

Abstract
Metalloproteinase 9 (MMP-9) is able to degrade collagen IV, an important component of blood–brain barrier (BBB). Expression of MMPs, especially MMP-9, correlates with BBB disruption during central nervous system inflammation. Propofol has been reported to have anti-inflammation effects. In this study, we investigated the effects of propofol on TNF-α-induced MMP-9 expression in human cerebral microvascular endothelial cells (hCMEC/D3 cells) and explored the underlying mechanisms. The hCMEC/D3 cells were treated with propofol (25 μM), followed by TNF-α (25 ng/mL). We showed that TNF-α treatment markedly increased MMP-9 expression and decreased collagen IV expression in hCMEC/D3 cells, which was blocked by pretreatment with propofol. TNF-α-induced downregulation of collagen IV was also reversed by MMP-9 knockdown with siRNA. We revealed that TNF-α upregulated MMP-9 expression in hCMEC/D3 cells through activation of Ca2+/CAMK II/ERK/NF-κB signaling pathway; co-treatment with inhibitors of CaMK II (KN93), ERK (LY3214996), NF-κB (PDTC) or Ca2+chelator (BAPTA-AM) abrogated the effect of TNF-α on MMP-9 expression. We further established an in vitro BBB model by co-culturing of hCMEC/D3 cells and human astrocytes for 6 days and measuring trans-endothelial electrical resistance (TEER) to reflect the BBB permeability. TNF-α treatment markedly decreased TEER value, which was attenuated by pretreatment with propofol (25 μM) or MMP-9 knockdown with siRNA. In conclusion, propofol inhibits TNF-α-induced MMP-9 expression in hCMEC/D3 cells via repressing the Ca2+/CAMKII/ERK/NF-κB signaling pathway. TNF-α-impaired BBB integrity could be reversed by propofol, and propofol attenuates the inhibitory effect of TNF-α on collagen IV.
Keywords: blood-brain barrier, propofol; TNF-α, MMP-9, hCMEC/D3 cells
Introduction
With the continuous development of medical technology, perioperative mortality and severe surgical complications are now well controlled. However, postoperative cognitive dysfunction (POCD) is still an important and difficult issue that challenges surgeons and anesthesiologists. POCD is a recognized postoperative neurological complication. It has been reported that ~15% of patients over the age of 60 have a measurable decline in cognitive function as a result of anesthesia and surgery [1]. However, the pathogenic mechanisms of POCD remain unknown. The current evidence suggests that neuroinflammation and peripheral inflammation are involved in POCD [1]. The human blood–brain barrier (BBB), consisting mostly of cerebral microvascular endothelial cells, is highly sensitive to inflammatory stimuli. An inflammatory insult may disrupt the structure of the BBB and increase capillary permeability [2]. The damaged BBB may allow additional toxins, macrophages, and inflammation factors to affect hippocampal neurons and ultimately lead to POCD [3].
In many neuroinflammatory diseases, the expression of matrix metalloproteinases (MMPs), especially MMP-9, is associated with BBB disruption. It has been reported that increased serum MMP-9 levels in elderly patients after general anesthesia are involved in the pathological process of POCD [4]. Studies have documented that the degradation of collagen IV in the basal lamina coincides with the expression of MMP-9 [5, 6]. The expression of MMP-9 is also involved in apoptosis and neuroinflammation [7, 8]. Accordingly, MMP-9 may play a crucial role in inflammation-induced BBB damage.
In the perioperative period, the plasma levels of inflammatory factors, such as TNF-α, IL-1β, IL-6 and IL-8, are increased as a result of surgical trauma. TNF-α, one of the most important inflammatory cytokines, plays a crucial role in inflammation of the central nervous system. In addition, it has been demonstrated that TNF-α can induce the expression of MMP-9 in endothelial cells [9].
Propofol (2,6-diisopropylphenol), one of the most commonly used intravenous anesthetic agents, has been reported to have anti-inflammatory effects in the central nervous system [10]. It has been reported that propofol can inhibit the levels of TNF-α, IL-1β, IL-6, and IL-8 in cerebrospinal fluid [10]. It has also been reported that propofol can attenuate TNF-α-induced HT22 cell apoptosis [11]. In addition, it has been revealed that the incidence of POCD was remarkably lower in patients receiving propofol anesthesia compared to patients who received sevoflurane or isoflurane anesthesia intraoperatively [12]. Previous studies have demonstrated that propofol can decrease the secretion of IL-6, IL-8, and TNF-α in vascular endothelial cells [13]. However, whether propofol can attenuate the noxious effect of inflammatory cytokines on endothelial cells is largely unknown.
Based on the above scientific background, we hypothesized that propofol would attenuate inflammation-induced BBB disruption. In this study, we investigated the effects of propofol on TNF-α-induced MMP-9 expression in hCMEC/D3 cells and sought to reveal the underlying mechanisms.
Materials and Methods
Cell culture and reagents
The human cerebral microvascular endothelial cell (hCMEC) line, D3 clone, was purchased from Shanghai GuanDao Biological Engineering Company and maintained in Dulbecco’s modified Eagle’s medium (DMEM; HyClone, Australia). The human astrocytes were obtained from Prof. Zhen Wang (Fudan University Shanghai Cancer Center, Shanghai, China) and maintained in DMEM. Culture media were supplemented with 10% fetal bovine serum (Gibco, Australia), 100 U/mL penicillin, and 100 mg/mL streptomycin, and the cells were cultured in a humidified 5% CO2 atmosphere at 37 °C. The cells were passaged and cultured upon reaching 80%–90% confluence, and the cell medium was changed every day.
Propofol (Sigma, St. Louis, MO, USA) was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich). We pretreated cells with 25 μM for 2 h in all the related experiments. The ERK inhibitor LY3214996 (5 μM) and the NF-κB inhibitor PDTC (20 μM) were purchased from Sigma-Aldrich. KN93 (10 μM), an inhibitor of calmodulin-dependent protein kinase II (CaMKII), and BAPTA-AM (10 μM), the chelator of Ca2+, were purchased from Selleck (Shanghai, China). These doses of inhibitors were shown to be effective by previous studies [14–17]. BAPTA-AM, KN93, LY3214996, and PDTC were dissolved in DMSO. The final concentration of DMSO was adjusted to 0.01% for each solution to avoid possible nonspecific effects. TNF-α powder was obtained from PeproTech China and dissolved in serum-free DMEM. We treated cells with 25 ng/mL TNF-α for 8 h in all the related experiments.
Establishment of an in vitro BBB model
An in vitro BBB model was built by coculturing hCMEC/D3 cells and human astrocytes on opposing sides of 24-well cell culture inserts with 3.0 μm pores (Sigma-Aldrich, Shanghai, China). The inserts were coated with poly-L-lysine on the underside and type I collagen on the upper side. Human astrocytes were seeded at a density of 15 000 cells/cm2 on the underside of the insert, and hCMEC/D3 cells were seeded at a density of 25 000 cells/cm2 on the upper side. Subsequently, the inserts were cultured in endothelial basal medium containing 10% FBS. The media were replenished every 2–3 days, and Transepithelial electrical resistance (TEER) was measured every day to assess the integrity of the in vitro BBB model. In this study, the in vitro BBB model was ready for experiments after 6 days of coculturing, and thereafter, all experimental procedures were performed in serum-free media.
Measurement of TEER
TEER was measured using an epithelial voltmeter (World Precision Instruments, FL, USA.). In brief, after the respective treatments, the medium was aspirated from the upper chamber, and the cell culture inserts were transferred to a new well and washed with phenol red-free DMEM. TEER was analyzed using an epithelial voltmeter (World Precision Instruments, FL, USA), and values were expressed as Ω·cm2. According to previous findings [18], when the TEER reached over 100 Ω·cm2, the model was considered valid. Each assay was performed in five replicates, and the values of TEER are presented as the mean ± SD.
Western blot analysis
Whole-cell extracts, nuclear extracts and cytoplasmic extracts were prepared using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo, Waltham, Massachusetts, USA) according to the manufacturer’s instructions. After the designed treatments, the cell extracts were collected and lysed with RIPA lysis buffer (Beyotime Biotechnology, Suzhou, China) supplemented with protease and phosphatase inhibitors (Roche, Rotkreuz, Switzerland). The lysates were mixed with 5 × loading buffer and boiled for 5 min at 100 °C. Equivalent amounts of protein in each sample were separated by 10% or 12% SDS-polyacrylamide gel and transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA) using a semidry electroblotting system. The membranes were blocked with 5% skim milk in phosphate-buffered saline-Tween 20 (PBST) for 1 h at room temperature and then incubated with the appropriate primary antibody at a dilution of 1:1000 at 4 °C overnight. The primary antibodies included antibodies against MMP-9 (CST, Massachusetts, USA), p-NF-κB (Ser536) (CST, Massachusetts, USA), NF-κB (CST, Massachusetts, USA), p-ERK (CST, Massachusetts, USA), ERK (CST, Massachusetts, USA), p-CaMKII (CST, Massachusetts, USA), CaMKII (CST, Massachusetts, USA), collagen IV (Proteintech, Wuhan, China), and β-actin (Proteintech, Wuhan, China). Next, the membranes were washed with PBST and incubated with secondary antibodies conjugated with horseradish peroxidase (HRP) for 1 h at room temperature. After the membranes were washed again, the signals were detected using a LAS-4000 mini-CCD camera (GE Healthcare). β-Actin was used as an internal control, and the intensity of each protein band was normalized to that of β-actin. For nuclear protein, histone H3 was used as an internal control. Each assay was performed in five replicates.
Detection of intracellular Ca2+
Intracellular free Ca2+ concentration measurements were performed using a Fluo-3 AM Fluorescence Kit (Beyotime Biotechnology, Shanghai, China). hCMEC/D3 cells were seeded at an appropriate density in a 6-well plate. After the designated treatments, the cells were washed with PBS twice before the Fluo-3 AM fluorescence probes (0.5 μM) were added to each well. The cells were incubated with the probes for 1 h in a humidified 5% CO2 atmosphere at 37 °C in the dark. Then, the cells were washed with serum-free medium 3 times to remove the extracellular Fluo-3 AM that might enter the cells. The intensity of Fluo-3 fluorescence was detected using flow cytometry at an excitation wavelength of 488 nm according to the manufacturer’s instructions. The intracellular Ca2+ concentration was proportional to the fluorescence intensity. Each assay was performed in five replicates, and the levels of intracellular Ca2+ are presented as the mean ± SD.
Transient transfection with siRNAs
siRNAs against MMP-9 were synthesized by GenePharma (Shanghai, China). The sequence of siRNA-1 against MMP-9 was 5′-GACCTGGGCAGATTCCAAA-3′, and that of siRNA-2 was 5′- GGAAACCCTGCCAGTTTCCATTCAT-3′. The sequence of the control siRNA was 5′-GATCTGGACACCCATGACT-3′. MMP-9 siRNA-1 and 2 and the control siRNA were transfected into cells with the reagent Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, USA) according to the manufacturer’s instructions. Briefly, Lipofectamine 2000 and siRNA were incubated with Opti-MEM medium (Invitrogen) for 5 min separately. Then, Lipofectamine 2000 was mixed with siRNA for 20 min at room temperature before the mixture was added into the cells. After being cultured for 6 h, cells were washed and resuspended in fresh media for 24 h.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cultured cells using TRIzol Reagent (Tiangen, Beijing, China). Total RNA was reverse transcribed with TaKaRa PrimeScript® RT Reagent Kit (TaKaRa, Beijing, China) with gDNA eraser. Quantitative real-time PCR was carried out using SYBR Green Master Mix (TaKaRa, Beijing, China). All of the reactions were run in triplicate. Real-time PCR analysis of MMP-9 and β-actin expression was conducted with the following primers (Sangon, Shanghai, China): MMP-9-F 5′-TTTGACAGCGACAAGAAGTG-3′, MMP-9-R 5′-CAGGGCGAGGACCATAGAGG-3′, β-actin-F 5′-CGGGAAGCTTGTCATCAATGG-3′, β-actin-R 5′- GGCAGTGATGGCATGGACTG-3′.
Immunofluorescence analysis
The hCMEC/D3 cells were seeded onto glass coverslips in 24-well plates at 1 × 104 cells/well. Cells were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature after dealing with corresponding agents. Then, the cells were permeabilized with 0.1% Triton X-100 for 10 min, followed by blocking with 5% BSA for 1 h. Next, the cells were washed and incubated with primary polyclonal antibody against collagen IV (Proteintech, Wuhan, China) overnight at 4 °C. Then, the cells were incubated in Alexa Fluor 488-conjugated AffiniPure goat anti-rabbit IgG (Proteintech, Wuhan, China) for 2 h at room temperature protected from light. DNA was counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Negative controls were conducted in all cases without primary antibody. Photomicrographs were captured by a Leica TCS SP5II microscope at the same light intensity. Five fields from each section were imaged randomly. Each assay was performed in five replicates.
Statistical analysis
Data were expressed as the mean ± SD, and results were obtained from at least 5 separate experiments. Sample size (n) represents the number of replicate experiments, which were performed with different batches of cell cultures. Differences between groups were determined by one-way analysis of variance (ANOVA) followed by the Newman–Keuls test. The statistical program InStat (GraphPad Software, San Diego, CA, USA) was used for the statistical analyses. All results were considered statistically significant at a value of P < 0.05.
Results
MMP-9 expression was induced by TNF-α in hCMEC/D3 cells
As shown in Fig. 1a, we treated hCMEC/D3 cells with different concentrations of TNF-α (1, 5, 10, 25, 50 ng/mL) for 8 h and observed that 25 ng/mLTNF-α could greatly induce MMP-9 expression. Then, we treated cells with 25 ng/mL TNF-α for different times (2, 4, 8, 12, 24 h) and found that 8 h treatment with TNF-α could significantly induce MMP-9 expression (Fig. 1b). Thus, we treated cells with 25 ng/mL TNF-α for 8 h in follow-up experiments to explore the mechanism of TNF-α-induced MMP-9 expression in hCMEC/D3 cells.
Fig. 1.

TNF-α could induce MMP-9 expression in hCMEC/D3 cells. a The cells were treated with TNF-α (25 ng/mL) for different times (2, 4, 8, 12, 24 h). Left panel is the image of a representative Western blot for MMP-9; right panel is the plot of normalized ratios of optical densities. b The cells were incubated with different concentrations of TNF-α (5, 10, 25, and 50 ng/mL) for 8 h. The left side shows the image of a representative Western blot for MMP-9; the right panel is the plot of normalized ratios of optical densities. β-Actin served as an internal loading control. Data were expressed as the mean ± SD. *P < 0.05, compared with the control group. Each assay was performed in five replicates
TNF-α-mediated MMP-9 expression was attenuated by propofol in hCMEC/D3 cells
As shown in Fig. 2a, we pretreated hCMEC/D3 cells with different concentrations of propofol (10, 25, 50, 100 μM) for 2 days followed by TNF-α (25 ng/mL, 8 h) treatment and found that 25 μM propofol pretreatment significantly inhibited the effect of TNF-α on the expression of MMP-9 in hCMEC/D3 cells. As shown in Fig. 2b, we confirmed that the solvent for propofol, DMSO, had no effect on TNF-α-induced MMP-9 expression. Therefore, 25 μM propofol pretreatment for 2 h was used in the following experiments to investigate the mechanisms of propofol against TNF-α-induced MMP-9 expression.
Fig. 2.

Propofol could inhibit TNF-α-induced MMP-9 expression in hCMEC/D3 cells. a The cells were pretreated with different concentrations of propofol (10, 25, 50, 100 μM) for 2 h, followed by TNF-α treatment (25 ng/mL, 8 h). The upper panel shows the protein expression of MMP-9 in hCMEC/D3 cells. The lower panel is the normalized ratio of optical densities. b DMSO served as a solvent for propofol. The upper panel shows the protein expression of MMP-9 in hCMEC/D3 cells treated with TNF-α, propofol and DMSO (0.01%). The lower panel shows the normalized ratios of optical densities, as shown by histogram. β-Actin served as an internal loading control. Data were repressed as the mean ± SD. *P < 0.05, compared with the control group. #P < 0.05, compared with TNF-α treatment group. NS = no significance. Each assay was performed in five replicates
Propofol attenuated the effect of TNF-α on the integrity of the in vitro BBB model
As shown in Fig. 3a, the integrity of the in vitro BBB model was examined by measuring TEER after coculturing hCMEC/D3 cells and human astrocytes for different days (1, 2, 3, 4, 5, 6 and 7 days). We found that TEER reached a peak value of 140 Ω·cm2 after 6 days of coculturing hCMEC/D3 cells and human astrocytes, suggesting the successful establishment of an in vitro BBB model. As shown in Fig. 3b, we detected that TNF-α (25 ng/mL, 8 h) could impair the integrity of the in vitro BBB model, which was attenuated by propofol (25 μM). Importantly, 25 μΜ propofol or DMSO alone had no effect on BBB integrity. As shown in Fig. 3c, we found that TNF-α-impaired BBB integrity was mitigated by si-MMP-9-1 and si-MMP-9-2 in hCMEC/D3 cells, suggesting that the effect of TNF-α on TEER was dependent on MMP-9. As shown in Fig. 3d, we found that siRNA and propofol alone had no effect on BBB integrity. The efficacy of siRNA against MMP-9 has also been confirmed in Fig. 4c, d.
Fig. 3.

Propofol, via repression of the expression of MMP-9, could attenuate the effect of TNF-α on the integrity of the in vitro BBB model. a Evaluation of the in vitro BBB model integrity by measuring TEER over the course of 7 days coculturing of hCMEC/D3 cells and human astrocytes. The panel is the value of TEER that measured every day over the course of 7 days. Data were repressed as the mean ± SD. *P < 0.05, compared with Day 1. Each assay was performed in five replicates. b Evaluation of TEER in the in vitro BBB model after treatment. Data are expressed as the mean ± SD. *P < 0.05, compared with the control group. #P < 0.05, compared with the TNF-α treatment group. Each assay was performed in five replicates. c Evaluation of TEER in the in vitro BBB model that was measured after each treatment. Please note that the control group was treated with siRNA-NC. Data are expressed as the mean ± SD. *P < 0.05, compared with the control group. #P < 0.05, compared with the TNF-α treatment group. Each assay was performed in five replicates. d Evaluation of TEER in an in vitro BBB model that was measured after each treatment. Data are expressed as the mean ± SD. Please note that the control group was treated with siRNA-NC. *P < 0.05, compared with the control group. Each assay was performed in five replicates
Fig. 4.

Propofol inhibited the effect of TNF-α on collagen IV via decreasing the expression of MMP-9 in hCMEC/D3 cells. a, b si-MMP-9-1 served as the siRNA against MMP-9. The hCMEC/D3 cells were treated with TNF-α, propofol and si-MMP-9-1. The protein content of collagen IV in hCMEC/D3 cells was detected by Western blotting and immunofluorescence. c, d The mRNA and protein expression of MMP-9 in hCMEC/D3 cells treated with siRNA against MMP-9 (si-MMP-9-1 and si-MMP-9-2). Please note that the control group was treated with siRNA-NC. Data were repressed as the mean ± SD. *P < 0.05, compared with the control group. β-Actin served as an internal loading control. Each assay was performed in five replicates
Propofol, via inhibiting the expression of MMP-9, attenuated the effect of TNF-α on collagen IV
As shown in Fig. 4a, b, TNF-α decreased the level of collagen IV in hCMEC/D3 cells, while propofol attenuated this effect. The effect of TNF-α on collagen IV was also reversed by MMP-9 knockdown (Fig. 4a, b). To diminish MMP-9 expression, we treated cells with siRNA (si-MMP-9-1, si-MMP-9-2) against MMP-9. To examine the efficacy of siRNA against MMP-9, we measured MMP-9 expression by Western blot and RT-PCR. As shown in Fig. 4c, d, we confirmed that si-MMP-9-1 and si-MMP-9-2 all significantly reduced the expression of MMP-9 at both the protein and mRNA levels.
NF-κB was involved in the effects of propofol and TNF-α on MMP-9 expression in hCMEC/D3 cells
It has been reported that the expression of MMP-9 was induced by transcription factors such as NF-κB [19]. Thus, we examined whether NF-κB phosphorylation was involved in the inhibitory effect of propofol on TNF-α-induced MMP-9 expression. As shown in Fig. 5a, the phosphorylation of NF-κB was induced by TNF-α and was modulated by propofol in hCMEC/D3 cells, while the expression of NF-κB was not changed. Furthermore, TNF-α caused the translocation of NF-κB from the cytosol to the nuclei, and this effect was inhibited by propofol. As shown in Fig. 5b, we found that PDTC (20 μM), an inhibitor of NF-κB, markedly attenuated TNF-α-induced MMP-9 expression.
Fig. 5.

The phosphorylation of NF-κB was involved in the effects of propofol and TNF-α on MMP-9 expression in hCMEC/D3 cells. a Left panel is the protein expression of p-NF-κB, total NF-κB, nuclear NF-κB, cytoplasmic NF-κB and MMP-9 in hCMEC/D3 cells treated with TNF-α and propofol; right panel is the plot of normalized ratios of optical densities. b hCMEC/D3 cells were treated with TNF-α, propofol, and NF-κB inhibitor (PDTC, 20 μM). Left panel is the protein expression of p-NF-κB, NF-κB and MMP-9; right panel is the plot of normalized ratios of optical densities. Data were repressed as the mean ± SD. *P < 0.05, compared with the control group. #P < 0.05, compared with TNF-α treatment group. β-Actin served as an internal loading control. Histone H3 served as an internal loading control for nuclear extract protein. Each assay was performed in five replicates
ERK was involved in propofol- and TNF-α-mediated NF-κB phosphorylation and MMP-9 expression in hCMEC/D3 cells
NF-κB phosphorylation has been shown to be regulated by ERK in vascular endothelial cells [20]. As shown in Fig. 6a, the phosphorylation of ERK was induced by TNF-α and was inhibited by propofol in hCMEC/D3 cells, while the expression of ERK was not changed. We found that LY3214996 (5 μM), an inhibitor of ERK, significantly attenuated the effects of TNF-α on NF-κB phosphorylation and MMP-9 expression (Fig. 6b).
Fig. 6.

The phosphorylation of ERK was involved in the effects of propofol and TNF-α on NF-κB phosphorylation and MMP-9 expression in hCMEC/D3 cells. a Left panel is the protein expression of p-ERK, ERK and MMP-9 in hCMEC/D3 cells treated with TNF-α and propofol; right panel is the plot of normalized ratios of optical densities. b The hCMEC/D3 cells were treated with TNF-α, propofol and an ERK inhibitor (LY3214996, 5 μM). Left panel is the protein expression of p-ERK, p-NF-κB, ERK, NF-κB and MMP-9; right panel is the plot of normalized ratios of optical densities. Data were repressed as the mean ± SD. *P < 0.05, compared with the control group. #P < 0.05, compared with TNF-α treatment group. β-Actin served as an internal loading control. Each assay was performed in five replicates
CaMKII was involved in the effects of propofol and TNF-α on the phosphorylation of ERK and NF-κB and the expression of MMP-9 in hCMEC/D3 cells
It has been demonstrated that the phosphorylation of ERK could be regulated by the CaMKII signaling pathway [21]. As shown in Fig. 7a, the phosphorylation of CaMKII was induced by TNF-α and was attenuated by propofol in hCMEC/D3 cells, while the expression of CaMKII was not changed. We found that KN93 (10 μM), an inhibitor of CaMKII, markedly decreased the phosphorylation of CaMKII, ERK and NF-κB and the expression of MMP-9 (Fig. 7b).
Fig. 7.

The phosphorylation of CaMKII was involved in the effects of propofol and TNF-α on the phosphorylation of ERK and NF-κB and the expression of MMP-9 in hCMEC/D3 cells. a Left panel is the protein expression of p-CaMKII, CaMKII and MMP-9 in hCMEC/D3 cells that were treated with TNF-α and propofol; right panel is the plot of normalized ratios of optical densities. b The hCMEC/D3 cells were treated with TNF-α, propofol and the inhibitor of CaMKII (KN93, 10 μM). Left panel is the protein expression of p-CaMKII, p-ERK, p-NF-κB, CaMKII, ERK, NF-κB and MMP-9; right panel is the plot of normalized ratios of optical densities. Data were repressed as the mean ± SD. *P < 0.05, compared with the control group. #P < 0.05, compared with TNF-α treatment group. β-Actin served as an internal loading control. Each assay was performed in five replicates
Intracellular Ca2+ was involved in the effects of propofol and TNF-α on the phosphorylation of CaMKII, ERK, and NF-κB and the expression of MMP-9 in hCMEC/D3 cells
It is generally recognized that the phosphorylation of CaMKII was induced by intracellular Ca2+. It has also been reported that the Ca2+ signaling pathway is involved in the expression of MMP-9 [22]. Thus, we examined the effects of TNF-α and propofol on intracellular Ca2+ in hCMEC/D3 cells. As shown in Fig. 8a, TNF-α increased the intracellular Ca2+ concentration in hCMEC/D3 cells, while propofol inhibited this effect. Then, we found that BAPTA-AM (10 μM), the Ca2+ chelator, could significantly attenuate the phosphorylation of CaMKII, ERK and NF-κB and the expression of MMP-9 (Fig. 8b). Furthermore, we found that treatment with PDTC, LY3214996, KN93, or BAPTA-AM alone had no effect on MMP-9 expression in hCMEC/D3 cells (Fig. 8c).
Fig. 8.

Intracellular Ca2+ concentration was involved in the effects of propofol and TNF-α on the phosphorylation of CaMKII, ERK, and NF-κB and the expression of MMP-9 in hCMEC/D3 cells. TNF-α (25 ng/mL, 8 h) increased the concentration of intracellular Ca2+, while propofol (25 μM) attenuated the effect of TNF-α. a hCMEC/D3 cells were treated with TNF-α and propofol. The concentration of intracellular Ca2+ was detected using a Fluo-3 AM Fluorescence Kit. b The hCMEC/D3 cells were treated with TNF-α, propofol and a Ca2+ chelator (BAPTA-AM, 10 μM). Left panel is the protein expression of p-CaMKII, p-ERK, p-NF-κB, CaMKII, ERK, NF-κB and MMP-9; right panel is the plot of normalized ratios of optical densities. β-Actin served as an internal loading control. c hCMEC/D3 cells were treated with TNF-α, PDTC, LY3214996, KN93, and BAPTA-AM. Left panel is the protein expression of MMP-9; right panel is the plot of normalized ratios of optical densities. Data were repressed as the mean ± SD. *P < 0.05, compared with the control group. #P < 0.05, compared with TNF-α treatment group. β-Actin served as an internal loading control. Each assay was performed in five replicates
Discussion
In the present study, we detected that propofol could inhibit the expression of MMP-9, which was induced by TNF-α. More importantly, we indicated that the Ca2+/CaMKII/ERK/NF-κB signaling pathway was involved in the effect of TNF-α and propofol on MMP-9 expression.
It has been reported that disruption of the BBB is involved in early POCD [3]. The BBB mainly consists of cerebral microvascular endothelial cells, astrocytes, pericytes, and the basal lamina. Human cerebral microvascular endothelial cells, which are derived from pluripotent stem cells and form the lumen of capillaries, are the most important component of the BBB. hCMEC/D3 cells, an established model of the human BBB in vitro, are suitable for studying the molecular mechanisms of BBB disruption at the cellular level [23, 24]. It has been reported that hCMEC/D3 cells maintain the barrier properties of the human BBB in vitro, even in the absence of abluminal astrocytes [25]. Thus, we chose hCMEC/D3 cells as the BBB model in vitro to investigate the molecular mechanisms of BBB disruption at the cellular level.
The impairment of the BBB is an early and representative event in the inflammation of the central nervous system, which was induced by extrinsic or intrinsic stimuli, including TNF-α, IL-1β, and IL-6 [26]. TNF-α is the most important inflammatory cytokine after surgical trauma and plays a crucial role in central nervous system inflammation. Studies have demonstrated that TNF-α could induce an inflammatory response in endothelial cells [20]. This study revealed that TNF-α could induce inflammatory mediators, IL-6, vascular cell adhesion molecule-1, and inducible nitric oxide synthase expression in endothelial cells [20]. We chose TNF-α as the main stimulus to mimic the in vivo inflammation environment.
MMPs, a type of zinc ion-dependent endopeptidase, could regulate cell function and activate extracellular matrix degradation, decreasing cell adhesion, and inducing apoptosis. In recent decades, MMP-2 and MMP-9 have been widely studied. The expression of MMP-2 and MMP-9 were involved in BBB disruption and neuroinflammation [27, 28]. One study found that TNF-α led to an increased expression of MMP-2 and MMP-9 in human umbilical vein endothelial cells [29]. Interestingly, we found that TNF-α could notably increase MMP-9 expression, while the expression of MMP-2 was not altered in hCMEC/D3 cells (data not shown). Accordingly, we inferred that TNF-α may induce MMP-2 expression in certain types of endothelial cells but not in hCMEC/D3 cells.
The anti-inflammatory effects of propofol have been uncovered in clinical trials, which revealed that propofol could inhibit the release of inflammation factors, including TNF-α, IL-1β and IL-6 after intracranial surgery [10]. Studies also demonstrated that propofol was able to decrease the levels of TNF-α, IL-6 and IL-8 during cardiopulmonary bypass [30]. Recently, a study demonstrated that propofol could alleviate intracerebral hemorrhage in rats by inhibiting inflammatory factor release and up-regulating MMP-9 in the brain [31]. However, whether propofol could attenuate the noxious effects of these inflammation factors remains unclear. The plasma concentration of propofol ranges from 5 to 50 μM during the induction and maintenance of general anesthesia [32]. Accordingly, we examined the effects of propofol within this range. In line with the study of Han D [31], we found that 25 μM propofol, which is within the clinical range of plasma concentrations, could protect against TNF-α-induced impairment of BBB integrity and significantly decrease TNF-α-induced MMP-9 expression.
It has been reported that MMP-9 can degrade type IV collagen, which is an important component of the BBB and plays a key role in maintaining the neural microenvironment [5]. Therefore, we explored whether TNF-α could decrease the level of collagen IV in hCMEC/D3 cells. As predicted, we indicated that TNF-α could inhibit the level of collagen IV in hCMEC/D3 cells, while propofol could attenuate this effect. In this study, we also explored whether the effects of TNF-α and propofol on collagen IV are due to regulating its synthesis or degradation. We found that MMP-9 silencing also reversed the effect of TNF-α on collagen IV as propofol. Therefore, we inferred that propofol could attenuate the effects of TNF-α on collagen IV by inhibiting MMP-9 expression. Moreover, we also indicated that TNF-α-induced collagen IV decrease is due to its degradation rather than synthesis.
The Ca2+ signaling pathway has been reported to participate in BBB disruption and neuroinflammation [33, 34]. As a second messenger, intracellular Ca2+, which is induced by many extracellular stimuli, plays a key role in regulating cell function. One study demonstrated that the increase in intracellular Ca2+ could decrease the transendothelial electrical resistance of an in vitro BBB model [35]. Therefore, we examined the effect of TNF-α and propofol on the concentration of intracellular Ca2+ in hCMEC/D3 cells. As shown in Fig. 8a, we confirmed that propofol could inhibit the increase in intracellular Ca2+ concentration in hCMEC/D3 cells. Furthermore, we showed that the chelator of Ca2+ could inhibit the expression of MMP-9 as propofol did. Thus, we inferred that Ca2+ was involved in the effect of TNF-α and propofol on MMP-9 expression. However, we could not determine whether the increased intracellular Ca2+ came from extracellular sources or organelles, such as mitochondria and the endoplasmic reticulum. We investigated this issue in our follow-up experiments.
It is known that intracellular Ca2+ can affect cell functions by activating CaMKII. As a direct target of Ca2+ signaling, CaMKII has been reported to be related to MMP-9 expression [36]. Moreover, it was previously shown that TNF-α may induce Ca2+ influx and activate CaMKII in macrophages [37] and cardiomyocytes [38]. Consistently, we found that TNF-α could induce the phosphorylation of CaMKII, while calcium chelator could repress it. Accordingly, we believed that TNF-α-induced CaMKII activation was mediated through increased intracellular calcium concentration and that the Ca2+/CaMKII signaling pathway was involved in the effects of TNF-α and propofol on MMP-9 expression.
The ERK pathway is also reportedly related to BBB damage and neuroinflammation [39–41]. Moreover, it has been demonstrated that the phosphorylation of CaMKII can activate ERK [21]. Our study showed that TNF-α significantly increased the phosphorylation of ERK in hCMEC/D3 cells, while propofol repressed it. We also observed that an inhibitor of CaMKII could inhibit the phosphorylation of ERK. Furthermore, an inhibitor of ERK could mimic the effect of propofol on MMP-9 expression. Thus, we inferred that the Ca2+/CaMKII/ERK signaling pathway was involved in the effect of TNF-α and propofol on MMP-9 expression. It has been reported that p38 MAPK is also activated by TNF-α and is involved in MMP-9 expression [42]. Although we did not examine the phosphorylation and expression of p38 MAPK in this study, we found that the inhibitor of ERK could almost completely depress TNF-α-induced MMP-9 expression. Thus, we inferred that compared with p38 MAPK, ERK plays a more important role in TNF-α-induced MMP-9 expression.
An increasing amount of evidence has confirmed that the expression of MMP-9 is induced by transcription factors such as NF-κB [19, 43, 44]. It has also been reported that the phosphorylation of ERK can activate the NF-κB pathway in endothelial cells [45]. In this study, we found that TNF-α induced the phosphorylation of NF-κB, while propofol attenuated this effect. Then, we detected that the inhibitor of NF-κB could significantly decrease the expression of MMP-9 as propofol did. These results also indicated that the inhibitor of ERK could inhibit the phosphorylation of NF-κB, as propofol did. It has been reported that AP-1 was also activated by ERK and was involved in TNF-α-induced MMP-9 expression [46]. In another ongoing study, we found that although TNF-α could induce the phosphorylation of c-jun and c-fos, the inhibitor of AP-1 had no effect on MMP-9 expression (data not shown). Therefore, we believed that AP-1 was not involved in TNF-α-induced MMP-9 expression in hCMEC/D3 cells. Taken together, our results revealed that TNF-α could induce the expression of MMP-9 via the Ca2+/CaMKII/ERK/NF-κB signaling pathway.
Although it is well known that the sedative effect of propofol is mediated through the gamma-aminobutyric acid (GABA) receptor, recent findings have indicated that propofol may also act on the N-methyl-D-aspartate (NMDA) receptor. It has been found that propofol inhibits intracellular calcium overload and Ca2+/CaMKII as well as ERK, thus attenuating the malignant potential of pancreatic cancer, and the effect was mediated by inhibition of NMDA receptors [47]. In another study, it was reported that propofol protected against LPS-mediated inflammation in primary microglial cells, and the protective effect was mediated by inhibition of NMDA receptors and the Ca2+/CaMKII pathway [48]. An animal study also showed that propofol inhibited the NMDA receptor/ERK signaling pathway, exerting preventive analgesia in a rat model of inflammatory pain [49]. In combining these data with our current findings, although we did not examine which receptor is responsible for the effect of propofol in this study, we believe that it is mediated by inhibition of NMDA receptors. This needs to be confirmed by further research.
Furthermore, although the neuroprotective effect of propofol has been widely reported, the neurotoxicity effect of propofol has also been demonstrated in animal studies. A study revealed that propofol may disrupt BBB permeability by inducing the expression of heat shock protein 72 (HSP72) and albumin in the developing mouse brain [50]. We realized that the protective effects of propofol and the mechanism found in this in vitro study should be confirmed in animal models, and we are planning to examine the effect of propofol against TNF-α-mediated MMP-9 expression and BBB impairment in the mouse model. Propofol may play different roles at different stages of brain development. In summary, our study revealed that propofol inhibited TNF-α-induced MMP-9 expression by repressing the Ca2+/CaMKII/ERK/NF-κB signaling pathway in hCMEC/D3 cells (Fig. 9).
Fig. 9.
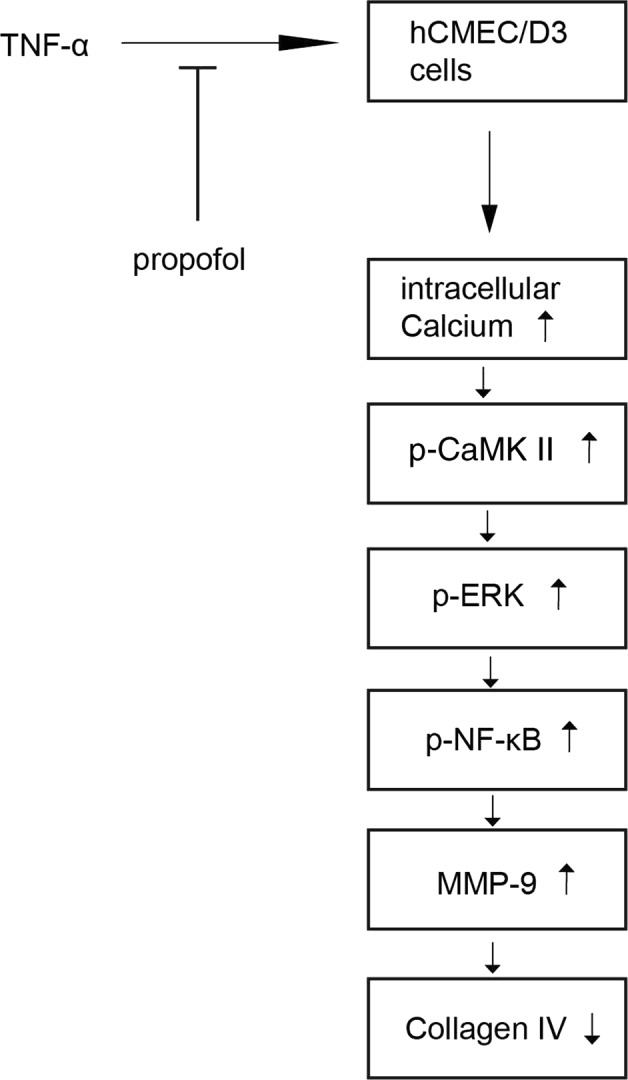
The proposed mechanism of the protective effect of propofol against TNF-α-induced MMP-9 expression in hCMEC/D3 cells
Acknowledgements
We thank Zhen Wang (Fudan University Shanghai Cancer Center, Shanghai, China) for providing human astrocytes and for her assistance with technical aspects of performing the experiments. This work was supported by the Shanghai Municipal Commission of Health and Family Planning Foundation (No. 201540080) and by National Key R&D Program of China (No. 2018YFC2001900-04).
Author contributions
XWD performed the experiments and wrote the paper. XS performed the experiments and the data analysis. XFS performed the experiments and wrote the paper. YL performed the experiments and analyzed the data. JQW performed the experiments and analyzed the data. ZRS designed the study and modified the manuscript. CHM designed the study and modified the manuscript. JWC designed the study and modified the manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Zhi-rong Sun, Email: sunrongsun@aliyun.com.
Chang-hong Miao, Email: miao_chh@126.com.
Jia-wei Chen, Email: jiawei_chen@hotmail.com.
References
- 1.Skvarc DR, Berk M, Byrne LK, Dean OM, Dodd S, Lewis M, et al. Post-operative cognitive dysfunction: an exploration of the inflammatory hypothesis and novel therapies. Neurosci Biobehav Rev. 2018;84:116–33. doi: 10.1016/j.neubiorev.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 2.Theoharides TC, Tsilioni I, Patel AB, Doyle R. Atopic diseases and inflammation of the brain in the pathogenesis of autism spectrum disorders. Transl Psychiatry. 2016;6:e844. doi: 10.1038/tp.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He HJ, Wang Y, Le Y, Duan KM, Yan XB, Liao Q, et al. Surgery upregulates high mobility group box-1 and disrupts the blood-brain barrier causing cognitive dysfunction in aged rats. CNS Neurosci Ther. 2012;18:994–1002. doi: 10.1111/cns.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie H, Huang D, Zhang S, Hu X, Guo J, Wang Z, et al. Relationships between adiponectin and matrix metalloproteinase-9 (MMP-9) serum levels and postoperative cognitive dysfunction in elderly patients after general anesthesia. Aging Clin Exp Res. 2016;28:1075–9. doi: 10.1007/s40520-015-0519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamamura R, Nagatsuka H, Siar CH, Katase N, Naito I, Sado Y, et al. Comparative analysis of basal lamina type IV collagen alpha chains, matrix metalloproteinases-2 and -9 expressions in oral dysplasia and invasive carcinoma. Acta Histochem. 2013;115:113–9. doi: 10.1016/j.acthis.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Sillat T, Saat R, Pollanen R, Hukkanen M, Takagi M, Konttinen YT. Basement membrane collagen type IV expression by human mesenchymal stem cells during adipogenic differentiation. J Cell Mol Med. 2012;16:1485–95. doi: 10.1111/j.1582-4934.2011.01442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang CM, Hsieh HL, Lin CC, Shih RH, Chi PL, Cheng SE, et al. Multiple factors from bradykinin-challenged astrocytes contribute to the neuronal apoptosis: involvement of astroglial ROS, MMP-9, and HO-1/CO system. Mol Neurobiol. 2013;47:1020–33. doi: 10.1007/s12035-013-8402-1. [DOI] [PubMed] [Google Scholar]
- 8.Yang CC, Hsiao LD, Yang CM, Lin CC. Thrombin enhanced matrix Metalloproteinase-9 expression and migration of SK-N-SH Cells via PAR-1, c-Src, PYK2, EGFR, Erk1/2 and AP-1. Mol Neurobiol. 2017;54:3476–91. doi: 10.1007/s12035-016-9916-0. [DOI] [PubMed] [Google Scholar]
- 9.Ma L, Guan YQ, Du ZD. Salvianolic acid B down-regulates matrix metalloproteinase-9 activity and expression in tumor necrosis factor-alpha-induced human coronary artery endothelial cells. Chin Med J. 2015;128:2658–63. doi: 10.4103/0366-6999.166037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berger M, Ponnusamy V, Greene N, Cooter M, Nadler JW, Friedman A, et al. The effect of propofol vs. isoflurane anesthesia on postoperative changes in cerebrospinal fluid cytokine levels: results from a randomized trial. Front Immunol. 2017;8:1528. doi: 10.3389/fimmu.2017.01528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Z, Lu Y, Wang J, Ding X, Chen J, Miao C. The protective effect of propofol against TNF-alpha-induced apoptosis was mediated via inhibiting iNOS/NO production and maintaining intracellular Ca(2 + ) homeostasis in mouse hippocampal HT22 cells. Biomed Pharmacother. 2017;91:664–72. doi: 10.1016/j.biopha.2017.04.110. [DOI] [PubMed] [Google Scholar]
- 12.Geng YJ, Wu QH, Zhang RQ. Effect of propofol, sevoflurane, and isoflurane on postoperative cognitive dysfunction following laparoscopic cholecystectomy in elderly patients: A randomized controlled trial. J Clin Anesth. 2017;38:165–71. doi: 10.1016/j.jclinane.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Tang J, Chen X, Tu W, Guo Y, Zhao Z, Xue Q, et al. Propofol inhibits the activation of p38 through up-regulating the expression of annexin A1 to exert its anti-inflammation effect. PLoS ONE. 2011;6:e27890. doi: 10.1371/journal.pone.0027890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang M, Zhang L, Liu Z, Zhou J, Pan Q, Fan J, et al. AGO1 may influence the prognosis of hepatocellular carcinoma through TGF-beta pathway. Cell Death Dis. 2018;9:324. doi: 10.1038/s41419-018-0338-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao MZ, Mao WZ, Ma GL, Li Y. NF-kappab inhibitor PDTC enhances tumor necrosis factor alpha-induced apoptosis of gastric cancer cell SGC-7901. Zhonghua Wei Chang Wai Ke Za Zhi. 2013;16:578–82. [PubMed] [Google Scholar]
- 16.Wu Q, Zhao Y, Duan W, Liu Y, Chen X, Zhu M. Propofol inhibits high glucose-induced PP2A expression in human umbilical vein endothelial cells. Vasc Pharmacol. 2017;91:18–25. doi: 10.1016/j.vph.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Hsu SS, Jan CR, Liang WZ. The investigation of the pyrethroid insecticide lambda-cyhalothrin (LCT)-affected Ca(2 + ) homeostasis and -activated Ca(2 + )-associated mitochondrial apoptotic pathway in normal human astrocytes: The evaluation of protective effects of BAPTA-AM (a selective Ca(2 + ) chelator) Neurotoxicology. 2018;69:97–107. doi: 10.1016/j.neuro.2018.09.009. [DOI] [PubMed] [Google Scholar]
- 18.Daniels BP, Cruz-Orengo L, Pasieka TJ, Couraud PO, Romero IA, Weksler B, et al. Immortalized human cerebral microvascular endothelial cells maintain the properties of primary cells in an in vitro model of immune migration across the blood brain barrier. J Neurosci Methods. 2013;212:173–9. doi: 10.1016/j.jneumeth.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song ZB, Ni JS, Wu P, Bao YL, Liu T, Li M, et al. Testes-specific protease 50 promotes cell invasion and metastasis by increasing NF-kappaB-dependent matrix metalloproteinase-9 expression. Cell Death Dis. 2015;6:e1703. doi: 10.1038/cddis.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang D, Xiao CX, Su ZH, Huang MW, Qin M, Wu WJ, et al. -)-7(S)-hydroxymatairesinol protects against tumor necrosis factor-alpha-mediated inflammation response in endothelial cells by blocking the MAPK/NF-kappaB and activating Nrf2/HO-1. Phytomedicine. 2017;32:15–23. doi: 10.1016/j.phymed.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Ha TT, Burwell ST, Goodwin ML, Noeker JA, Heggland SJ. Pleiotropic roles of Ca( + 2)/calmodulin-dependent pathways in regulating cadmium-induced toxicity in human osteoblast-like cell lines. Toxicol Lett. 2016;260:18–27. doi: 10.1016/j.toxlet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu CY, Hsieh HL, Sun CC, Yang CM. IL-1beta induces MMP-9 expression via a Ca2 + -dependent CaMKII/JNK/c-JUN cascade in rat brain astrocytes. Glia. 2009;57:1775–89. doi: 10.1002/glia.20890. [DOI] [PubMed] [Google Scholar]
- 23.Dauchy S, Miller F, Couraud PO, Weaver RJ, Weksler B, Romero IA, et al. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochem Pharmacol. 2009;77:897–909. doi: 10.1016/j.bcp.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Ohtsuki S, Ikeda C, Uchida Y, Sakamoto Y, Miller F, Glacial F, et al. Quantitative targeted absolute proteomic analysis of transporters, receptors and junction proteins for validation of human cerebral microvascular endothelial cell line hCMEC/D3 as a human blood-brain barrier model. Mol Pharm. 2013;10:289–96. doi: 10.1021/mp3004308. [DOI] [PubMed] [Google Scholar]
- 25.Cucullo L, Couraud PO, Weksler B, Romero IA, Hossain M, Rapp E, et al. Immortalized human brain endothelial cells and flow-based vascular modeling: a marriage of convenience for rational neurovascular studies. J Cereb Blood Flow Metab. 2008;28:312–28. doi: 10.1038/sj.jcbfm.9600525. [DOI] [PubMed] [Google Scholar]
- 26.Sibson NR, Blamire AM, Perry VH, Gauldie J, Styles P, Anthony DC. TNF-alpha reduces cerebral blood volume and disrupts tissue homeostasis via an endothelin- and TNFR2-dependent pathway. Brain. 2002;125:2446–59. doi: 10.1093/brain/awf256. [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Su P, Meng S, Aschner M, Cao Y, Luo W, et al. Role of matrix metalloproteinase-2/9 (MMP2/9) in lead-induced changes in an in vitro blood-brain barrier model. Int J Biol Sci. 2017;13:1351–60. doi: 10.7150/ijbs.20670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu JZ, Wang XK, Cao Y, Li DZ, Wu TD, Zhang T, et al. Tetramethylpyrazine facilitates functional recovery after spinal cord injury by inhibiting MMP2, MMP9, and vascular endothelial cell apoptosis. Curr Neurovasc Res. 2017;14:110–6. doi: 10.2174/1567202614666170313114115. [DOI] [PubMed] [Google Scholar]
- 29.Lee YJ, Hwang SM, Kang DG, Kim JS, Lee HS. Effect of Gastrodia elata on tumor necrosis factor-alpha-induced matrix metalloproteinase activity in endothelial cells. J Nat Med. 2009;63:463–7. doi: 10.1007/s11418-009-0352-6. [DOI] [PubMed] [Google Scholar]
- 30.Samir A, Gandreti N, Madhere M, Khan A, Brown M, Loomba V. Anti-inflammatory effects of propofol during cardiopulmonary bypass: a pilot study. Ann Card Anaesth. 2015;18:495–501. doi: 10.4103/0971-9784.166451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han D, Li S, Xiong Q, Zhou L, Luo A. Effect of propofol on the expression of MMP-9 and its relevant inflammatory factors in brain of rat with intracerebral hemorrhage. Cell Biochem Biophys. 2015;72:675–9. doi: 10.1007/s12013-015-0516-9. [DOI] [PubMed] [Google Scholar]
- 32.Mathy-Hartert M, Deby-Dupont G, Hans P, Deby C, Lamy M. Protective activity of propofol, Diprivan and intralipid against active oxygen species. Mediat Inflamm. 1998;7:327–33. doi: 10.1080/09629359890848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen JT, Lin YL, Chen TL, Tai YT, Chen CY, Chen RM. Ketamine alleviates bradykinin-induced disruption of the mouse cerebrovascular endothelial cell-constructed tight junction barrier via a calcium-mediated redistribution of occludin polymerization. Toxicology. 2016;368-369:142–51. doi: 10.1016/j.tox.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 34.Lerdkrai C, Asavapanumas N, Brawek B, Kovalchuk Y, Mojtahedi N, Olmedillas Del Moral M, et al. Intracellular Ca(2 + ) stores control in vivo neuronal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2018;115:E1279–e88. doi: 10.1073/pnas.1714409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park J, Fan Z, Kumon RE, El-Sayed ME, Deng CX. Modulation of intracellular Ca2 + concentration in brain microvascular endothelial cells in vitro by acoustic cavitation. Ultrasound Med Biol. 2010;36:1176–87. doi: 10.1016/j.ultrasmedbio.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dai L, Zhuang L, Zhang B, Wang F, Chen X, Xia C, et al. DAG/PKCdelta and IP3/Ca(2)( + )/CaMK IIbeta operate in parallel to each other in PLCgamma1-driven cell proliferation and migration of human gastric adenocarcinoma cells, through Akt/mTOR/S6 pathway. Int J Mol Sci. 2015;16:28510–22. doi: 10.3390/ijms161226116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tano JY, Lee RH, Vazquez G. Involvement of calmodulin and calmodulin kinase II in tumor necrosis factor alpha-induced survival of bone marrow derived macrophages. Biochem Biophys Res Commun. 2012;427:178–84. doi: 10.1016/j.bbrc.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang GJ, Wang HX, Yao YS, Guo LY, Liu P. The role of Ca2 + /calmodulin-dependent protein kinase II and calcineurin in TNF-alpha-induced myocardial hypertrophy. Braz J Med Biol Res. 2012;45:1045–51. doi: 10.1590/S0100-879X2012007500121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang LF, Li X, Gao YB, Wang SM, Zhao L, Dong J, et al. Activation of VEGF/Flk-1-ERK pathway induced blood-brain barrier injury after microwave exposure. Mol Neurobiol. 2015;52:478–91. doi: 10.1007/s12035-014-8848-9. [DOI] [PubMed] [Google Scholar]
- 40.Fan H, Wu PF, Zhang L, Hu ZL, Wang W, Guan XL, et al. Methionine sulfoxide reductase A negatively controls microglia-mediated neuroinflammation via inhibiting ROS/MAPKs/NF-kappaB signaling pathways through a catalytic antioxidant function. Antioxid Redox Signal. 2015;22:832–47. doi: 10.1089/ars.2014.6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng D, Wang B, Ma Y, Shi W, Tao K, Zeng W, et al. The Ras/Raf/Erk pathway mediates the subarachnoid hemorrhage-induced apoptosis of hippocampal neurons through phosphorylation of p53. Mol Neurobiol. 2016;53:5737–48. doi: 10.1007/s12035-015-9490-x. [DOI] [PubMed] [Google Scholar]
- 42.Tsai CL, Chen WC, Lee IT, Chi PL, Cheng SE, Yang CM. c-Src-dependent transactivation of PDGFR contributes to TNF-alpha-induced MMP-9 expression and functional impairment in osteoblasts. Bone. 2014;60:186–97. doi: 10.1016/j.bone.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 43.Huang Q, Lan F, Wang X, Yu Y, Ouyang X, Zheng F, et al. IL-1beta-induced activation of p38 promotes metastasis in gastric adenocarcinoma via upregulation of AP-1/c-fos, MMP2 and MMP9. Mol Cancer. 2014;13:18. doi: 10.1186/1476-4598-13-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin WS, Hong Y, Lee HW, Lee ST. Catalytically defective receptor protein tyrosine kinase PTK7 enhances invasive phenotype by inducing MMP-9 through activation of AP-1 and NF-kappaB in esophageal squamous cell carcinoma cells. Oncotarget. 2016;7:73242–56. doi: 10.18632/oncotarget.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong Y, Cheng CF, Luo YZ, Tian CW, Yang H, Liu BR, et al. C-reactive protein stimulates RAGE expression in human coronary artery endothelial cells in vitro via ROS generation and ERK/NF-kappaB activation. Acta Pharm Sin. 2015;36:440–7. doi: 10.1038/aps.2014.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen YJ, Chang LS. NFkappaB- and AP-1-mediated DNA looping regulates matrix metalloproteinase-9 transcription in TNF-alpha-treated human leukemia U937 cells. Biochim Biophys Acta. 2015;1849:1248–59. doi: 10.1016/j.bbagrm.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 47.Chen X, Wu Q, You L, Chen S, Zhu M, Miao C. Propofol attenuates pancreatic cancer malignant potential via inhibition of NMDA receptor. Eur J Pharmacol. 2017;795:150–9. doi: 10.1016/j.ejphar.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 48.Wu Q, Zhao Y, Chen X, Zhu M, Miao C. Propofol attenuates BV2 microglia inflammation via NMDA receptor inhibition. Can J Physiol Pharmacol. 2018;96:241–8. doi: 10.1139/cjpp-2017-0243. [DOI] [PubMed] [Google Scholar]
- 49.Qiu Q, Sun L, Wang XM, Lo ACY, Wong KL, Gu P, et al. Propofol produces preventive analgesia via GluN2B-containing NMDA Receptor/ERK1/2 Signaling Pathway in a rat model of inflammatory pain. Mol Pain. 2017;13:1744806917737462. doi: 10.1177/1744806917737462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma HS, Ponten E, Gordh T, Eriksson P, Fredriksson A, Sharma A. Propofol promotes blood-brain barrier breakdown and heat shock protein (HSP 72 kd) activation in the developing mouse brain. CNS Neurol Disord Drug Targets. 2014;13:1595–603. doi: 10.2174/1871527313666140806122906. [DOI] [PubMed] [Google Scholar]