

Abstract
c-Myc plays an important role in cell proliferation, differentiation, and cell apoptosis. FasL/Fas pathway is a key regulator of cell apoptosis. This study was aimed to investigate the effects of c-Myc on the FasL/Fas pathway in ischemia-reperfusion (I/R)-induced renal injury. Rats were objected to bilateral renal ischemia for 60 min and reperfused for 24 or 48 h. NRK-52E cells were treated with hypoxia-reoxygenation (H/R) or FasL. Immunohistochemistry was used to identify the distribution of c-Myc. Cell apoptosis was assessed by TUNEL staining. Ad-c-Myc and recombinant pcDAN 3.0 were used to overexpress c-Myc and c-FLIP, respectively. ChIP assay and luciferase assay were used to detect the binding of c-Myc to c-FLIP promoter. In I/R rats, c-Myc was increased significantly and mainly located in renal tubular epithelial cells; meanwhile, c-FLIP was decreased, cleaved caspase-8, cleaved caspase-3 and TUNEL-positive staining cells were increased. Treatment of I/R rats with c-Myc inhibitor 10058-F4 significantly attenuated the decrease in c-FLIP, the increase in cleaved caspase-8, cleaved caspase-3, TUNEL-positive cells, Scr and BUN in I/R rats. In NRK-52E cells, hypoxia and reoxygen induced the increase in c-Myc and decrease in c-FLIP. ChIP and luciferase assay results indicated that c-Myc binds to the promoter region of c-FLIP gene. Overexpression of c-Myc markedly decreased c-FLIP. Overexpression of c-FLIP inhibited the increase in cleaved caspase-8 and caspase-3 induced by FasL. Data indicated that c-Myc is increased in kidneys of I/R rats and negatively regulates the expression of c-FLIP, then enhanced FasL-induced cell apoptosis in I/R stress.
Keywords: c-Myc, c-FLIP, FasL/Fas, ischemia-reperfusion, tubular cell apoptosis
Introduction
Acute kidney injury (AKI) is a major risk factor in clinic [1], which accounts for 25–40% mortality worldwide [2, 3]. Ischemia-reperfusion (I/R)-induced renal injury is the leading cause of AKI [4]. Previous studies have suggested that the renal tubular epithelial cell apoptosis, necrosis, and interstitial inflammation are all involved in I/R-induced renal injury. However, the underlying mechanisms remain to be fully elucidated.
Renal tubule is the initial target for the I/R-induced renal injury [5]. Activation of FasL/Fas-related cell apoptosis pathway is essential for I/R-induced cell apoptosis and organ damage [6–10]. Activation of Fas receptor by Fas ligand (FasL) recruits and activates caspase-8 by binding to FADD (Fas-associating protein with a novel death domain). Recent studies have shown that the activation of Fas receptor contributes to tubular epithelial cell apoptosis and knockout on Fas receptor protects the kidney against I/R-induced injury [11–15].
Cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (c-FLIP), an inactive homolog of procaspase-8, is a master negative antiapoptotic regulator for FasL-induced apoptosis [16, 17]. Once c-FLIP binds to FADD, it inhibits the recruitment and activation of caspase-8. It has been demonstrated that c-FLIP is highly expressed in renal tubule, and involved in interleukin (IL)-2-induced renal tubular epithelial cell apoptosis [18]. However, the role of c-FLIP in I/R-induced renal injury has not been investigated.
c-Myc is originally identified as an oncogene and is accepted as a reprogramming factor. As an important nuclear transcriptional factor, c-Myc regulates cell proliferation, differentiation, cell cycle as well as apoptosis [19]. Our previous study has proved that c-Myc contributes to renal fibroblast activation by inducing ITGAV-mediated TGF-β signaling [20]. A recent study determined that the c-Myc was upregulated in the kidney of I/R mice; however, the exact effect of c-Myc in I/R-induced renal injury remains to be clarified [21].
In this study, our data confirmed that c-Myc is upregulated in the kidney suffering from I/R and contributes to I/R-induced renal tubular cell apoptosis. c-Myc promotes FasL/Fas-mediated renal tubular cell apoptosis by repressing the expression of negative regulator c-FLIP.
Materials and methods
Reagents
c-Myc inhibitor 10058-F4 was purchased from Selleck (Selleck Chemicals, Shanghai, China). Fetal bovine serum (FBS) was acquired from Gibco (Grand Island, NY, USA). The BCA Protein Assay Kit was from Shenergy Biocolor BioScience and Technology (Shanghai, China). The c-Myc antibody was purchased from Abcam Biotechnology (Cambridge, MA, USA). The c-FLIP, Fas, FasL, cleaved caspase-8, and cleaved caspase-3 antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Adenoviral vector harboring c-Myc gene and empty vector were purchased from HanBio, Shanghai, China. The anti-actin antibody, IgG, Enhanced Chemiluminescence (ECL) detection kit were obtained from Beyotime Institute of Biotechnology (Haimen, China).
Animals
Sprague Dawley (SD) rats (5 weeks) were purchased from Shanghai SLAC Laboratory Animal (Shanghai, China). Rats were housed in a specific pathogen-free facility for 3–4 weeks to accommodate the new environment. The individuals with body weight between 250 and 280 g were recruited in the experiment. All animal experiments were performed according to the criteria of the Medical Laboratory Animal Administrative Committee of Shanghai and the Guide for Care and Use of Laboratory Animals of Fudan University. The protocols were approved by the Ethics Committee for Experimental Research, Shanghai Medical College, Fudan University.
I/R model
Rat bilateral renal ischemia models were conducted as previously described [22]. Rats were anesthetized with chloral hydrate and then dorsal incision was performed to expose the kidneys. Both renal pedicles were bluntly dissected. A nontraumatic microvascular clamp was placed across each renal pedicle for 60 min. Kidney was kept well hydrated with warm sterile saline until the end of renal pedicle clamping. After ischemia for 60 min, the clamp was gently removed and the incision was closed. Sham rats underwent identical surgical procedures except that the clamp was not applied. The success of the AKI model was confirmed by an increase in serum creatinine concentration along with renal histologic damage at 24 h after ischemia.
Cell culture
NRK-52E cells were obtained from the Institute of Biochemistry and Cell Biology (Shanghai, China) and cultured in Dulbecco's Modified Eagle Medium (DMEM) (Sigma, St Louis, MO, USA) supplemented with 10% FBS (Gibco, Grand Island, NY, USA) and 50 IU/mL penicillin. Cells were cultured at 37 °C in a 20% O2–5% CO2 atmosphere. NRK-52E cells were cultured overnight and grown to 50–60% confluence. Vector expressing c-FLIP plasmid and scrambled plasmid was purchased from HanBio (Shanghai, China). When 50−60% confluence was reached, NRK-52E cells were transfected with the plasmids using Lipofectamine 2000 (InvivoGen, Shanghai, China) according to the manufacturer’s protocol.
Western blotting
Protein extracts of total renal tissue (50 mg) or NRK-52E cells were prepared as previously described [23]. Briefly, kidney tissues or NRK-52E cells were homogenized in RIPA lysis buffer on ice. Protein concentration was examined using a BCA Protein Assay Kit (Shenergy Biocolor BioScience and Technology, Shanghai, China) according to the manufacturer’s instructions. Whole lysates were mixed with Sodium dodecylsulfate loading buffer and heated at 100 °C, then the proteins were separated on 10–20% polyacrylamide gels and transferred to polyvinylidene fluoride (PVDF) membranes.
Assessment of renal morphological changes
Kidneys were fixed in 4% paraformaldehyde and embedded in paraffin. Sections (4 μm) were stained with hematoxylin-eosin (H&E) and periodic acid-silver nitrate (PAS).
Fluorescence confocal microscopy
After overexpressing c-Myc for 24 h by adenoviral vector (HanBio, Shanghai, China), NRK-52E cells were fixated with 4% paraformaldehyde for 15 min at room temperature, and then the cells were incubated with primary antibodies against c-Myc and c-FLIP overnight at 4 °C. After being washed by PBS for three times, the cells were incubated with secondary antibodies for 1 h followed by incubation with 4',6-diamidino-2-phenylindole (DAPI) for 5 min. Cells were imaged under a confocal laser scanning biological microscope (Leica, Wetzlar, Germany).
TUNEL staining
Twenty-four or forty-eight hours after ischemia-reperfusion, kidneys were perfusion fixed with 4% paraformaldehyde, sectioned at 4 μm, and the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) reaction was performed according to the manufacturer’s protocol.
Statistical analysis
Column graphs were drawn by using GraphPad Prism 6.0 (San Diego, CA, USA). ANOVA was used to determine if differences among mean values reached statistical significance. t test was used for comparisons between groups. P value < 0.05 was considered as statistically significant.
Results
c-Myc is upregulated in rat kidney after I/R in vivo and renal tubular epithelial cell following H/R in vitro
To examine the effect of I/R on the expression of c-Myc in kidney, real-time quantitative PCR, Western blotting, and immunohistochemistry were used to identify the expression of c-Myc. In I/R rats, both c-Myc mRNA and protein level were significantly increased after 24 h of reperfusion (Fig. 1a, b). As shown in Fig. 1c, c-Myc-positive signal was relatively weak in control rats and was markedly induced in I/R kidneys. The distribution of c-Myc was mainly located in the nucleus of renal tubular epithelial cell. To validate the observation in I/R rats, cultured renal tubular epithelial cells (NRK-52E) were treated with hypoxia for 6 h following reoxygenation for 3 or 6 h (H/R). As shown in Fig. 1d, H/R treatment increased c-Myc expression in a time-dependent manner. These data show that the expression of c-Myc can be induced by either I/R in vivo or H/R in vitro.
Fig. 1.

c-Myc is upregulated in the kidneys of I/R rats and H/R-treated NRK-52E cells. SD rats were subjected to 60 min ischemia and 24 or 48 h reperfusion. a RT-PCR was performed to assay the levels of c-Myc mRNA in the kidneys (six rats per group). b Western blotting was performed to visualize c-Myc expression in kidneys (six rats per group). c The levels of c-Myc were visualized using immunohistologic analysis in the kidneys of control and 24 h reperfusion respectively (six rats per group). Bar = 100 μm. Original magnification ×200 or ×400. d The protein levels of c-Myc were assayed by Western blotting in NRK-52E cells with or without 6 h hypoxia and 3 or 6 h reoxygenation treatment (three experiments per group). Data are presented as the mean ± SEM. **P < 0.01 versus control
FasL/Fas-mediated apoptosis pathway is activated in I/R rats and FasL-treated NRK-52E cells
Western blotting was used to confirm the activation of FasL/Fas pathway in I/R rats and FasL-treated renal tubular epithelial cells. As shown in Fig. 2a, FasL was significantly increased in rat kidney after I/R; meanwhile, the cleaved caspase-8 and cleaved caspase-3, the downstream effectors of FasL/Fas apoptosis pathway, were also significantly increased in I/R rats (Fig. 2a). To validate the results in vitro, NRK-52E cells were exposed to recombinant FasL protein. As shown in Fig. 2b, both cleaved caspase-8 and cleaved caspase-3 were significantly increased in NRK-52E cells after FasL (0.125 μg/mL) treatment for 24 or 48 h. These data show that FasL/Fas-mediated apoptosis pathway is activated in I/R rats.
Fig. 2.

FasL/Fas-mediated apoptosis pathway is activated in kidneys of I/R rats and FasL-treated NRK-52E cells. a Western blotting was performed to visualize the protein levels of FasL, cleaved caspase-8, and cleaved caspase-3 in kidneys (six rats per group). b The protein levels of cleaved caspase-8 and cleaved caspase-3 were assayed by Western blotting in NRK-52E cells with or without FasL (0.125 μg/mL) treatment for 24 or 48 h (three experiments per group). Data are presented as the mean ± SEM. **P < 0.01 versus control
Inhibition of c-Myc protects against I/R-induced renal injury
To identify the role of c-Myc in I/R-induced renal injury, c-Myc inhibitor was applied to I/R rats. c-Myc inhibitor 10058-F4 or its vehicle corn oil was administrated by gavage to the rats and then subjected to sham operation or bilateral renal ischemia for 60 min and reperfusion for 24 h. 10058-F4 treatment significantly attenuated renal dysfunction which exhibited as the increases in blood urea nitrogen and serum creatinine after I/R were blunted (Fig. 3a, b). H&E and PAS staining showed that 10058-F4 treatment relieved renal tubular damage in I/R rats (Fig. 3c, d). Furthermore, the TUNEL staining results indicated that 10058-F4 treatment significantly ameliorated the increase in tubular cell apoptosis after I/R (Fig. 4a, b). These results suggest that the elevated c-Myc is involved in I/R-induced renal injury. Inhibition of c-Myc exhibits a protective effect in I/R-induced renal damage.
Fig. 3.

Inhibition of c-Myc protects against I/R-induced renal injury. Control and I/R (24 h) rats received vehicle and 10058-F4 (12.5 mg/kg body weight), respectively, via oral gavage 6 h before I/R surgery and gavage every 6 h during reperfusion. a, b Serum creatinine and blood urea nitrogen were assayed using ELISA kit (six rats per group). c, d H&E and PAS were performed to visualize renal injury (six rats per group). Representative sections of H&E- and PAS-stained kidneys. Original magnification ×200 or ×400. Data are presented as the mean ± SEM. **P < 0.01 versus control, #P < 0.05 versus I/R
Fig. 4.

Inhibition of c-Myc protects against I/R-induced renal tubular apoptosis. Control and I/R (24 h) rats received vehicle and 10058-F4 (12.5 mg/kg body weight), respectively, via oral gavage 6 h before I/R surgery and gavage every 6 h during reperfusion. a TUNEL staining of kidney cortex sections were performed to assay renal tubular epithelial cell apoptosis (six rats per group). Representative sections of TUNEL-stained kidneys. Original magnification ×200 or ×400. b Quantification of apoptotic cells by TUNEL staining in Control, I/R, and I/R + 10058-F4. Data are presented as the mean ± SEM. **P < 0.01 versus control, #P < 0.05 versus I/R
Decrease in c-FLIP is responsible for FasL-induced cell apoptosis
As shown in Fig. 5a, the protein level of c-FLIP (55 kDa) was decreased in kidneys after I/R. To validate the observation in I/R rats, cultured renal tubular epithelial cells (NRK-52E) were treated with hypoxia for 6 h following reoxygenation for 3 or 6 h (H/R). c-FLIP was decreased after H/R treatment for 6 h (Fig. 5b). To further confirm whether c-FLIP was involved in FasL-induced cell apoptosis, c-FLIP expression plasmid was transfected into NRK-52E cells. As shown in Fig. 5c, c-FLIP level was increased significantly after the transfection of recombinant pcDNA3.0 plasmid harboring c-FLIP for 24 h. Overexpression of c-FLIP significantly attenuated FasL-induced cell apoptosis (Fig. 5d), which indicating as that the increases in cleaved caspase-8 and cleaved caspase-3 after FasL exposure were significantly blunted. Additionally, to identify whether inhibition of c-Myc affects FasL-induced cell apoptosis in vitro, NRK-52E cells were pretreated with c-Myc inhibitor 10058-F4 before FasL treatment. 10058-F4 significantly attenuated FasL-induced increases in cleaved caspase-8 and cleaved caspase-3 (Fig. 5e). These data show that c-FLIP is involved in I/R-induced renal injury and FasL-induced cell apoptosis.
Fig. 5.

c-FLIP is decreased in kidneys of I/R rats and the decrease in c-FLIP is responsible for FasL-induced cell apoptosis. a Western blotting was performed to visualize the protein levels of c-FLIP in kidneys of rats (six rats per group). b The protein levels of c-FLIP were assayed by Western blotting in NRK-52E cells with or without 6 h hypoxia and 3 or 6 h reoxygenation treatment (three experiments per group). c NRK-52E cells were transfected with c-FLIP overexpression plasmid for 24 h. The protein levels of c-FLIP were assayed by Western blotting (three experiments per group). d The protein levels of cleaved caspase-8 and cleaved caspase-3 were assayed by Western blotting in FasL (0.125 μg/mL)-treated NRK-52E cells with or without pretreatment with c-FLIP expression plasmid (three experiments per group). e The protein levels of cleaved caspase-8 and cleaved caspase-3 were assayed by Western blotting in FasL (0.125 μg/mL)-treated NRK-52E cells with or without pretreatment with c-Myc inhibitor 10058-F4 (three experiments per group). Data are presented as the mean ± SEM. *P < 0.05 versus control, **P < 0.01 versus control or pcDNA 3.0. #P < 0.05 versus FasL. PcDNA 3.0, empty plasmid vector; c-FLIP plasmid, an plasmid harboring c-FLIP gene
c-Myc binds to the promoter region of c-FLIP gene
Using the JASPAR2018 database, bioinformatics analysis result showed that there were at least seven putative c-Myc binding sites in c-FLIP promoter region (Fig. 6a). The binding site showed the highest score was selected in the follow-up study (Score 14.7483, Fig. 6a). Chromatin immunoprecipitation assay result showed that a significant enrichment of the chromatin region was detected in Anti-c-Myc group compared with the IgG control (Fig. 6b, c). To further identify the effect of c-Myc on c-FLIP expression, a reconstructed luciferase vector harboring wild-type (c-FLIP WT) promoter region was transferred into NRK-52E cells 1 day after transfection with empty adenoviral vector (Ad-Control) or adenoviral vector harboring c-Myc gene (Ad-c-Myc). Cotransfection of an Ad-c-Myc significantly suppressed luciferase activity. Mutation of c-FLIP promoter region by deleting c-Myc binding site (c-FLIP mut) abolished the effect of Ad-c-Myc cotransfection (Fig. 6d). These data indicate that nuclear transcriptional factor c-Myc is able to bind to the promoter region of c-FLIP and negatively regulates the expression of c-FLIP.
Fig. 6.

c-Myc binds to the promoter region of c-FLIP gene. a c-Myc binding sites in c-FLIP promoter predicted by JASPAR 2018. b Schematic diagram of the binding site of c-Myc in c-FLIP promoter. Red words indicate c-Myc binding motif. c c-Myc bound to the proximal promoter of the c-FLIP gene as determined by chromatin immunoprecipitation assay. Cross-linked nuclear extracts of NRK-52E cells transduced with c-Myc virus vectors (Ad-c-Myc) were immunoprecipitated using either an anti-c-Myc antibody or control IgG. The regions of the c-Myc binding sites were quantified and normalized to IgG pulldown. d Luciferase expression analysis using wild-type (c-FLIP WT) and mutant c-FLIP promoter regions (c-FLIP mut) in NRK-52E cells 1 day after transfection with empty vector (Ad-Control) or Ad-c-Myc. Mutant c-FLIP promoter was constructed with a 10-bp deletion of the binding site. Data are presented as the mean ± SEM. **P < 0.01 versus IgG. ***P < 0.001 versus Ad-Control
c-Myc promotes I/R-induced renal injury by suppressing c-FLIP expression
Ad-c-Myc or Ad-Con was transferred into renal epithelial cells. Western blotting results showed that Ad-c-Myc transfection significantly increased the protein level of c-Myc and decreased c-FLIP in NRK-52E cells. Immunofluorescence staining revealed that the accumulation of c-FLIP protein in cytoplasm was much lower in Ad-c-Myc group than in the Ad-Con group. Ad-c-Myc transfection decreased the protein level of c-FLIP (Fig. 7a, b). Finally, the effect of c-Myc on c-FLIP was tested in I/R rats. As shown in Fig. 8, c-Myc inhibitor 10058-F4 attenuated the decrease of c-FLIP in I/R rats. Meanwhile, the increases in cleaved caspase-8 and cleaved caspase-3 in I/R rats were blunted by 10058-F4 treatment.
Fig. 7.
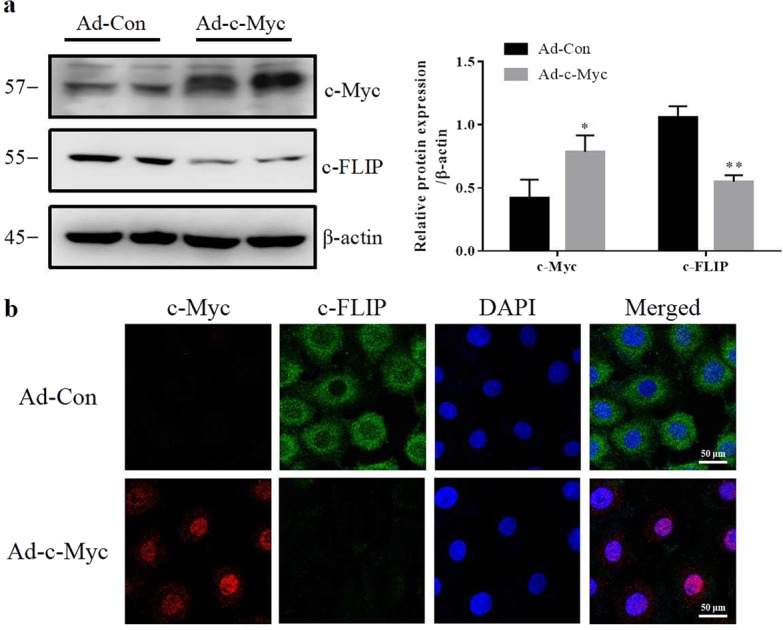
Overexpression of c-Myc deregulates the protein level of c-FLIP. NRK-52E cells were treated with recombinant adenoviruses expressing c-Myc for 24 h. a Western blotting was performed to visualize the c-Myc and c-FLIP expression (three experiments per group). b Levels of c-Myc and c-FLIP were visualized using immunofluorescence (three experiments per group). Data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 versus Ad-con. Ad-c-Myc, an adenoviral vector harboring c-Myc gene; Ad-Con, empty adenovirus vector
Fig. 8.
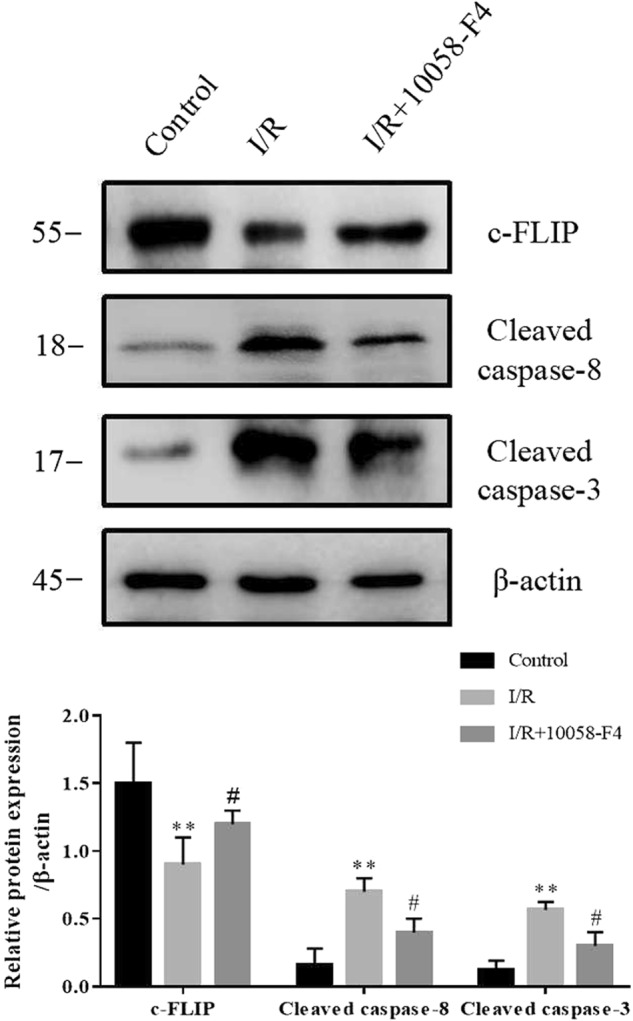
c-Myc promotes I/R-induced renal injury by suppressing c-FLIP expression. The protein levels of c-FLIP, cleaved caspase-8 and cleaved caspase-3 were assayed by Western blotting in kidneys of rats with or without of c-Myc inhibitor treatment (six rats per group). Data are presented as the mean ± SEM. **P < 0.01 versus control, #P < 0.05 versus I/R
Discussion
I/R is one of the most common causes of AKI. [1]. Previous studies have reported that various pathological processes are involved in I/R-induced renal injury, such as tubular epithelial cell injury, microvascular dysfunction, and inflammation. Death receptor-mediated signaling pathway is responsible for cell apoptosis. It has been proved that the activation of membrane-bound Fas receptors is an important cause of ischemic-induced tissue injury [13, 15]. In this study, we investigated the effect of FasL/Fas-mediated apoptosis pathway in I/R-induced renal injury and its regulating mechanism.
Transcription factor c-Myc is an important regulator in maintaining cell homeostasis. Previous study has reported that ischemia or hypoxia induces the increase in c-Myc in the cardiovascular system [24]. In this study, our data indicated that c-Myc was upregulated in kidneys of I/R rats and was mainly located in the nucleus of renal tubular epithelial cells. In cultured renal tubular epithelial cells, H/R treatment also induced an increase in c-Myc expression. Inhibition of c-Myc attenuated I/R-induced renal tubular epithelial cell apoptosis and renal injury. These results suggest that elevated c-Myc is involved in I/R-induced AKI.
c-FLIP is a master antiapoptotic regulator of tumor necrosis factor-α (TNF-α), FasL, and TNF-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis pathway [16, 25]. FasL/Fas-mediated pathway is an important pathway in initiating I/R-induced cell apoptosis [8, 13, 26]. c-FLIP inhibits activation of caspase-8 by competitively binding to the DED domain of FADD and then attenuates cell apoptosis. Three c-FLIP splice variants, c-FLIPL (55 kDa), c-FLIPS (26 kDa), and c-FLIPR (25 kDa), have been identified and all of them function as apoptosis inhibitors. In the experiment, only c-FLIPL (55 kDa) was identified to be expressed in rat kidney. This result was consistent with the previous observation [8, 13, 26]. In the experiments, our result showed that the expression of c-FLIP was decreased in I/R rat kidneys and the decrease in c-FLIP was associated with I/R-induced renal injury. The decrease in c-FLIP, a negative suppressor in FasL/Fas-mediated apoptosis pathway, enhanced the effect of FasL by facilitating the activation of downstream effector caspase-8 and then promoted renal tubular cell apoptosis.
As a transcriptional factor, c-Myc is able to target a variety of genes and elicits different effects in regulating cellular function [19]. c-Myc is originally identified as an oncogene and considered to promote cell proliferation. However, a number of studies have shown that c-Myc can not only stimulate cell proliferation, but also promotes cell apoptosis [27–32]. Among all the genes regulated by c-Myc, some are prosurvival whereas others may be prodeath. For example, p53, one target gene of c-Myc [33–36], contributes to renal tubular epithelial cell apoptosis in renal injury [37, 38]. In different pathological conditions, c-Myc was also observed to be able to regulate different signaling pathways, including c-Myc-p21 Cip1 pathway and c-Myc-p19ARF-p53 pathway [39, 40]. In this study, we observed that c-Myc was able to bind to the promoter region of c-FLIP and negatively regulates the expression of c-FLIP. Overexpression of c-Myc suppressed the expression of c-FLIP, a negative regulator of FasL/Fas-mediated cell apoptosis pathway, thereby promoting cell apoptosis.
In summary, we identified that c-Myc is significantly elevated in kidney of I/R rats and serves as a positive regulator in I/R-induced renal tubular epithelial cell apoptosis. As a transcriptional factor, c-Myc negatively regulates the expression of c-FLIP, a suppressor in FasL/Fas-mediated apoptosis pathway, thus enhancing FasL-induced cell apoptosis in I/R stress (Fig. 9). This study provided novel evidence in recognizing I/R-induced AKI.
Fig. 9.
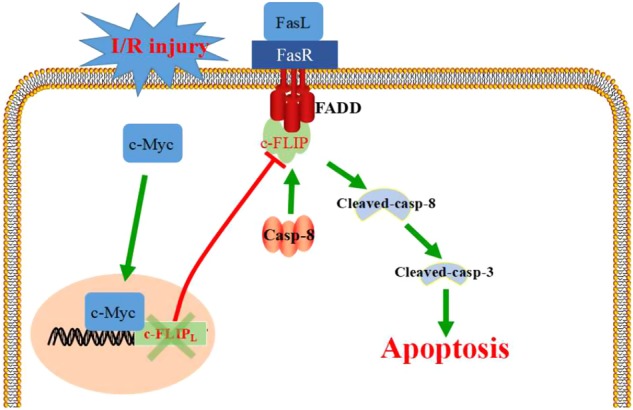
Schematic diagram of this study. c-Myc promotes tubular cell apoptosis in I/R-induced renal injury by negatively regulating c-FLIP and enhanced FasL/Fas-mediated apoptosis pathway. FasL fas ligand, FasR fas receptor, Casp-8 caspase-8, FADD Fas-associating protein with a novel death domain
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China Nos. 81670664, 81470591 to Li-min Lu, No. 81100531 to Wei Zhang and No. 81770718 to Xiao-xia Wang.
Author contributions
L-m L, J L, H X, X-x W, W Z, D X, and B W designed the research project; D X, B W, P-p C, and F Y performed the experiments; Y-z W, N-j M, Z-l Z, L Z, Q C, and H-y X analyzed the data; L-m L, D X, and B W wrote the manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Hong Xue, Email: xuehong@fudan.edu.cn.
Wei Zhang, Email: wzhang@shmu.edu.cn.
Li-min Lu, Email: lulimin@shmu.edu.cn.
References
- 1.Negi Shigeo, Koreeda Daisuke, Kobayashi Sou, Yano Takuro, Tatsuta Koichi, Mima Toru, Shigematsu Takashi, Ohya Masaki. Acute kidney injury: Epidemiology, outcomes, complications, and therapeutic strategies. Seminars in Dialysis. 2018;31(5):519–527. doi: 10.1111/sdi.12705. [DOI] [PubMed] [Google Scholar]
- 2.Kanagasundaram NS. Pathophysiology of ischaemic acute kidney injury. Ann Clin Biochem. 2015;52:193–205. doi: 10.1177/0004563214556820. [DOI] [PubMed] [Google Scholar]
- 3.Hoste EA, Clermont G, Kersten A, Venkataraman R, Angus DC, De Bacquer D, et al. RIFLE criteria for acute kidney injury are associated with hospital mortality in critically ill patients: a cohort analysis. Crit Care. 2006;10:R73. doi: 10.1186/cc4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Requiao-Moura LR, Durao JMS, Matos AC, Pacheco-Silva A. Ischemia and reperfusion injury in renal transplantation: hemodynamic and immunological paradigms. Einst (Sao Paulo) 2015;13:129–35. doi: 10.1590/S1679-45082015RW3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang YL, Qiao SK, Wang RY, Guo XN. NGAL attenuates renal ischemia/reperfusion injury through autophagy activation and apoptosis inhibition in rats. Chem Biol Interact. 2018;289:40–46. doi: 10.1016/j.cbi.2018.04.018. [DOI] [PubMed] [Google Scholar]
- 6.Date T, Mochizuki S, Belanger AJ, Yamakawa M, Luo Z, Vincent KA, et al. Differential effects of membrane and soluble Fas ligand on cardiomyocytes: role in ischemia/reperfusion injury. J Mol Cell Cardiol. 2003;35:811–21. doi: 10.1016/S0022-2828(03)00139-1. [DOI] [PubMed] [Google Scholar]
- 7.Zheng M, Song D, Luo Z, Lu Z, Wu Y, Wang W. Effect of puerarin on expression of Fas/FasL mRNA in pulmonary injury induced by ischemia-reperfusion in rabbits. Nat Prod Commun. 2015;10:253–56. [PubMed] [Google Scholar]
- 8.Liu XM, Yang ZM, Liu XK. Fas/FasL induces myocardial cell apoptosis in myocardial ischemia-reperfusion rat model. Eur Rev Med Pharmacol Sci. 2017;21:2913–18. [PubMed] [Google Scholar]
- 9.Yin XH, Han YL, Zhuang Y, Yan JZ, Li C. Geldanamycin inhibits Fas signaling pathway and protects neurons against ischemia. Neurosci Res. 2017;124:33–39. doi: 10.1016/j.neures.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 10.Filep JG. Attenuation of lung ischemia-reperfusion injury: silencing the Fas gene. Crit Care Med. 2016;44:1619–20. doi: 10.1097/CCM.0000000000001687. [DOI] [PubMed] [Google Scholar]
- 11.Ko GJ, Jang HR, Huang Y, Womer KL, Liu M, Higbee E, et al. Blocking Fas ligand on leukocytes attenuates kidney ischemia-reperfusion injury. J Am Soc Nephrol. 2011;22:732–42. doi: 10.1681/ASN.2010010121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nogae S, Miyazaki M, Kobayashi N, Saito T, Abe K, Saito H, et al. Induction of apoptosis in ischemia-reperfusion model of mouse kidney: possible involvement of Fas. J Am Soc Nephrol. 1998;9:620–31. doi: 10.1681/ASN.V94620. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Mu Y, Zhou X, Ji H, Gao X, Cai WW, et al. SIRT2-mediated FOXO3a deacetylation drives its nuclear translocation triggering FasL-induced cell apoptosis during renal ischemia reperfusion. Apoptosis. 2017;22:519–30. doi: 10.1007/s10495-016-1341-3. [DOI] [PubMed] [Google Scholar]
- 14.Hamar P, Song E, Kokeny G, Chen A, Ouyang N, Lieberman J. Small interfering RNA targeting Fas protects mice against renal ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2004;101:14883–88. doi: 10.1073/pnas.0406421101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Del RM, Imam A, DeLeon M, Gomez G, Mishra J, Ma Q, et al. The death domain of kidney ankyrin interacts with Fas and promotes Fas-mediated cell death in renal epithelia. J Am Soc Nephrol. 2004;15:41–51. doi: 10.1097/01.ASN.0000104840.04124.5C. [DOI] [PubMed] [Google Scholar]
- 16.Safa AR. c-FLIP, a master anti-apoptotic regulator. Exp Oncol. 2012;34:176–84. [PMC free article] [PubMed] [Google Scholar]
- 17.Bagnoli M, Canevari S, Mezzanzanica D. Cellular FLICE-inhibitory protein (c-FLIP) signalling: a key regulator of receptor-mediated apoptosis in physiologic context and in cancer. Int J Biochem Cell Biol. 2010;42:210–13. doi: 10.1016/j.biocel.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 18.Du C, Guan Q, Yin Z, Zhong R, Jevnikar AM. IL-2-mediated apoptosis of kidney tubular epithelial cells is regulated by the caspase-8 inhibitor c-FLIP. Kidney Int. 2005;67:1397–409. doi: 10.1111/j.1523-1755.2005.00217.x. [DOI] [PubMed] [Google Scholar]
- 19.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/MCB.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen Y, Miao N, Wang B, Xu J, Gan X, Xu D, et al. c-Myc promotes renal fibrosis by inducing integrin alphav-mediated transforming growth factor-beta signaling. Kidney Int. 2017;92:888–99. doi: 10.1016/j.kint.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 21.Rogers NM, Zhang ZJ, Wang JJ, Thomson AW, Isenberg JS. CD47 regulates renal tubular epithelial cell self-renewal and proliferation following renal ischemia reperfusion. Kidney Int. 2016;90:334–47. doi: 10.1016/j.kint.2016.03.034. [DOI] [PubMed] [Google Scholar]
- 22.Yilmaz S, Paavonen T, Hayry P. Chronic rejection of rat renal allografts. II. The impact of prolonged ischemia time on transplant histology. Transplantation. 1992;53:823–27. doi: 10.1097/00007890-199204000-00023. [DOI] [PubMed] [Google Scholar]
- 23.Miao N, Wang B, Xu D, Wang Y, Gan X, Zhou L, et al. Caspase-11 promotes cisplatin-induced renal tubular apoptosis through a caspase-3-dependent pathway. Am J Physiol Ren Physiol. 2018;314:F269–79. doi: 10.1152/ajprenal.00091.2017. [DOI] [PubMed] [Google Scholar]
- 24.Sun Z, Shen L, Sun X, Tong G, Sun D, Han T, et al. Variation of NDRG2 and c-Myc expression in rat heart during the acute stage of ischemia/reperfusion injury. Histochem Cell Biol. 2011;135:27–35. doi: 10.1007/s00418-010-0776-9. [DOI] [PubMed] [Google Scholar]
- 25.Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274:1541–48. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- 26.Zhang JF, Shi LL, Zhang L, Zhao ZH, Liang F, Xu X, et al. MicroRNA-25 negatively regulates cerebral ischemia/reperfusion injury-induced cell apoptosis through Fas/FasL pathway. J Mol Neurosci. 2016;58:507–16. doi: 10.1007/s12031-016-0712-0. [DOI] [PubMed] [Google Scholar]
- 27.Patel JH, McMahon SB. BCL2 is a downstream effector of MIZ-1 essential for blocking c-MYC-induced apoptosis. J Biol Chem. 2007;282:5–13. doi: 10.1074/jbc.M609138200. [DOI] [PubMed] [Google Scholar]
- 28.Järvinen K, Hotti A, Santos L, Nummela P, Hölttä E. Caspase-8, c-FLIP, and caspase-9 in c-Myc-induced apoptosis of fibroblasts. Exp Cell Res. 2011;317:2602–15. doi: 10.1016/j.yexcr.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 29.Morrish F, Giedt C, Hockenbery D. c-MYC apoptotic function is mediated by NRF-1 target genes. Genes Dev. 2003;17:240–55. doi: 10.1101/gad.1032503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maclean KH, Keller UB, Rodriguez-Galindo C, Nilsson JA, Cleveland JL. c-Myc augments gamma irradiation-induced apoptosis by suppressing Bcl-XL. Mol Cell Biol. 2003;23:7256–70. doi: 10.1128/MCB.23.20.7256-7270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieminen AI, Partanen JI, Klefstrom J. c-Myc blazing a trail of death: coupling of the mitochondrial and death receptor apoptosis pathways by c-Myc. Cell Cycle. 2007;6:2464–72. doi: 10.4161/cc.6.20.4917. [DOI] [PubMed] [Google Scholar]
- 32.Juin P, Hunt A, Littlewood T, Griffiths B, Swigart LB, Korsmeyer S, et al. c-Myc functionally cooperates with Bax to induce apoptosis. Mol Cell Biol. 2002;22:6158–69. doi: 10.1128/MCB.22.17.6158-6169.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai MS, Jin Y, Gallegos JR, Lu H. Balance of Yin and Yang: ubiquitylation-mediated regulation of p53 and c-Myc. Neoplasia. 2006;8:630–44. doi: 10.1593/neo.06334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–69. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boone DN, Qi Y, Li Z, Hann SR. Egr1 mediates p53-independent c-Myc-induced apoptosis via a noncanonical ARF-dependent transcriptional mechanism. Proc Natl Acad Sci USA. 2011;108:632–37. doi: 10.1073/pnas.1008848108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong X, O’Kelly J, Xie D, Mori A, Lemp N, McKenna R, et al. Cyr61 suppresses the growth of non-small-cell lung cancer cells via the beta-catenin-c-myc-p53 pathway. Oncogene. 2004;23:4847–55. doi: 10.1038/sj.onc.1207628. [DOI] [PubMed] [Google Scholar]
- 37.Kelly KJ, Plotkin Z, Vulgamott SL, Dagher PC. P53 mediates the apoptotic response to GTP depletion after renal ischemia-reperfusion: protective role of a p53 inhibitor. J Am Soc Nephrol. 2003;14:128–38. doi: 10.1097/01.ASN.0000040596.23073.01. [DOI] [PubMed] [Google Scholar]
- 38.Ying Y, Kim J, Westphal SN, Long KE, Padanilam BJ. Targeted deletion of p53 in the proximal tubule prevents ischemic renal injury. J Am Soc Nephrol. 2014;25:2707–16. doi: 10.1681/ASN.2013121270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F, et al. Myc represses thep21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci USA. 2001;98:4510–15. doi: 10.1073/pnas.081074898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Madapura HS, Salamon D, Wiman KG, Lain S, Klein E, Nagy N. cMyc-p53 feedback mechanism regulates the dynamics of T lymphocytes in the immune response. Cell Cycle. 2016;15:1267–75. doi: 10.1080/15384101.2016.1160975. [DOI] [PMC free article] [PubMed] [Google Scholar]