

Abstract
The pathogenesis of Alzheimer's disease (AD) is characterized by both accumulation of β-amyloid (Aβ) plaque and formation of neurofibrillary tangles in the brain. Recent evidence shows that autophagy activation may potently promote intracellular Aβ clearance. Thus targeting autophagy becomes a promising strategy for discovery of drug leads against AD. In the present study, we established a platform to discover autophagy stimulator and screened the lab in-house FDA-approved drug library. We found that anti-parasitic drug nitazoxanide (NTZ) was an autophagy activator and could efficiently improve learning and memory impairments in APP/PS1 transgenic mice. In BV2 cells and primary cortical astrocytes, NTZ stimulated autophagy and promoted Aβ clearance by inhibiting both PI3K/AKT/mTOR/ULK1 and NQO1/mTOR/ULK1 signaling pathways; NTZ treatment attenuated LPS-induced inflammation by inhibiting PI3K/AKT/IκB/NFκB signaling. In SH-SY5Y cells and primary cortical neurons, NTZ treatment restrained tau hyperphosphorylation through inhibition of PI3K/AKT/GSK3β pathway. The beneficial effects and related signaling mechanisms from the in vitro studies were also observed in APP/PS1 transgenic mice following administration of NTZ (90 mg·kg−1·d−1, ig) for 100 days. Furthermore, NTZ administration decreased Aβ level and senile plaque formation in the hippocampus and cerebral cortex of APP/PS1 transgenic mice, and improved learning and memory impairments in Morris water maze assay. In conclusion, our results highlight the potential of NTZ in the treatment of AD.
Keywords: Alzheimer’s disease, nitazoxanide, autophagy, inflammation, APP/PS1 transgenic mice
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that is characterized by irreversible neuronal death and consequent damages to learning and memory abilities. Senile plaques formed by abnormal deposition of β-amyloid (Aβ) and harmful neurofibrillary tangles that result from the hyperphosphorylation of tau protein are two hallmarks of AD [1]. To date, there is a lack of clinical drugs that are effective in the treatment of AD, although there are some drugs that can alleviate the symptoms of the disease. AD has caused substantial problems in society.
In recent years, several pharmaceutical companies, including Merck and Eli Lilly, have heavily invested in drug research against AD [2], and some drug candidates were developed by targeting Aβ, including α-secretase activators [3], β-secretase inhibitors [4], γ-secretase inhibitors [2], Aβ-aggregation inhibitors [5] and Aβ clearance vaccines [6]. Although some agents have entered into clinical trials, none of them have passed the trials [7].
Autophagy is a regulated mechanism for cells to disassemble unnecessary or dysfunctional components [8]. Clinical studies have shown that intracellular autophagy gradually declines with age, leading to the tendency that elderly individuals are more vulnerable to suffering diseases [7]. Autophagic damage occurs in AD [9], and autophagy activation promotes the intracellular clearance of Aβ [10]. In addition, autophagy stimulation can suppress tau hyperphosphorylation [11]. Therefore, autophagy is believed to be a promising target for the drug discovery against AD.
The discovery of autophagy agonists has been attractive in drug research, and the intriguing example is that the immunosuppressive agent rapamycin was discovered to be active in delaying the pathological progress of neurodegenerative diseases, including AD, through the activation of autophagy despite its clinical side effects of immunosuppression [12]. Currently, the role of autophagy agonists in anti-AD study has been widely accepted.
Furthermore, considering the advantage of determining new functions from approved drugs in accelerating the speed of discovery and development of new drugs and saving R&D costs, the exploration of new indications against approved drugs has become a promising strategy in contemporary drug discovery [13]. It is believed that targeting approved drugs is one of the most efficient and economical strategies for new drug development. To date, increasing results from approved drugs have been reported in anti-AD drug research [14]. For example, dasatinib, an antitumor drug candidate in phase III [15], and the antidiabetic drug metformin showed therapeutic potential in treating or preventing AD [16].
With the above-mentioned facts, we established a platform to discover autophagy stimulators by screening the lab in-house FDA-approved drug library, and the clinical anti-parasite drug nitazoxanide (NTZ) [17] was ultimately identified. We found that NTZ was capable of promoting Aβ clearance and inhibiting tau hyperphosphorylation, while effectively ameliorated the learning and memory impairments of APP/PS1 transgenic AD model mice [18]. Moreover, the mechanism that underlies the effects of NTZ has been intensively investigated. Our results have highlighted the potential of NTZ in the treatment of AD.
Materials and methods
Materials
All cell culture reagents were purchased from Gibco (Invitrogen, USA). SC79 and β-Lapachone were purchased from Selleck (USA).
Cell cultures
SH-SY5Y cells were cultured in Dulbecco’s modified Eagle’s medium and Ham’s F-12 (DMEM/F12) supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin-streptomycin (PS). BV2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) that contained 10% FBS and 100 unit/mL PS. All cells were cultured in a humidified incubator with 5% CO2 at 37 °C.
Primary cortical astrocyte culture
Primary cortical astrocytes were prepared according to a published approach [19]. Briefly, the mouse brains were removed from the embryos (embryonic day 18) and maintained in ice-cold D-Hanks buffer (5.4 mM KCl, 0.41 mM KH2PO4, 138 mM NaCl, 4.5 mM NaHCO3, 0.22 mM Na2HPO4 pH 7.4). The meninges and capillaries were separated from the brains, and the cerebral cortices were minced into small pieces and digested with D-Hanks buffer that contained 0.125% trypsin and 200 U/mL Dnase (Sigma-Aldrich, USA) for 15 min at 37 °C. The dissociated cells were seeded in a 75-cm2 flask coated with poly-D-lysine (PDL) (Sigma-Aldrich) at a density of 200,000 cells/cm2 using DMEM that contained 10% FBS and 50 U/mL PS. After 7 d, the flask was agitated in a shanking incubator overnight at 37 °C, and adhered cells were collected and selected by Ara-C (Cytosine β-D-arabinofuranoside, Sigma-Aldrich) treatment and identified as astrocytes using GFAP and DAPI staining.
Primary neuronal cell culture
The preparation of primary cortical neurons was performed according to a published approach, and the isolation procedure was similar to that of primary astrocytes. The isolated neurons were seeded at a density of 500,000 cells/mL over PDL-coated cell culture plates. After 6 h, the medium was replaced by neurobasal medium supplemented with 2% B27, 0.5 mM L-glutamine (Sigma-Aldrich), and 50 U/mL PS. Cultures were maintained in a humidified incubator with 5% CO2 at 37 °C.
Confocal laser scanning microscopy assay
Stimulation of NTZ on autophagy was investigated using a tagRFP-mWasabi-LC3 translocation assay [20], in which BV2 cells were transfected with tagRFP-mWasabi-LC3 plasmid via an adenovirus (Biovecter, China). After 6 h, the cells were treated with 5, 10 or 20 μM NTZ for 24 h, followed by immobilization with 4% paraformaldehyde for 15 min. An Olympus Fluoview FV1000 confocal microscope (Olympus, Japan) was used to detect the absorbance of fluorescence.
Western blot
BV2 cells, SH-SY5Y cells, primary astrocytes and primary neurons were treated with different concentrations of NTZ (5, 10, and 20 μM) and subsequently lysed with RIPA buffer (Thermo, USA) that contained a protease and phosphatase inhibitor cocktail (Thermo). BCA protein assay kits (Thermo) were used to detect the protein concentration of the samples. The samples were mixed with 2 × SDS-PAGE sample buffer (25% SDS, 62.5 mM Tris-HCl, pH 6.8, 25% glycerol, 0.5 M DTT and 0.1% Bromophenol Blue) and then boiled for 15 min at 99 °C in an Eppendorf ThermoMixer.
The cerebral cortices of four mice per group were homogenized with RIPA buffer (Thermo) that contained a protease and phosphatase inhibitor cocktail (Thermo) by a hand-hold motor and were maintained on ice for 1 h to completely lyse the cells. The homogenates were then centrifuged by 20,000 × g at 4 °C for 30 min. The supernatants were collected, and the protein concentration was determined using a BCA protein assay kit. Equal amounts of lysates (4 mg/mL protein) were mixed with × 2 SDS-PAGE sample buffers and subsequently boiled for 15 min at 99 °C in an Eppendorf ThermoMixer.
For Western blot assays, cells or tissue extracts were separated using SDS-PAGE and transferred to polyvinylidene difluoride membrane filters (GE, USA). After incubation with the corresponding antibodies overnight, the blots were visualized using a Dura detection system (Thermo). Antibodies against p-JNK, JNK, p-PI3K, PI3K, p-AKT, AKT, p-mTOR, mTOR, p-ULK1, ULK1, p-P70S6K, P70S6K, p62, LC3, IL-1β, iNOS and TNF-α and GAPDH were obtained from Cell Signaling Technology (USA).
ELISA assay
ELISA assay was carried out according to a published approach [19]. Samples of BV2 cells, the hippocampus or the cortex were homogenized with 5 M guanidine hydrogen chloride, followed by the addition of a protease inhibitor cocktail (Thermo) using a hand-hold motor, and then boiled for 15 min at 99 °C in an Eppendorf ThermoMixer [21]. The homogenates were subsequently centrifuged at 12,000 × g for 1 min. The supernatants were collected, and the levels of Aβ40 and Aβ42 were determined by Aβ40/42 ELISA kits (Invitrogen, USA) according to the protocols.
Aβ clearance assay
The intracellular Aβ clearance was evaluated according to the Landreth approach in BV2 cells and astrocytes [22]. Briefly, BV2 cells or astrocytes were seeded in 12-well plates and cultured with various concentrations of NTZ (5, 10, and 20 μM) for 24 h, followed by the addition of 2 μg/mL soluble Aβ40/42 and incubation for 3 h. The culture was abandoned, and BV2 cells were homogenized with 5 M guanidine hydrogen chloride with a complete protease inhibitor cocktail (Thermo) for 30 min and boiled for 15 min at 99 °C in an Eppendorf ThermoMixer. Aβ40/42 ELISA kits were used to identify the intracellular Aβ peptide, and the protein concentration was determined using BCA protein assay kits; all Aβ levels were normalized to the total protein concentration.
Immunofluorescence
In Aβ clearance assay, BV2 cells were cultured with various concentrations of NTZ (5, 10, and 20 μM) for 24 h and then incubated with Aβ40 for 3 h, followed by immobilization with 4% paraformaldehyde for 15 min. BV2 cells were subsequently immunostained according to the steps of immunofluorescence. An Olympus Fluoview FV1000 confocal microscope (Olympus, Japan) was used to detect the fluorescence.
Animal experiments
All animal experiments were carried out according to the Institutional Ethical Guidelines of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences, on animal care.
APP/PS1 transgenic mice [B6C3-Tg (APPswe, PS1dE9)], 6 months of age, were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Prior to the experiment, the genotypes of the mice were determined by tail biopsies with Tg-negative mice as a negative control to confirm APP/PS1 DNA sequences [23]. The mice were fed in standard conditions of 12 h of light/day at a room temperature at 22 °C for 6 to 7 months [24]. In our work, 20 male APP/PS1 transgenic mice were randomly divided into two groups with 10 mice per group, and nontransgenic mice were used as the negative control group. NTZ was dissolved in 0.9% sodium chloride that contained 2% Tween 80. The two groups of APP/PS1 transgenic mice were administered 90 mg NTZ/kg per day or vehicle via intragastric administration, and the non-transgenic group was administered vehicle through intragastric administration. After 100 d of administration, the Morris water maze (MWM) assay was performed to determine the learning ability of the mice. Following the completion of the MWM test, all mice were euthanized, and the brains were removed and bisected in the midsagittal plane. The left sides of the brains were fixed in 4% paraformaldehyde, and the right sides were stored at −80 °C.
Real-time PCR
Cells or brains were treated with TRIzol Reagent to obtain the total mRNA according to the protocols [25]. Complementary DNA synthesis was performed with oligo-dT primers via the instructions of the reverse-PCR kit (TaKaRa Bio, Japan), and real-time PCR was carried out using SYBR Premix Ex Taq on a DNA Engine Opticon system (TaKaRa Bio). The following primers were used (5′→3′):
MGAPDH-F: ACCACAGTCCATGCCATCAC;
MGAPDH-R: TCCACCACCCTGTTGCTGTA;
MIL-1β-F: GCAGCAGCACATCAACAAGAGC;
MIL-1β-R: TGTCCTCATCCTGGAAGGTCCACG;
MTNF-α-F: CCCACGTCGTAGCAAACCACCA;
MTNF-α-R: CCATTGGCCAGGAGGGCGTTG;
MiNOS-F: CCTCCTCCACCCTACCAAGT;
MiNOS-R: CACCCAAAGTGCTTCAGTCA.
MWM assay
The MWM test was carried out as previously described [19]. At the training trials, the mice were trained to find the invisible submerged platform in a circular pool (120 cm in diameter, 50 cm deep) filled with milk-tinted water using various visual cues located on the pool walls; training was performed for 3 trials per day for 6 consecutive days. For each trial, the mice were given 90 s to find the invisible platform. Each mouse was allowed to stay on the platform for 15 s, if the mouse found the platform within the given time. If the mouse failed to find the platform within the given time, they were manually placed on the platform and maintained there for 15 s. On the last day, a probe trial was performed by removing the platform, and the animals were allowed to swim for 90 s to search for it. All data were collected for the animal performance analysis. For data analysis, the pool was divided into four equal quadrants formed by imaging lines, which intersected the center of the pool at right angles, and the quadrants were termed north, south, east and west.
Statistical analysis
The significant differences between the multiple treatments and controls were analyzed using a one-way ANOVA followed by Dunnett’s post hoc tests or t tests. P values less than 0.05 were considered statistically significant. Data were reported as the means ± SEM, and the error bars in the graphs represent the SEM.
Results
NTZ was an autophagy activator and promoted Aβ clearance
NTZ promoted Aβ clearance
To screen for autophagy activators against our lab in-house commercial FDA-approved drug library, we initially used an ELISA assay to screen out the agent able to stimulate Aβ clearance in BV2 cells and primary astrocytes, which had been reported to play a central role as a moderator of Aβ clearance and degradation in the brain [26]. Finally, the anti-parasite drug NTZ (Fig. 1a) was found to efficiently enhance Aβ40/42 clearance in BV2 cells (Figs. 1b, d) and primary astrocytes (Figs. 1c, e).
Fig. 1.

NTZ promoted Aβ clearance by stimulating autophagy. a Structure of NTZ. b–e BV2 cells or astrocytes were treated with different concentrations of NTZ (20, 10, 5, or 0 μM) for 24 h, followed by the addition of 2 μg/mL soluble Aβ40/Aβ42 and incubation for 3 h. Aβ40/Aβ42 level was detected by ELISA assay (t test, *P < 0.01; **P < 0.05; ***P < 0.001 vs DMSO group). f Aβ40 level in the supernatant was detected by ELISA assay. g Immunofluorescence images of BV2 cells treated with NTZ and 2 μg/mL soluble Aβ40. h Quantitative results of Figures g. i BV2 cells were treated with DMSO, NTZ (20 μM), CQ (20 μM) and NTZ (20 μM) in combination with CQ (20 μM), and 2 μg/mL soluble Aβ40 was added into the cells and incubated for 3 h. Aβ40 level was detected by ELISA assay. (one-way ANOVA, **P < 0.01 vs DMSO group; #P < 0.05 vs NTZ combined with CQ; &P < 0.01 vs DMSO group). All data were obtained from three independent experiments and are presented as the mean ± SEM
An immunofluorescence assay was subsequently applied to detect Aβ40 level in cells to further investigate the stimulation of NTZ on Aβ clearance. As shown in Figs. 1g, h, NTZ dose-dependently reduced the Aβ40 level in BV2 cells. To exclude the possibility that the decrease of the Aβ level in cells by NTZ treatment was due to the inhibition of Aβ uptake, we also detected the Aβ40 level in the supernatant by ELISA assay. As shown in Fig. 1f, the Aβ40 level in the supernatant of the wells with BV2 cells significantly decreased compared to the wells without cells; however, there was no significant change in the Aβ40 level in the supernatant under different concentrations of NTZ treatment, which thus indicated that NTZ did not affect the uptake of Aβ40 in BV2 cells.
NTZ promoted Aβ clearance by stimulating autophagy
We subsequently attempted to investigate whether the previously described NTZ-induced promotion of Aβ clearance resulted from autophagy activation in BV2 cells. In the assay, the known autophagy inhibitor chloroquine (CQ, [27]) was applied, and the cells were treated with DMSO, NTZ (20 μM), CQ (20 μM), and NTZ (20 μM) in combination with CQ (20 μM), while the level of intracellular Aβ was evaluated using an ELISA assay.
As expected, CQ as an autophagy inhibitor suppressed Aβ40 clearance, which resulted in a higher Aβ40 level than that of the DMSO group (Fig. 1i), and NTZ enhanced Aβ40 clearance, which led to a lower Aβ40 level than the DMSO group (Fig. 1i). However, treatment of CQ clearly antagonized the NTZ-induced enhancement of Aβ40 clearance (lower Aβ40 level), which resulted in a higher Aβ40 level than the NTZ group (Fig. 1i). Thus, all results have implied that NTZ promoted Aβ clearance by activating autophagy.
We subsequently verified the promotion of NTZ on autophagy by examining the key autophagy-related proteins in BV2 cells and primary astrocytes. Notably, Unc51-like kinase 1 (ULK1) is the initiator of the autophagy process, and its activated phosphorylation inhibits autophagy [28], while P62 is an autophagy substrate that characterizes the activation of autophagy [29]. Moreover, microtubule-associated protein light chain 3 (LC3) is a central protein of autophagosomes and is responsible for substrate selection and autophagosome biogenesis [30]. LC3 is widely used as a marker of autophagosomes. In cells, there are two forms of LC3: LC3-I and LC3-II. When autophagy is activated, LC3-I may convert into the membrane-bound form of LC3-II to initiate the formation of autophagosomes [31]. As shown in Figs. 2a–d, NTZ dose-dependently decreased the levels of p-ULK1 and P62 and enhanced the LC3-II level in both BV2 cells and primary astrocytes. This result indicates that NTZ stimulated autophagy in cells.
Fig. 2.

NTZ stimulated autophagy. a–d BV2 cells (a) or primary astrocytes (c) were cultured with different concentrations of NTZ (20, 10, 5, or 0 μM) for 24 h, and phosphorylated ULK1 and protein levels of LC3II and p62 were detected by Western blot assay. b, d Quantitative results of (a) and (c). GAPDH was used as a loading control in Western blot assays (one-way ANOVA. n = 3. *P < 0.05; **P < 0.01; ***P < 0.001 vs DMSO group). e CLSM images of BV2 cells transiently expressing tagRFP-mWasabi-LC3 (green and red puncta indicate mWasabi and tagRFP, respectively. Scale bar: 5 μm). All data were obtained from three independent experiments and are presented as the mean ± SEM
Moreover, to further investigate the activation of NTZ on autophagy, we also used tagRFP-mWasabi fluorescence-tagged LC3 (tagRFP-mWasabi-LC3) to monitor the change of autophagic flux in BV2 cells with NTZ treatment [32]. In the assay, when autophagy and autophagosome formation initiated, both red (tagRFP) and green (mWasabi) signals were perceived and merged as yellow. When autophagosomes fused with lysosomes, the environment inside the autolysosomes was acidic [20], thus quenching the fluorescent signal of mWasabi with less effect on tagRFP. At this time, only the red (tagRFP) signal was perceived. In our experiment, the cells were pretransfected with the expression vector encoding tandem tagRFP-mWasabi-LC3 for 24 h and incubated with different concentrations of NTZ for 24 h. As shown in Fig. 2e, NTZ could effectively increase both yellow and red puncta in BV2 cells compared with the control cells. Thus, this result indicated that NTZ could stimulate autophagic flux in BV2 cells.
Taken together, these results suggested that NTZ motivated autophagy to promote Aβ clearance in BV2 cells and primary astrocytes.
NTZ stimulated autophagy involving PI3K/AKT/mTOR and NQO1/mTOR signaling pathways
NTZ stimulated autophagy involving PI3K/AKT/mTOR signaling
Given that we have determined the capability of NTZ in stimulating autophagy, we subsequently explored the related mechanism that underlies the effect of NTZ.
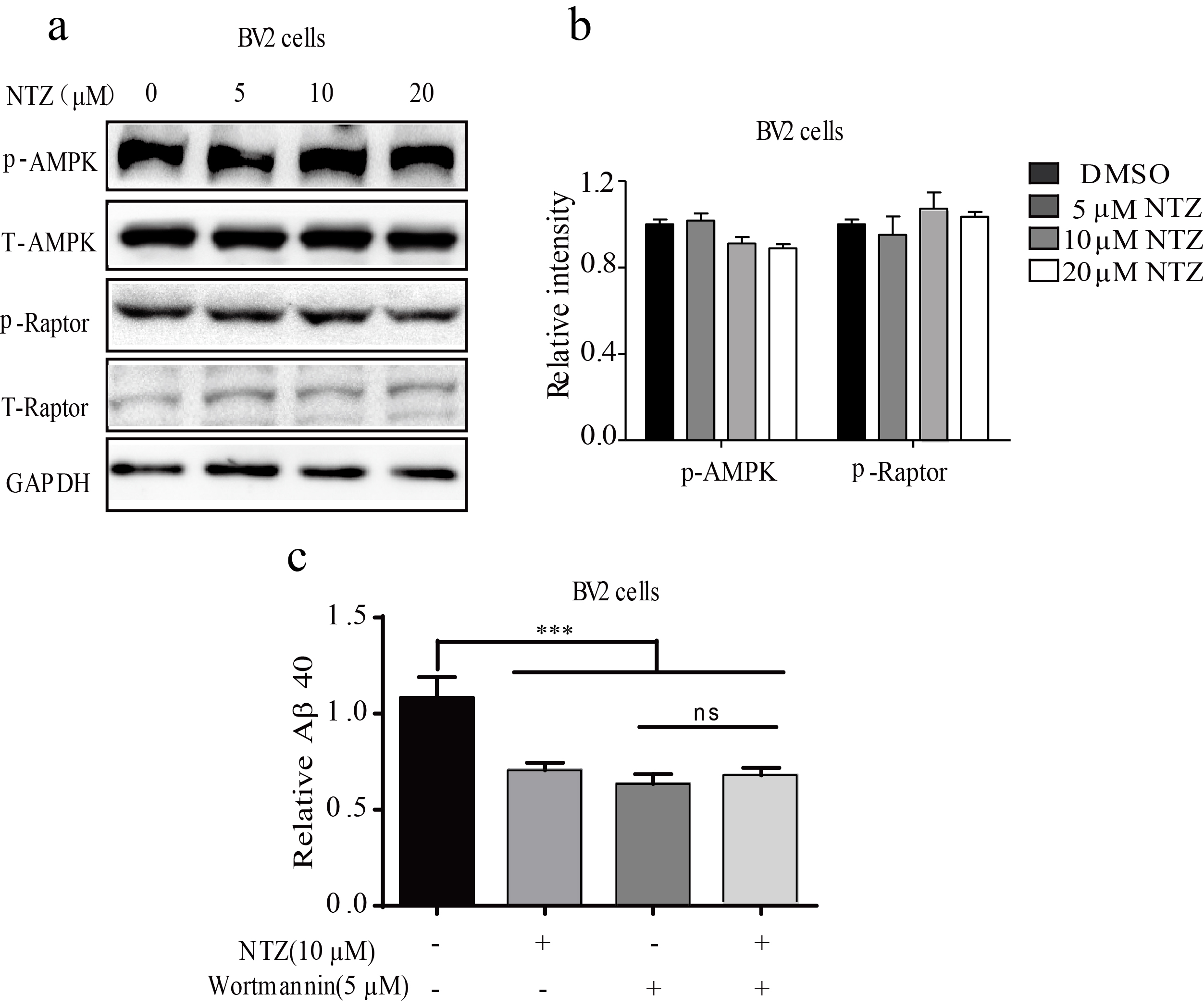
According to the published result, mTOR signaling activation phosphorylates ULK1 to disrupt autophagy [33], and mTOR phosphorylation is regulated by PI3K/AKT and AMPK/Raptor signaling [34]. With these facts, we investigated whether NTZ might regulate these two signaling pathways in response to its autophagy promotion in BV2 cells or primary astrocytes via Western blot assay. As indicated in Figs. 3a–d, NTZ treatment decreased the phosphorylation levels of PI3K, AKT, mTOR and P70S6K (the substrate of mTOR [35]) and had no effects on the phosphorylation level of AMPK or Raptor (Supplementary Figures S1a and b).
Fig. 3.

NTZ stimulated autophagy by inhibiting PI3K/AKT/mTOR and NQO1/mTOR signaling pathways. a–d BV2 cells (a) or astrocytes (b) were treated with different concentrations of NTZ (20, 10, 5 or 0 μM) for 24 h, and phosphorylation levels of PI3K, AKT and mTOR, as well as protein level of P70S6K were detected by Western blot assay. c, d Quantitative results of (a, b). GAPDH was used as loading control in Western blot assays. (one-way ANOVA. n = 3. *P < 0.05; **P < 0.01; ***P < 0.001 vs DMSO group). e–f BV2 cells (e) were cultured with DMSO, NTZ (20 μM), SC79 (5 μM) and NTZ (20 μM) in combination with SC79 (5 μM) for 24 h, and phosphorylation levels of PI3K, AKT and mTOR were detected by Western blot assay. f quantitative results of (e). GAPDH was used as a loading control in Western blot assays (one-way ANOVA. n = 3. *P < 0.01; ***P < 0.001 vs DMSO group; *P < 0.01; ***P < 0.001 vs NTZ combined with SC79). g BV2 cells were cultured with DMSO, NTZ (20 μM), β-lapachone (2 μM), and NTZ (20 μM) in combination with β-lapachone (2 μM), and 2 μg/mL soluble Aβ40 was added into the cells and incubated for 3 h. Aβ40 level was detected by ELISA assay (t test, ***P < 0.001 vs DMSO group; #P < 0.05 vs NTZ combined with β-lapachone). h–i BV2 cells (h) were cultured with different concentrations of NTZ (20, 10, 5 or 0 μM) for 24 h, and phosphorylation levels of PI3K, AKT or mTOR were detected by Western blot. i quantitative results of (h). GAPDH was used as a loading control in Western blot assays (one-way ANOVA. n = 3. ***P < 0.001; *P < 0.05 vs DMSO group; &&&P < 0.001; &P < 0.05 vs β-lapachone). All data were obtained from three independent experiments and are presented as the mean ± SEM
PI3K inhibitor wortmannin [36] was subsequently applied to the assay to prove that NTZ promoted Aβ clearance (activated autophagy) through PI3K/AKT signaling pathway. As shown in Supplementary Figure S1c, NTZ (10 μM) or wortmannin (5 μM) could promote Aβ40 clearance in BV2 cells, while cotreatment of NTZ with wortmannin failed to have a preferable ability in promoting Aβ40 clearance compared with the NTZ treatment result in the cells. These results thus suggested that NTZ promoted Aβ40 clearance via the PI3K-dependent signaling pathway.
In addition, AKT activator SC79 [37] was applied to verify that PI3K/AKT signaling is responsible for the regulation of NTZ against mTOR. In the assay, BV2 cells were treated with DMSO, NTZ (20 μM), SC79 (5 μM), or NTZ (20 μM) in combination with SC79 (5 μM), and the phosphorylation levels of PI3K, AKT and mTOR were detected by Western blot. As indicated in Figs. 3e, f, SC79 stimulated phosphorylated AKT and mTOR and effectively reversed the NTZ-induced suppression of the phosphorylation levels of AKT and mTOR; however, it had no effects on phosphorylated PI3K.
Therefore, these results demonstrated that NTZ promoted autophagy involving PI3K/AKT/mTOR signaling.
NTZ stimulates autophagy involving NQO1/mTOR signaling
It is noted that NTZ has previously been reported to be capable of enhancing autophagy by inhibiting NQO1 signaling in Mycobacterium tuberculosis [38]. Thus, we wondered whether NTZ also inhibited NQO1 signaling to stimulate autophagy in neuron-like cells.
Thus, we used β-lapachone (an activator of NQO1 [39]) to evaluate the potential of NTZ in inhibiting NQO1 via ELISA in BV2 cells. In the assay, the cells were treated with DMSO, NTZ (20 μM), β-lapachone (2 μM), or NTZ (20 μM) in combination with β-lapachone (2 μM). As expected, the results in Fig. 3g demonstrated that β-lapachone effectively reversed the NTZ-induced decrease in the Aβ40 level, which implies the potential of β-lapachone in weakening Aβ40 clearance and antagonizing the NTZ-induced promotion of autophagy. Thus, we presumed that NTZ might also activate autophagy involving the regulation of NQO1 signaling in BV2 cells. To verify this presumption, a Western blot assay was carried out to detect the effect of β-lapachone in reversing the NTZ-induced decreases in the levels of P-PI3K, P-AKT and P-mTOR. As indicated in Figs. 3h, i, β-lapachone could antagonize the NTZ-induced decrease in phosphorylated mTOR, while there was no influence on P-PI3K or P-AKT. Thus, these results demonstrated that NTZ stimulated autophagy involving NQO1/mTOR signaling.
Taken together, NTZ stimulates autophagy involving the regulation of PI3K/AKT/mTOR and NQO1/mTOR signaling pathways.
NTZ restrains tau hyperphosphorylation involving PI3K/AKT/GSK3β signaling
NTZ suppressed phosphorylation levels of PI3K, AKT, and GSK3β
We have determined that NTZ as an autophagy activator promoted Aβ clearance; thus, we subsequently investigated whether NTZ also exhibited activity in suppressing tau hyperphosphorylation, another hallmark of AD [40]. In the assay, we chose SH-SY5Y and primary neuron cells for the tau-related study because SH-SY5Y cells belong to a neuroblastoma cell line that closely resembles primary neurons, while BV2 cells and primary astrocytes are considered more suitable for Aβ clearance related assays [41].
According to the published reports, two main kinases, glycogen synthase kinase 3β (GSK3β) and cyclin-dependent kinase 5 (CDK5), are highly responsible for tau phosphorylation [42], and GSK3β is regulated by PI3K/AKT signaling [43]. As NTZ has been determined to be active in repressing the phosphorylation levels of PI3K and AKT in BV2 cells and astrocytes, we subsequently investigated whether it also exhibited these effects in SH-SY5Y and primary neuron cells. As expected, the results indicated that NTZ could also inhibit the phosphorylation of PI3K or AKT in both SH-SY5Y and primary neuron cells (Figs. 4a–d).
Fig. 4.

NTZ reduced tau hyperphosphorylation by inhibiting GSK3β activity. a–l SH-SY5Y (a, e, i) or neuronal (c, g, k) cells were cultured with different concentrations of NTZ (20, 10, 5 or 0 μM) for 24 h, and phosphorylation levels of PI3K, AKT, GSK3β and Tau were detected by Western blot. b, f, j Quantitative results of (a, e, and i). d, h, l Quantitative results of Figures c, g and k. GAPDH was used as a loading control in Western blot assays. (one-way ANOVA. n = 3. *P < 0.05; **P < 0.01; ***P < 0.001 vs DMSO group). All data were obtained from three independent experiments and are presented as the mean ± SEM
It is reported that GSK3β activity is activated when the residue Tyr216 is phosphorylated and inhibited when Ser9 phosphorylated [44]. With these facts, we performed a Western blot assay to investigate the phosphorylation level of GSK3β for the two previously described residues to evaluate the GSK3β enzymatic activity in response to NTZ treatment. As shown in Figs. 4e-h, NTZ decreased GSK3β phosphorylation at Tyr216, while it had no effects on the phosphorylation at Ser9 in both SH-SY5Y and primary neuronal cells. These results thus suggested that NTZ suppressed GSK3β activity.
We subsequently examined the potential regulation of NTZ against CDK5 enzyme in SH-SY5Y and primary neuron cells. According to the literature reports, CDK5 alone is an inactive catalytic subunit and can be activated by p35 or p25 (the cleaved product of p35) [45]. As indicated in Supplementary Figure S2a-d, NTZ had no effects on CDK5, p25 or p35, which thus implies that NTZ rendered no influence on CDK5 activity.
NTZ restrained tau phosphorylation
As NTZ has been determined to restrain GSK3β activity, we subsequently investigated its suppression against tau hyperphosphorylation in cells. As expected, the results indicated that NTZ efficiently decreased tau phosphorylation at multiple sites, including Ser396, 199 and Tyr231 [46], in both SH-SY5Y and primary neuron cells (Figs. 4i–l). Moreover, the level of total tau did not change with different concentrations of NTZ, which thus implied that NTZ-inhibited tau phosphorylation might not occur via autophagy.
We subsequently investigated whether PI3K/AKT pathway was required for the regulation of NTZ against tau phosphorylation. As shown in Supplementary Figures S2e-f, NTZ (10 μM) or wortmannin (5 μM, PI3K inhibitor) could decrease the phosphorylation of tau, while the cotreatment of NTZ with wortmannin failed to further suppress the phosphorylation of tau, thereby implying that NTZ reduced tau phosphorylation via PI3K/AKT signaling pathway.
Taken together, NTZ suppressed tau phosphorylation involving PI3K/AKT/GSK3β signaling pathway.
NTZ attenuated LPS-induced inflammation involving PI3K/AKT/IκB/NFκB pathway
NTZ inhibited NFκB signaling via PI3K/AKT/IκB pathway
It is reported that the regulation of IκB phosphorylation involves PI3K/AKT signaling, and phosphorylation and degradation are two essential steps for NFκB activation, while P65, the subunit of NFκB, could translocate into the nucleus evoking the transcription of pro-inflammatory mediators [47]. Thus, the capability of NTZ in inhibiting PI3K/AKT signaling also encouraged us to evaluate its potential in suppressing inflammation, which is also closely related to AD [48]. In the assay, lipopolysaccharide (LPS) [47] was used as an inducer of inflammation in BV2 cells.
As indicated in the Western blot results (Figs. 5a, b), LPS increased the phosphorylation levels of PI3K, AKT and IκB, decreased the protein level of total IκB, and induced nuclear translocation of the NFκB subunit p65 in BV2 cells, while NTZ treatment effectively reversed the LPS-induced activation of P-PI3K, P-AKT and IκB and inhibited the nuclear translocation of NFκB/p65. These results thus suggested that NTZ attenuated NFκB activation involving PI3K/AKT/IκB pathway.
Fig. 5.

NTZ reduced LPS-stimulated inflammation involving suppression of PI3K/Akt/NFκB signaling. a, b BV2 cells (a) were cultured with different concentrations of NTZ (20, 10, 5 or 0 μM) and 1 mg/mL LPS for 24 h, and phosphorylation levels of PI3K, AKT and IκB and protein level of NFκB in cytosol and nuclear fraction were detected by Western blot. b Quantitative results of Figure a. GAPDH and Lamin B1 were used as loading controls in Western blot assays (one-way ANOVA. n = 3. *P < 0.05; **P < 0.01; ***P < 0.001 vs LPS group). c RT-PCR assay was used to test the effects of NTZ on transcription of inflammatory cytokines with 1 mg/mL LPS, including TNF-α, IL-1β, and iNOS (one-way ANOVA. n = 3. *P < 0.05; **P < 0.01; ***P < 0.001 vs LPS group). d, e BV2 cells (d) were cultured with different concentrations of NTZ (20, 10, 5 or 0 μM) for 24 h with 1 mg/mL LPS, and protein levels of TNF-α, IL1 and iNOS were detected by Western blot. e quantitative results of (d). GAPDH was used as a loading control in Western blot assays (one-way ANOVA. n = 3. *P < 0.05; **P < 0.01; ***P < 0.001 vs LPS group). All data were obtained from three independent experiments and are presented as the mean ± SEM
NTZ inhibited the expression of pro-inflammatory cytokines
Given the determined activity of NTZ in repressing NFκB/p65 nuclear translocation, we subsequently examined its potential inhibition against pro-inflammatory cytokines, including IL-1β, iNOS and TNF-α [49], by real-time PCR and Western blot in BV2 cells. As indicated in Figs. 5c–e, LPS treatment induced increases in both the mRNA and protein levels of IL-1β, iNOS and TNF-α compared to the control group. Moreover, NTZ antagonized these LPS-induced promotions on these pro-inflammatory cytokines. Thus, these results showed that NTZ attenuated LPS-induced inflammation involving PI3K/AKT/IκB/NFκB pathway.
NTZ improved learning and memory impairments in APP/PS1 transgenic mice
As we have determined that NTZ was capable of promoting Aβ clearance and suppressing tau hyperphosphorylation and inflammation in cells, we subsequently evaluated its efficacy in ameliorating learning and memory impairments in APP/PS1 transgenic mice using the MWM test [50] because APP/PS1 transgenic mice expressing chimeric mouse/human Swedish mutant APP and mutant human presenilin 1 protein have been widely used as AD model mice for research [24].
As indicated in Figs. 6a, b, in the training test, the latencies to find the platform for the vehicle-administered transgenic mice (transgenic vehicle group) were longer than those for the vehicle-administered nontransgenic mice (nontransgenic vehicle group). However, the latencies of the transgenic mice administered 90 mg/kg NTZ (transgenic NTZ group) were clearly decreased compared with those for the transgenic vehicle group. These results thus suggested that NTZ administration ameliorated the space learning ability of APP/PS1 transgenic mice.
Fig. 6.

NTZ effectively improved learning and memory impairments in APP/PS1 transgenic mice. MWM test was used to evaluate the effect of NTZ on APP/PS1 transgenic mice. a Representative tracing graphs in the training trials. b Escape latency during platform trials (two-way ANOVA, n = 10. *P < 0.01; ***P < 0.001 compared with TV). c Representative tracing graphs in the probe trials. d Crossing times of the platform in the probe trials (one-way ANOVA, n = 10. *P < 0.01; ***P < 0.001 compared with TV). NV, nontransgenic vehicle group; TV, transgenic AD model vehicle group, NTZ, transgenic AD model with NTZ administration (90 mg·kg−1·d−1) group. Values indicate the mean ± SEM
As demonstrated in Figs. 6c, d, in the probe trial assay, the transgenic vehicle group crossed over the hidden location of the platform less frequently than the nontransgenic vehicle group, while the transgenic NTZ group crossed over the hidden location of the platform more frequently than the transgenic vehicle group. Thus, these results demonstrated that NTZ improved the memory ability of the transgenic mice.
In the assay, we selected three doses of NTZ (10, 30 and 90 mg/kg) for the in vivo assay, and no positive effects were obtained for the 10 and 30 mg/kg NTZ groups (data not shown).
NTZ reduced Aβ level and senile plaque formation in APP/PS1 transgenic mice
NTZ reduced Aβ level
We subsequently evaluated the inhibition of NTZ against the Aβ40/42 level in both the hippocampus and cerebral cortex in the transgenic mice using an ELISA assay. As shown in Figs. 7a–d, the Aβ40/42 levels of the transgenic vehicle group were expectedly higher than those of the nontransgenic vehicle group, and the transgenic NTZ group mice had a lower Aβ40/42 level in the hippocampus or cerebral cortex than in the transgenic vehicle group.
Fig. 7.

NTZ reduced Aβ level and senile plaque formation in APP/PS1 transgenic mice. a, d ELISA assay was used to evaluate Aβ40/42 levels in APP/PS1 transgenic mice (t-test, n = 4. **P < 0.01; ***P < 0.001 compared with TV). e–f Representative micrographs of Thioflavine S-stained amyloid plaques in the hippocampus and cortex of APP/PS1 transgenic mice, scale bar: 100 μm. f Statistical analysis of images e. (t test, ***P < 0.001 compared with TV). NV, nontransgenic vehicle group; TV, transgenic vehicle group, NTZ, transgenic mice administration with NTZ administration (90 mg·kg−1·d−1) group. Values indicate the mean ± SEM
NTZ suppressed senile plaque formation
As we have determined the inhibition of NTZ against Aβ40/42 in vivo, we subsequently investigated the potential of NTZ in restraining senile plaque formation in the transgenic mice via the Thioflavine S staining assay, with senile plaque stained in green. As shown in Figs. 7e, f, the senile plaques in the hippocampus or cerebral cortex of the nontransgenic vehicle group were less than in the transgenic vehicle mice, while 90 mg/kg NTZ administration clearly decreased the senile plaque formation in the transgenic mice.
NTZ promoted autophagy involving PI3K/AKT/mTOR signaling in APP/PS1 transgenic mice
Given that NTZ enhanced autophagy involving PI3K/AKT/mTOR and NQO1/mTOR signaling pathways in response to Aβ clearance promotion in BV2 and primary astrocytes, we subsequently assessed these effects in APP/PS1 transgenic mice.
NTZ promoted autophagy
We initially investigated the promotion of NTZ on autophagy in the transgenic mice by Western blot. As indicated in Figs. 8a–c, the transgenic vehicle group had higher protein levels of p62 and phosphorylated ULK1 and a lower protein level of LC3II than those in the nontransgenic vehicle group, which thus suggested the suppression of the autophagy process in transgenic mice [24]. However, NTZ administration clearly alleviated the protein levels of p62 and phosphorylated ULK1, while it promoted LC3II protein level (Figs. 8a–c), thus implying that NTZ activated autophagy in APP/PS1 transgenic mice.
Fig. 8.

NTZ promoted autophagy involving PI3K/AKT/mTOR signaling in APP/PS1 transgenic mice. a–c Phosphorylation levels of PI3K, AKT, mTOR, ULK1 and P70S6K and protein levels of p62 and LC3 in the cortices of the transgenic mice. b, c Quantitative results of Figure a. GAPDH was used as a loading control in Western blot assays (one-way ANOVA. n = 4. *P < 0.05; **P < 0.01; ***P < 0.001 vs TV). NV, nontransgenic vehicle group, TV, transgenic vehicle group, NTZ, transgenic mice with NTZ administration (90 mg·kg−1·d−1) group. Values indicate the mean ± SEM
NTZ inhibited PI3K/AKT/mTOR signaling
We subsequently examined the inhibition of NTZ against the PI3K/AKT/mTOR pathway by Western blot assay in vivo. As expected, the transgenic vehicle group mice had higher levels of P-PI3K, P-AKT, P-mTOR and P-P70S6K than the nontransgenic vehicle group mice (Figs. 8a–c), while NTZ administration clearly reversed these effects as indicated in the transgenic NTZ group mice.
In addition, we also attempted to examine the inhibition of NTZ against NQO1 activity in vivo, but failed (data not shown). This failure might be due to the complicated ingredients released from the crushed brains, which influenced the enzymatic assay against NQO1 activity.
Taken together, these results have demonstrated that NTZ stimulated autophagy involving the inhibition of PI3K/AKT/mTOR pathway in APP/PS1 transgenic mice.
NTZ reduced GSK3β activity and Tau hyperphosphorylation in APP/PS1 transgenic mice
Similar to the cell-based assay in examining the inhibition of NTZ against GSK3β, we also investigated the phosphorylation levels of GSK3β at Tyr216 and Ser 9 [44] to evaluate the GSK3β activity in the cortex of the transgenic mice. As indicated in Figs. 9a, b, NTZ administration efficiently decreased the level of GSK3β phosphorylation at Tyr216, while it had no effects on GSK3β phosphorylation at Ser9 as in the case of the cell-based assay, which demonstrated that NTZ administration inhibited GSK3β activity in the transgenic mice.
Fig. 9.

NTZ reduced Tau hyperphosphorylation by inhibiting GSK3β activity in APP/PS1 transgenic mice. a–d Phosphorylation and protein levels of GSK3β (a) and Tau (c) were assessed by Western blot. b, d quantitative results of (a) and (c). GAPDH was used as a loading control in Western blot assays (one-way ANOVA. n = 4. *P < 0.05; **P < 0.01; ***P < 0.001 vs TV). NV, nontransgenic vehicle group, TV, transgenic vehicle group, NTZ, transgenic mice with NTZ administration (90 mg·kg−1·d−1) group. Values indicate the mean ± SEM
A Western blot assay was subsequently conducted to detect the suppression of NTZ against tau hyperphosphorylation in the cortex of the transgenic mice. As expected, NTZ administration restrained tau phosphorylation at multiple sites, including Ser396, 199 and Tyr231 (Figs. 9c, d), similar to the results of the cell-based assay (Figs. 3i–l). These results thus indicated that NTZ administration reduced tau hyperphosphorylation in APP/PS1 transgenic mice.
Furthermore, we also investigated the protein levels of CDK5 and p35/p25 by Western blot in the cortex of the mice according to the cell-based assay. As indicated in Supplementary Figures S3e-f, NTZ administration rendered no influence on these three protein levels. This result thus demonstrated that NTZ did not inhibit CDK5 enzymatic activity, consistent with the cell-based assay results.
NTZ inhibits pro-inflammatory mediators in APP/PS1 transgenic mice
According to a published report, Aβ mediates inflammatory activation in APP/PS1 transgenic mice [51]. As we have determined the activities of NTZ in promoting Aβ clearance as an autophagy activator and suppressing the LPS-induced excessive expression of pro-inflammatory mediators in BV2 cells, we subsequently investigated the potential anti-inflammatory activity of NTZ in the cortex of the transgenic mice.
As indicated in Figs. 10a–c, the real-time PCR and Western blot results both showed that the mRNA and protein levels of IL-1β, iNOS and TNF-α in the transgenic vehicle group mice were higher than those in the nontransgenic vehicle group mice, indicative of the activation of inflammation in transgenic mice. NTZ administration efficiently inhibited the expressions of these pro-inflammatory mediators at both the mRNA and protein levels.
Fig. 10.

NTZ reduced inflammation in APP/PS1 transgenic mice. a RT-PCR assay was used to assess the effects of NTZ on the transcription of inflammatory cytokines in APP/PS1 transgenic mice (one-way ANOVA, Dunnett’s multiple comparison test. n = 3. *P < 0.05; **P < 0.01; ***P < 0.001 vs TV). b–c Protein levels of inflammatory cytokines in APP/PS1 transgenic mice were assessed by Western blot. c Quantitative results of (b). GAPDH was used as a loading control in Western blot assays (one-way ANOVA. n = 4. *P < 0.05; **P < 0.01 vs TV). NV, nontransgenic vehicle group, TV, transgenic vehicle group, NTZ, transgenic mice with NTZ administration (90 mg·kg−1·d−1) group. Values indicate the mean ± SEM
Discussion
AD is a neurodegenerative disease with hallmarks of memory recession and behavior ability impairment [52]. The current drugs for AD treatment can only alleviate the symptoms of the patient’s illness [53].
It is reported that both extracellular and intracellular accumulation of Aβ initiate a cascade of events leading to neurodegeneration [54] in AD patients. Although Aβ is mainly produced by neurons, microglia and astrocytes have also been reported to function potently in Aβ clearance and degradation. In addition, astrocytes play key roles in the homoeostasis, defense and regeneration of the central nervous system [55]. In the pathogenesis of AD, Aβ and its precursor APP, as well as AD-related genes inhibit autophagy, and promoting autophagy in response to the promotion of Aβ clearance by astrocytes is believed to be a promising strategy for the drug discovery against AD. Autophagy is regulated by various signaling pathways, including PI3K/AKT/mTOR and AMPK/Raptor signaling. Moreover, the activity of PI3K/AKT/mTOR signaling is markedly enhanced in cells or transgenic mice that overexpress APP, leading to the inhibition of autophagy [56]. In the current work, we found that NTZ promoted autophagy by inhibiting PI3K/AKT/mTOR signaling.
In addition, NTZ has previously been discovered to inhibit NQO1 activity, and we determined that NQO1/mTOR signaling was also involved in NTZ-stimulated autophagy. Given the report that NQO1 is highly expressed in the brains of patients with AD [57], we tentatively suppose that excessive activation of NQO1 may inhibit autophagy, leading to the accumulation of Aβ. Furthermore, published reports have also indicated that NQO1 overexpression is caused by Aβ-induced oxidative stress, and NQO1 mutation-induced inactivation (C609T) can increase the risk of AD in the Chinese population [58]. Thus, these results indicate the complicated role of NQO1 in the pathogenesis of AD.
In addition to Aβ accumulation, the hyperphosphorylation of tau protein is a key pathological feature of AD. In our work, we determined that NTZ decreased the phosphorylation of P216-GSK3β in response to GSK3β inhibition by the PI3K/AKT signaling pathway. It has been reported that P216-GSK3β level is high in the cortex and hippocampus of transgenic mice or AD patients [44], and GSK3β activity regulation involves PI3K/AKT signaling via the phosphorylation of GSK3β at Ser9 or Tyr216 [59]. It has also been reported that PI3K inhibitors, wortmannin or LY294002, decrease tau phosphorylation [36, 60], which is consistent with our results. However, there are also other reports that demonstrated wortmannin causes tau phosphorylation [61, 62]. Thus, these results have implied the complexity of wortmannin as a PI3K inhibitor in regulating tau phosphorylation.
In AD, neuroinflammation is a significant contributor to disease progression [63]. Aβ accumulation causes excessive activation of astrocytes and increases transcription and protein levels of inflammatory factors [64], while the occurrence of inflammation could further aggravate tau phosphorylation. NSAIDs (nonsteroidal anti-inflammatory drugs) have been shown to be effective in ameliorating AD symptoms [65]. In our work, NTZ was found to exhibit an anti-inflammatory effect by inhibiting the nuclear entry of NFκB and reducing the transcription of inflammatory factors. Therefore, these results have addressed the potential multifunction of NTZ in the treatment of AD.
In summary, we reported that NTZ can stimulate autophagy and promote Aβ clearance by inhibiting both PI3K/AKT/mTOR/ULK1 and NQO1/mTOR/ULK1 signaling pathways. NTZ repressed tau hyperphosphorylation by inhibiting PI3K/AKT/GSK3β pathway and attenuated LPS-induced inflammation by inhibiting PI3K/AKT/IκB/NFκB signaling. Moreover, NTZ efficiently improved the learning and memory impairments of APP/PS1 transgenic mice (Fig. 11). These results have highlighted the potential of nitazoxanide in the treatment of AD.
Fig. 11.
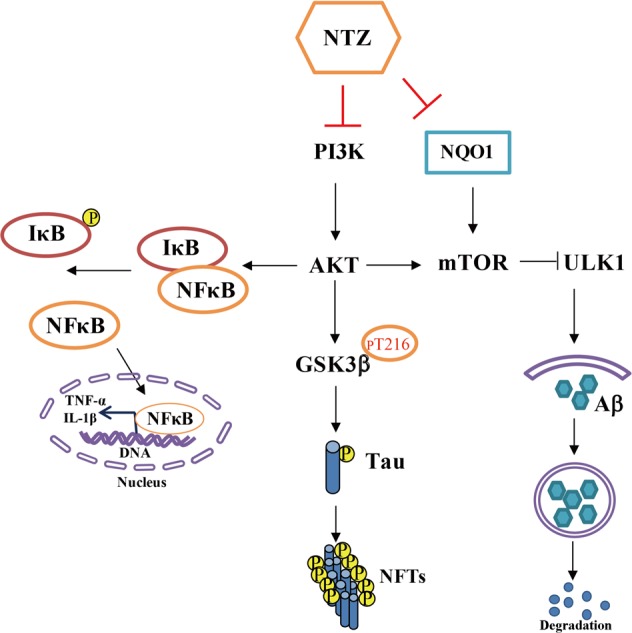
A proposed model illustrating the mechanism that underlies NTZ in the amelioration of AD pathology. NTZ stimulated autophagy and promoted Aβ clearance by inhibiting both PI3K/AKT/mTOR/ULK1 and NQO1/mTOR/ULK1 signaling pathways. NTZ was capable of repressing tau hyperphosphorylation involving inhibition of PI3K/AKT/GSK3β pathway and attenuating LPS-induced inflammation by inhibiting PI3K/AKT/IκB/NFκB signaling
Supplementary information
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number 81473141), NSFC-TRF collaboration projects (grant number NSFC81561148011), the Key Laboratory of Receptor Research of the Chinese Academy of Sciences (grant number SIMM1606YZZ-04), Personalized Medicines: Molecular Signature-based Drug Discovery and Development, Strategic Priority Research Program of the Chinese Academy of Sciences (grant number XDA12040303) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (Integration of Chinese and Western Medicine).
Author contributions
LF, ZYZ, JC, XWG, YHZ, and XS designed the research; LF, JLL, JYW, XH, ZYZ, and JL performed the research; XXQ, FM, JZ, and JL synthesized the compounds; LF, JR, JMY, and XS wrote the paper.
Competing interests
The authors declare no competing interests.
Contributor Information
Jian Li, Email: jianli@ecust.edu.cn.
Xu Shen, Email: xshen@njucm.edu.cn.
Supplementary information
The online version of this article (10.1038/s41401-019-0220-1) contains supplementary material, which is available to authorized users.
References
- 1.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–44. doi: 10.1212/WNL.34.7.939. [DOI] [PubMed] [Google Scholar]
- 2.Oehlrich D, Berthelot DJC, Gijsen HJM. gamma-secretase modulators as potential disease modifying anti-Alzheimer's drugs. J Med Chem. 2011;54:669–98. doi: 10.1021/jm101168r. [DOI] [PubMed] [Google Scholar]
- 3.Lichtenthaler SF. Alpha-secretase in Alzheimer's disease: molecular identity, regulation and therapeutic potential. J Neurochem. 2011;116:10–21. doi: 10.1111/j.1471-4159.2010.07081.x. [DOI] [PubMed] [Google Scholar]
- 4.Jeppsson F, et al. Discovery of AZD3839, a potent and selective BACE1 inhibitor clinical candidate for the treatment of Alzheimer disease. J Biol Chem. 2012;287:41245–57. doi: 10.1074/jbc.M112.409110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bukhari SN, Jantan I. Synthetic curcumin analogs as inhibitors of beta amyloid peptide aggregation: potential therapeutic and diagnostic agents for Alzheimer's disease. Mini Rev Med Chem. 2015;15:1110–21. doi: 10.2174/138955751513150923101841. [DOI] [PubMed] [Google Scholar]
- 6.Sterner RM, Takahashi PY, Yu Ballard AC. Active vaccines for Alzheimer disease treatment. J Am Med Dir Assoc. 2016;17:e11–5. doi: 10.1016/j.jamda.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 7.Doody RS, et al. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013;369:341–50. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- 8.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 9.Carroll B, Hewitt G, Korolchuk VI. Autophagy and ageing: implications for age-related neurodegenerative diseases. Essays Biochem. 2013;55:119–31. doi: 10.1042/bse0550119. [DOI] [PubMed] [Google Scholar]
- 10.Kizilarslanoglu MC, Ulger Z. Role of autophagy in the pathogenesis of Alzheimer disease. Turk J Med Sci. 2015;45:998–1003. doi: 10.3906/sag-1407-75. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Mandelkow E. Degradation of tau protein by autophagy and proteasomal pathways. Biochem Soc Trans. 2012;40:644–52. doi: 10.1042/BST20120071. [DOI] [PubMed] [Google Scholar]
- 12.Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and tau effects on cognitive impairments. J Biol Chem. 2010;285:13107–20. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 14.Lukiw WJ. Amyloid beta (Aβ) peptide modulators and other current treatment strategies for Alzheimer's disease (AD). Expert Opin Emerg Drugs. 2012;17:43–60. [DOI] [PMC free article] [PubMed]
- 15.Wang X, Roy A, Hochhaus A, Kantarjian HM, Chen TT, Shah NP. Differential effects of dosing regimen on the safety and efficacy of dasatinib: retrospective exposure-response analysis of a Phase III study. Clin Pharmacol Adv Appl. 2013;5:85–97. doi: 10.2147/CPAA.S42796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weintraub D, et al. Time to response to citalopram treatment for agitation in Alzheimer disease. Am J Geriatr Psychiatry. 2015;23:1127–33. doi: 10.1016/j.jagp.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamamoto Y, Hakki A, Friedman H, Okubo S, Shimamura T, Hoffman PS, et al. Nitazoxanide, a nitrothiazolide antiparasitic drug, is an anti-Helicobacter pylori agent with anti-vacuolating toxin activity. Chemotherapy. 1999;45:303–12. doi: 10.1159/000007200. [DOI] [PubMed] [Google Scholar]
- 18.Qiao J, Wang J, Wang H, Zhang Y, Zhu S, Adilijiang A, et al. Regulation of astrocyte pathology by fluoxetine prevents the deterioration of Alzheimer phenotypes in an APP/PS1 mouse model. Glia. 2016;64:240–54. doi: 10.1002/glia.22926. [DOI] [PubMed] [Google Scholar]
- 19.Zhu Z, Yan J, Jiang W, Yao XG, Chen J, Chen L, et al. Arctigenin effectively ameliorates memory impairment in Alzheimer's disease model mice targeting both beta-amyloid production and clearance. J Neurosci. 2013;33:13. doi: 10.1523/JNEUROSCI.4790-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–60. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 21.Zhu ZY, Li CJ, Wang X, Yang ZY, Chen J, Hu LH, et al. 2,2',4'-Trihydroxychalcone from Glycyrrhiza glabra as a new specific BACE1 inhibitor efficiently ameliorates memory impairment in mice. J Neurochem. 2010;114:374–85. doi: 10.1111/j.1471-4159.2010.06751.x. [DOI] [PubMed] [Google Scholar]
- 22.Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malumbres M, Mangues R, Ferrer N, Lu S, Pellicer A. Isolation of high molecular weight DNA for reliable genotyping of transgenic mice. Biotechniques. 1997;22:1114–9. doi: 10.2144/97226st03. [DOI] [PubMed] [Google Scholar]
- 24.Reiserer RS, Harrison FE, Syverud DC, McDonald MP. Impaired spatial learning in the APP(Swe) + PSEN1 Delta E9 bigenic mouse model of Alzheimer's disease. Genes Brain Behav. 2007;6:54–65. doi: 10.1111/j.1601-183X.2006.00221.x. [DOI] [PubMed] [Google Scholar]
- 25.Rio DC, Ares M, Jr., Hannon GJ, Nilsen TW. Purification of RNA using TRIzol (TRI reagent) Cold Spring Harb Protoc. 2010;2010:prot5439. doi: 10.1101/pdb.prot5439. [DOI] [PubMed] [Google Scholar]
- 26.Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. Plos One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73:3–7. doi: 10.1158/0008-5472.CAN-12-2464. [DOI] [PubMed] [Google Scholar]
- 28.Wong PM, Puente C, Ganley IG, Jiang XJ. The ULK1 complex Sensing nutrient signals for autophagy activation. Autophagy. 2013;9:124–37. doi: 10.4161/auto.23323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. Febs J. 2015;282:4672–8. doi: 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- 30.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 31.Huang R, Xu Y, Wan W, Shou X, Qian J, You Z, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57:456–66. doi: 10.1016/j.molcel.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 32.Kononenko NL, Classen GA, Kuijpers M, Puchkov D, Maritzen T, Tempes A, et al. Retrograde transport of TrkB-containing autophagosomes via the adaptor AP-2 mediates neuronal complexity and prevents neurodegeneration. Nat Commun. 2017;8:14819. doi: 10.1038/ncomms14819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 35.Lafay-Chebassier C, Paccalin M, Page G, Barc-Pain S, Perault-Pochat MC, Gil R, et al. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer's disease. J Neurochem. 2005;94:215–25. doi: 10.1111/j.1471-4159.2005.03187.x. [DOI] [PubMed] [Google Scholar]
- 36.Jo H, Mondal S, Tan D, Nagata E, Takizawa S, Sharma AK, et al. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc Natl Acad Sci USA. 2012;109:10581–6. doi: 10.1073/pnas.1202810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lam KK, Zheng X, Forestieri R, Balgi AD, Nodwell M, Vollett S, et al. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012;8:e1002691. doi: 10.1371/journal.ppat.1002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu DR, Lee JN, Oh GS, Kim HJ, Kim MS, Lee SH. The inhibitory effect of beta-lapachone on RANKL-induced osteoclastogenesis. Biochem Biophys Res Commun. 2017;482:1073–9. doi: 10.1016/j.bbrc.2016.11.160. [DOI] [PubMed] [Google Scholar]
- 39.Lloret A, Fuchsberger T, Giraldo E, Vina J. Molecular mechanisms linking amyloid beta toxicity and Tau hyperphosphorylation in Alzheimer's disease. Free Radic Biol Med. 2015;83:186–91. doi: 10.1016/j.freeradbiomed.2015.02.028. [DOI] [PubMed] [Google Scholar]
- 40.Li L, McBride DW, Doycheva D, Dixon BJ, Krafft PR, Zhang JH, et al. G-CSF attenuates neuroinflammation and stabilizes the blood-brain barrier via the PI3K/Akt/GSK-3beta signaling pathway following neonatal hypoxia-ischemia in rats. Exp Neurol. 2015;272:135–44. doi: 10.1016/j.expneurol.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, et al. Astrocyte senescence as a component of Alzheimer's disease. PLoS One. 2012;7:e45069. [DOI] [PMC free article] [PubMed]
- 42.Hung AS, Liang Y, Chow TC, Tang HC, Wu SL, Wai MS, et al. Mutated tau, amyloid and neuroinflammation in Alzheimer disease-A brief review. Prog Histochem Cytochem. 2016;51:1–8. doi: 10.1016/j.proghi.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 43.Chen K, Wang N, Diao Y, Dong W, Sun Y, Liu L, et al. Hydrogen-rich saline attenuates brain injury induced by cardiopulmonary bypass and inhibits microvascular endothelial cell apoptosis via the PI3K/Akt/GSK3beta signaling pathway in rats. Cell Physiol Biochem. 2017;43:1634–47. doi: 10.1159/000484024. [DOI] [PubMed] [Google Scholar]
- 44.Reis CR, Chen PH, Srinivasan S, Aguet F, Mettlen M, Schmid SL. Crosstalk between Akt/GSK3 beta signaling and dynamin-1 regulates clathrin-mediated endocytosis. Embo J. 2015;34:2132–46. doi: 10.15252/embj.201591518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu SL, Wang C, Jiang T, Tan L, Xing A, Yu JT. The role of Cdk5 in Alzheimer's disease. Mol Neurobiol. 2016;53:4328–42. doi: 10.1007/s12035-015-9369-x. [DOI] [PubMed] [Google Scholar]
- 46.Kruger L, Mandelkow EM. Tau neurotoxicity and rescue in animal models of human Tauopathies. Curr Opin Neurobiol. 2016;36:52–8. doi: 10.1016/j.conb.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 47.Saponaro C, Cianciulli A, Calvello R, Dragone T, Iacobazzi F, Panaro MA. The PI3K/Akt pathway is required for LPS activation of microglial cells. Immunopharm Immunot. 2012;34:858–65. doi: 10.3109/08923973.2012.665461. [DOI] [PubMed] [Google Scholar]
- 48.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14:463–77. doi: 10.1038/nri3705. [DOI] [PubMed] [Google Scholar]
- 50.D'Hooge R, De Deyn PP. Applications of the Morris water maze in the study of learning and memory. Brain Res Brain Res Rev. 2001;36:60–90. doi: 10.1016/S0165-0173(01)00067-4. [DOI] [PubMed] [Google Scholar]
- 51.Lopez-Gonzalez I, Schluter A, Aso E, Garcia-Esparcia P, Ansoleaga B, LL F, et al. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol. 2015;74:319–44. doi: 10.1097/NEN.0000000000000176. [DOI] [PubMed] [Google Scholar]
- 52.Mufson EJ, Ikonomovic MD, Counts SE, Perez SE, Malek-Ahmadi M, Scheff SW, et al. Molecular and cellular pathophysiology of preclinical Alzheimer's disease. Behav Brain Res. 2016;311:54–69. doi: 10.1016/j.bbr.2016.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Briggs R, Kennelly SP, O'Neill D. Drug treatments in Alzheimer's disease. Clin Med. 2016;16:247–53. doi: 10.7861/clinmedicine.16-3-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ries M, Sastre M. Mechanisms of abeta clearance and degradation by glial cells. Front Aging Neurosci. 2016;8:160. doi: 10.3389/fnagi.2016.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodriguez-Arellano JJ, Parpura V, Zorec R, Verkhratsky A. Astrocytes in physiological aging and Alzheimer's disease. Neuroscience. 2016;323:170–82. doi: 10.1016/j.neuroscience.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 56.LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist Updates. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raina AK, Templeton DJ, Deak JC, Perry G, Smith MA. Quinone reductase (NQO1), a sensitive redox indicator, is increased in Alzheimer's disease. Redox Report. 1999;4:23–7. doi: 10.1179/135100099101534701. [DOI] [PubMed] [Google Scholar]
- 58.Luo JR, Li S, Qin X, Peng QL, Liu YQ, Yang S, et al. Association of the NQO1 C609T polymorphism with Alzheimer's disease in Chinese populations: a meta-analysis. Int J Neurosci. 2016;126:19. doi: 10.3109/00207454.2015.1004573. [DOI] [PubMed] [Google Scholar]
- 59.Kanno T, Tsuchiya A, Tanaka A, Nishizaki T. Combination of PKC epsilon activation and PTP1B inhibition effectively suppresses a beta-induced GSK-3 beta activation and tau phosphorylation. Mol Neurobiol. 2016;53:4787–97. doi: 10.1007/s12035-015-9405-x. [DOI] [PubMed] [Google Scholar]
- 60.Zhou FT, Chen SR, Xiong JP, Li YH, Qu LN. Luteolin reduces zinc-induced tau phosphorylation at Ser262/356 in an ROS-dependent manner in SH-SY5Y cells. Biol Trace Elem Res. 2012;149:273–9. [DOI] [PubMed]
- 61.Takashima A, Noguchi K, Michel G, Mercken M, Hoshi M, Ishiguro K, et al. Exposure of rat hippocampal neurons to amyloid beta peptide (25–35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci Lett. 1996;203:33–6. doi: 10.1016/0304-3940(95)12257-5. [DOI] [PubMed] [Google Scholar]
- 62.Tsujio I, Tanaka T, Kudo T, Nishikawa T, Shinozaki K, Grundke-Iqbal I, et al. Inactivation of glycogen synthase kinase-3 by protein kinase C delta: implications for regulation of tau phosphorylation. FEBS Lett. 2000;469:111–7. doi: 10.1016/S0014-5793(00)01234-5. [DOI] [PubMed] [Google Scholar]
- 63.Lopez-Rodriguez AB, Hennessy E, Murray C, Lewis A, de Barra N, Fagan S, et al. Microglial and astrocyte priming in the APP/PS1 model of Alzheimer's disease: increased vulnerability to acute inflammation and cognitive deficits. 2018.
- 64.Blach-Olszewska Z, Zaczynska E, Gustaw-Rothenberg K, Avila-Rodrigues M, Barreto GE, Leszek J, et al. The innate immunity in Alzheimer disease- relevance to pathogenesis and therapy. Curr Pharm Des. 2015;21:3582–8. doi: 10.2174/1381612821666150710144829. [DOI] [PubMed] [Google Scholar]
- 65.Lichtenstein MP, Carriba P, Masgrau R, Pujol A, Galea E. Staging anti-inflammatory therapy in Alzheimer's disease. Front Aging Neurosci. 2010;2:142. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.