

Abstract
Magnesium lithospermate B (MLB) is an active component of Salvia miltiorrhiza Radix, a traditional Chinese herb used in treating cardiovascular diseases. In this study, we investigated the protective effects of MLB against inflammation-induced endothelial dysfunction in vitro and in vivo, and the underlying mechanisms. Endothelial dysfunction was induced in human dermal microvascular endothelial cells (HMEC-1) in vitro by lipopolysaccharide (LPS, 1 μg/mL). We showed that pretreatment with MLB (10–100 μM) dose-dependently inhibited LPS-induced upregulation of inflammatory cytokines ICAM1, VCAM1, and TNFα, which contributed to reduced leukocytes adhesion and attenuation of endothelial hyperpermeability in HMEC-1 cells. SD rats were injected with LPS (10 mg/kg, ip) to induce endothelial dysfunction in vivo. We showed that pretreatment with MLB (25–100 mg/kg, ip) dose-dependently restored LPS-impaired endothelial-dependent vasodilation in superior mesenteric artery (SMA), attenuated leukocyte adhesion in mesenteric venules and decreased vascular leakage in the lungs. We further elucidated the mechanisms underlying the protective effects of MLB, and revealed that MLB pretreatment inhibited NF-κB activation through inhibition of IκBα degradation and subsequent phosphorylation of NF-κB p65 in vitro and in vivo. In HMEC-1 cells, MLB pretreatment activated the nuclear factor erythroid-2-related factor 2 (Nrf2) pathway. Knockdown of Nrf2 with siRNA abolished the inhibitory effects of MLB on IκBα degradation and ICAM1 up-regulation, which were mimicked by PKC inhibition (Gö6983) or PI3K/Akt inhibition (LY294002). In summary, our results demonstrate that MLB inhibits NF-κB activation through PKC- and PI3K/Akt-mediated Nrf2 activation in HMEC-1 cells and protects against LPS-induced endothelial dysfunction in murine model of acute inflammation.
Keywords: magnesium lithospermate B, endothelium, human dermal microvascular endothelial cells, rat superior mesenteric artery rings, lipopolysaccharide, inflammation, Nrf2, PKC, PI3K/Akt
Introduction
The endothelium lies at the interface between blood and tissue, where it regulates vascular permeability, vascular tone, and the basal trafficking of hematopoietic cells by sensing and transducing signals from both blood and tissues. Various stimuli can lead to endothelial dysfunction, which has been associated with atherogenesis, diabetic vasculopathy, heart failure, sepsis, insulin resistance, and hypertension [1]. Endothelial cells (ECs) act as both target and effector cells in the innate immune system during inflammation [2, 3]. Recent advances have revealed the pivotal role of endothelial dysfunction in the progression of systemic inflammation, indicating the potential of the endothelium as a therapeutic target for inflammation control.
Vascular cells express an extensive repertoire of antioxidant genes that are regulated by nuclear factor erythroid-2-related factor-2 (Nrf2). Nrf2 is recognized as a critical transcriptional activator of antioxidant responses and has been suggested to possess potent anti-inflammatory properties [4]. Under basal conditions, Nrf2 is suppressed by Kelch-like ECH-associated protein 1 (Keap1), which targets Nrf2 for ubiquitination and proteasomal degradation. Following stimulation with antioxidants, shear stress, or electrophilic agents, Nrf2 dissociates from Keap1, escapes proteasome degradation, translocates to the nucleus, binds to the ARE (AU-rich element) sequence, and activates the transcriptional expression of its target genes. Nrf2 has also been found to regulate endothelium functions, such as angiogenesis, nitric oxide (NO) production, and blood–barrier function. Global deletion of Nrf2 or specific deletion of Nrf2 in ECs can result in a reduction of vascular density as well as endothelial cell sprouting [5]. Activation of Nrf2 has been shown to maintain eNOS in the coupled state by ensuring a stoichiometric BH4:eNOS ratio, which subsequently improves NO bioavailability [6]. Pharmacological activation of Nrf2 has been proven to be effective at protecting the blood–brain barrier after brain injury, a process that can be blocked by the disruption of Nrf2 signaling with ARE-containing decoy oligonucleotides or loss of the nrf2 gene [7]. These protective effects on the endothelium are mainly attributed to the antioxidative features of Nrf2. Accumulating evidence supports the anti-inflammatory role of Nrf2 and its downstream antioxidative genes in vascular inflammation models. Pharmacological activation of Nrf2 and expression of constitutively active Nrf2 in EC have proven to be effective at suppressing the proinflammatory state of EC [4, 8].
Magnesium lithospermate B (MLB) is the most abundant, active, hydrophilic component of Salvia miltiorrhiza Radix, a traditional Chinese herb mainly used to treat cardiovascular diseases. The benefits of MLB on the cardiovascular system have been addressed in numerous studies, most of which have focused on ischemia-reperfusion and cardiomyopathy. Some studies have reported that MLB protects EC against injuries induced by glucose [9], oxLDL [10], hypoxia [11, 12], or vasospasm [13, 14]. Previous work from our laboratory demonstrated that MLB inhibits Ca2+ influx and stimulates NO production in EC [15, 16], a mechanism that contributes, at least in part, to the vasoprotective effects of MLB. Only a few of these studies provided in vivo evidence of MLB-induced improvement of endothelial function.
Studies have shown that the hydrophilic bioactive components from Salvia miltiorrhiza Radix can activate the Nrf2 pathway in various cell types and tissues [17–22]. Among them, previous studies in vascular smooth muscle cells (VSMCs) [23, 24] and pancreatic β-cells [25] have demonstrated that MLB protects against hyperglycemia-induced proliferation and migration of VSMCs and cytokine-induced apoptosis of β-cells through the Nrf2-ARE pathway.
From these observations, we hypothesized that MLB protects ECs against inflammation-induced dysfunction and through Nrf2 activation. The aim of this study was to test our hypothesis in a lipopolysaccharide (LPS)-induced model of acute inflammation.
Materials and methods
Chemicals and reagents
MLB (99.7% purity) was isolated from S. miltiorrhiza in our laboratory. MCDB-131 medium, L-glutamine, fetal bovine serum (FBS), RPMI-1640 medium, and Lipofectamine 3000 were purchased from Thermo Fisher Scientific (Carlsbad, CA, USA). LPS (from Escherichia coli 055: B5), anti-β-actin antibody, and Evans blue dye (EBD) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Anti-GAPDH, anti-p65, anti-p-p65 (Ser536), anti-IκBα, anti-Akt, and anti-p-Akt (Ser473) antibodies were obtained from Cell Signaling Technologies (Danvers, MA, USA). Anti-HO1, anti-Nrf2, anti-p-Nrf2 (Ser40), anti-ICAM1, anti-eNOS, and anti-iNOS antibodies were purchased from Abcam (Cambridge, UK).
Cell culture
Human dermal microvascular endothelial cells (HMEC-1) were maintained in culture dishes containing MCDB-131 medium supplemented with 10% FBS (v/v), 100 ng/mL human epidermal growth factor (Peprotech, London, UK), 1.461 g/L L-glutamine and 1 μg/mL hydrocortisone (Sigma). Human monocyte-derived THP-1 cells were maintained in RPMI-1640 supplemented with 10% FBS (v/v). Cells were incubated at 37 °C in a humidified 5% CO2 atmosphere.
RNA extraction and quantitative real-time PCR
HMEC-1 total RNA was extracted using TRIzol reagent (Thermo Fisher) and reverse-transcribed into cDNA (Takara, Dalian, China). Quantitative real-time polymerase chain reaction (PCR) was performed on a CFX Real-Time PCR Detection System (Bio-Rad) using SYBR Green Supermix (Bio-Rad, CA, USA) to determine ICAM1 (intercellular adhesion molecule 1), VCAM1 (vascular cell adhesion molecule 1), and TNFα (tumor necrosis factor-α) mRNA levels in HMEC-1 cells. All values were normalized against the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). ΔΔCT method [26]. The following PCR primer pairs were used: ICAM1, 5′-GTG CAA GAA GAT AGC CAA CCA A-3′ (sense) and 5′-TGC CAG TTC CAC CCG TTC-3′ (antisense); VCAM1, 5′-TAC ATA TCA CCC AAG AAT ACAG-3′ (sense) and 5′-TTC CCA ATC AAA TTA ACT CC-3′ (antisense); TNFɑ, 5′-TCA ACC TCC TCT CTG CCA TC-3′ (sense) and 5′-CCA AAG TAG ACC TGC CCA GA-3′ (antisense); GAPDH, 5′-AAG AAG GTG GTG AAG CAG G-3′ (sense), and 5′-AGG TGG AGG AGT GGG TGT CG-3′ (antisense).
Western blot analysis
Total protein from HMEC-1 cells was extracted by homogenization in ice-cold RIPA buffer supplemented with a protease inhibitor cocktail (Merck, Argentina; USA). Lung and aortic tissues were washed three times with ice-cold phosphate-buffered saline (PBS) to remove residual blood and then homogenized and lysed in ice-cold RIPA buffer. Protein samples (20 μg/lane) were separated by SDS-PAGE, blotted onto PVDF membranes, blocked with 5% non-fat milk or bovine serum albumin, and incubated with primary antibodies at 4 °C overnight. The membranes were then washed three times with Tris-buffered saline containing 0.1% Tween-20 (TBST) and incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody for 2 h at room temperature. After rinsing three times in TBST, immunolabeled proteins were visualized using Clarity Western ECL Blotting substrates (Bio-Rad). Information regarding the primary antibodies used in this study is shown in Table 1.
Table 1.
Primary antibody information
| Name | Cat. no. | Dilution factor |
|---|---|---|
| Anti-GAPDH antibody | 2118 | 1:5000 |
| Anti-β-actin antibody | A5441 | 1:5000 |
| Anti-p65 antibody | 8242 | 1:1000 |
| Anti-p-p65 (Ser536) antibody | 3033 | 1:1000 |
| Anti-IκBα antibody | 4812 | 1:1000 |
| Anti-Akt antibody | 9272 | 1:1000 |
| Anti-p-Akt (Ser473) antibody | 4060 | 1:1000 |
| Anti-HO1 antibody | ab68477 | 1:1000 |
| Anti-Nrf2 antibody | ab62352 | 1:1000 |
| Anti-p-Nrf2 (Ser40) antibody | ab76026 | 1:1000 |
| Anti-eNOS antibody | ab199956 | 1:500 |
| Anti-iNOS antibody | ab3523 | 1:250 |
RNA interference
HMEC-1 cells were transfected at approximately 50%–60% confluency using the Lipofectamine 3000 reagent according to the manufacturer’s instructions (Invitrogen, Life Science Technologies, CA, USA). Small interfering RNA (siRNA) against human Nrf2 (forward, 5′-AGA UAU AGA UCU UGG AGU AdTdT-3′; reverse, 5′-UAC UCC AAG AUC UAU AUC UdTdT-3′) and scrambled siRNA (negative control) was purchased from Biomics Co., Ltd. (Jiangsu, China). Cells were transfected with Nrf2 siRNA (50 nM) or scrambled siRNA (50 nM) for 6 h. After transfection, cells were cultured in fresh medium for an additional 48 h before MLB treatment.
In vitro leukocyte adhesion
HMEC-1 cells plated on 6-well plates were treated with 100 μM MLB for 12 h and then stimulated with 1 μg/mL LPS for 6 h. THP-1 mononuclear cells (2 × 105 cells/mL) were labeled with 2.5 μg/mL calcein AM (Sigma) in RPMI-1640 medium. After washing twice with PBS, calcein AM-labeled THP-1 cells were resuspended in fresh MCDB-131 medium and then added at 105 cells/well onto an HMEC-1 monolayer and incubated for 30 min. Adherent THP-1 cells from five randomly selected fields were counted under a fluorescence microscope (Olympus, Tokyo, Japan) [27].
In vitro EC permeability
In vitro fluorescein isothiocyanate (FITC)-dextran permeability assays were performed as previously described [28]. In brief, HMEC-1 cells were seeded onto Transwell inserts (0.4-μm pore size; Corning Costar, Tewksbury, MA) coated with Matrigel (Invitrogen, Life Sciences) and grown to confluence. After the indicated treatments, 10 μg/mL FITC-dextran (Sigma-Aldrich) solution was added to the inserts. After allowing 20 min for diffusion, the medium from the lower chamber was collected and analyzed on a fluorescence plate reader.
Animals
Male Sprague-Dawley rats (200–300 g) were purchased from the Shanghai Experimental Animal Center. All experimental procedures used in this study were approved by the Animal Care and Use Committee of Shanghai Institute, University of Chinese Academy of Sciences, and are in compliance with ARRIVE guidelines [29, 30]. All animals were housed in a temperature-controlled room under a 12-h light–dark cycle with free access to standard chow and water. Animals were held in animal facilities for at least 1 week before the experiments.
The pharmacological effects of MLB on inflammation-induced endothelial dysfunction were evaluated by first pretreating animals with different concentrations of MLB for 2 h and then stimulating with LPS (10 mg/kg, i.p.) for 4 h. Three sets of in vivo experiments were carried out to comprehensively evaluate endothelial function. For evaluations of leukocyte–endothelium adhesion, animals were randomly divided into four groups: control, LPS only, 50 mg/kg MLB + LPS, and 100 mg/kg MLB + LPS (n = 5 animals/group). For vascular tone recordings and permeability assays, animals were divided into five groups: control, LPS only, 25 mg/kg MLB + LPS, 50 mg/kg MLB + LPS, and 100 mg/kg MLB + LPS (n = 5 animals/group).
In vivo leukocyte adhesion
Real-time leukocyte–endothelium interactions in rat mesenteric veins were observed as reported previously [31]. Four hours after LPS induction, animals were anesthetized with sodium pentobarbital (60 mg/kg, i.p.) and retro-orbitally injected with 100 μL of rhodamine 6G (1 mg/mL) to label leukocytes and platelets in vivo. To observe the mesenteric microcirculation, the abdomen was opened along the midline, and the mesentery was carefully drawn out of the abdominal cavity. Single, unbranched veins without obvious bends and with diameters ranging from 200 to 300 μm were selected. Blood cell–endothelial interactions were recorded for 1 min (n = 4 veins/mouse) by intravital microscopy. Adherent leukocytes, defined as those showing no visible movement for 30 s, were counted in a 0.04 mm2 field. The images were processed using Image J software.
Isolation of rat SMA and measurement of vascular tone
Isolation of rat SMA and measurement of vascular tone were performed as reported previously [15]. After the indicated treatments, animals were anesthetized with sodium pentobarbital, and their mesenteric beds were exposed. The SMA were separated from surrounding tissues, dissected out, and placed in ice-cold Krebs solution. All adherent adipose and connective tissues were then carefully removed without damaging the endothelial layer. Each artery was cut into four rings (approximately 2 mm in length) and mounted in 20-mL bath chambers filled with Krebs solution (pH 7.4) at 37 °C and aerated with a 95% O2/5% CO2 gas mixture. The vascular tone was recorded and analyzed using an integrated myograph system (PowerLab 4/20, AD Instruments, Sydney, Australia). During the initial 1-h equilibration, artery rings were stretched until the resting tension remained steady at 0.5 g. Artery rings were evaluated for a reproducible contractile response to 30 mM KCl. After pre-contraction with 1 μM norepinephrine, acetylcholine (ACh) concentration–response curves were constructed by the cumulative application of ACh (1 nM to 10 μM) to the artery rings at 2-min intervals. For the sodium nitroprusside (SNP)-induced vasodilation assay, SMA rings were perfused with 1.80 mg/mL sodium deoxycholate (Sigma-Aldrich) in saline for 30 s to remove endothelium as described previously [15] and then rinsed with Krebs for 40 min. The removal of endothelium was confirmed by the lack of vasodilatory response to 1 nM ACh. After pre-contraction with 1 µM norepinephrine, SNP (Sigma-Aldrich) concentration–response curves were then constructed by the cumulative application of SNP (1 nM to 10 µM) to the artery rings at 2 min intervals.
Blood samples were collected from the abdominal aorta before dissection of the SMA and then centrifuged to obtain serum. The lungs, mesenteric bed, and aorta were collected after dissection of the SMA, washed three times in ice-cold saline, frozen in liquid nitrogen, and stored at −80 °C for further evaluation.
Nitrite/nitrate assay
Total NO production in serum and tissue samples was determined by measuring the concentration of nitrite/nitrate using the modified Griess reaction method, according to the manufacturer’s instructions (Beyotime Institute of Biotechnology, Jiangsu, China).
Lung permeability assay
Lung permeability was measured using the well-characterized EBD extravasation assay [32]. EBD (30 mg/kg) was administered by tail vein injection 2.5 h after LPS injection. After an additional 1.5 h, tissues were perfused, removed, dried at 55 °C for 24 h, and weighed. EBD was extracted by homogenizing tissue in formamide (0.1 g tissue/mL formamide) and incubating for 16 h. After centrifugation (12 000 × g, 30 min), supernatants were collected, and the absorbance was measured at 620 nm. Formamide extracts from rats that did not receive EBD injection were prepared in the same way to generate an Evans blue standard curve.
Statistics
For each experiment, the values were expressed as the mean ± SEM. The differences among groups were statistically assessed by analysis of variance (ANOVA) using GraphPad Prism software (version 7.0). P-values < 0.05 were considered statistically significant.
Results
MLB suppresses the LPS-induced expression of inflammatory cytokines in HMEC-1 cells
Elevated expression of inflammatory cytokines is a hallmark of NF-κB activation in response to LPS and contributes to the initiation and progression of endothelial dysfunction. To determine whether MLB affects the inflammation-associated increases in cytokines, we measured mRNA levels of ICAM1, VCAM1, and TNFα in HMEC-1 cells. LPS stimulation significantly increased ICAM1, VCAM1, and TNFα mRNA levels; this effect was inhibited in a concentration-dependent manner by MLB (Fig. 1a). In line with the observed changes in mRNA levels, pretreatment with MLB caused a concentration-dependent inhibition of LPS-induced upregulation of ICAM1 protein (Fig. 1b). These results indicate that MLB exerts an inhibitory effect on the LPS-induced expression of cytokines.
Fig. 1.
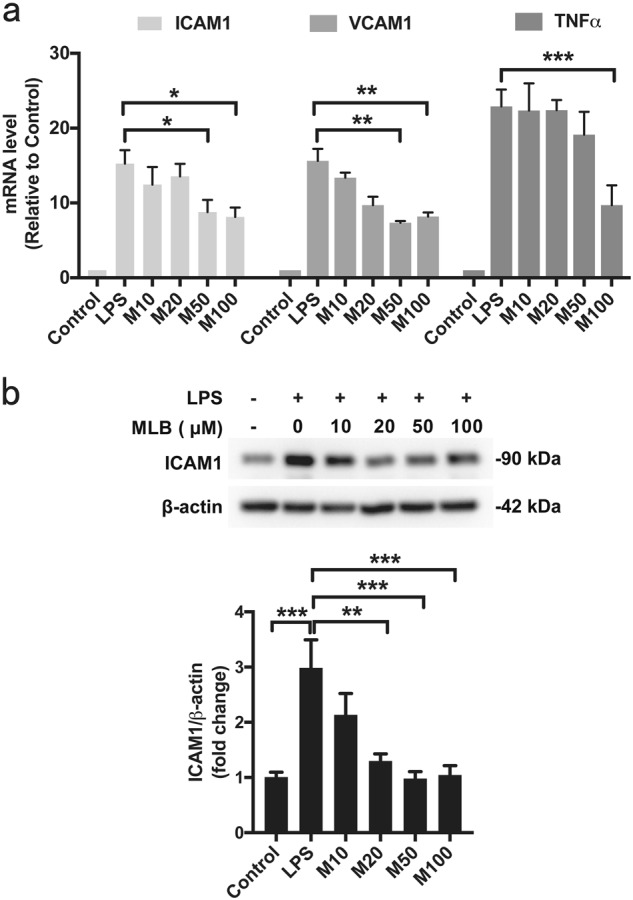
a Inhibitory effects of MLB on LPS-stimulated ICAM1, VCAM1, and TNFα mRNA expression. HMEC-1 cells were pretreated with increasing concentrations of MLB (10–100 μM) for 12 h and then stimulated with 1 μg/mL LPS for 2 h. b Inhibitory effect of MLB on LPS-induced ICAM1 protein expression. Data are presented as means ± SEM of five independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001)
MLB decreases leukocyte adhesion to ECs in vitro and in vivo
The endothelium regulates leukocyte trafficking through the expression of adhesion molecules and chemokines/cytokines, which are vital to the development of vascular inflammation. To determine whether the attenuation of cytokine expression affects the process of leukocyte adhesion, we evaluated leukocyte–endothelial interactions both in vitro and in vivo.
After the indicated treatments, confluent HMEC-1 cells were incubated with calcein AM-labeled THP-1 cells, and cell adherence to HMEC-1 monolayers was assessed. LPS stimulation for 6 h resulted in an approximately 1.8-fold increase in adherent THP-1 cells to HMEC-1 monolayers; this effect was attenuated by pretreatment with 100 μM MLB (Fig. 2a, c). In vivo recordings of leukocyte–endothelial interactions in mesenteric venules yielded results consistent with the in vitro assays (Fig. 2b, c). Four hours after LPS injection with or without MLB pretreatment, the number of adherent leukocytes in the mesenteric venules was counted. The number of adherent leukocytes was significantly increased in the LPS-treated group (18.5 ± 2.72/0.04 mm2) compared with that in the control group (1.2 ± 0.20/0.04 mm2). The number of adherent leukocytes in mesenteric venules after LPS stimulation was significantly reduced by pre-administration of 50 mg/kg MLB (9.45 ± 0.49/0.04 mm2) or 100 mg/kg MLB (8.25 ± 1.39/0.04 mm2). Taken together, these results indicate that, by inhibiting the expression of adhesion molecules, MLB reduces leukocyte–endothelial interactions during inflammation.
Fig. 2.

a Representative images of THP-1 adhesion to an HMEC-1 monolayer. HMEC-1 cells were treated with 100 μM MLB for 12 h and then stimulated with 1 μg/mL LPS for 6 h. The number of adherent THP-1 cells per microscopic field was determined by fluorescence microscopy. Scale bar = 500 μm. b Representative images of leukocyte adhesion to endothelium in mesenteric venules observed by intravital fluorescence microscopy. Yellow squares represent 0.04-mm2 counting fields. Representative videos of the above results are included in the supplementary information. Scale bar = 100 μm. c Statistical analyses of leukocyte adhesion in vitro and in vivo. For in vitro adhesion experiments, the total number of adherent THP-1 cells was counted in five randomly selected optical fields per well. Data are expressed as means ± SEM of five independent experiments. For in vivo adhesion experiments, adherent leukocytes, defined as those with no visible movement for 30 s, were counted in a 0.04-mm2 field. Data are expressed as means ± SEM (n = 5 animals/group, *P < 0.05
MLB restores endothelial-dependent vasodilation in mesenteric arteries by increasing NO bioavailability
Endothelial-dependent vasodilation was induced by cumulative addition of different concentrations of ACh (1 nM to 10 μM) to norepinephrine-precontracted SMA. SMA segments from the LPS-challenged group displayed a significantly diminished vasodilatory response to ACh. Administration of MLB at a single concentration of 50 or 100 mg/kg progressively restored ACh-induced vasodilation (Fig. 3a), while no improvements were observed in SNP-induced vasodilation in denuded vessels (Supporting Information 2). Further analyses revealed a 31.3-fold increase in EC50 in the LPS-challenged group (3.409 ± 1.89 μM) compared with that of the controls (0.109 ± 0.019 μM), which was completely abrogated by 50 and 100 mg/kg MLB (0.095 ± 0.028 and 0.113 ± 0.029 μM, respectively) (Fig. 3b).
Fig. 3.

a Endothelial-dependent vasodilatory responses to ACh in SMA segments harvested from Control, LPS-only, and MLB (25, 50, 100 mg/kg) + LPS groups (n = 5 animals/group). b EC50 values from concentration–response curves. c MLB pretreatment did not affect total nitrite/nitrate levels in serum (n = 5 animals/group). d MLB elevated nitrite/nitrate levels in aortic and mesenteric tissues (n = 3 for mesenteric tissues in the control group, n = 5 for all the other groups). e MLB pretreatment attenuated LPS induced eNOS protein decrease and iNOS protein induction in both aortic tissues (n = 5 animals/group) and SMAs. SMAs from five animals in each group were pooled together to get enough protein for Western blot. Data are expressed as mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001)
ACh-induced endothelial-dependent vasodilation is mainly mediated by NO. Accordingly, we evaluated the nitrite/nitrate concentration in the serum and vascular tissues as a surrogate measure of NO. LPS stimulation resulted in a significant increase in nitrite/nitrate levels in serum compared with that in the controls. MLB pretreatment exerted no inhibitory effect on LPS-induced serum NO production (Fig. 3c). However, the results from aortic tissue revealed a decrease in NO bioavailability after LPS stimulation that was partially rescued by pretreatment with 100 mg/kg MLB. In mesenteric tissues, basal NO production was much lower than that in the aorta and was not further affected by LPS. Interestingly, 100 mg/kg MLB significantly increased mesenteric nitrite/nitrate levels compared with those in the LPS-only group (Fig. 3d). LPS stimulation resulted in decreased eNOS and increased iNOS protein in both aortic tissues and SMA, which were abrogated by the MLB pretreatment (Fig. 3e).
Together, these results suggest that MLB is capable of preventing LPS-induced impairment in endothelial-dependent vasodilation. Moreover, this process may be attributable to improvements in NO bioavailability in the aorta and mesenteric tissues.
MLB suppresses endothelial hyperpermeability in vitro and in vivo
The endothelium constitutes a semi-selective barrier between the intravascular and extravascular compartments, and increased endothelial permeability is thought to be a significant contributor to tissue edema during sepsis. In vitro permeability assays showed that LPS stimulation increased the passage of FITC-dextran across an HMEC-1 monolayer. Notably, this increase was completely abolished by pretreatment with 100 μM MLB, which alone did not affect the HMEC-1 monolayer permeability (Fig. 4a). To assess vascular permeability in vivo, we measured EBD extravasation in the lung, heart, and ear after LPS challenge. LPS treatment induced a significant increase in vascular leakage in the lung that was rescued by MLB pretreatment (Fig. 4b). No increase in vascular leakage was observed in the heart, liver, or ear (Supplementary Information 3).
Fig. 4.
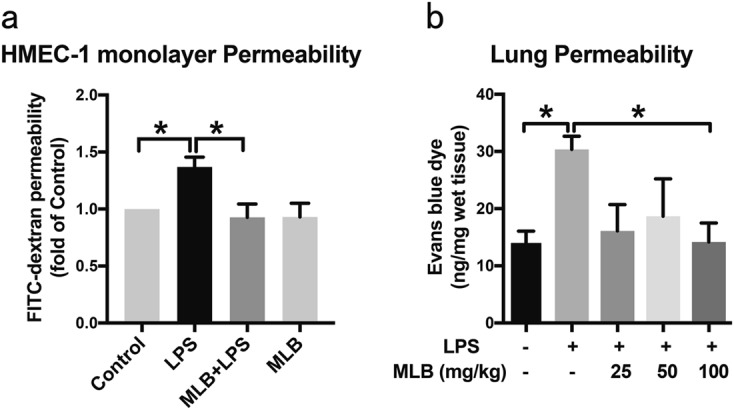
Effects of MLB on LPS-induced vascular leakage in vitro and in vivo. a MLB blunted LPS-induced FITC-dextran permeability in confluent HMEC-1 monolayers. Data are expressed as means ± SEM of five independent experiments. b MLB rescued LPS-induced endothelial leakage in the lung in a concentration-dependent manner. Data are expressed as means ± SEM (n = 5 animals/group; *P < 0.05)
MLB protects ECs from LPS-induced activation through inhibition of the NF-κB pathway
Endothelial dysfunction in response to LPS is mainly mediated by NF-κB [33]. Accordingly, we examined the effects of MLB on LPS-induced NF-κB activation. In HMEC-1 cells, MLB (10–100 μM) caused a concentration-dependent inhibition of IκBα protein degradation and prevented LPS-induced Ser536 phosphorylation of NF-κB p65, which are hallmarks of NF-κB activation (Fig. 5a). Western blot analysis of IκBα protein levels in the lung and aortic tissues also revealed partial inhibition of NF-κB activation (Fig. 5b), suggesting that MLB exerts an inhibitory effect on the NF-κB pathway both in vitro and in vivo.
Fig. 5.

MLB inhibits LPS-induced NF-κB activation in vitro and in vivo. a Effects of MLB on LPS-induced Ser536 phosphorylation of p65 and IκBα degradation. HMEC-1 cells were pretreated with 100 μM MLB for 12 h and then stimulated with 1 μg/mL LPS for 0.5 h. Data are expressed as means ± SEM of five independent experiments. b Lung and aorta expression of IκBα protein in the control, LPS-only and 100 mg/kg MLB + LPS groups. Data are expressed as means ± SEM, n = 5 animals/group (*P < 0.05, **P < 0.01, ***P < 0.001)
Nrf2 mediates the anti-inflammatory effects of MLB
Next, we sought to elucidate the mechanisms underlying the inhibitory effects of MLB on NF-κB activation. Western blot analyses demonstrated that MLB induces a concentration-dependent activation of the Nrf2 pathway, characterized by increased phosphorylation of Nrf2 at Ser40 and upregulation of the Nrf2 target, HO1 (Fig. 6a). siRNA-mediated Nrf2 knockdown resulted in approximately a 55% reduction in Nrf2 at both the mRNA and protein levels. Disrupting this pathway with Nrf2 siRNA significantly decreased HO1 expression and abrogated the inhibitory effect of MLB on IκBα degradation (Fig. 6b). This, in turn, led to an increase in downstream ICAM1 production (Fig. 6c), indicating an indispensable role for Nrf2 in MLB-mediated NF-κB inhibition. Basal ICAM1 mRNA levels were also higher in Nrf2 siRNA-transfected cells, although this difference did not reach statistical significance. Collectively, these data suggest that the anti-inflammatory effects of MLB are mediated by Nrf2 activation.
Fig. 6.

Nrf2 mediates the anti-inflammatory effects of MLB. a Western blot showing the effects of MLB on Nrf2 phosphorylation and HO1 expression (n = 5). b Western blot showing the effects of siRNA-mediated Nrf2 knockdown on stabilization of IκBα by MLB (n = 5). c siRNA-mediated Nrf2 knockdown abrogated the reduction in ICAM1 mRNA induced by MLB pretreatment (n = 6). HMEC-1 cells were transfected with Nrf2 siRNA for 48 h, pretreated with MLB for 12 h, and then stimulated with LPS. Data are expressed as means ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001)
The PKC and PI3K/Akt pathways are involved in the anti-inflammatory effects of MLB
Previous studies have revealed that the PKC pathway is responsible for phosphorylation of Nrf2 at Ser40 [34, 35]. Therefore, we employed the pan-PKC inhibitor Gö6983 to determine whether MLB activates Nrf2 through the PKC pathway. As expected, Gö6983 (10 μM) prevented MLB-induced Nrf2 phosphorylation at Ser40 and abrogated HO1 upregulation (Fig. 7a). Pretreatment with Gö6983 also blunted the anti-inflammatory effects of MLB on HMEC-1 cells (Fig. 7b).
Fig. 7.

MLB activates Nrf2 through the PKC pathway. a Western blot analyses of MLB-induced Ser40 phosphorylation of Nrf2 and HO1 expression with and without Gö6983 pretreatment (n = 5). b The effect of Gö6983 on MLB-induced IκBα stabilization (n = 5). c Gö6983 prevented the inhibitory effect of MLB on ICAM1 mRNA levels (n = 5). HMEC-1 cells were incubated with 100 μM MLB for 12 h in the presence or absence of Gö6983 (10 μM) or GF109203X (10 μM, data not shown), and then stimulated with LPS (1 μg/mL). Protein and total RNA samples were extracted 0.5 and 2 h after LPS stimulation, respectively, for detection of IκBα protein and evaluation of ICAM1 mRNA levels. Data are expressed as mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001)
It is well established that the PI3K/Akt pathway supports optimal EC survival during various pathological conditions and may contribute to the activation of Nrf2 [36, 37]. To investigate the involvement of PI3K/Akt in MLB-mediated endothelial protection, we first confirmed that MLB causes a concentration-dependent activation of the PI3K/Akt pathway (Fig. 8a). Pharmacological inhibition of PI3K with LY294002 reduced MLB-mediated Nrf2 phosphorylation at Ser40 and HO1 upregulation (Fig. 8b). LY294002 also reduced the MLB-mediated inhibition of IκBα degradation and ICAM1 mRNA upregulation (Fig. 8c, d), suggesting the involvement of the PI3K/Akt pathway in the protective action of MLB.
Fig. 8.

PI3K/Akt is involved in the anti-inflammatory effects of MLB. a MLB promoted phosphorylation of Akt in a concentration-dependent manner. b LY294002 reversed MLB-mediated Nrf2 activation. c MLB-induced stabilization of IκBα was attenuated by LY294002. d LY294002 prevented the inhibitory effects of MLB on ICAM1 mRNA levels. HMEC-1 cells were incubated with 100 μM MLB for 12 h in the presence or absence of LY294002 (10 μM) and then stimulated with LPS (1 μg/mL). Protein and total RNA samples were extracted 0.5 and 2 h after LPS stimulation, respectively, for detection of IκBα protein and evaluation of ICAM1 mRNA level. The results are representative of five independent experiments. Data are expressed as means ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001)
Discussion
Acute inflammation leads to endothelial dysfunction, and the resulting vascular damage ultimately causes organ damage. The results from the present study demonstrate for the first time that MLB potently protects ECs from LPS-induced dysfunction in HMEC-1 cells and a murine model of acute inflammation. Mechanistic studies revealed that MLB inhibits the NF-κB pathway through PKC- and PI3K/Akt-mediated activation of Nrf2.
It is well recognized that endothelial dysfunction contributes to the progression of inflammation and subsequent organ damage. In a transgenic murine model of sepsis, EC-specific blockade of NF-κB was shown to be sufficient to improve survival, reduce leukocyte infiltration, decrease vascular leakage, and inhibit coagulation [32, 33, 38] in experimental sepsis models. In another study, an E-selectin-specific NF-κB inhibitor developed to target activated ECs selectively reduced extravasation of inflammatory cells in an experimental model of rheumatoid arthritis [39]. Here, we provided in vitro and in vivo evidence that administration of a single dose of MLB reduces LPS-induced leukocyte adhesion, restores EC-dependent vasodilation, and maintains endothelial barrier function through inhibition of NF-κB, indicating improved endothelial function. Interestingly, LPS-induced systemic NO production was not affected by the MLB pretreatment. These observations rule out the possibility that MLB protects endothelial function by inhibiting systemic inflammation, which agrees with a report by Xu et al. in which the EC-specific blockade of NF-κB was shown to contribute minimally to systemic inflammation [32]. However, whether repeated administration or long-term MLB treatment would affect systemic inflammation will require further investigation.
The pharmacological effects of MLB on vascular function have been demonstrated in various experimental models. In murine models of diabetes, chronic administration of MLB was shown to attenuate the diabetic impairment of endothelial-dependent vasodilation. Further mechanistic studies on human aortic endothelial cells (HAECs) revealed the involvement of the PI3K/Akt-Nrf2 pathway [9], as supported by our findings in HMEC-1 cells. Chang et al. revealed that MLB exerts an opposing effect on vasoconstriction in a murine vasospasm model that could be blocked by the eNOS inhibitor, L-NAME, indicating the participation of eNOS [13, 14]. Our previous reports revealed that ex vivo incubation with MLB activated the large-conductance, Ca2+-dependent potassium (BKCa) channel in VSMCs and induced NO release by ECs, both of which contributed to the subsequent vasodilation of SMA [15]. In the present study, the SMA was isolated after LPS challenge in the absence or presence of MLB, an experimental strategy that should reflect the in vivo effects of MLB on the endothelium of SMA.
In most organs, ECs form a dynamic barrier between the vasculature and interstitial space that allows the selective exchange of certain molecules. Disruption of endothelial barrier function can cause excessive leakage of fluid and proteins from the vasculature and lead to severe organ edema and inflammatory responses, even causing an increase in the mortality rate due to sepsis [40]. Some studies have suggested that restoring the endothelial barrier function alleviates symptoms and improves outcomes in murine sepsis models [41, 44]. Clinically, vascular hyperpermeability is most evident in the pulmonary microcirculation and results in acute lung injury. In this study, we observed a significant increase in permeability in lung tissues, but not in the liver, heart, or ear, reflecting the different properties of different vascular beds [45].
Studies have shown that Nrf2 exerts protective effects against endothelial injuries in various pathological models. The most well-studied is the diabetic mellitus animal model [36, 46, 48] in which hyperglycemia was shown to decrease endothelium-dependent vasodilation and can be reversed by pharmacological or genetic activation or overexpression of Nrf2. In a high-salt-diet-induced endothelial dysfunction model, Nrf2 deficiency abolished low-dose Ang II-induced endothelium protection, as characterized by an increased vasodilation response to ACh [49]. In addition, studies have also implicated a protective role of Nrf2 on ECs in the development of atherosclerosis. As mentioned before, Nrf2 in ECs can be activated by laminar flow [50, 51], which has been shown to be a critical regulator of endothelial function. Atheroprotective vessel regions under steady laminar flow demonstrated greater Nrf2 activation [52] and lower NF-κB activation [53] compared with that in atherosusceptible vessel regions, which may be responsible for the elevated eNOS protein expression in atheroprotective regions.
Cellular events that occur during inflammation are strongly associated with redox balance. Studies have shown that LPS-induced NF-κB activation dramatically increases reactive oxygen species (ROS) production in various cell types, which in turn exacerbates and prolongs inflammation. As a master regulator of the redox balance, Nrf2 has long been considered a potential anti-inflammatory target. Studies have shown that Nrf2 deficiency results in enhanced activation of NF-κB and increased mortality in experimental sepsis models [54], whereas Nrf2-activated antioxidant cascades exert anti-inflammatory effects in various experimental models. Our data revealed that transient knockdown of Nrf2 resulted in a trend toward increased ICAM1 mRNA in control and LPS-treated cells compared with that in cells transfected with scrambled siRNA. In the MLB + LPS treatment group, siRNA-mediated Nrf2 knockdown abrogated the reduction in ICAM1 induced by MLB pretreatment, confirming the indispensable role of Nrf2 in MLB-induced NF-κB inhibition.
Several signaling pathways, including extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), protein kinase C (PKC), and PI3K/Akt have been implicated in Nrf2 activation. Among these, members of the PKC family were the first kinases implicated in inducing Nrf2 activation [34, 35]. The PI3K/Akt pathway has been reported to augment Nrf2 transcription [55] and enhance Nrf2 activity by inhibiting Gsk3β (glycogen synthase kinase 3 beta) [36, 56] and disrupting Keap1–Nrf2 interactions [50]. In the present study, both the pan-PKC inhibitor Gö6983 and PI3K/Akt inhibitor LY294002 reversed MLB-induced inhibition of NF-κB, indicating the involvement of the PKC and PI3K/Akt pathways.
One limitation of the present study is that we did not identify which PKC isoform is responsible for MLB-mediated Nrf2 activation. The PKC family is a group of serine/threonine kinases with at least 12 members with various functions, which has only been partially understood. Few studies have reported the PKC isoforms involved in Nrf2 activation. In ECs, the augmented activity of PKC ε has been proven to induce Nrf2 activation and protect ECs against TNFα-induced apoptosis [57]. In bone marrow multipoint mesenchymal stromal cells, PKC δ was reported to interact with CR6-interacting factor I (CRIFI) to induce Nrf2 Ser40 phosphorylation [58]. In our previous work, we used two PKC inhibitors, Gö6983 (10 µM) and GF109203X 10 μM, data not shown, to evaluate the role of PKC in MLB-induced NF-κB inhibition. Surprisingly, though both Gö6983 and GF109203X are pan-PKC inhibitors, Gö6983 successfully blocked MLB-induced NF-κB inhibition (evaluated by the ICAM1 mRNA level), while GF109203X did not. According to previous studies [59], Gö6983 blocks the activity of PKC α, β, γ, δ, and ζ with an IC50 of 7, 7, 6, 10, and 60 nM, respectively, and GF109203X blocks the activity of PKC α, β, γ, δ, ε, and ζ with an IC50 of 8.4, 18, 20, 210, 132, and 580 nM, respectively. Based on these data, we can presume that Gö6983 has higher inhibitory effects on PKC γ, δ, and ζ, which may be one possible explanation for the different results we obtained with the two inhibitors, but the exact isoform required for Nrf2 activation requires further investigation.
In summary, our study presents multiple lines of evidence to support the conclusion that MLB treatment profoundly ameliorates LPS-induced endothelial dysfunction in a murine sepsis model through inhibition of the NF-κB pathway, identifying MLB as a potential candidate for the treatment of endothelial dysfunction.
Electronic supplementary material
{kind=link}
Acknowledgements
We thank Xiao-yu Zhou, Jing Zhao, Zi-ying Shen, and Wen-wei Xu for their technical assistance. We would also like to express our special gratitude and deep condolence for our mentor, Professor Yi-ping Wang, who passed away on 11 April 2018, for his tremendous mentoring and support all along.
Author contributions
FG, YPW, and LJX designed the experiments and wrote the manuscript; FG performed the experiments and the data analyses. JML, CX, HHL, and YLL participated in the data analyses of this manuscript. All authors have read, revised, and approved the submitted version of this manuscript.
Competing interests
The authors declare no competing interests.
Electronic supplementary material
The online version of this article (10.1038/s41401-018-0189-1) contains supplementary material, which is available to authorized users.
References
- 1.Daiber A, Steven S, Weber A, Shuvaev VV, Muzykantov VR, Laher I, et al. Targeting vascular (endothelial) dysfunction. Br J Pharmacol. 2017;174:1591–619. doi: 10.1111/bph.13517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Backer D, Orbegozo Cortes D, Donadello K, Vincent JL. Pathophysiology of microcirculatory dysfunction and the pathogenesis of septic shock. Virulence. 2014;5:73–9. doi: 10.4161/viru.26482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascon GA, et al. The endothelium in sepsis. Shock. 2016;45:259–70. doi: 10.1097/SHK.0000000000000473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, et al. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol. 2006;290:H1862–70. doi: 10.1152/ajpheart.00651.2005. [DOI] [PubMed] [Google Scholar]
- 5.Wei Y, Gong J, Thimmulappa RK, Kosmider B, Biswal S, Duh EJ. Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc Natl Acad Sci USA. 2013;110:E3910–8. doi: 10.1073/pnas.1309276110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heiss EH, Schachner D, Werner ER, Dirsch VM. Active NF-E2-related factor (Nrf2) contributes to keep endothelial NO synthase (eNOS) in the coupled state: role of reactive oxygen species (ROS), eNOS, and heme oxygenase (HO-1) levels. J Biol Chem. 2009;284:31579–86. doi: 10.1074/jbc.M109.009175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao J, Moore AN, Redell JB, Dash PK. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J Neurosci. 2007;27:10240–8. doi: 10.1523/JNEUROSCI.1683-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zakkar M, Van der Heiden K, Luong le A, Chaudhury H, Cuhlmann S, Hamdulay SS, et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler Thromb Vasc Biol. 2009;29:1851–7. doi: 10.1161/ATVBAHA.109.193375. [DOI] [PubMed] [Google Scholar]
- 9.Kim SH, Kim SH, Choi M, Lee Y, Kim YO, Ahn DS, et al. Natural therapeutic magnesium lithospermate B potently protects the endothelium from hyperglycaemia-induced dysfunction. Cardiovasc Res. 2010;87:713–22. doi: 10.1093/cvr/cvq089. [DOI] [PubMed] [Google Scholar]
- 10.Au-Yeung KK, Choy PC, Zhu DY, Siow YL. Magnesium tanshinoate B protects endothelial cells against oxidized lipoprotein-induced apoptosis. Can J Physiol Pharmacol. 2007;85:1053–62. doi: 10.1139/Y07-096. [DOI] [PubMed] [Google Scholar]
- 11.Luo WB, Wang YP. Magnesium lithospermate B inhibits hypoxia-induced calcium influx and nitric oxide release in endothelial cells. Acta Pharmacol Sin. 2001;22:1135–42. [PubMed] [Google Scholar]
- 12.Luo WB, Dong L, Wang YP. Effect of magnesium lithospermate B on calcium and nitric oxide in endothelial cells upon hypoxia/reoxygenation. Acta Pharmacol Sin. 2002;23:930–6. [PubMed] [Google Scholar]
- 13.Chang CZ, Wu SC, Kwan AL, Hwang SL, Howng SL. Magnesium lithospermate B alleviates the production of endothelin-1 through an NO-dependent mechanism and reduces experimental vasospasm in rats. Acta Neurochir (Wien) 2011;153:2211–7. doi: 10.1007/s00701-011-1082-6. [DOI] [PubMed] [Google Scholar]
- 14.Chang CZ, Wu SC, Kwan AL. Magnesium lithospermate B, an active extract of Salvia miltiorrhiza, mediates sGC/cGMP/PKG translocation in experimental vasospasm. Biomed Res Int. 2014;2014:272101. doi: 10.1155/2014/272101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang HF, Chen XQ, Hu GY, Wang YP. Magnesium lithospermate B dilates mesenteric arteries by activating BKCa currents and contracts arteries by inhibiting K(V) currents. Acta Pharmacol Sin. 2010;31:665–70. doi: 10.1038/aps.2010.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Zhang J, Zha R, Hu M, Wang Y. Magnesium lithospermate B decreases [Ca(2+)]i in endothelial cells by inhibiting K(+) currents. Eur J Pharmacol. 2011;650:285–9. doi: 10.1016/j.ejphar.2010.09.028. [DOI] [PubMed] [Google Scholar]
- 17.Joe Y, Zheng M, Kim HJ, Kim S, Uddin MJ, Park C, et al. Salvianolic acid B exerts vasoprotective effects through the modulation of heme oxygenase-1 and arginase activities. J Pharmacol Exp Ther. 2012;341:850–8. doi: 10.1124/jpet.111.190736. [DOI] [PubMed] [Google Scholar]
- 18.Lin M, Zhai X, Wang G, Tian X, Gao D, Shi L, et al. Salvianolic acid B protects against acetaminophen hepatotoxicity by inducing Nrf2 and phase II detoxification gene expression via activation of the PI3K and PKC signaling pathways. J Pharmacol Sci. 2015;127:203–10. doi: 10.1016/j.jphs.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Tongqiang L, Shaopeng L, Xiaofang Y, Nana S, Xialian X, Jiachang H, et al. Salvianolic acid b prevents iodinated contrast media-induced acute renal injury in rats via the PI3K/Akt/Nrf2 pathway. Oxid Med Cell Longev. 2016;2016:7079487. doi: 10.1155/2016/7079487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu B, Cao B, Zhang D, Xiao N, Chen H, Li GQ, et al. Salvianolic acid B protects against paraquat-induced pulmonary injury by mediating Nrf2/Nox4 redox balance and TGF-beta1/Smad3 signaling. Toxicol Appl Pharmacol. 2016;309:111–20. doi: 10.1016/j.taap.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 21.Marin V, Montero-Julian FA, Gres S, Boulay V, Bongrand P, Farnarier C, et al. The IL-6-soluble IL-6Ralpha autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: an experimental model involving thrombin. J Immunol. 2001;167:3435–42. doi: 10.4049/jimmunol.167.6.3435. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Liu YY, Jiang Q, Li KR, Zhao YX, Cao C, et al. Salvianolic acid A protects RPE cells against oxidative stress through activation of Nrf2/HO-1 signaling. Free Radic Biol Med. 2014;69:219–28. doi: 10.1016/j.freeradbiomed.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 23.Hur KY, Seo HJ, Kang ES, Kim SH, Song S, Kim EH, et al. Therapeutic effect of magnesium lithospermate B on neointimal formation after balloon-induced vascular injury. Eur J Pharmacol. 2008;586:226–33. doi: 10.1016/j.ejphar.2008.02.072. [DOI] [PubMed] [Google Scholar]
- 24.Hur KY, Kim SH, Choi MA, Williams DR, Lee YH, Kang SW, et al. Protective effects of magnesium lithospermate B against diabetic atherosclerosis via Nrf2-ARE-NQO1 transcriptional pathway. Atherosclerosis. 2010;211:69–76. doi: 10.1016/j.atherosclerosis.2010.01.035. [DOI] [PubMed] [Google Scholar]
- 25.Lee BW, Chun SW, Kim SH, Lee Y, Kang ES, Cha BS, et al. Lithospermic acid B protects beta-cells from cytokine-induced apoptosis by alleviating apoptotic pathways and activating anti-apoptotic pathways of Nrf2-HO-1 and Sirt1. Toxicol Appl Pharmacol. 2011;252:47–54. doi: 10.1016/j.taap.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD, et al. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. Toxicol Appl Pharmacol. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Roth Flach RJ, Skoura A, Matevossian A, Danai LV, Zheng W, Cortes C, et al. Endothelial protein kinase MAP4K4 promotes vascular inflammation and atherosclerosis. Nat Commun. 2015; 6: 8995. [DOI] [PMC free article] [PubMed]
- 28.Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem. 2009;284:21036–46. doi: 10.1074/jbc.M109.016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–9. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGrath JC, Lilley E. Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol. 2015;172:3189–93. doi: 10.1111/bph.12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herr N, Mauler M, Bode C, Duerschmied D. Intravital microscopy of leukocyte–endothelial and platelet–leukocyte interactions in mesenterial veins in mice. J Vis Exp. 2015:e53077. [DOI] [PMC free article] [PubMed]
- 32.Xu H, Ye X, Steinberg H, Liu SF. Selective blockade of endothelial NF-kappaB pathway differentially affects systemic inflammation and multiple organ dysfunction and injury in septic mice. J Pathol. 2010;220:490–8. doi: 10.1002/path.2666. [DOI] [PubMed] [Google Scholar]
- 33.Ding J, Song D, Ye X, Liu SF. A pivotal role of endothelial-specific NF-kappaB signaling in the pathogenesis of septic shock and septic vascular dysfunction. J Immunol. 2009;183:4031–8. doi: 10.4049/jimmunol.0900105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci USA. 2000;97:12475–80. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–82. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- 36.Dai X, Yan X, Zeng J, Chen J, Wang Y, Chen J, et al. Elevating CXCR7 improves angiogenic function of EPCs via Akt/GSK-3beta/Fyn-mediated Nrf2 activation in diabetic limb ischemia. Circ Res. 2017;120:e7–23. doi: 10.1161/CIRCRESAHA.117.310619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zenkov NK, Kozhin PM, Chechushkov AV, Martinovich GG, Kandalintseva NV, Menshchikova EB. Mazes of Nrf2 regulation. Biochemistry (Mosc) 2017;82:556–64. doi: 10.1134/S0006297917050030. [DOI] [PubMed] [Google Scholar]
- 38.Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med. 2008;205:1303–15. doi: 10.1084/jem.20071393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sehnert B, Burkhardt H, Wessels JT, Schröder A, May MJ, Vestweber D, et al. NF-κB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-κB in immune-mediated diseases. Proc Natl Acad Sci USA. 2013;110:16556–61. doi: 10.1073/pnas.1218219110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou PC, Filbin MR, Wang H, Ngo L, Huang DT, Aird WC, et al. Endothelial permeability and hemostasis in septic shock: results from the ProCESS Trial. Chest. 2017;152:22–31. doi: 10.1016/j.chest.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, Herzog C, Kaushal GP, Gokden N, Mayeux PR. Actinonin, a meprin A inhibitor, protects the renal microcirculation during sepsis. Shock. 2011;35:141–7. doi: 10.1097/SHK.0b013e3181ec39cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holthoff JH, Wang Z, Patil NK, Gokden N, Mayeux PR. Rolipram improves renal perfusion and function during sepsis in the mouse. J Pharmacol Exp Ther. 2013;347:357–64. doi: 10.1124/jpet.113.208520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Z, Sims CR, Patil NK, Gokden N, Mayeux PR. Pharmacologic targeting of sphingosine-1-phosphate receptor 1 improves the renal microcirculation during sepsis in the mouse. J Pharmacol Exp Ther. 2015;352:61–6. doi: 10.1124/jpet.114.219394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng Y, Adamson RH, Curry FR, Tarbell JM. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am J Physiol Heart Circ Physiol. 2014;306:H363–72. doi: 10.1152/ajpheart.00687.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res. 2007;100:174–90. doi: 10.1161/01.RES.0000255690.03436.ae. [DOI] [PubMed] [Google Scholar]
- 46.Li FY, Lam KS, Tse HF, Chen C, Wang Y, Vanhoutte PM, et al. Endothelium-selective activation of AMP-activated protein kinase prevents diabetes mellitus-induced impairment in vascular function and reendothelialization via induction of heme oxygenase-1 in mice. Circulation. 2012;126:1267–77. doi: 10.1161/CIRCULATIONAHA.112.108159. [DOI] [PubMed] [Google Scholar]
- 47.Tian S, Ge X, Wu K, Yang H, Liu Y. Ramipril protects the endothelium from high glucose-induced dysfunction through CaMKKbeta/AMPK and heme oxygenase-1 activation. J Pharmacol Exp Ther. 2014;350:5–13. doi: 10.1124/jpet.114.212928. [DOI] [PubMed] [Google Scholar]
- 48.Jimenez R, Toral M, Gomez-Guzman M, Romero M, Sanchez M, Mahmoud AM, et al. The role of Nrf2 signaling in PPARbeta/delta-mediated vascular protection against hyperglycemia-induced oxidative stress. Oxid Med Cell Longev. 2018;2018:5852706. doi: 10.1155/2018/5852706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Priestley JRC, Kautenburg KE, Casati MC, Endres BT, Geurts AM, Lombard JH. The NRF2 knockout rat: a new animal model to study endothelial dysfunction, oxidant stress, and microvascular rarefaction. Am J Physiol Heart Circ Physiol. 2016;310:H478–87. doi: 10.1152/ajpheart.00586.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA., Jr Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ Res. 2007;101:723–33. doi: 10.1161/CIRCRESAHA.107.152942. [DOI] [PubMed] [Google Scholar]
- 51.McSweeney SR, Warabi E, Siow RC. Nrf2 as an endothelial mechanosensitive transcription factor: going with the flow. Hypertension. 2016;67:20–9. doi: 10.1161/HYPERTENSIONAHA.115.06146. [DOI] [PubMed] [Google Scholar]
- 52.Kim M, Kim S, Lim JH, Lee C, Choi HC, Woo CH. Laminar flow activation of ERK5 protein in vascular endothelium leads to atheroprotective effect via NF-E2-related factor 2 (Nrf2) activation. J Biol Chem. 2012;287:40722–31. doi: 10.1074/jbc.M112.381509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA. 2000;97:9052–7. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–95. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 56.Kaidanovich-Beilin O, Woodgett JR. GSK-3: functional insights from cell biology and animal models. Front Mol Neurosci. 2011;4:40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mylroie H, Dumont O, Bauer A, Thornton CC, Mackey J, Calay D, et al. PKCepsilon-CREB-Nrf2 signalling induces HO-1 in the vascular endothelium and enhances resistance to inflammation and apoptosis. Cardiovasc Res. 2015;106:509–19. doi: 10.1093/cvr/cvv131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen L, Ran Q, Xiang Y, Xiang L, Chen L, Li F, et al. Co-activation of PKC-delta by CRIF1 modulates oxidative stress in bone marrow multipotent mesenchymal stromal cells after irradiation by phosphorylating NRF2 Ser40. Theranostics. 2017;7:2634–48. doi: 10.7150/thno.17853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Way KJ, Chou E, King GL. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci. 2000;21:181–7. doi: 10.1016/S0165-6147(00)01468-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.