLC system and analyzed by off-line radiodetection and Triple TOF 5600+ MS.
Urine
A 20-mL portion of the pooled urine samples was reduced to dryness under a stream of nitrogen at 40 °C and reconstituted in acetonitrile/water (50/50, 300 µL). A 100-µL aliquot was injected into the HPLC system and analyzed by off-line radiodetection and Triple TOF 5600+ MS.
Plasma
Pooled plasma samples were extracted by adding acetonitrile (6 mL) to plasma (3 mL). After the mixture was vortexed (1 min) and centrifuged (3500× g, 5 min at room temperature), the remaining supernatant was reduced to dryness under a stream of nitrogen at 40 °C. The residues were reconstituted in acetonitrile/water (50/50, 300 µL), and a 100-µL aliquot was injected into the HPLC system and analyzed by off-line radiodetection and Triple TOF 5600+ MS.
Pharmacokinetic analysis
Pharmacokinetic parameters of the total radioactivity, pyrotinib, SHR150980, SHR151468, and SHR151136 in plasma were derived using standard non-compartmental methods with Phoenix WinNonlin Professional Version 7.0 (Pharsight Corporation, Mountain View, CA, USA). The following pharmacokinetic parameters were assessed: maximum concentration (Cmax); time to reach Cmax (Tmax); elimination half-life time (t1/2); area under the concentration-time curve (AUC) from time 0 to 48 h (AUC0–48 h); AUC from time 0 to time of the last quantifiable concentration (AUC0-last); AUC from time 0 to infinite time (AUC0–∞) calculated as the sum of AUC0-last and Clast/λz, in which Clast is the last observed quantifiable concentration and λz is the elimination half-life time to last quantifiable concentration; apparent volume of distribution (Vz/F) based on the terminal phase calculated as D/(λz × AUC0–∞); and total drug clearance (CL/F) after extravascular administration calculated as D/AUC0–∞.
Safety evaluation
Safety and tolerability were evaluated throughout the study, which ranged from the time of obtaining informed consent to the end of the study or early withdrawal assessment. The physical examination, vital signs, clinical laboratory investigations (hematology, serum chemistry, urinalysis, and stool analysis), and twelve-lead electrocardiography occurred before the administration of the study drug and at the end of the study assessment (after collection of the final fecal sample). The adverse effect severity was graded according to the criteria of CTCAE 4.03 [11].
Incubation of [14C]pyrotinib with human plasma, serum albumin or α1-acid glycoprotein and hydrolysis of human plasma by HCl
Briefly, [14C]pyrotinib (200 ng/mL) was incubated with human plasma, serum albumin (45 mg/mL) (Sigma-Aldrich, St. Louis, MO, USA) or α1-acid glycoprotein (0.70 mg/mL) (Sigma-Aldrich) at 37 °C [12–14]. After incubation for 0, 2, 5, 8, and 24 h, sample aliquots were removed, added to 2 volumes of acetonitrile, vortexed for 1 min, and centrifuged at 14,000× g for 5 min. The total radioactivity of the supernatant was determined using a Packard Tri-Carb 3100TR liquid scintillation counter to examine the extraction recovery of [14C]pyrotinib after incubation with human plasma, serum albumin, or α1-acid glycoprotein.
After incubation with [14C]pyrotinib for 24 h, a 0.2-mL aliquot of human plasma was also transferred into 1 mL of HCl solution (2 mol/L) and incubated at 90 °C for 2 h under stirring. At the end of the incubation, the mixture was neutralized by the addition of 1 mL of 2 mol/L NaOH. Then, a 100-μL aliquot was injected into the HPLC system and analyzed by on-line radiodetection (AIM Research Co., Hockessin, DE, USA) and Triple TOF 5600+ MS.
Reversibility of pyrotinib covalent binding with plasma proteins
The reversibility of pyrotinib covalent binding with plasma proteins was evaluated according to the method reported by Chandrasekaran et al [15]. After pyrotinib (20 μg/mL) was incubated for 0 and 6 h with human plasma at 37 °C, aliquots (50 μL) were removed from the samples and analyzed to determine the concentration of pyrotinib using the LC-MS/MS method. After the 6-h incubation, human plasma was also transferred and extracted with methyl-tert-butyl ether (MTBE) (twice with 4.5 volumes). Then, aliquots (100 μL) of the aqueous layer (plasma protein residue) were suspended in 10 mL of phosphate buffer (0.1 M, pH 7.5) and sonicated for 10 min. The mixtures were incubated with gentle shaking at 37 °C to release the pyrotinib that was covalently bound to human plasma protein. After suspension for 0 and 16 h, aliquots (50 μL) were removed from the samples and analyzed to determine the concentration of pyrotinib using the LC-MS/MS method.
Results
Safety
Two out of six volunteers in the safety analysis set experienced at least one adverse effect, which was occult blood in the stool with grade 1 severity. No clinically relevant changes were observed by physical examination, clinical laboratory analyses, or vital sign measurements.
Pharmacokinetics
The mean concentration-time profiles of total radioactivity, pyrotinib, SHR150980, SHR151468, and SHR151136 in plasma are presented in Fig. 2, and the related pharmacokinetic parameters are summarized in Table 1. After a single oral dose of 402 mg of [14C]pyrotinib, the median time to maximum plasma concentration (tmax) for radioactivity, pyrotinib, SHR150980, SHR151468, and SHR151136 was ~4 h, and the maximum plasma concentration (Cmax) was 396, 99.0, 16.3, 10.2, and 43.6 ng eq./mL, respectively. The exposure of total radioactivity (AUC0–∞) was 18200 h × ng eq./mL. Pyrotinib, SHR150980, SHR151468, and SHR151136 accounted for only 10.9, 1.9, 1.0, and 3.0%, respectively, of the total plasma radioactivity based on geometric mean AUC0–∞ ratios. The mean plasma terminal half-life for radioactivity was 47.9 h, which was increased compared with that for pyrotinib at 29.3 h. The mean apparent total clearance and apparent volume of distribution for pyrotinib were 207 L/h and 8570 L, respectively, compared with 22.7 L/h and 1460 L, respectively, for the total plasma radioactivity.
Fig. 2.
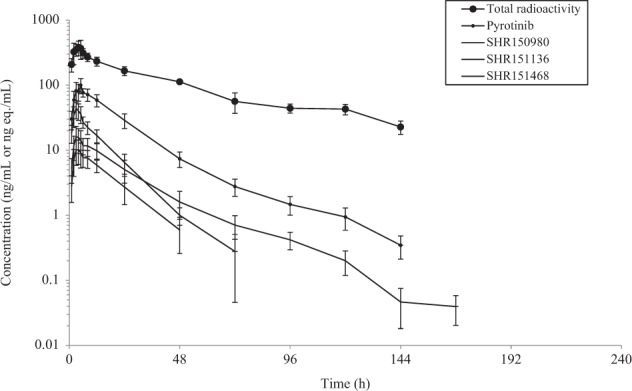
Mean (+SD) concentration-time profiles of radioactivity, pyrotinib, SHR150980, SHT151468, and SHR151136 in plasma in six healthy male volunteers after a single oral dose of 402 mg of [14C]pyrotinib
Table 1.
Summary of plasma pyrotinib, SHR150980, SHR151468, SHR151136, and radioactivity pharmacokinetic parameters in six healthy male volunteers after a single oral 402-mg dose of [14C]pyrotinib
| Radioactivity | Pyrotinib | SHR150980 | SHR151468 | SHR151136 | ||
|---|---|---|---|---|---|---|
| AUC0–∞ h × ng eq./mL | Geometric mean (Geometric CV, %) (range) | 18200 (25.7) (13200–26300) | 1970 (18.9) (1510–2500) | 352 (33.3) (210–510) | 179 (30.5) (124–284) | 553 (20.2) (463–783) |
| AUC0–last h × ng eq./mL | Geometric mean (Geometric CV, %) (range) | 12900 (34.0) (8220–18400) | 1940 (18.9) (1490–2470) | 339 (34.5) (199–504) | 173 (30.9) (119–276) | 546 (19.9) (457–769) |
| AUC0–48 h h × ng eq./mL | Geometric mean (Geometric CV, %) (range) | 9020 (13.8) (7670–11100) | 1730 (19.3) (1340–2290) | 292 (33.3) (174–458) | 170 (29.7) (119–276) | 533 (20.6) (457–769) |
| Cmax ng eq./mL | Geometric mean (Geometric CV, %) (range) | 396 (27.2) (296–557) | 99.0 (22.3) (85.1–151) | 16.3 (38.2) (8.96–28.6) | 10.2 (36.8) (5.70–17.2) | 43.6 (26.9) (32.8–69.1) |
| tmax, h | Mean (range) | 3.7 (3.0–5.0) | 4.5 (3.0–5.0) | 3.7 (3.0–5.0) | 4.3 (3.0–8.0) | 3.5 (3.0–5.0) |
| t1/2, h | Mean (range) | 47.9 (25.5–76.2) | 29.3 (17.0–39.2) | 32.2 (16.3–55.3) | 10.4 (8.1–13.4) | 9.75 (7.1–11.7) |
| CL/F, liters/h | Mean (range) | 22.7 (15.3–30.5) | 207 (161–266) | N/A | N/A | N/A |
| Vz/F, liters | Mean (range) | 1460 (1120–1720) | 8570 (4370–107000) | N/A | N/A | N/A |
| RAUC, 0–48 h%a | Geometric mean (range) | N/A | 19.2 (14.8–22.9) | 3.2 (2.3–4.6) | 1.9 (1.6–2.8) | 5.9 (5.1–7.7) |
| RAUC, 0–∞ %a | Geometric mean (range) | N/A | 10.9 (9.5–11.8) | 1.9 (1.1–2.5) | 1.0 (0.6–1.2) | 3.0 (2.6–3.5) |
AUC area under the concentration-time curve, CL/F apparent total clearance, Cmax maximum drug concentration, CV coefficient of variation, N/A not applicable, t1/2 terminal half-life, tmax time to maximum concentration, Vz/F apparent volume of distribution during terminal phase
aRatio of AUC0–48 h or AUC0–∞ plasma pyrotinib, SHR150980, SHR151468, and SHR151136 to total plasma radioactivity
Mass balance
In healthy volunteers (n = 6) after a single 402-mg oral dose of [14C]pyrotinib, the mean total radioactivity recovery was 92.6% (range 85.4–95.5%). Fecal excretion was the predominant route of elimination, accounting for a mean of 90.9% (range 84.1–93.6%) of the administered dose. The majority of the radioactivity in feces was excreted within 48 h after the dose, accounting for a mean of 81.5% (range 76.1–84.4%) of the administered dose. Urinary excretion was minimal (mean 1.7%; range 1.3–2.3%) (Table 2).
Table 2.
Percentage of cumulative radioactivity recovered from six healthy male volunteers after a single 402-mg oral dose of [14C]pyrotinib
| Time period | Feces | Urine (% dose) | Total | |
|---|---|---|---|---|
| 0–24 h | Mean (range) | 51.9 (33.4–71.8) | 0.8 (0.7–1.1) | 52.6 (34.1–72.9) |
| 0–48 h | Mean (range) | 81.5 (76.1–84.4) | 1.1 (0.9–1.6) | 82.6 (77.2–85.4) |
| 0–72 h | Mean (range) | 85.5 (80.5–87.3) | 1.2 (1.0–1.7) | 86.7 (81.6–89.0) |
| 0–96 h | Mean (range) | 87.9 (81.4–91.4) | 1.3 (1.1–1.8) | 89.2 (82.6–92.7) |
| 0–120 h | Mean (range) | 89.1 (82.3–92.4) | 1.4 (1.2–1.9) | 90.5 (83.5–93.9) |
| 0–144 h | Mean (range) | 89.8 (83.2–92.7) | 1.5 (1.2–2.0) | 91.3 (84.4–94.3) |
| 0–168 h | Mean (range) | 90.4 (83.4–93.2) | 1.6 (1.3–2.0) | 92 (84.7–94.9) |
| 0–192 h | Mean (range) | 90.6 (83.4–93.4) | 1.6 (1.3–2.1) | 92.2 (84.7–95.2) |
| 0–216 h | Mean (range) | 90.8 (83.8–93.6) | 1.7 (1.3–2.3) | 92.5 (85.1–95.5) |
| 0–240 h | Mean (range) | 90.9 (84.1–93.6) | 1.7 (1.3–2.3) | 92.6 (85.4–95.5) |
Metabolite profiles in plasma, urine, and feces
The extraction procedures for plasma and feces were optimized to obtain the maximum recovery of radioactivity. The use of acetonitrile/methanol washes ensured that greater than 95% of pyrotinib and SHR150980, SHR151468, and SHR151136 spiked into control fecal homogenates could be recovered. However, the mean recovery of total radioactivity in feces postdose was only 39.3%. Similarly, the recovery of total radioactivity in plasma postdose was ~50% (Table 3), whereas greater than 90% of pyrotinib and SHR150980, SHR151468, and SHR151136 spiked into control plasma could be recovered using acetonitrile as the extraction solvent. This unextractable radioactivity was attributable to covalently bound materials [16–19]. The supernatants were analyzed by HPLC radiochromatography and high-solution MS for metabolite identification. Representative radiochromatograms are presented in Fig. 3. Three primary metabolic pathways for pyrotinib were distinguished based on the nature of the identified metabolites: O-depicoline (M1), the oxidation of pyrrolidine (M5) and the oxidation of pyridine (M6-1, M6-2, M6-3, and M6-4). Additional metabolites (M2, M7-1, M7-2, M7-3, M7-4, M7-5, and M8) were formed by combinations or further secondary metabolism of these main metabolites. In addition, some phase II metabolites, including cysteine adducts (M9-1, M9-2, M10-1, and M10-2) and N-acetylcysteine adducts (M12-1 and M12-2), were also detected (Fig. 4).
Table 3.
Levels of covalently bound radioactivity in plasma
| Time | Total radioactivity (plasma) | Covalent bound radioactivity (plasma pellet) |
|---|---|---|
| h | ng eq./mL | ng eq./mL (%) a |
| 0 | BLQ | – |
| 1.00 | 207 | – |
| 2.00 | 324 | 95.8 (29.3) |
| 3.00 | 351 | – |
| 4.00 | 378 | – |
| 5.00 | 368 | 122 (33.2) |
| 6.00 | 311 | – |
| 8.00 | 269 | 94.0 (34.9) |
| 12.0 | 232 | 101 (43.5) |
| 24.0 | 166 | 84.1 (50.7) |
| 48.0 | 112 | 73.0 (65.2) |
| C max | 378 | 122 (32.3) |
| t max | 4 | 5 |
| AUC0-48 h | 9020 | 4132 (45.8) |
| AUC0-∞ | 21700 | 12648 (58.3) |
apercentage of covalently bound radioactivity in plasma pellets accounting for total plasma radioactivity; Cmax, maximum drug concentration; tmax, time to maximum concentration; AUC area under the concentration-time curve
Fig. 3.

Representative radiochromatograms of pyrotinib and metabolites in pooled plasma (a), urine (b), and feces (c)
Fig. 4.

Proposed metabolic pathways of pyrotinib in humans. (), percentage of metabolite in feces accounting for the dose administered (N.D., not detected by radiochromatography; asterisk M2 and M6-5 were coeluted by HPLC)
In feces, the major drug-related materials identified included pyrotinib, M5, M1, and M2, which accounted for 22.4, 17.3, 4.18, and 12.0%, respectively, of the peak area of the radiochromatogram of pooled feces. It should be noted that M2 and M6-5 were coeluted by HPLC. However, M2 was identified as the major metabolite by MS analysis. The concentrations of the phase II metabolites were very low at less than 1% of the peak area of the radiochromatogram of pooled feces. Combined with the radioactivity extraction efficiency (39.3%) and the mass balance study results demonstrating that 92.6% of the administered dose was excreted via feces, the major drug-related materials pyrotinib, M5, M1, and M2 accounted for 8.00%, 6.18%, 1.49%, and 4.29% of the dose, respectively. In urine, the major drug-related materials identified included pyrotinib, M6-2, M1, and M2, which accounted for 0.852%, 0.179%, 0.127%, and 0.099% of the administered dose, respectively.
In addition to pyrotinib, the metabolites identified in plasma by MS analysis included M1, M2, M4, M5, M6-1, M6-2, and M6-5. However, only pyrotinib and M5 were detected by radiochromatography because most of the metabolites were generally below the detection limit of the radiodetector. Therefore, the metabolite profiles in plasma could only be qualitatively compared. The unextracted radioactivity in human plasma accounted for 29.3%, 33.2%, 34.9%, 43.5%, 50.7%, and 65.2% of total plasma activity at 2, 5, 8, 12, 24, and 48 h, respectively. Based on the AUC0–∞ ratios, unextracted radioactivity accounted for ~58.3% of the total plasma radioactivity (Table 3).
Covalent binding of [14C]pyrotinib to human plasma protein, serum albumin, or α1-acid glycoprotein and hydrolysis of human plasma by HCl
To explore the reason for the incomplete recovery of radioactivity from human plasma after the oral administration of [14C]pyrotinib, [14C]pyrotinib was incubated with human plasma, serum albumin, or α1-acid glycoprotein at 37 °C for different amounts of time and extracted with acetonitrile. As shown in Table 4, the percentages of free [14C]pyrotinib (unbound, radioactivity recovery in acetonitrile extracts) in human plasma were 97.4%, 91.8%, 69.6%, and 46.7%, following incubation for 2, 5, 8, and 24 h, respectively. Similarly, the percentages of free [14C]pyrotinib in serum albumin were 90.0%, 85.0%, 80.5%, and 61.5% and the percentages of free [14C]pyrotinib in α1-acid glycoprotein were 82.5%, 81.5%, 78.5% and 78.0% following incubation for 2, 5, 8, and 24 h, respectively. The percent of covalent binding of pyrotinib with protein increased with increasing incubation time. After a 24-h incubation, the percentages of pyrotinib undergoing covalent binding with human plasma protein, serum albumin, and α1-acid glycoprotein were 53.3%, 38.5%, and 22.0%, respectively, suggesting that pyrotinib was mainly covalently bound with serum albumin and α1-acid glycoprotein to a less extent. Furthermore, these results revealed that the covalent binding of pyrotinib was independent on enzymes [17, 19].
Table 4.
Extractable radioactivity following incubation of 200 ng/mL [14C]pyrotinib with human plasma, serum albumin, or α1-acid glycoprotein at 37°C for different amounts of time
| Time | Plasma | Serum albumin | α1-acid glycoprotein |
|---|---|---|---|
| (h) | ng eq./mL (%)a | ng eq./mL (%)a | ng eq./mL (%)a |
| 0 | 204 (102) | 202 (101) | 194 (97.2) |
| 2 | 195 (97.4) | 180 (90.0) | 165 (82.5) |
| 5 | 184 (91.8) | 170 (85.0) | 163 (81.5) |
| 8 | 139 (69.6) | 161 (80.5) | 157 (78.5) |
| 24 | 93.4 (46.7) | 123 (61.5) | 156 (78.0) |
apercentage of extractable radioactivity accounting for the radioactivity added into human plasma, serum albumin, or α1-acid glycoprotein
To investigate the binding site, after [14C]pyrotinib-treated human plasma was directly hydrolyzed by HCl at 90 °C for 2 h, the sample was analyzed by HPLC radiochromatography (Fig. 5a). In addition to amide hydrolyzed pyrotinib (the latest eluted) and pyrotinib, the radioactive peaks identified by mass spectral data were a group of pyrotinib adducts that included pyrotinib-lysine (m/z 731) and pyrotinib adducts of peptides Gly-Lys (m/z 788), Lys-Ala (m/z 802), Gly-Lys-Ala (m/z 859), and Lys-Ala-Ser (m/z 889). The mass spectral data, including accurate molecular masses and the diagnostic product ions of these adducts, are summarized in Table 5. Of note, protonated [14C]pyrotinib (m/z 585) is a product ion for all peptide adducts listed in the table, which is evidence that pyrotinib was bound to these peptides. Proposed general fragmentation pathways of these pyrotinib peptide/lysine adducts are presented in Fig. 5b and include the cleavage of the α,β C-C of the carbonyl group to fragment II; cleavage of the C-N bond of the peptide to pyrotinib to protonated pyrotinib I; and subsequent cleavage of the amide bond of pyrotinib to a fragment III.
Fig. 5.

Radiochromatogram of human plasma incubated with [14C]pyrotinib for 24 h after HCl hydrolysis (a) and proposed fragmentation pathways of [14C]pyrotinib peptide adducts (b). AA, the amino acid residues
Table 5.
Mass spectral data of radioactive [14C]pyrotinib peptide adducts obtained by HCl hydrolysis of human plasma incubated with [14C]pyrotinib for 24 h
| [14C]Pyrotinib adducts | [M+H]+, m/z | Characteristic fragment ions, m/z a |
|---|---|---|
| Lys* | 731.3240 | 585.2160 (I), 242.1836 (II), 138.0908 (III) |
| Gly-Lys* | 788.3448 | 585.2174 (I), 299.2056 (II), 138.0905 (III) |
| Lys*-Ala | 802.3630 | 585.2218 (I), 313.2217 (II), 138.0904 (III) |
| Gly-Lys*-Ala | 859.3845 | 585.2240 (I), 370.2442 (II), 138.0902 (III) |
| Lys*-Ala-Ser | 889.3903 | 585.2160 (I), 400.2536 (II), 138.0886 (III) |
* the AA residue to which pyrotnib is bound
aI, II and III in the brackets indicate the fragmentation pathway of product ion formation (see Figure 5b)
Reversibility of pyrotinib covalent binding with plasma proteins
The protein obtained from human plasma treated with pyrotinib after exhaustive extraction with MTBE was subsequently reconstituted with phosphate buffer (pH 7.4) and incubated at 37 °C to assess the reversibility of pyrotinib binding with plasma protein. The time course for the recovery of pyrotinib in plasma is presented in Table 6. After a 6-h incubation with human plasma, the extractable pyrotinib concentration was 16.6 µg/mL. The results suggested that the amount of extractable pyrotinib was 83.0%, and 17.0% remained in the protein. After resuspension of the treated protein in phosphate buffer (pH 7.4), the amount of extractable pyrotinib from protein was only 0.336 µg/mL at time 0, and this amount increased to 7.15 µg/mL after incubation for 16 h at 37 °C, suggesting that 21.0% of covalently bound pyrotinib was released.
Table 6.
Time course of pyrotinib recovery after incubation with human plasma and resuspension with phosphate buffer, pH 7.4
| Incubation time (h) | Concentration (µg/mL) | Recovery (%)a |
|---|---|---|
| 0 | 20.5 | 103 |
| 6 | 16.6 | 83.0 |
| Reconstitution time (h) | Concentration (µg/mL) | Recovery (%)b |
|---|---|---|
| 0 | 0.0336 | 0.988 |
| 16 | 0.715 | 21.0 |
apercentage of extractable pyrotinib accounting for pyrotinib added into human plasma
bpercentage of pyrotinib recovered after resuspension with phosphate buffer accounting for pyrotinib covalently bound with human plasma
Discussion
This study investigated the pharmacokinetic characteristics, the elimination routes and the metabolic profile of [14C]pyrotinib in healthy volunteers. After the oral administration of pyrotinib, most of the recovered dose was present in feces (90.9%), with urinary excretion representing a minor elimination pathway (1.72%). The overall recovery of 92.6% of the radioactive dose indicates a complete mass balance.
The values obtained for the time to reach the maximum plasma concentration, the maximum plasma concentration, the area under the plasma concentration-time curve and terminal half-life for pyrotinib in this healthy volunteer study were comparable with those observed in cancer patients [2]. The exposures of pyrotinib and its major metabolites, including SHR150980, SHR151468, and SHR151136, accounted for ~10.9%, 1.9%, 1.0%, and 3.0%, respectively, of total plasma radioactivity based on AUC0–∞ ratios. Approximately 58.3% of total radioactivity was attributed to the radioactivity covalently bound to human plasma protein.
Data from the in vitro experiments indicated that [14C]pyrotinib, when incubated in human plasma at 37 °C, could not be extracted completely. The incomplete recovery of pyrotinib from plasma was caused by the covalent binding of pyrotinib with human plasma protein, which did not require the bioactivation of pyrotinib and occurred independently of enzymes. These results are not surprising because pyrotinib contains an α,β-unsaturated amide moiety, which was pharmacologically designed to undergo Michael addition to covalently bind to the conserved, solvent-exposed cysteine residue present in some ErbB family members (Cys773 in epidermal growth factor receptor and Cys805 in Her-2) [1]. Similarly, the reactivity of the α,β-unsaturated amide entity renders pyrotinib covalently bound to other proteins in vivo, as suggested by the incomplete recovery of pyrotinib from human plasma after incubation. Furthermore, a group of pyrotinib adducts, including pyrotinib-lysine and pyrotinib adducts of peptides Gly-Lys, Lys-Ala, Gly-Lys-Ala, and Lys-Ala-Ser, was detected in the HCl digestion mixture of the incubated plasma. Wang et al reported that neratinib covalently bound to the Lys190 residue of albumin, the most abundant protein in plasma. This findings was identified by a series of methods, including SDS-PAGE, LC/MS, and the synthetic standard neratinib-lysine [17]. Therefore, based on the identification of these pyrotinib peptide/lysine adducts and the similarity in the structures of pyrotinib and neratinib, it could be proposed that pyrotinib covalently binds to the amino acid residue Lys190 of human serum albumin via Michael addition.
It could be hypothesized that this covalently bound radioactivity is eliminated via two pathways. One pathway is excretion as pyrotinib peptide/lysine adducts following catabolism of the albumin tether. In this context, the elimination of covalently bound radioactivity would depend mostly on the catabolism of tethered albumin molecules. In fact, the half-life of the radioactivity (47.9 h, ~2 d) was considerably shorter than the half-life of albumin in humans (~19 d) [20, 21]. Furthermore, no pyrotinib peptide/lysine adducts were detected in human plasma after the oral administration of pyrotinib. These results indicate that the covalently bound radioactivity was eliminated by the way that covalently bound pyrotinib was dissociated from albumin followed metabolism [22, 23], which is reasonable because Michael additions are reversible [24]. Furthermore, pyrotinib was released from its protein adducts when it was suspended in phosphate buffer (pH 7.4), as observed the in vitro incubation experiment. These results indicated that covalently bound pyrotinib could dissociate from the human plasma protein and be metabolized by oxidation and excreted via feces.
In summary, after a single oral dose of 402 mg of [14C]pyrotinib in humans, pyrotinib and its three major metabolites, SHR150980, SHR151468, and SHR151136, accounted for ~16.8% of the total plasma radioactivity. Approximately 58.3% of total radioactivity was attributed to the unextractable radioactivity, which appeared to be derived from pyrotinib covalently bound to human plasma protein. Covalent binding occurred independently of enzymes, and the amino acid residue Lys190 of human serum albumin was proposed to covalently bind to pyrotinib via Michael addition. Finally, the covalently bound drug-related materials appeared to dissociate from albumin and be excreted via feces following oxidative metabolism.
Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (No 81521005), the Strategic Priority Research Program of the Chinese Academy of Sciences (No XDA12050306), the National Key New Drug Creation Special Programs (No 2017ZX09304-021), Jiangsu Provincial Medical Youth Talent (QNRC2016714), National Natural Science Foundation of China (No.81773820), the Jiangsu Provincial Medical Talent (ZDRCA2016048) and the Suzhou Key Laboratory of Drug Clinical Research and Personalized Medicine (SZS201719).
Author contribution
JM, XYC, DFZ, and LYM were responsible for study design and data analyses. JM, X-YL, SMA, HZ, SDY, YFZ, MXC, XYZ, YL, LY, and XLD conducted the study. YFZ conducted data analyses. JM and DFZ were responsible for writing the manuscript.
Competing interests
The authors declare that there is no competing interests.
Contributor Information
Li-yan Miao, Email: miaolysuzhou@163.com.
Da-fang Zhong, Email: dfzhong@simm.ac.cn.
References
- 1.Li X, Yang CY, Wan H, Zhang G, Feng J, Zhang L, et al. Discovery and development of pyrotinib: a novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. Eur J Pharm Sci. 2017;110:51–61. doi: 10.1016/j.ejps.2017.01.021. [DOI] [PubMed] [Google Scholar]
- 2.Ma F, Li Q, Chen SS, Zhu WJ, Fan Y, Wang JY, et al. Phase I study and biomarker analysis of pyrotinib, a novel irreversible Pan-ErbB receptor tyrosine kinase inhibitor, in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol. 2017;35:3105–12. doi: 10.1200/JCO.2016.69.6179. [DOI] [PubMed] [Google Scholar]
- 3.Zhu YT, Li L, Zhang G, Wan H, Yang CY, Diao XX, et al. Metabolic characterization of pyrotinib in humans by ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry. J Chromatogr B. 2016;1033–1034:117–27. doi: 10.1016/j.jchromb.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 4.Johnson DS, Weerapana E, Cravatt BF. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med Chem. 2010;2:949–64. doi: 10.4155/fmc.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–17. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 6.Kalgutkar AS, Dalvie DK. Drug discovery for a new generation of covalent drugs. Expert Opin Drug Discov. 2012;7:561–81. doi: 10.1517/17460441.2012.688744. [DOI] [PubMed] [Google Scholar]
- 7.Mah R, Thomas JR, Shafer CM. Drug discovery considerations in the development of covalent inhibitors. Bioorg Med Chem Lett. 2014;24:33–39. doi: 10.1016/j.bmcl.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Hossam M, Lasheen DS, Abouzid KAM. Covalent EGFR inhibitors: binding mechanisms, synthetic approaches, and clinical profiles. Arch Pharm Chem Life Sci. 2016;349:573–93. doi: 10.1002/ardp.201600063. [DOI] [PubMed] [Google Scholar]
- 9.Cheng H, Nair SK, Murray BW. Recent progress on third generation covalent EGFR inhibitors. Bioorg Med Chem Lett. 2016;26:1861–8. doi: 10.1016/j.bmcl.2016.02.067. [DOI] [PubMed] [Google Scholar]
- 10.Lacy S, Hsu B, Miles D, Dana A, Wang R, Nguye L. Metabolism and disposition of cabozantinib in healthy male volunteers and pharmacologic characterization of its major metabolites. Drug Metab Dispos. 2015;43:1190–207. doi: 10.1124/dmd.115.063610. [DOI] [PubMed] [Google Scholar]
- 11.US Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03. Bethesda, MD: National Institutes of Health; 2010.
- 12.Imbs DC, Paludetto MN, Négrier S, Powell H, Lafont T, White-Koning M, et al. Determination of unbound fraction of pazopanib in vitro and in cancer patients reveals albumin as the main binding site. Invest New Drugs. 2016;34:41–8. doi: 10.1007/s10637-015-0304-9. [DOI] [PubMed] [Google Scholar]
- 13.Yeggoni DP, Manidhar DM, Suresh Reddy C, Subramanyam R. Investigation of binding mechanism of novel 8-substituted coumarin derivatives with human serum albumin and α-1-glycoprotein. J Biomol Struct Dyn. 2016;34:2023–36. doi: 10.1080/07391102.2015.1104264. [DOI] [PubMed] [Google Scholar]
- 14.Smith SA, Gagnon S, Waters NJ. Mechanistic investigations into the species differences in pinometostat clearance: impact of binding to alpha-1-acid glycoprotein and permeability-limited hepatic uptake. Xenobiotica. 2017;47:185–93. doi: 10.3109/00498254.2016.1173265. [DOI] [PubMed] [Google Scholar]
- 15.Chandrasekaran A, Shen L, Lockhead S, Oganesian A, Wang JY, Scatina J. Reversible covalent binding of neratinib to human serum albumin in vitro. Drug Metab Lett. 2010;4:220–7. doi: 10.2174/187231210792928206. [DOI] [PubMed] [Google Scholar]
- 16.Zhang D, Krishna R, Wang L, Zeng J, Mitroka J, Dai R, et al. Metabolism, pharmacokinetics, and protein covalent binding of radiolabeled MaxiPost (BMS-204352) in humans. Drug Metab Dispos. 2005;33:83–93. doi: 10.1124/dmd.104.001412. [DOI] [PubMed] [Google Scholar]
- 17.Wang JY, Li-Chan XX, Atherton J, Deng L, Espina R, Yu L, et al. Characterization of HKI-272 covalent binding to human serum albumin. Drug Metab Dispos. 2010;38:1083–93. doi: 10.1124/dmd.110.032292. [DOI] [PubMed] [Google Scholar]
- 18.Scheers E, Leclercq L, Jong JD, Bode N, Bockx M, Laenen A, et al. Absorption, metabolism, and excretion of oral 14C radiolabeled ibrutinib: an open-label, phase I, single-dose study in healthy men. Drug Metab Dispos. 2015;43:289–97. doi: 10.1124/dmd.114.060061. [DOI] [PubMed] [Google Scholar]
- 19.Dickinson PA, Cantarini MV, Collier J, Frewer P, Martin S, Pickup K, et al. Metabolic disposition of osimertinib in rats, dogs, and humans: insights into a drug designed to bind covalently to a cysteine residue of epidermal growth factor receptor. Drug Metab Dispos. 2016;44:1201–12. doi: 10.1124/dmd.115.069203. [DOI] [PubMed] [Google Scholar]
- 20.Schreiber G, Urban J, Zahringer J, Reutter W, Frosch U. The secretion of serum protein and the synthesis of albumin and total protein in regenerating rat liver. J Biol Chem. 1970;246:4531–8. [PubMed] [Google Scholar]
- 21.Yang B, Kim JC, Seong J, Tae G. Comparative studies of the serum half-life extension of a protein via site-specific conjugation to a species-matched or -mismatched albumin. Biomater Sci. 2018;6:2092–2100. doi: 10.1039/C8BM00456K. [DOI] [PubMed] [Google Scholar]
- 22.Narita N, Morohashi A, Tohyama K, Takeuchi T, Tagawa Y, Kondo T, et al. Mechanism for covalent binding of MLN3126, an oral chemokine C-C motif receptor 9 antagonist, to serum albumins. Drug Metab Dispos. 2018;46:204–13. doi: 10.1124/dmd.117.078782. [DOI] [PubMed] [Google Scholar]
- 23.Reed RG, Davison LK, Burrington CM, Peters T., Jr. Non-resolving jaundice: bilirubin covalently attached to serum albumin circulates with the same metabolic half-life as albumin. Clin Chem. 1988;34:1992–4. [PubMed] [Google Scholar]
- 24.Johansson MH. Reversible michael additions: covalent inhibitors and prodrugs. Mini Rev Med Chem. 2012;12:1330–44. doi: 10.2174/13895575112091330. [DOI] [PubMed] [Google Scholar]